Introduction
Hepatocellular carcinoma (HCC) is the sixth most
frequently diagnosed cancer and the third most common leading cause
of cancer-related death worldwide, and the burden of this
devastating cancer is expected to increase in coming years
(1,2). The majority of patients are diagnosed
at advanced stage and thus not in a position for curative
treatments (3), which have very
limited options, and the only systemic therapy currently approved
is sorafenib, an oral tyrosine kinase inhibitor drug targeting
multiple signalling pathways. The objective response rate (ORR),
however, was poor (4). Cisplatin
is a widely used anticancer agent and has a broad range of
antitumor activity, which is likely a result of inhibition of
replication by cisplatin-DNA adducts and induction of apoptosis
(5). HCC is considered a poor
responder to chemotherapy and a standard systemic treatment does
not exist for patients for whom sorafenib is unsuitable or
unavailable. The reason could be the frequently observed
development of multidrug tumor resistance, which is related to the
high expression of the multidrug-resistance gene (6–9). The
progression of cancer is no longer recognized as an independent
event which only relates to the genetic mutation and uncontrollable
growth of cancer cells. New therapeutic strategies are urgently
needed, and these will most likely result from a better
understanding of the cell biology of HCC.
The interaction between tumor cells and their
microenvironment has been recognized to fundamentally affect tumor
progression (10–12). Physiologically, the surrounding
microenvironment constitutes an important barrier to cell
transformation and notably modulate cell growth (13). Under cancer conditions, the tumor
microenvironment experiences drastic changes including the
recruitment and the activation of stromal cells and the remodeling
of extracellular matrix (ECM), may influence the phenotype of tumor
cells and provide a selective pressure for tumor initiation,
progression and metastasis (14,15).
The remodeling of tumor microenvironment is one of
the hallmarks of HCC pathogenesis (16). The majority of HCC patients have an
established background of chronic liver disease (17), and the presence of liver cirrhosis
is the main risk factor for the development of HCC (18,19).
In addition, HCC is the leading cause of death among patients with
cirrhosis (20). Hepatic stellate
cells (HSCs), the versatile mesenchymal cells in the liver
parenchyma, are vital to the liver’s response to inflammation.
Activation of HSCs is recognized as a central event in the
development of hepatic fibrosis and subsequent, cirrhosis (21). In response to liver injury,
quiescent HSCs become activated and convert into highly
proliferative myofibroblast-like cells, which express inflammatory
and fibrogenic mediators responsible for ECM accumulation within
the microenvironment, and may affect proliferation, invasiveness
and metastasis of tumor cells, and facilitates hepatic
tumorigenesis (21–24). However, the detailed mechanisms
responsible for these effects remain unclear.
Tumor mass is a balance between cell proliferation
and death, and factors affecting this balance have a profound
effect on tumor growth. Regulation of apoptosis in tumor cells
remains poorly understood. Furthermore, few studies demonstrate the
effects of HSCs on HCC from the chemotherapy-resistance
perspective.
The aim of our study here was to examine if HSCs
could provide a survival signal, or block a death signal, thereby
protecting HCC cells from chemotherapy-induced apoptosis.
Materials and methods
Reagents
Cisplatin was purchased from Sigma-Aldrich
(Shanghai) Trading Co., Ltd. (Shanghai, China). RNAiso Plus
reagent, Primescript™ reverse transcription (RT) reagent kit and
SYBR® Premix Ex Taq™ II were obtained from Takara
Biotechnology Co. (Dalian, China). WST-8 Cell Counting Kit-8
(CCK-8), Hoechst 33258 staining kit, RIPA lysis buffer and
bicinchoninic acid (BCA) were purchased from Beyotime Biotechnology
(Haimen, China). Monoclonal antibody raised in rabbit against Bax
(ab32503), Bcl-2 (ab136285) and monoclonal antibody raised in mouse
against p53 (ab28), β-actin (ab8224) were obtained from Abcam Inc.
(Cambridge, UK). Horseradish peroxidase (HRP)-conjugated goat
anti-rabbit or anti-mouse IgG was purchased from Zhongshan Golden
Bridge Biotech (Beijing, China).
Cell lines and cell culture
HepG2 human hepatoma cells (Cell Collection of the
Chinese Academy of Sciences, Shanghai, China) and HSCs, LX-2
(kindly provided by Professor Hong Chen, Lanzhou University First
Hospital, Lanzhou University, Lanzhou, China) were maintained by
Dulbecco’s modified Eagle’s medium (DMEM) (HyClone Laboratories
Inc., Logan, UT, USA) supplemented with heat-inactivated 10% fetal
bovine serum (FBS) (HyClone Laboratories Inc.), 100 U/ml penicillin
and 100 μg/ml streptomycin (North China Pharmaceutical Co., Inc.,
Shijiazhuang, China) in a humidified atmosphere containing 5%
CO2 at 37°C. The cells in mid-log phase were used in the
experiments.
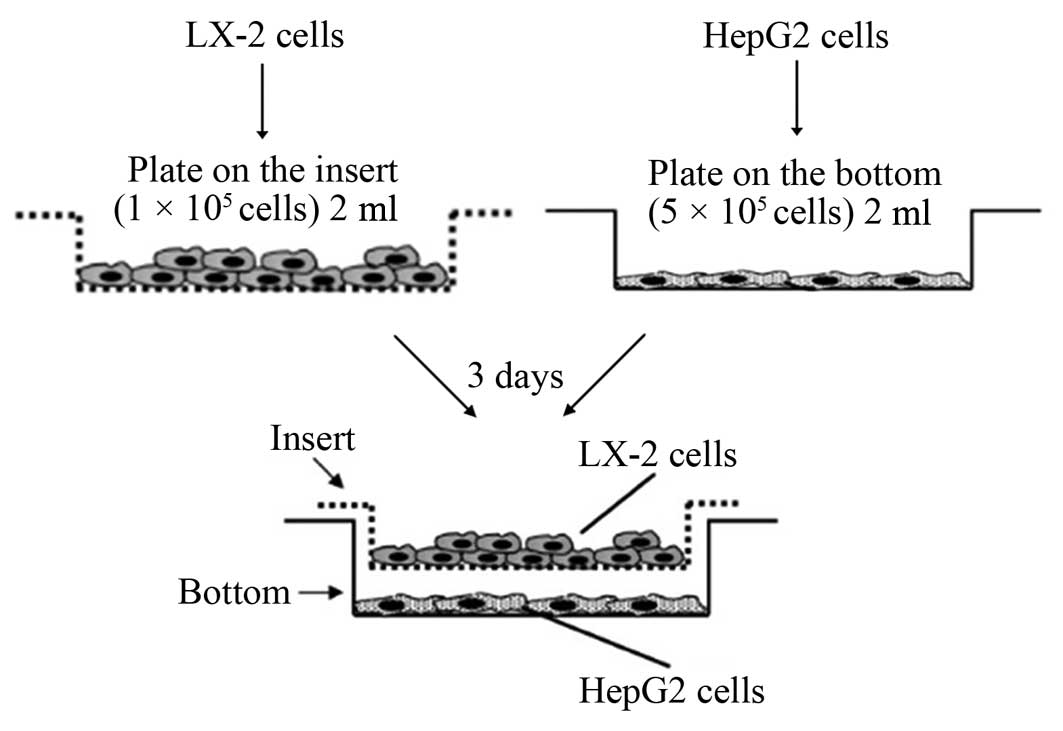
The model used in the experiments described below is
based on the co-culture of HepG2 cells with LX-2 cells. Details for
the co-culture model are shown in Fig.
1. Cells were co-incubated using cell culture inserts of 1.0-mm
pore size (Merck Chemicals Co., Ltd, Shanghai, China) to separate
both cell populations; this allows study of the effect of mediators
from LX-2 cells on HepG2 cells, which resembles the physiological
situation in the liver. The ratio of HepG2 cells to LX-2 cells was
5:1, similar to that of parenchymal: nonparenchymal cells in liver
(25). HepG2 cells, after
co-culturing for 3 days, were carefully removed from the 6-well
plates. They were washed twice with PBS, and the cells were removed
using 0.25% trypsin and collected by centrifugation at 1000 rpm for
5 min. At this time all additions were made. In addition, the
designation HepG2 refers to data found in HepG2 cells incubated
with an empty insert; HepG2/lX-2 refers to results obtained in
HepG2 cells after co-culture with the LX-2 cells.
Cytotoxicity assay
The viability of the cultured cells was analyzed
using a CCK-8 colorimetric assay as described previously (26). In pilot experiments we tested
cisplatin with varying concentrations to obtain an optimal
concentration of cisplatin and chose a similar OD value to ensure
the effect of cisplatin were comparable. The cells were seeded in
96-well flat bottom microtiter plates at a density of
5×103 cells per well, allowed to adhere in 5%
CO2 incubator at 37°C, and appropriate concentrations of
cisplatin ranging from 0.5 to 32.0 μg/ml (5) were then added. Control group and zero
adjustment wells were also set. After incubation for 24–72 h, CCK-8
solution (10 μl) was added to each well, the cells were incubated
at 37°C for 1 h and then the absorbance at a test wavelength of 450
nm was measured using an automatic multiwell spectrophotometer
(PowerWave X, Bio-Tek Instruments Inc., Winooski, VT, USA). At
least three independent experiments were performed.
Flow cytometry for cell cycle
analysis
Cell cycle distribution was analyzed by flow
cytometry. HepG2 cells were treated with cisplatin alone or
pretreated with LX-2 cells for 3 days before incubation with
cisplatin for 24 h. Cells were harvested by trypsinization and
washed with cold PBS, followed by centrifugation at 2000 rpm for 10
min. The pellet of cells was resuspended in 70% cold ethanol and
stored at 4°C overnight. Cells were washed twice with PBS. The
pellet was resuspended in PBS containing 20 μg/ml RNase A, 3.4
mmol/l sodium citration and 1% Triton X-100 in the dark, incubated
at room temperature for 30 min, stained with PI (50 μg/ml), and
analyzed by EPICS XL flow cytometry (Beckman Coulter, Brea, CA,
USA). Each assay was performed in triplicate.
Morphologic observation
HepG2 cells were equally seeded in 24-well plate
(Costar 3524, Corning Inc., Corning, NY, USA) and then treated with
cisplatin alone or pretreated with LX-2 cells for 3 days before
incubation with cisplatin for 24 h. The cells were washed gently
three times with PBS. The morphology of HepG2 and HepG2/lX-2 cells
was observed under an inverted phase contrast microscope (Olympus,
Tokyo, Japan).
Nuclear staining analysis by Hoechst
33258
Nuclear morphology of apoptotic cells was also
examined after nuclear staining using Hoechst 33258 staining kit.
Briefly, after the drug treatment, the cells were fixed with 4%
paraformaldehyde for 30 min and washed three times with PBS. Then
cells were stained with 10 mg/l Hoechst 33258 in the dark at room
temperature for 10 min. Morphologic changes in apoptotic nuclei
were observed under fluorescence microscope. The cells with
condensed chromatin and shrunken nuclei were classified as
apoptotic cells. Apoptosis was assessed by counting the number of
apoptotic cells in five random fields per slide. At least three
slides were counted.
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR) assay
Total RNA was isolated from the cultured cells using
RNAiso Plus reagent following the recommendation of the
manufacturer, then reverse transcribed into cDNA using the
Primescript reverse transcription (RT) Master Mix according to
manufacturer’s instructions. Reverse transcription reaction was
performed at 37°C for 15 min followed by 85°C for 5 sec. qRT-PCR
amplifications were performed using a standard protocol and
undertaken with Applied Biosystems 7500/7500 Fast Real-time PCR
Software (Applied Biosystems, Foster, CA, USA) using the SYBR
Premix Ex Taq II. Reactions were carried out in a 20-μl reaction
volume. The thermal cycle conditions were as follows: 95°C for 30
sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 34 sec.
The primers used in the PCR reactions are described in Table I. The level of gene expression was
normalized with GAPDH. All primers were synthesized by Takara
Biotechnology Co. Amplification was performed at least thrice in
three independent experiments. Data were analyzed according to the
comparative Ct method.
 | Table IPrimer sequences used to detect Bax,
Bcl-2 and p53 for qPCR. |
Table I
Primer sequences used to detect Bax,
Bcl-2 and p53 for qPCR.
| Gene | Primer
sequence | Product size
(bp) |
|---|
| Bax | F:
5′-ATCCAAGACCAGGGTGGTT-3′
R: 5′-ATCTGGAAGAAGATGGGCTG-3′ | 125 |
| Bcl-2 | F:
5′-GGCCTCTGTTTGATTTCTCC-3′
R: 5′-AGTGAAGTCAACATGCCTGC-3′ | 116 |
| p53 | F:
5′-TACATCTGGCCTTGAAACCA-3′
R: 5′-TTTCTACAGTTGGGCAGCTG-3′ | 119 |
| GAPDH | F:
5′-ATCATCCCTGCCTCTACTGG-3′
R: 5′-GTGTCAGTGGTGGACCTGAC-3′ | 122 |
Western blotting
Western blots were performed and analyzed as
previously described (27).
Treated cells were washed with ice-cold PBS and then lysed using
RIPA lysis buffer supplemented with 1 mM PMSF. After centrifugation
at 12,000 rpm for 15 min at 4°C, the supernatant was collected and
the protein concentration was measured by the BCA protein assay.
Forty micrograms of protein per sample were loaded on
SDS-polyacrylamide gel (10%) and transferred to polyvinylidene
fluoride (PVDF) membranes. The blots were blocked with 5% skimmed
milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1
h at room temperature and then incubated at 4°C overnight with
primary antibodies against Bax (1:1000), Bcl-2 (1:800) and p53
(1:1000). β-actin (1:4000) was used as a loading control. After
being washed in TBST three times, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibody (1:10,000) for
1 h at room temperature. The bands were visualized with the
SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher
Scientific, Inc., Rockford, IL, USA) and imaged using a VersaDoc
Imaging System (Bio-Rad Laboratories Co., Ltd. Hercules, CA, USA).
Densitometric analysis was performed using Quantity One Software
v4.62 (Bio-Rad Laboratories Co.) and the results were presented as
the mean of three independent experiments.
Statistical analysis
Values are shown as mean ± standard deviation (SD)
of at least three independently performed experiments to avoid
possible variation. All statistical analysis were carried out using
SPSS 19.0 (IBM, Armonk, NY, USA). Statistical significance was
determined with Student’s t-test and one-way ANOVA followed by the
Bonferroni correction. A P-value of <0.05 was considered
statistically significant.
Results
LX-2 cells protect cisplatin-induced
cytotoxicity in HepG2 cells
In order to evaluate the effects of LX-2 cells on
cisplatin-induced cytotoxicity in HepG2 cells, HepG2 cells were
pretreated with LX-2 cells for 3 days and subsequently treated with
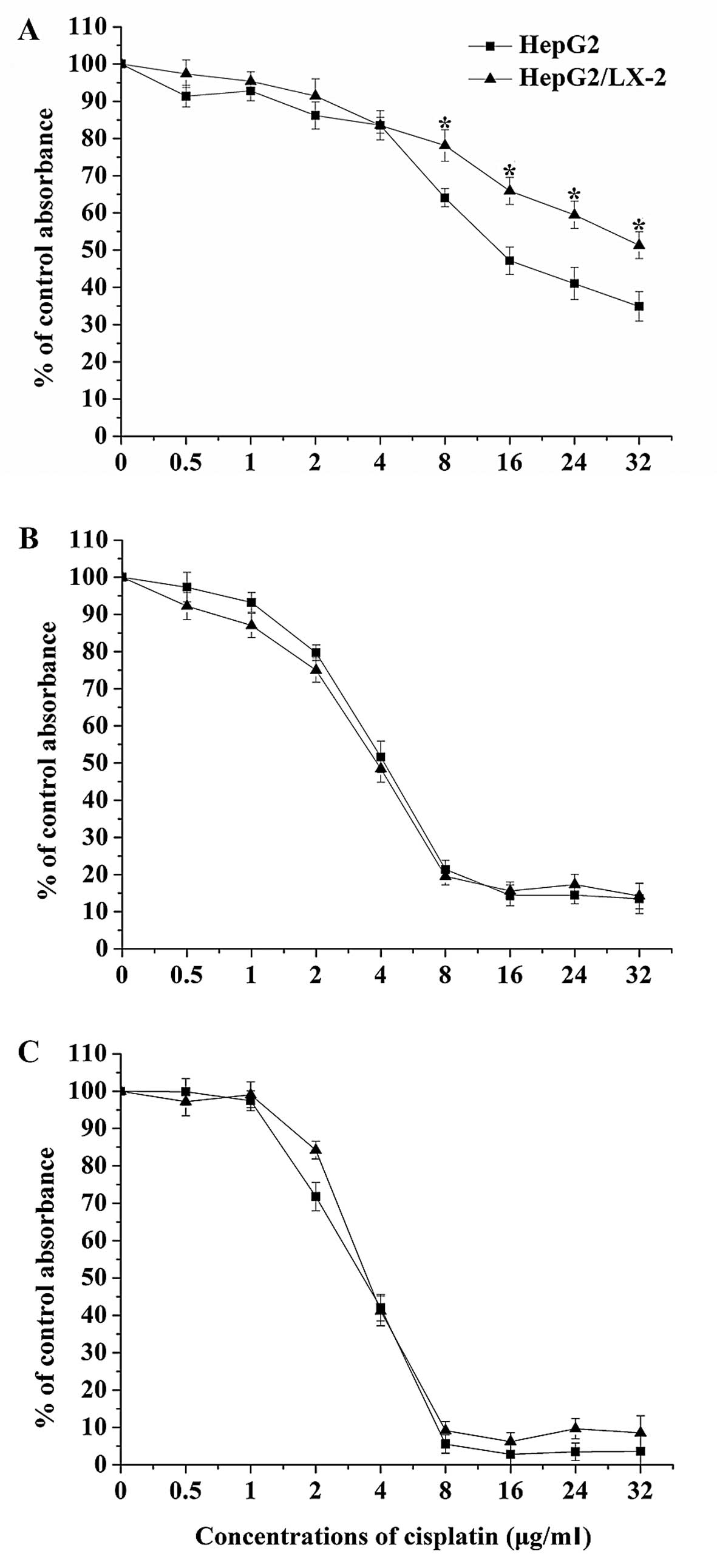
increasing concentrations cisplatin. As shown in Fig. 2, the results showed that cisplatin
inhibited the proliferation of HepG2 and HepG2/lX-2 cells in a
time- and concentration-dependent manner. However, LX-2 cells
protected HepG2 cells from cell death induced by cisplatin,
significantly in the presence of 8.0–32.0 μg/ml cisplatin for 24 h.
IC50 defined as the drug concentrations resulting in 50%
loss of cell viability relative to the untreated cells was
determined for HepG2 and HepG2/lX-2 cells from Fig. 2 (Table II). The IC50 doses for
cisplatin in HepG2 and HepG2/lX-2 cells were determined to be
16.09±1.52 μg/ml and 38.23±3.65 μg/ml, respectively. Thus,
cisplatin was more than 2.4 times more potent in HepG2 cells than
in HepG2/lX-2 cells. Therefore, based on above results, 16.0 μg/ml
(low concentration) and 32.0 μg/ml (high concentration) of
cisplatin at 24 h were used to stand for appropriate and high
doses, respectively, in the following experiments.
| Table IICisplatin cytotoxicity in HepG2 and
HepG2/lX-2 cells. |
Table II
Cisplatin cytotoxicity in HepG2 and
HepG2/lX-2 cells.
| Cisplatin | | HepG2 | HepG2/lX-2 |
|---|
| 24 h |
IC50 | 16.09±1.52 | 38.23±3.65a |
LX-2 cells suppress cisplatin-induced
apoptosis in HepG2 cells
To further evaluate the effects of LX-2 cells on
cisplatin-induced apoptosis in HepG2 cells, HepG2 cells were
pretreated with LX-2 cells for 3 days, and then incubated them with
cisplatin high (32 μg/ml) and low (16 μg/ml) concentrations. As
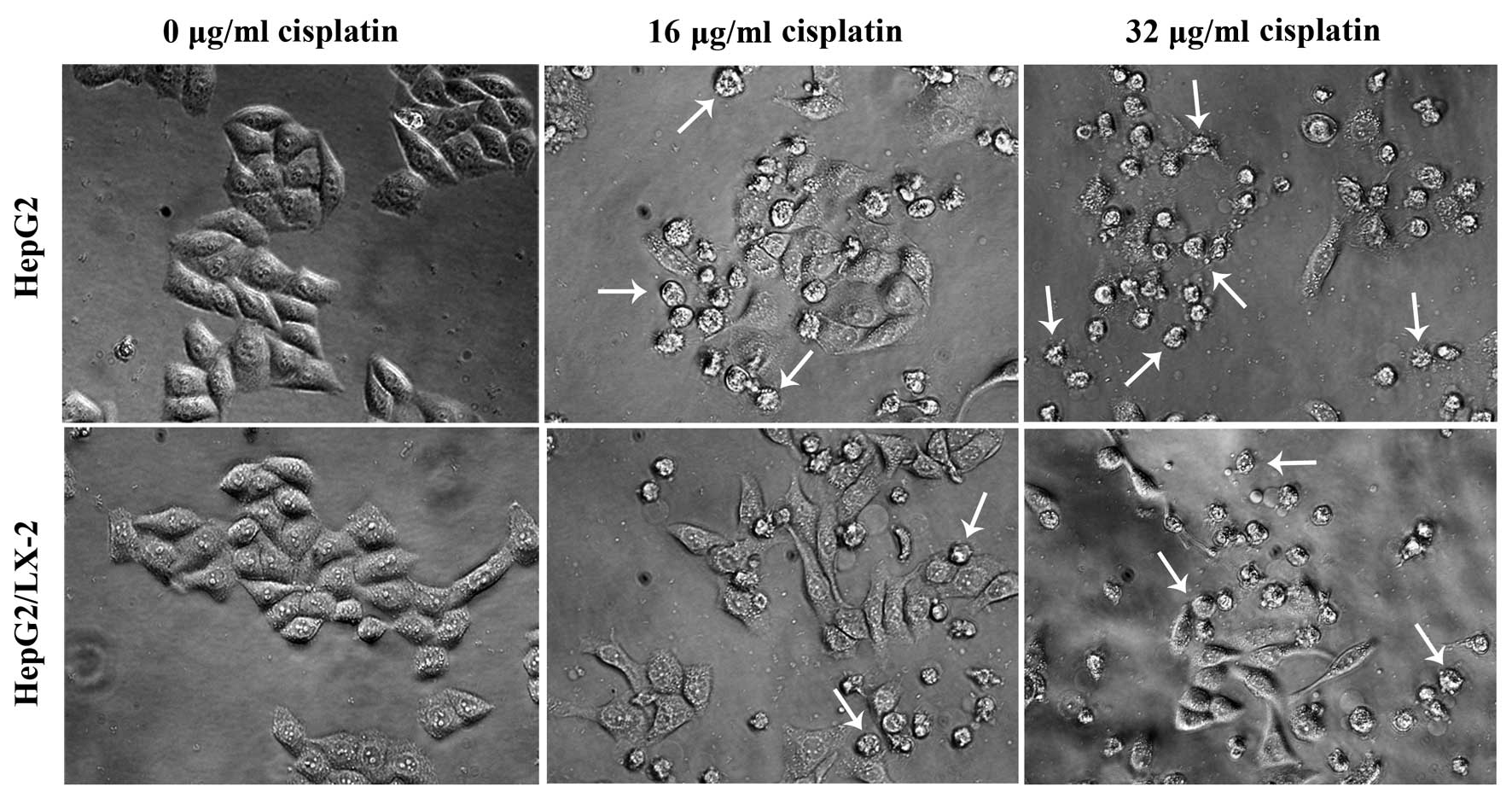
shown in Fig. 3, after exposure to
cisplatin for 24 h, many of the cells exhibited cytoplasmic
shrinkage and either detached from each other or floated in the
medium. At high dose, there was a significant increase in the
percentage of rounded cells with progressive nuclear shrinkage. The
cells eventually became detached from the culture dish, and
cytoplasmic vacuolation and cellular fragmentation were clearly
seen. However, LX-2 cell pretreatment before cisplatin exposure to
HepG2 cells suppressed the morphological characteristics of
apoptosis induced by cisplatin, and increased the number of
surviving cells.
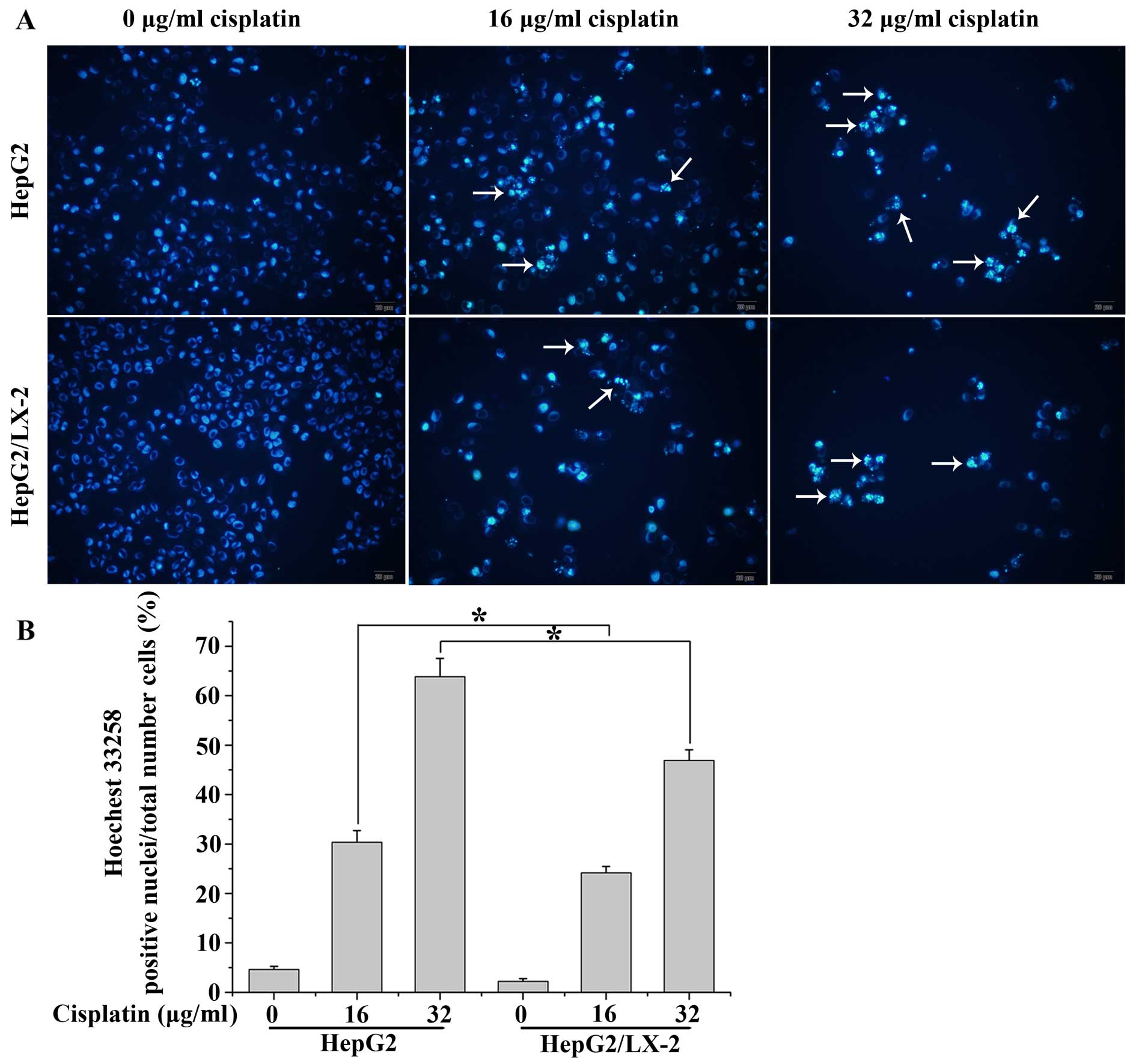
These results were further confirmed by a
morphological analysis using nuclear staining analysis by Hoechst
33258. The Hoechst fluorochrome was able to diffuse through intact
membranes of cells and stain DNA. As shown in Fig. 4A, nuclei morphological change was
observed by Hoechst 33258 staining, which illustrated that the
control cells exhibited uniformly dispersed chromatin, normal
organelle and intact cell membrane; exposed to cisplatin for 24 h,
cells featured typical characteristics of apoptosis, including the
condensation of chromatin, the shrinkage of nuclei and the
appearance of a few apoptotic bodies and ~30.4% underwent apoptosis
with cisplatin at the concentration 16.0 μg/ml, 63.7% with
cisplatin at the concentration 32.0 μg/ml; however, LX-2 cells
protected HepG2 cells from cell death induced by cisplatin, when
compared with those of the treated cells with cisplatin alone
(~24.2% underwent apoptosis with cisplatin at the concentration
16.0 μg/ml, 46.9% with cisplatin at the concentration 32.0 μg/ml)
(Fig. 4B).
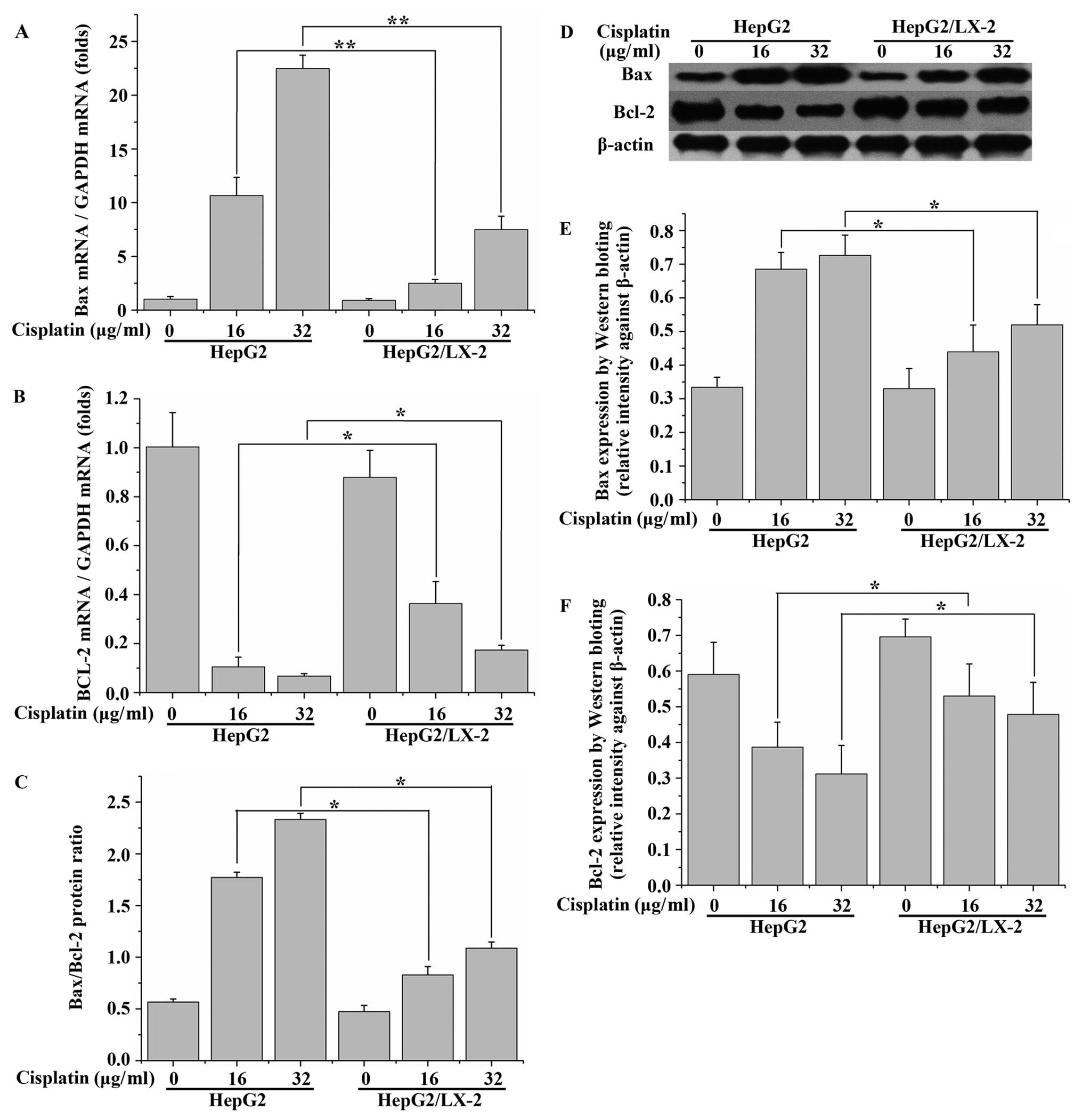
LX-2 cells reduce the Bax:Bcl-2 ratio in
cisplatin-treated HepG2 cells
It is thought that the ratio of the pro-apoptotic
protein Bax and the anti-apoptotic protein Bcl-2 plays a crucial
role in the control of the intrinsic pathway of apoptosis. We
therefore reasoned that the cell death inhibited by LX-2 cells in
HepG2 cells treated with cisplatin might be due to changes in this
ratio. The expression of Bcl-2 and Bax was probed by qRT-PCR and
western blot analysis. As shown in Fig. 5A, cisplatin upregulated the Bax
mRNA expression in HepG2 cells in a dose-dependent manner.
Following 24 h of treatment with 16.0 and 32.0 μg/ml cisplatin, the
Bax mRNA expression level increased nearly 10- and 20-fold,
respectively, however, pretreatment with LX-2 for 3 days induced a
marked reduction in Bax mRNA expression. The results showed a
reverse trend to that in Bcl-2 mRNA expression (Fig. 5B). These results were consistent
with those obtained from western blot analysis by use of anti-Bax
and anti-Bcl-2 antibodies (Fig.
5D–F). Treatment with 32.0 μg/ml cisplatin enhanced the ratio
of Bax to Bcl-2 approximately 4.1-fold, as compared with that for
the untreated cells. However, when the cells were cultured with
32.0 μg/ml cisplatin together with LX-2 cells, this ratio was
decreased to ~55% of that for the cisplatin-treated cells (Fig. 5C). These results further
demonstrated that LX-2 cells had the potential to suppress
cisplatin-induced apoptosis in HepG2 cells.
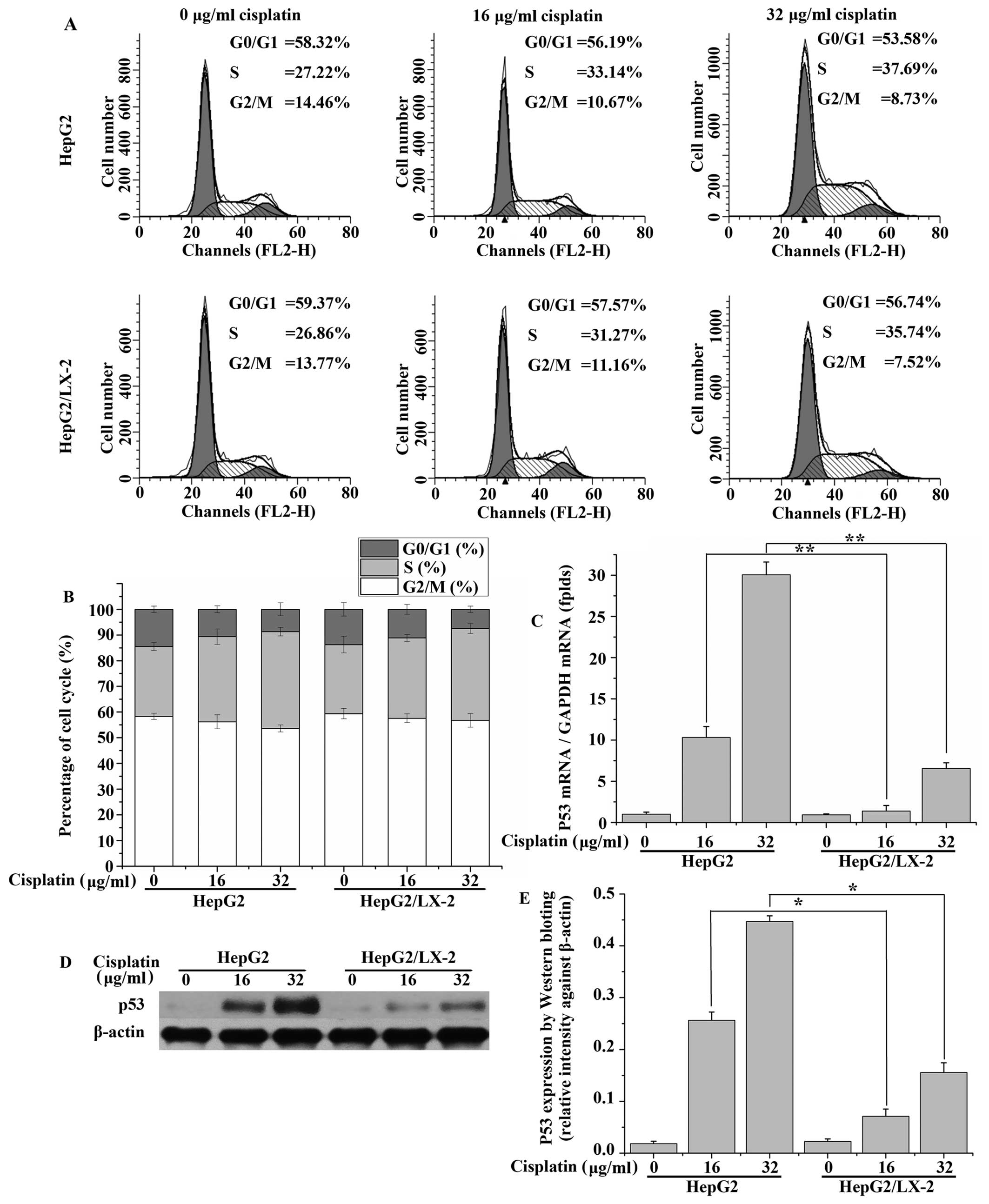
LX-2 cells suppress the expression of p53
and cell cycle arrest in cisplatin-treated HepG2 cells
Cisplatin induces p53 activation in HepG2 cells. p53
level was elevated at 24 h after treated with cisplatin compared
with control cells (Fig. 6C). LX-2
cells abolished this elevation of p53 induced by cisplatin. This
suggests that LX-2 cells may play a critical role in cell-induced
resistance to the effects of anticancer drugs which upregulate p53.
The same trend was seen in the protein expression levels of p53
(Fig. 6D–E). Cell cycle analysis
showed that cisplatin caused G0/G1-S cell cycle arrest. The cell
population in the S phase increased in cells exposed to 32.0 μg/ml
cisplatin for 24 h (37.69%) compared to cells without cisplatin
treatment (27.22%). LX-2 cells slightly suppress this increase
induced by cisplatin (Fig. 6A and
B).
Discussion
The results of our present study demonstrate that
LX-2 cells protect HepG2 cells against cisplatin-induced
cytotoxicity and its protective effects occur via the
anti-apoptosis pathway in HepG2 cells. Flow cytometry showed that
cisplatin arrested HepG2 cells in G0/G1-S phase of cell cycle,
whereas LX-2 cells retarded the progression of cell cycle (Fig. 3), which may be one of the
mechanisms underlying the protective effect of LX-2 cells on HepG2
cells.
The role of the microenvironment during the
initiation and progression of carcinogenesis is now realized to be
of critical importance, both for enhanced understanding of
fundamental cancer biology, as well as exploiting this source of
relatively new knowledge for improved molecular diagnostics and
therapeutics (28,29). The tumor microenvironment,
developed from Paget’s ‘seed and soil’ theory (30), which is a changing concept that
defines the behavior of cancer not only by the tumor cells alone,
but also by the surrounding microenvironment that the tumor cells
need for survival (29). It has
long been established that patients with chronic liver disease, and
particularly those with cirrhosis, have an increased risk of
developing hepatocellular carcinoma (19). Although the importance of the
association of HCC with cirrhosis is still obscure, such an
association provides a means to identify patients at high risk for
HCC. The cross-talk between HCC cells and the surrounding tumor
microenvironment is believed to play a pivotal role in modulating
the biological behaviour of the tumor, likely affecting a different
clinical outcome. Cisplatin is a widely used anticancer agent and
has a broad range of antitumor activity (31). However, its curative effect is
limited due to chemoresistance of HCC, which remains a major
clinical obstacle (32,33). Thus, it is urgent to advance our
understanding of mechanisms of chemoresistance and improve the
efficacy of the current treatment strategies.
It has been demonstrated that various stroma cell
types are recruited to neoplasms, where they substantially promote
the proliferation, invasiveness and metastatic potential of cancer
cells. HSCs belong to one of the most important stroma cell types
in the liver tumor environment. Previous study demonstrated that
activated HSCs promoted tumorigenicity of HCC, stimulating
proliferation and migration of three different human HCC cell lines
in vitro and promote growth and invasiveness of human HCC
cells in nude mice (22). However,
the detailed molecular mechanisms underlying the chemopreventive
effects of HSCs remained unclear. In this study, a novel activity
of HSCs was identified in HepG2 cells, namely the protective
effects from chemotherapy-induced apoptosis. In line with our data,
we demonstrate the effects of HSCs on HCC from the
chemotherapy-resistance perspective, which shows potential as a new
cancer treatment modality.
In this study, cisplatin induced apoptosis of HepG2
cells in a time- and dose-dependent manner (Fig. 2). Consistent with the ability of
cisplatin to kill HepG2 cells via apoptotic processes, cisplatin
upregulated expression of the proapoptosis gene Bax, in a
dose-dependent manner, whereas LX-2 cells inhibited this process
(Fig. 5A), indicating that LX-2
cells were likely to protect HepG2 cells from death via restraining
the apoptosis pathway. Apoptosis is the process of cell death
characterized by cell shrinkage, nuclear condensation, DNA
fragmentation, expression of apoptosis-related genes, and
activation of the caspase cascade. The pro-apoptotic factor Bax
resides in the cytosol, in contrast, the anti-apoptotic Bcl-2 is
localized in the outer mitochondrial membrane, and this protein
inhibits the release of cytochrome c. Translocation of Bax to the
mitochondrial membrane might lead to a loss of mitochondrial
membrane potential and an increase in mitochondrial permeability.
Cisplatin downregulated expression of the anti-apoptosis gene
Bcl-2, in a dose-dependent manner, whereas LX-2 cells inhibited
this change (Fig. 5B). In this
study, abundant cytoplasmic vacuoles were observed in
cisplatin-treated HepG2 cells under an inverted phase contrast
microscope (Fig. 3). When LX-2
cells were applied, the number of apoptotic cells decreased.
Hoechst 33258 staining demonstrated independent evidence supporting
the conclusion that LX-2 cells has an anti-apoptotic effect on
HepG2 cells induced by cisplatin (Fig.
4). The above-mentioned apoptotic features indicated that LX-2
cells have anti-apoptotic effects on HepG2 cells.
LX-2 cells also suppress the G1/G0-S cell cycle
arrest induced by cisplatin, most probably due to the decrease in
p53. p53 is implicated in DNA repair, apoptosis and cell cycle
control (34). Hepatoblastoma
derived HepG2 cells, which have wild-type p53 (35), were more sensitive to cisplatin,
suggesting that wild-type p53 might be one of the factors that
modulates the sensitivity to cisplatin (35). In the current study, we found that
cisplatin induces activation of p53. LX-2 cells decrease p53 levels
and suppress cell death.
In summary, the present study further supports the
impact of stromal activated HSCs on HCC progression in
vitro. These results demonstrate that HSCs partially protect
HepG2 cells against cisplatin-induced apoptosis and its protective
effects occur via decreasing the level of p53. LX-2 cells may
therefore play an important role in cell-induced resistance to the
effects of anticancer drugs by modulating the cellular levels of
p53. In further study, the precise molecular mechanism of the
protective effect by LX-2 cells against cisplatin-induced apoptosis
should be elucidated.
Acknowledgements
We wish to thank Dr Hongwen Zhu and Dr Yang Zhao
(Lanzhou University Second Hospital) for many helpful suggestions
and advice. This work was supported by the National Natural Science
Foundation of China (Grant ID: 31270532) and the Fundamental
Research Funds of the Central Universities, China (Grant ID:
lzujbky-2013-m04).
Abbreviations:
|
Bax
|
BCL2-associated X
|
|
BCA
|
bicinchoninic acid
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
CCK-8
|
WST-8 Cell Counting Kit-8
|
|
ECM
|
extracellular matrix
|
|
FBS
|
fetal bovine serum
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
HCC
|
hepatocellular carcinoma
|
|
HSCs
|
hepatic stellate cells
|
|
PBS
|
phosphate-buffered saline
|
|
PMSF
|
phenylmethanesulfonyl fluoride
|
|
PI
|
propidium iodide
|
|
SD
|
standard deviation
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yazici C, Niemeyer DJ, Iannitti DA and
Russo MW: Hepatocellular carcinoma and cholangiocarcinoma: An
update. Expert Rev Gastroenterol Hepatol. 8:63–82. 2014. View Article : Google Scholar
|
|
3
|
Patrikidou A, Sinapi I, Regnault H, Fayard
F, Bouattour M, Fartoux L, Faivre S, Malka D, Ducreux M and Boige
V: Gemcitabine and oxaliplatin chemotherapy for advanced
hepatocellular carcinoma after failure of anti-angiogenic
therapies. Invest New Drugs. 32:1028–1035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qin LF and Ng IO: Induction of apoptosis
by cisplatin and its effect on cell cycle-related proteins and cell
cycle changes in hepatoma cells. Cancer Lett. 175:27–38. 2002.
View Article : Google Scholar
|
|
6
|
Frenzel A, Grespi F, Chmelewskij W and
Villunger A: Bcl2 family proteins in carcinogenesis and the
treatment of cancer. Apoptosis. 14:584–596. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei W, Chua MS, Grepper S and So SK:
Blockade of Wnt-1 signaling leads to anti-tumor effects in
hepatocellular carcinoma cells. Mol Cancer. 8:762009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Venkatesan B, Prabhu SD, Venkatachalam K,
Mummidi S, Valente AJ, Clark RA, Delafontaine P and Chandrasekar B:
WNT1-inducible signaling pathway protein-1 activates diverse cell
survival pathways and blocks doxorubicin-induced cardiomyocyte
death. Cell Signal. 22:809–820. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Germano D and Daniele B: Systemic therapy
of hepatocellular carcinoma: Current status and future
perspectives. World J Gastroenterol. 20:3087–3099. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coulouarn C, Corlu A, Glaise D, Guénon I,
Thorgeirsson SS and Clément B: Hepatocytestellate cell cross-talk
in the liver engenders a permissive inflammatory microenvironment
that drives progression in hepatocellular carcinoma. Cancer Res.
72:2533–2542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trimboli AJ, Cantemir-Stone CZ, Li F,
Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H,
Chong JL, et al: Pten in stromal fibroblasts suppresses mammary
epithelial tumours. Nature. 461:1084–1091. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nelson CM and Bissell MJ: Of extracellular
matrix, scaffolds, and signaling: Tissue architecture regulates
development, homeostasis, and cancer. Annu Rev Cell Dev Biol.
22:287–309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Polyak K, Haviv I and Campbell IG:
Co-evolution of tumor cells and their microenvironment. Trends
Genet. 25:30–38. 2009. View Article : Google Scholar
|
|
16
|
Hao H, Liu M, Wu P, Cai L, Tang K, Yi P,
Li Y, Chen Y and Ye D: Lipoxin A4 and its analog suppress
hepatocellular carcinoma via remodeling tumor microenvironment.
Cancer Lett. 309:85–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sherman M: Hepatocellular carcinoma:
Epidemiology, surveillance, and diagnosis. Semin Liver Dis.
30:3–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: From genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bruix J and Sherman M; American
Association for the Study of Liver Diseases. Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alazawi W, Cunningham M, Dearden J and
Foster GR: Systematic review: Outcome of compensated cirrhosis due
to chronic hepatitis C infection. Aliment Pharmacol Ther.
32:344–355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar
|
|
22
|
Amann T, Bataille F, Spruss T, Mühlbauer
M, Gäbele E, Schölmerich J, Kiefer P, Bosserhoff AK and Hellerbrand
C: Activated hepatic stellate cells promote tumorigenicity of
hepatocellular carcinoma. Cancer Sci. 100:646–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang BB, Cheng JY, Gao HH, Zhang Y, Chen
ZN and Bian H: Hepatic stellate cells in
inflammation-fibrosis-carcinoma axis. Anat Rec (Hoboken).
293:1492–1496. 2010. View
Article : Google Scholar
|
|
24
|
Jia YL, Shi L, Zhou JN, Fu CJ, Chen L,
Yuan HF, Wang YF, Yan XL, Xu YC, Zeng Q, et al: Epimorphin promotes
human hepatocellular carcinoma invasion and metastasis through
activation of focal adhesion kinase/extracellular signal-regulated
kinase/matrix metalloproteinase-9 axis. Hepatology. 54:1808–1818.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nieto N and Cederbaum AI: Increased
Sp1-dependent transactivation of the LAMgamma 1 promoter in hepatic
stellate cells co-cultured with HepG2 cells overexpressing
cytochrome P450 2E1. J Biol Chem. 278:15360–15372. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang HY and Sun H: Up-regulation of Foxp3
inhibits cell proliferation, migration and invasion in epithelial
ovarian cancer. Cancer Lett. 287:91–97. 2010. View Article : Google Scholar
|
|
27
|
Guo LY, Li YM, Qiao L, Liu T, Du YY, Zhang
JQ, He WT, Zhao YX and He DQ: Notch2 regulates matrix
metallopeptidase 9 via PI3K/AKT signaling in human gastric
carcinoma cell MKN-45. World J Gastroenterol. 18:7262–7270. 2012.
View Article : Google Scholar
|
|
28
|
Mohla S and Witz IP: The 5th International
Conference on Tumor Microenvironment: Progression, therapy and
prevention. Versailles, France, October 20–24, 2009: conference
summary. Cancer Microenviron. 3:1–5. 2010. View Article : Google Scholar
|
|
29
|
Mbeunkui F and Johann DJ Jr: Cancer and
the tumor microenvironment: A review of an essential relationship.
Cancer Chemother Pharmacol. 63:571–582. 2009. View Article : Google Scholar
|
|
30
|
Paget S: The distribution of secondary
growths in cancer of the breast. 1889. Cancer Metastasis Rev.
8:98–101. 1989.PubMed/NCBI
|
|
31
|
Qayed M and Katzenstein HM: Dose-intensive
cisplatin for hepatoblastoma: Have you heard? Lancet Oncol.
14:791–792. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu N, Shen C, Luo Y, Xia L, Xue F, Xia Q
and Zhang J: Upregulated miR-130a increases drug resistance by
regulating RUNX3 and Wnt signaling in cisplatin-treated HCC cell.
Biochem Biophys Res Commun. 425:468–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang H, Tan G, Dong L, Cheng L, Li K, Wang
Z and Luo H: Circulating MiR-125b as a marker predicting
chemoresistance in breast cancer. PLoS One. 7:e342102012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gatz SA and Wiesmüller L: p53 in
recombination and repair. Cell Death Differ. 13:1003–1016. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pandit B and Gartel AL: Proteasome
inhibitors induce p53-independent apoptosis in human cancer cells.
Am J Pathol. 178:355–360. 2011. View Article : Google Scholar : PubMed/NCBI
|