Introduction
Non-small cell lung cancer (NSCLC) is one of the
most common malignancies with increasing incidence worldwide
(1–3). Despite the advances in early
detection and improvements in the treatment, long-term survival
from NSCLC still remains poor. Tumor relapse and metastasis are the
main factors influencing patient prognosis. The identification of
biomarkers that can predict the risk of recurrence and metastasis
is therefore clinically important.
The cystic fibrosis transmembrane conductance
regulator (CFTR) is a cAMP-activated anion channel which is
expressed ubiquitously in epithelial tissues. Germline mutations in
the gene encoding CFTR cause recessive cystic fibrosis (CF)
(4,5). Over the last three decades, long-term
survival rate for CF patients has significantly improved but an
elevated risk of cancer is being recognized to be associated with
survivorship. Intriguingly, an increased risk of cancer, primarily
of the gastrointestinal tract, has been reported in some, but not
all studies in the carriers of CFTR mutations (6–10).
Hypermethylation of the CFTR promoter is frequently seen in a
number of different tumor types, including lung cancer (11–13),
suggesting DNA methylation-mediated transcription silencing of CFTR
may influence cancer development (9,14,15).
Of note, both mutation and hypermethylation of CFTR have been
identified in NSCLC patients (12,16).
By analyzing a series of tumors from 296 lung cancer
patients we have shown that aberrant CFTR expression level is
significantly associated with NSCLC progression, metastasis and
poor prognosis. We have also shown that suppression of CFTR
promotes epithelial-mesenchymal transition (EMT) and metastasis
providing mechanistic basis for the role of CFTR in lung
cancer.
Materials and methods
Sample accrual
All samples were collected during surgery at the
Department of Surgery, the First Affiliated Hospital of Guangzhou
Medical University. One hundred and sixty-five tumor samples and 22
normal lung tissue were obtained from patients enrolled between
April 2007 and June 2009. An additional 131 tumor samples were
collected from patients recruited for the validation phase during
the period between June 2009 and June 2010. Tissue was snap-frozen
in liquid nitrogen and stored at −80°C. Diagnosis of lung cancer
was confirmed at the time of diagnosis after surgery and the
presence of tumor cells was verified by a pathologist (G.H.L.)
using H&E stained frozen sections in accordance with the World
Health Organization guidelines (17). Tumor stage was assigned according
to the American Joint Committee for Cancer criteria (18). The background samples were
confirmed free of tumor deposits. Clinical follow-up data on
patients were ascertained by review of medical records. The
follow-up time was up to 60 months. All human specimens and
correlative data were obtained according to a protocol reviewed and
approved by the local ethics committee, and all patients provided
signed informed consent.
Expression analysis of CFTR by real-time
PCR and immunochemical staining
Real-time reverse-transcriptase polymerase chain
reactions (RT-PCRs) with patient-derived cDNA were performed on a
StepOne Plus real-time PCR system (Applied Biosystems, Forster
City, USA). Briefly, RNA isolation was carried out using TRI
reagent from Sigma (St. Louis, MO, USA), according to the
manufacturer’s instructions. RNA (0.5 μg) was converted into cDNA
using the iScript™ cDNA Synthesis kit (Bio-Rad Laboratories, Hemel
Hemstead, UK). The levels of CFTR mRNA were quantified by TaqMan
Gene Expression Assays (Assay ID CFTR: Hs00357004_m1;
Applied Biosystems) using GAPDH as the internal control. All assays
were performed in triplicate. CFTR expression levels were
dichotomized by median values.
The tissue blocks were cut into 5-mm sections and
processed for IHC in accordance with a previously described
protocol (19).
Statistical analysis
Progression-free survival (PFS) was defined as the
minimum interval from the date of diagnosis to the date of tumor
recurrence, progression, the occurrence of a second malignancy,
death or the last follow-up. Overall survival (OS) was defined as
the interval from the date of diagnosis to the date of death or the
last follow-up. Living patients with local recurrence or metastasis
were considered as ‘in disease survival’.
The correlations between the CFTR levels and other
demographical and clinical features were assessed using
Mann-Whitney rank sum and Kruskal-Wallis one way analysis of
variance tests.
Patients were assigned to two equal sized groups as
defined by the median CFTR level. Kaplan-Meier survival curves and
log-rank tests were used to evaluate the differences in OS and PFS
between patients groups. Cox proportional hazards model was used to
estimate hazard ratios (HRs) and 95% CIs of CFTR levels at the
presence of other demographical and clinical parameters.
Survival analyses were carried out using the SPSS
statistical software (version 11; SPSS, Inc., Chicago, IL, USA).
All statistical tests were two-sided and any tests with P-value
<0.05 are considered statistically significant.
Cell lines, antibodies and reagents
The human lung adenocarcinoma cell lines A-549 and
H1299 were obtained from the American Type Culture Collection
(ATCC, Manassas, VA, USA). Antibodies were used against the
following: CFTR (Almone labs, Jerusalem, Israel); E-cadherin, uPA,
uPAR and vimentin (Santa Cruz Biotechnology, Santa Cruz, CA, USA);
GAPDH (Santa Cruz Biotechnology); CFTRinh-172 (Sigma).
Functional studies
CFTR channel function was blocked by CFTRinh-172 (10
μM). For wound healing assay, 1×106 cells/well cells
were seeded in 6-well plates and then pre-incubated for 24 h before
creating a ‘wound’ for the cell monolayer with a plastic tip. Cells
were then grown in culture medium with 1% FBS in the presence or
absence of 10 μM inh-172. The migration of cells was tracked and
recorded using a Time-lapse imaging system (Carl Zwiss) for 48 h.
Cell migration was determined by measuring distances between
parallel lines from initial sites to migrated sites. The procedure
was repeated three times. Cell invasion assay was performed using
modified Boyden chambers (8-μm), polycarbonate membranes (Corning
Incorp.) coated with 500 μg/ml Matrigel (BD Biosciences, San Jose,
CA, USA). A total of 20,000 cells were added to the Transwell
inserts over the top of the artificial basement membrane. Cell
growth rate was measured with an MTS proliferation assay. The
CellTiter 96 Aqueous One Solution Cell Proliferation assay
(Promega) was conducted according to the manufacturer’s
instructions.
Western blot analysis
Whole-cell extraction was conducted using RIPA
buffer [50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.1% SDS and 1%
nadeoxycholate (pH 7.4)] supplemented with protease inhibitors,
phenylmethylsulfonyl fluoride and pimix. Protein concentrations
were then measured by Bio-Rad protein assay kits (Bio-Rad
Laboratories, Hercules, CA, USA). Protein lysates were resolved by
SDS-PAGE and then transferred onto nitrocellulose membranes
(Hybond™-P; Amersham Biosciences, Piscataway, NJ, USA), blocked
with TBS containing 0.2% Tween-20 (TBST) and 5% non-fat dry milk
and incubated with primary antibodies at 4°C overnight. Antibody to
GAPDH was used as control for protein loading. After being washed
with TBST, membranes were incubated with secondary antibody in room
temperature for 1 h before exposure.
CFTR overexpression and knockdown
The pEGFPC3 plasmid expressing wild-type CFTR was
provided by Professor Tzyh-Chang Hwang (University of
Missouri-Columbia). For overexpression experiments, the A-549 cells
were transfected with 3 μg DNA and 6 μl Lipofectamine 2000
(Invitrogen, Camarillo, CA, USA). The transfected cells were
selected in full medium containing G418 (Calbiochem, Schwalbach,
Germany) at 1,200 μg/ml. To knock down CFTR expression, duplex
specific miRNAs to human CFTR was synthesized by utilizing Lift
Technologies. The siRNA sequence used was: 5′-TTG GAA AGG AGA CTA
ACA AG-3′. MiR expression vector, named as pcDNA™6.2-GW/EmGFP,
containing a double-stranded oligonucleotide (ds-oligo) encoding a
pre-miRNA sequence were established using BLOCK-iT™ Pol II miR RNAi
Expression Vector kits (Life Technologies, Rockville, MD, USA)
following the manufacturer’s protocol. Lentiviral particles were
produced by transient transfection of 293FT (Invitrogen) cells
using Lipofectamine 2000 (Invitrogen) reagent. Blasticidin at the
final concentration of 5 μg/ml was used to select the stable
clones.
Animal studies
Nude mice were provided by the Laboratory Animal
Service Center of the Chinese University of Hong Kong. Animals were
properly maintained in an air-conditioned room with controlled
temperature of 24±2°C and humidity of 55±15%, in a 12-h light/dark
cycle and were fed laboratory chow and water ad libitum. All
animal experiments were conducted in strict accordance with the
University Laboratory Animals Service Center guidelines on animal
experimentation with approval from the Animal Ethics Committee of
the University. Tumorigenicity was investigated by tumor xenograft
experiments. Briefly, female athymic balb/c nude mice, 6–8 weeks of
age, were injected with 10 μl suspension of CFTR knockdown cells or
vector control A-549 cells (~5×106) subcutaneously. Mice
injected with saline were used as sham controls. Tumor formation in
nude mice was monitored over approximately a 6-week period and
ratios of tumor weight to body weight were sacrificed. Tumor size,
animal health and behavior were measured and monitored twice every
week. Mice with a tumor size >1 cm in any dimension were
terminated. The tumor size was calculated according to the
following formula: 0.5234X [long diameter (short
diameter)2]. To develop the metastatic model, 6–8-week
old female balb/c nude mice were transplanted with 1×106
A-549 cells through the lateral tail vein under sterile conditions.
All mice were sacrificed 6 weeks after injection by perfusion.
Before the perfusion surgery, 100 mg/kg ketamine mixed with 10
mg/kg xylazine was used. Cold PBS (10 ml) for each mice was
perfused through right ventricle until lungs cleared of blood, then
10 ml of cold PFA was perfused to fix the tissue. Lungs were
collected, and further fixed in PFA overnight, and sectioned (5 μm)
in preparation for H&E staining for morphological
investigation. Number and size of tumor loci was countered and
calculated.
Results
Downregulation of CFTR expression in
NSCLC
A statistically significant lower CFTR transcript
level was observed in NSCLC tumor tissues than that in normal lung
tissues (Fig. 1A and Table I). Compared to other various NSCLC
subtypes, the level of CFTR was significantly higher in
adenocarcinoma patients (P=0.004; Fig.
1B). There were no significant differences between patients
with high and low CFTR expression levels as determined by age,
gender or smoking status (Table
I).
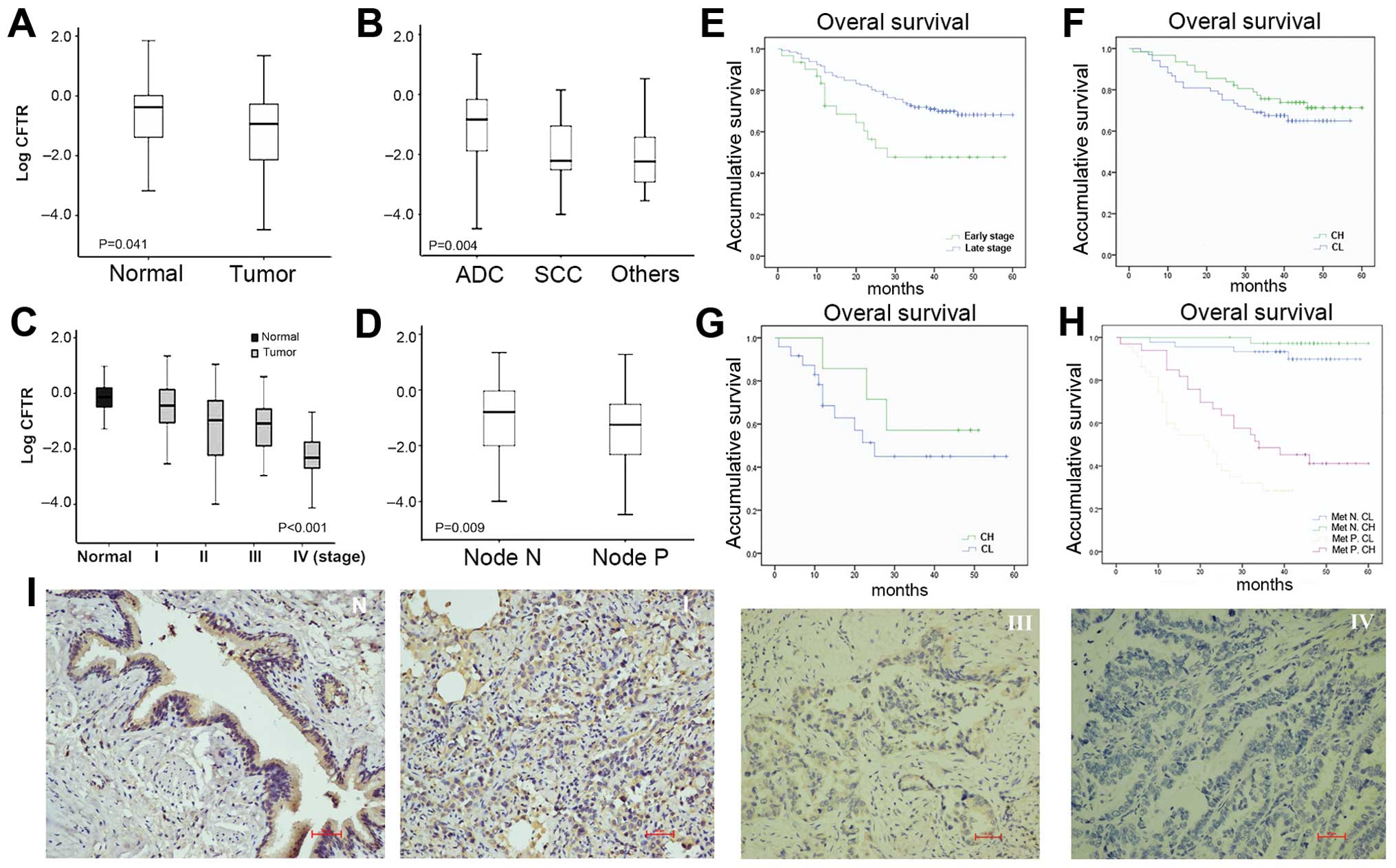 | Figure 1Expression of CFTR in NSCLC and its
association with disease progression and prognosis. CFTR mRNA
expression level (A) in tumor tissues and normal tissues; (B) in
adenocarcinoma, squamous cell carcinoma and other histology types;
(C) in different clinical stage progressions and (D) lymph node
metastasis. Mann-Whitney test was used to test the difference of
CFTR levels across strata. Kaplan-Meier survival curves for (E)
early- and late-stage patients; (F) patients groups stratified by
dichotomized CFTR expression levels at early-stage and (G)
late-stage of prognosis; and (H) patients groups stratified by
metastasis status and dichotomized CFTR levels. Met N. CH, group of
patients with high CFTR level and without metastasis; Met N. CL,
group of patients with low CFTR level and without metastasis; Met
P. CH, group of patients with metastasis but with high CFTR level;
Met P. CL, group of patients with low CFTR level and with
metastasis. (I) Representative images showed high expression of
CFTR by IHC in normal lung tissues (left, N), relatively strong
staining of CFTR in tumors with stage I (middle left, I) and low
expression of CFTR in tumors with stage III (middle right, III) and
negative expression of CFTR in stage IV tumor tissues (right, IV)
(scale bar, 100 μm). |
 | Table ICFTR expression and patient
characteristics. |
Table I
CFTR expression and patient
characteristics.
| Samples | CFTR | |
|---|
|
|
| |
|---|
|
Characteristics | N (%) | Median
(rangea) | P-value |
|---|
| Tissue type |
| Tumor | 165 (88.2) | 0.12 (0.55) | 0.04 |
| Normal | 22 (11.8) | 0.42 (1.11) | |
| Age (years) |
| ≤65 | 115 (61.5) | 0.09 (0.44) | 0.14 |
| >65 | 49 (26.2) | 0.19 (0.87) | |
| Gender |
| Female | 64 (34.2) | 0.13 (0.90) | 0.2 |
| Male | 102 (54.5) | 0.11 (0.36) | |
| Smoking status |
| Yes | 58 (31.0) | 0.09 (0.32) | 0.12 |
| No | 102 (54.5) | 0.13 (0.88) | |
| Histology |
| ADC | 139 (74.3) | 0.15 (0.69) | 0.004 |
| SCC | 17 (9.1) | 0.01 (0.11) | |
| Other | 9 (4.8) | 0.01 (1.72) | |
| Stage |
| I | 53 (28.3) | 0.36 (1.36) | <0.0001 |
| II | 46 (24.6) | 0.11 (0.61) | |
| IIIA | 33 (17.6) | 0.08 (0.27) | |
| IIIB | 9 (4.9) | | |
| IV | 24 (12.8) | 0.00 (0.02) | |
|
Differentiation |
| High | 117 (62.6) | 0.12 (0.84) | 0.95 |
| Low | 38 (20.3) | 0.14 (0.28) | |
| Tumor
nodularity |
| Unilateral
pulmonary | 158 (84.5) | 0.13 (0.68) | 0.08 |
| Bilateral
pulmonary | 7 (3.7) | 0.01 (0.14) | |
| Tumor size
(cm) |
| ≤2 | 27 (14.4) | 0.30 (0.83) | 0.09 |
| 2–5 | 97 (51.9) | 0.13 (0.61) | |
| >5 | 41 (21.9) | 0.04 (0.20) | |
| Pleura
involvement |
| Yes | 88 (47.1) | 0.09 (0.44) | 0.3 |
| No | 77 (41.2) | 0.13 (0.82) | |
| Lymph node
involvement |
| Yes | 92 (49.2) | 0.06 (0.31) | 0.01 |
| No | 73 (39.0) | 0.20 (0.94) | |
| Vascular
invasion |
| Yes | 6 (3.2) | 0.01 (1.71) | 0.3 |
| No | 159 (85.0) | 0.13 (0.58) | |
| Clinical
outcome |
| Alive | 109 (58.3) | 0.14 (0.66) | 0.42 |
| Death | 54 (28.9) | 0.05 (0.46) | |
| Time of survival
(months) |
| ≥36 | 94 (50.3) | 0.16 (0.68) | 0.04 |
| <36 | 69 (36.9) | 0.04 (0.47) | |
Lower CFTR expression is associated with
advanced disease in NSCLC
Those patients with low CFTR levels in general had
more advanced tumors. A gradual decrease in CFTR transcripts levels
was observed through stage I to IV patients (Fig. 1C and Table I). The protein levels of CFTR were
downregulated in advanced stage tumors compared to normal tissues
as assessed by immunohistochemistry (IHC) staining through a panel
of cohorts (Fig. 1I). A
significantly lower CFTR expression level was also found in
patients with lymph node metastasis (node P) compared to lymph
node-free patients (node N) (Fig.
1D and Table I). There was
evidence of downregulation of CFTR levels in patients with either
larger tumor, tumors with poorer differentiation, vascular or
pleural invasion, however, this difference was not statistically
significant.
Low CFTR gene expression is correlated
with poor prognosis and inferior survival
As CFTR expression appeared to be related to disease
progression, we analyzed its relationship with prognosis.
The association between CFTR expression levels and
disease progression was initially tested in 165 NSCLC patients
recruited to the discovery phase. Survival rates were compared
between patients groups defined by the median CFTR expression level
(0.15). Higher OS (median, 45; 95% CI, 42.9–47.0) and PFS (median,
41; 95% CI, 33.8–48.2) were observed in the patients with high CFTR
expression level (Fig. 1;
P<0.01), compared to lower OS (median, 36; 95% CI, 32.7–39.3)
and PFS (median, 30; 95% CI, 17.1–42.9). The effect of CFTR
expression remained highly significant after adjusted for age,
gender, smoking status and clinical stage, compatible with CFTR
expression levels being an independent prognosis predictor of
clinical stages (Table II). The
association between CFTR levels and prognosis were then analyzed
against early stage (stage I to IIIA) and late stage (stage IIIB to
IV) cancer patients. Although the clinical stage predicted
favorable OS (Fig. 1E), patients
with high CFTR levels (CH) had significantly better survival
regardless of early/late stage disease (Fig. 1F and G).
| Table IICox proportional hazard model. |
Table II
Cox proportional hazard model.
| Discovery | Replication | Fixed effect
meta-analysis |
|---|
|
|
|
|
|---|
| Factors | HR | 95% CI | P-value | HR | 95% CI | P-value | HR | 95% CI | P-value |
Phet |
|---|
| CFTR | 0.52 | 0.37–0.72 | <0.001 | 0.66 | 0.48–0.91 | 0.01 | 0.69 | 0.62–0.76 |
3.53×10−13 | 0.28 |
| Age | 1.24 | 0.87–1.76 | 0.23 | 1.09 | 0.46–2.58 | 0.85 | 0.91 | 0.79–1.04 | 0.17 | 0.18 |
| Gender | 1.11 | 0.74–1.66 | 0.61 | 1.37 | 0.77–2.44 | 0.29 | 0.88 | 0.76–1.02 | 0.09 | 0.92 |
| Smoking status | 0.86 | 0.56–1.31 | 0.48 | 1.42 | 0.79–2.56 | 0.25 | 0.82 | 0.71–0.95 | 0.01 | 0.37 |
| Clinical stage | 1.21 | 1.03–1.42 | 0.02 | 1.06 | 0.74–1.52 | 0.76 | 0.99 | 0.93–1.05 | 0.74 | 0.10 |
The association between CFTR levels and prognosis
was then validated in 131 NSCLC patients recruited between
2009–2010. The Kaplan-Meier survival and log-rank tests provided
evidence of improved OS for patients with higher CFTR levels albeit
not statistically significant.
Pooling the data from discovery and the validation
phase using an inverse variance meta-analysis approach showed a
significant association between CFTR level and prognosis
independent of clinical stages (HR, 0.69; 95% CI, 0.62–0.76;
P=3.53×10−13) (Table
II).
Notably four stage IV patients from both phases who
remained progression-free showed significantly higher CFTR levels
(mean ± SEM, 4.758±1.461) in comparison to those patients with
local recurrence, metastasis or those who died from lung cancer
(mean ± SEM, 0.0289±0.754; P=0.003). The surviving stage IV
patients had higher CFTR levels (1.611±0.863), compared to those
who died of the disease (0.030±0.996), indicated a particular role
of CFTR in late stage prognosis.
We analyzed OS stratified by CFTR expression levels
and metastasis status. Median OS rates were computed and compared
among the patient groups using log-rank testing (Fig. 1H). For patients without metastasis,
the median survival of high CFTR level group (Met N. CH) was 58
months while that of the low CFTR level group (Met N. CL) was 54
months. For the patients with metastasis, patients with high CFTR
levels (Met P. CH) experienced a considerably longer 41-month
median survival compared with the 24-month median survival of the
low CFTR expression group (Met P. CL) (Table III).
| Table IIILog-rank test on OS for CFTR with
metastasis. |
Table III
Log-rank test on OS for CFTR with
metastasis.
| Group | Patients (n) | Median OS
(months) | 95% CI | P-value |
|---|
| Metastasis (−) |
| Low CFTR | 44 | 54 | 51.3–57.9 | <0.0001 |
| High CFTR | 50 | 58 | 57.0–60.4 | |
| Total | 94 | 57 | 55.7–59.5 | |
| Metastasis (+) |
| Low CFTR | 49 | 24 | 20.6–29.4 | |
| High CFTR | 44 | 41 | 35.3–47.5 | |
| Total | 93 | 35 | 30.9–40.0 | |
| Overall | 187 | 47 | 43.9–49.8 | |
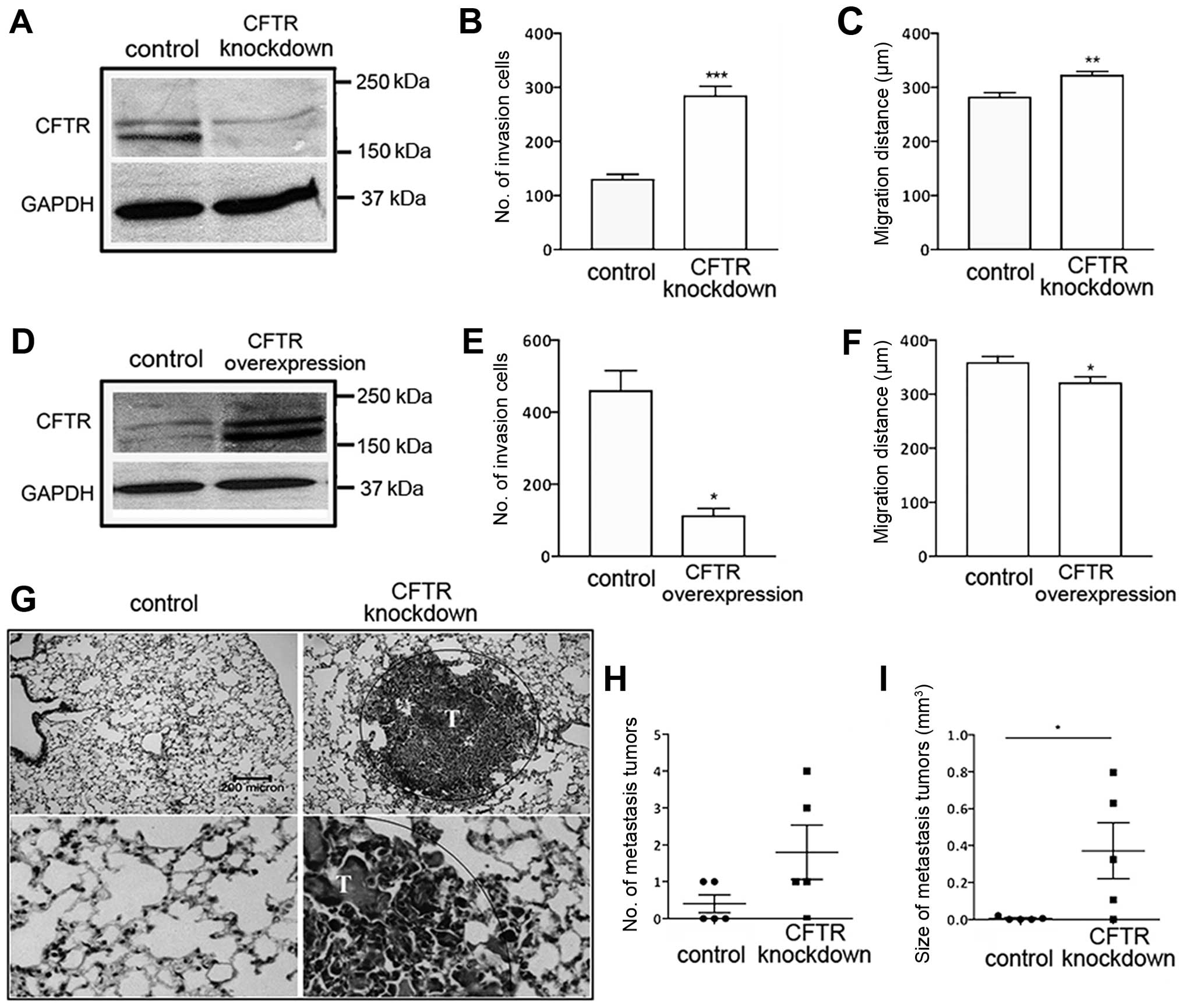
Manipulation of CFTR alters malignancy of
lung cancer in vitro and in vivo
The observed association between CFTR expression
level and NSCLC metastasis and prognosis prompted us to investigate
whether CFTR gene manipulation might affect the malignant
phenotype. The alteration of CFTR expression did not significantly
affect proliferation of NSCLC cell line A-549 (data not shown).
However, cell invasion and migration were significantly enhanced or
suppressed by knockdown or overexpression of CFTR, respectively
(Fig. 2B, C, E and F). The same
trend was seen in H1299 cells (data not shown).
To examine the role of CFTR in tumorigenicity in
vivo, we established xenograft models by subcutaneous injection
of A-549 cells transduced with shRNA targeting CFTR in mice. No
differences in primary tumor growth between control and
CFTR-knockdown A-549-injected mice were seen (data not shown),
however, 80% (4/5) of the mice injected with CFTR-knockdown cells
presented with lung metastasis in contrast to 40% (2/5) of the mice
injected with A-549-empty controls (Fig. 2G and H). Moreover, the tumor burden
in the vector and CFTR knockdown groups were significantly
different 6 weeks after the tumor cell inoculation (Fig. 2I).
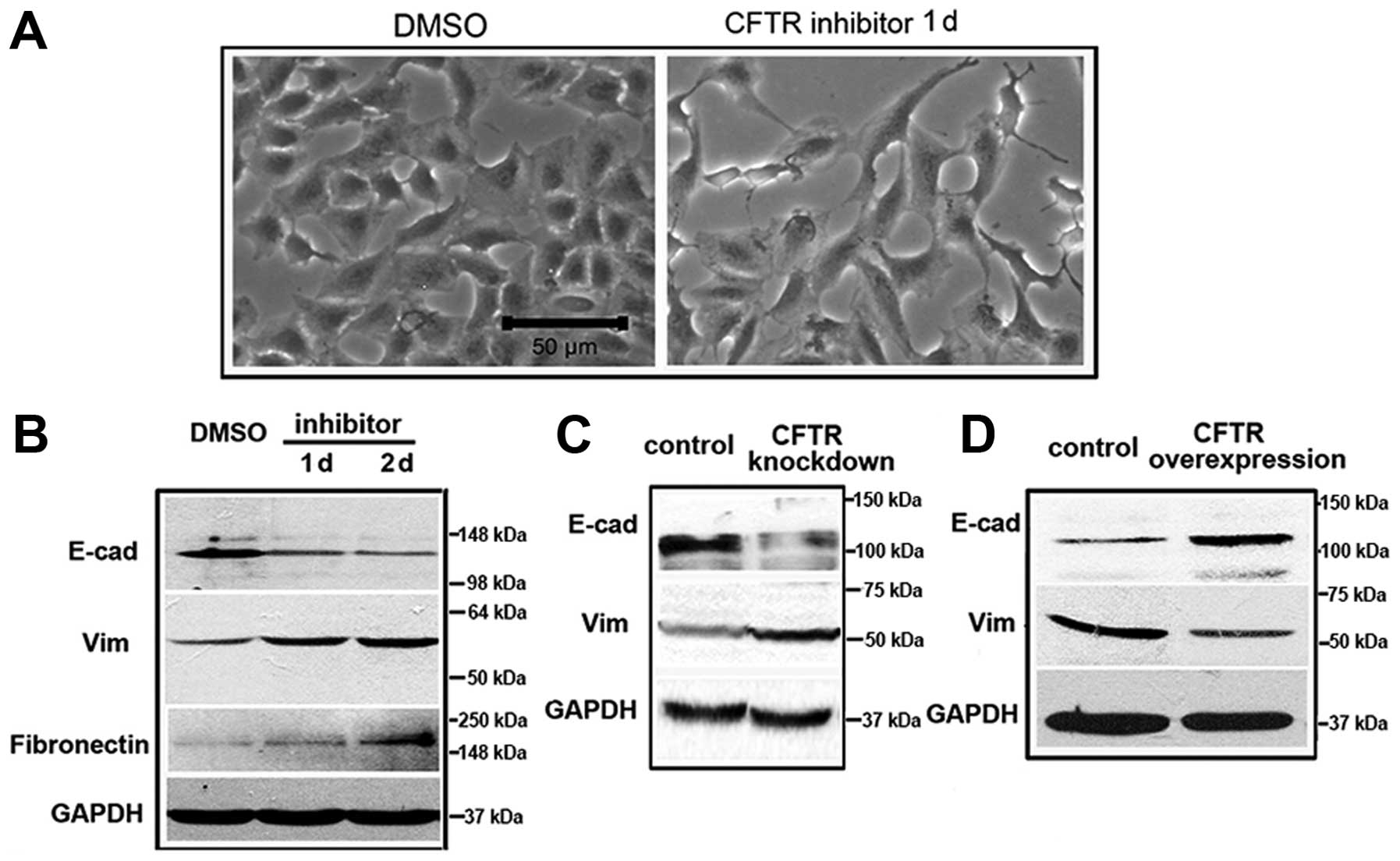
CFTR regulates EMT through the uPA/uPAR
pathway
Application of the CFTR activity inhibitor, inh-172
led A-549 cells to rapidly change their morphology into an
elongated fibroblast-like shape (Fig.
3A). This was reflected in downregulation of epithelial marker
expression including E-cadherin and upregulated mesenchymal markers
such as vimentin (Vim) and fibronectin (Fig. 3B), suggesting suppression of CFTR
function promotes EMT in lung cancer. The effect of CFTR on EMT in
lung cancer cells was also supported by the observation that CFTR
knockdown promoted EMT, whereas CFTR overexpression inhibited EMT
(Fig. 3C and D). It has been
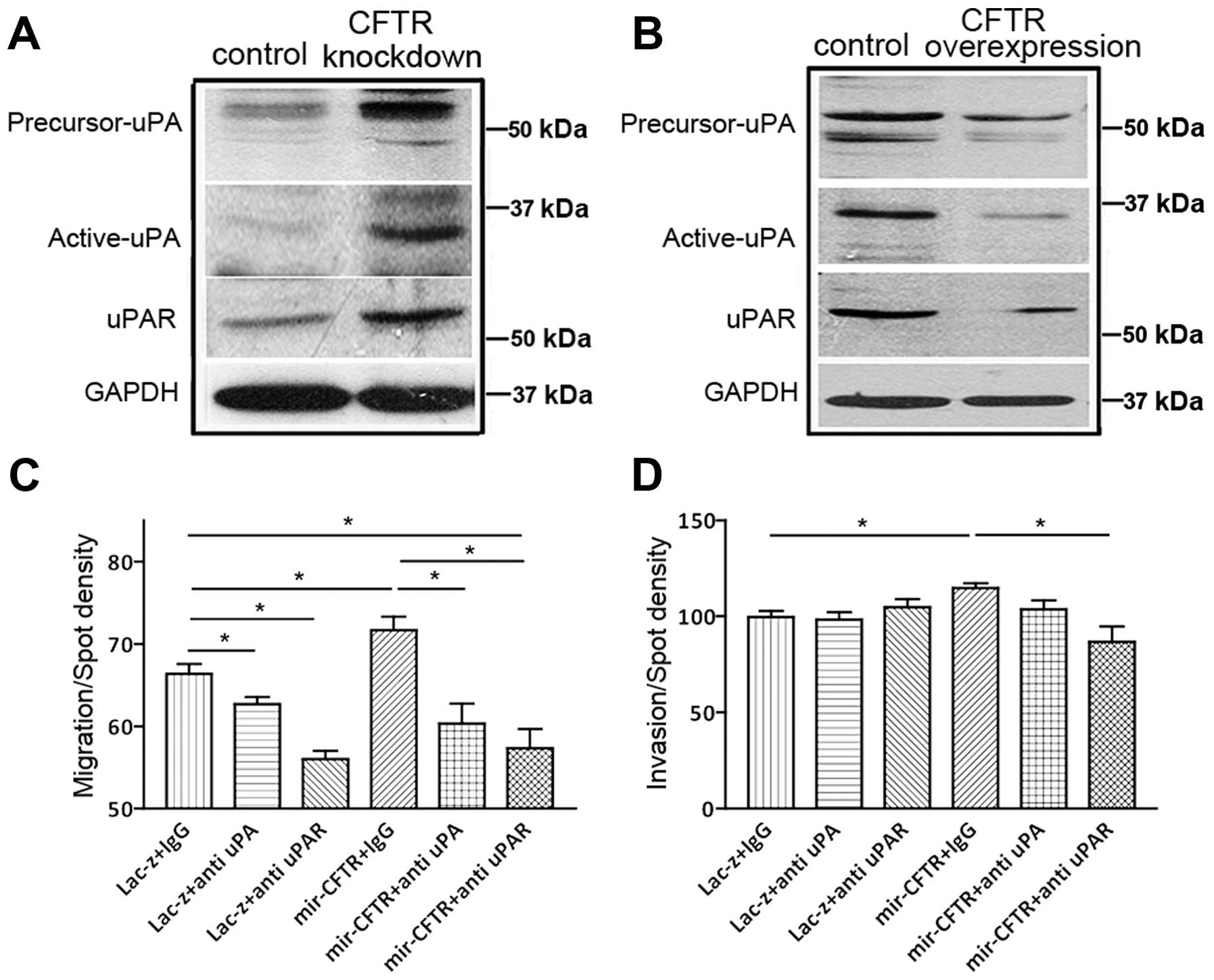
reported that CFTR is involved in the NF-κB mediated urokinase-type
plasminogen activator (uPA) pathway, which is associated with
cancer development (20–23). Given the central role of the
uPA/uPAR axis in EMT (24), we
reasoned that CFTR might be implicated in the regulation of EMT and
lung cancer metastasis through the uPA/uPAR pathway. As shown in
Fig. 4A and B, knockdown of CFTR
increased actived-uPA and uPAR protein expression, while
overexpression of CFTR inhibited actived-uPA and uPAR expression in
A-549 cells (Fig. 4A and B). The
activated level of uPA was accordingly altered by CFTR gene
manipulation, indicating that uPA activity is CFTR-dependent in
these cancer cells. Neutralization with uPA or uPAR antibodies in
CFTR-knockdown A-549 cells dramatically reversed the CFTR
knockdown-enhanced cell migration and invasion in these cells
(Fig. 4C and D), indicating that
upregulation of the uPA/uPAR axis activity is the major mechanism
leading to the observed increased malignancies induced by CFTR
knockdown.
Discussion
The recent discoveries of genomic alterations,
including mutation of EGFR, KRAS and ALK genes
in NSCLC, have suggested novel therapeutic targets for the
treatment of lung cancer (25–28).
A biomarker for the prediction of NSCLC recurrence and metastasis,
however, remains to be identified.
In the present study, CFTR was significantly
downregulated in NSCLC tumors and low expression of CFTR was
significantly correlated with NSCLC progression and metastasis
suggesting CFTR as a prognostic biomarker for lung cancer.
Importantly, our findings are consistent with CFTR having a strong
protective influence in NSCLC, especially in the context of
late-stage disease.
One of the key processes that occur during the
progression of tumor metastasis is EMT. In the present study, the
suppression of CFTR function leads to loss of epithelial markers,
whereas, overexpression of CFTR results in upregulation of
epithelial markers. This is in line with the observed changes of
EMT process and invasive phenotypes in lung cancer cell lines, and
is consistent with a metastasis-suppressing role of CFTR.
The present study also demonstrated that CFTR can
suppress lung cancer metastasis through the uPA system. The uPA
system has a multifunctional role in neoplastic evolution,
affecting cancer cell proliferation, tumor angiogenesis, adhesion
and migration (24). Furthermore,
increased expression of uPA, uPAR and PAI-1 has been documented in
many types of cancers (29). In
the present study, uPA expression was inversely associated with
CFTR expression in lung cancer cells. In addition, uPA or uPAR
neutralizing antibody reversed the enhanced cell migration and
invasion of CFTR knockdown. It is well established that CFTR is a
negative regulator of NF-κB (21–23),
and that NF-κB positively regulates uPA during cancer development
and progression (30,31). Therefore, it is possible that the
regulation of CFTR on the uPA pathways may be mediated through the
negative mediation of NF-κB. Since other EMT-inducing factors, such
as TNFα and HIF1α, have been demonstrated to downregulate CFTR
expression (32,33) and CFTR regulate EMT through
interaction with AF-6 and miR-193b in other cancer (34,35),
CFTR may act as a conjoint downstream effector in mediating the
effects of EMT inducers.
In the present study, we have provided evidence that
CFTR expression may be a biomarker for lung cancer metastasis and
prognosis. Moreover, since CFTR may also be a prognostic predictor
in breast cancer (36) it suggests
that CFTR may have a more generic function in cancer development,
both in vitro and in vivo functional studies and in
evaluation of CFTR expression. Of note, drugs that target specific
CFTR point mutations have shown therapeutic benefit in patients
with CF (37) and naturally
occurring polyphenol compound resveratrol may stimulate the
activity of CFTR (38). The
results of the present study warrant further investigation
exploring the utility of these agents in the management of advanced
lung cancer.
Acknowledgements
We are grateful to Professor Tzyh-Chang Hwang of the
Department of Biological Engineering, University of
Missouri-Columbia, USA, for providing CFTR-peGFPC3 and the control
plasmids. The present study was supported in part by National 973
projects (2013CB967403, 2013CB967404), the National Natural Science
Foundation of China (81201845), the Guangzhou Medical University
(2011A06) and the Research Grant Council of Hong Kong
(GRF-CUHK466413) and the Focused Investment Scheme of the Chinese
University of Hong Kong.
References
|
1
|
Chang S, Dai M, Ren JS, Chen YH and Guo
LW: Estimates and prediction on incidence, mortality and prevalence
of lung cancer in China in 2008. Zhonghua Liu Xing Bing Xue Za Zhi.
33:391–394. 2010.(In Chinese).
|
|
2
|
Chien CR and Chen TH: A Bayesian model for
age, period, and cohort effects on mortality trends for lung
cancer, in association with gender-specific incidence and
case-fatality rates. J Thorac Oncol. 4:167–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Subramanian J, Madadi AR, Dandona M,
Williams K, Morgensztern D and Govindan R: Review of ongoing
clinical trials in non-small cell lung cancer: a status report for
2009 from the ClinicalTrials.govurisimpleClinicalTrials.gov website.
J Thorac Oncol. 5:1116–1119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Collins FS: Cystic fibrosis: molecular
biology and therapeutic implications. Science. 256:774–779. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Riordan JR, Rommens JM, Kerem B, et al:
Identification of the cystic fibrosis gene: cloning and
characterization of complementary DNA. Science. 245:1066–1073.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McWilliams RR, Rabe KG, Olswold C, De
Andrade M and Petersen GM: Risk of malignancy in first-degree
relatives of patients with pancreatic carcinoma. Cancer.
104:388–394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McWilliams RR, Petersen GM, Rabe KG, et
al: Cystic fibrosis transmembrane conductance regulator (CFTR) gene
mutations and risk for pancreatic adenocarcinoma. Cancer.
116:203–209. 2010.
|
|
8
|
Neglia JP, FitzSimmons SC, Maisonneuve P,
et al: The risk of cancer among patients with cystic fibrosis.
Cystic Fibrosis and Cancer Study Group. N Engl J Med. 332:494–499.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Sun Z, Wu Y, et al: Cystic fibrosis
transmembrane conductance regulator gene mutation and lung cancer
risk. Lung Cancer. 70:14–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maisonneuve P, Marshall BC, Knapp EA and
Lowenfels AB: Cancer risk in cystic fibrosis: a 20-year nationwide
study from the United States. J Natl Cancer Inst. 105:122–129.
2013. View Article : Google Scholar
|
|
11
|
Mishra DK, Chen Z, Wu Y, Sarkissyan M,
Koeffler HP and Vadgama JV: Global methylation pattern of genes in
androgen-sensitive and androgen-independent prostate cancer cells.
Mol Cancer Ther. 9:33–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Son JW, Kim YJ, Cho HM, et al: Promoter
hypermethylation of the CFTR gene and clinical/pathological
features associated with non-small cell lung cancer. Respirology.
16:1203–1209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ding S, Gong BD, Yu J, et al: Methylation
profile of the promoter CpG islands of 14 ‘drug-resistance’ genes
in hepatocellular carcinoma. World J Gastroenterol. 10:3433–3440.
2004.PubMed/NCBI
|
|
14
|
Abraham EH, Vos P, Kahn J, et al: Cystic
fibrosis hetero- and homozygosity is associated with inhibition of
breast cancer growth. Nat Med. 2:593–596. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O’Connell MP, Fiori JL, Baugher KM, et al:
Wnt5A activates the calpain-mediated cleavage of filamin A. J
Invest Dermatol. 129:1782–1789. 2009. View Article : Google Scholar
|
|
16
|
Govindan R, Ding L, Griffith M, et al:
Genomic landscape of non-small cell lung cancer in smokers and
never-smokers. Cell. 150:1121–1134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brambilla E, Travis WD, Colby TV, Corrin B
and Shimosato Y: The new World Health Organization classification
of lung tumours. Eur Respir J. 18:1059–1068. 2001. View Article : Google Scholar
|
|
18
|
Edge SB and Compton CC: The American Joint
Committee on Cancer: the 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Ye L, Mansel RE and Jiang WG:
Potential prognostic value of repulsive guidance molecules in
breast cancer. Anticancer Res. 31:1703–1711. 2011.PubMed/NCBI
|
|
20
|
Smith SM, Vaughan JM, Donaldson CJ, et al:
Cocaine- and amphetamine-regulated transcript is localized in
pituitary lactotropes and is regulated during lactation.
Endocrinology. 147:1213–1223. 2006. View Article : Google Scholar
|
|
21
|
Vij N, Mazur S and Zeitlin PL: CFTR is a
negative regulator of NFkappaB mediated innate immune response.
PLoS One. 4:e46642009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
DiMango E, Ratner AJ, Bryan R, Tabibi S
and Prince A: Activation of NF-kappaB by adherent Pseudomonas
aeruginosa in normal and cystic fibrosis respiratory epithelial
cells. J Clin Invest. 101:2598–2605. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boncoeur E, Roque T, Bonvin E, et al:
Cystic fibrosis trans-membrane conductance regulator controls lung
proteasomal degradation and nuclear factor-kappaB activity in
conditions of oxidative stress. Am J Pathol. 172:1184–1194. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smith HW and Marshall CJ: Regulation of
cell signalling by uPAR. Nat Rev Mol Cell Biol. 11:23–36. 2010.
View Article : Google Scholar
|
|
25
|
Lipson D, Capelletti M, Yelensky R, et al:
Identification of new ALK and RET gene fusions from colorectal and
lung cancer biopsies. Nat Med. 18:382–384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kohno T, Ichikawa H, Totoki Y, et al:
KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 18:375–377.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takeuchi K, Soda M, Togashi Y, et al: RET,
ROS1 and ALK fusions in lung cancer. Nat Med. 18:378–381. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Strongin AY: Proteolytic and
non-proteolytic roles of membrane type-1 matrix metalloproteinase
in malignancy. Biochim Biophys Acta. 1803:133–141. 2010. View Article : Google Scholar :
|
|
30
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar
|
|
31
|
Sen R and Smale ST: Selectivity of the
NF-{kappa}B response. Cold Spring Harb Perspect Biol.
2:a0002572010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pruliere-Escabasse V, Fanen P, Dazy AC, et
al: TGF-beta 1 downregulates CFTR expression and function in nasal
polyps of non-CF patients. Am J Physiol Lung Cell Mol Physiol.
288:L77–L83. 2005. View Article : Google Scholar
|
|
33
|
Howe KL, Wang A, Hunter MM, Stanton BA and
McKay DM: TGFbeta down-regulation of the CFTR: a means to limit
epithelial chloride secretion. Exp Cell Res. 298:473–484. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xie C, Jiang XH, Zhang JT, et al: CFTR
suppresses tumor progression through miR-193b targeting urokinase
plasminogen activator (uPA) in prostate cancer. Oncogene.
32:2282–2291. 2013. View Article : Google Scholar
|
|
35
|
Sun TT, Wang Y, Cheng H, et al: Disrupted
interaction between CFTR and AF-6/afadin aggravates malignant
phenotypes of colon cancer. Biochim Biophys Acta. 1843:618–628.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang JT, Jiang XH, Xie C, et al:
Downregulation of CFTR promotes epithelial-to-mesenchymal
transition and is associated with poor prognosis of breast cancer.
Biochim Biophys Acta. 1833:2961–2969. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramsey BW, Davies J, McElvaney NG, et al:
A CFTR potentiator in patients with cystic fibrosis and the G551D
mutation. N Engl J Med. 365:1663–1672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang S, Yu BO, Sui Y, et al: CFTR chloride
channel is a molecular target of the natural cancer preventive
agent resveratrol. Pharmazie. 68:772–776. 2013.PubMed/NCBI
|