Introduction
Drug resistance is a major obstacle in successful
chemotherapy of metastatic breast cancer and most patients with
drug resistant metastatic cancer succumb to the disease. Therefore,
it is important to investigate the genetic basis of drug resistance
in metastatic cancer cells to design effective therapy of advanced
stage disease and improve their clinical outcome. Increased genetic
instability leads to progressive metastasis of tumors with
simultaneous acquisition of resistance to chemotherapeutic drugs.
Recent advances in understanding the mode of action of anticancer
drugs indicate that regardless of their diverse nature, most of
them elicit apoptosis in the target cells (1,2). The
genetic changes which inactivate apoptotic pathways during tumor
development make tumor cells resistant to drugs. Apoptosis is
associated with activation of a number of genes that mediate the
transition from quiescence to proliferative growth (3,4).
Premature activation of a subset of protein kinases, normally
active in mitosis, has been implicated as critical events in the
apoptosis process (5,6).
Cyclin proteins associating with and activating
cyclin dependent kinases are central to the control of cell
proliferation in eukaryotic cells. These proteins are involved at
critical nodal points, shared by pathways regulating both cellular
proliferation as well as apoptosis (7–13).
Phosphorylation of pRb by cyclin D/cdk4, cyclin E/cdk2 and/or
cyclin A/cdk2 during Gl/S transition releases the transcription
factor E2F1 which directs the timely expression of cell cycle
controlling genes regulating initiation and orderly progression of
S phase (14,15). Cyclin A kinase through binding and
phosphorylation of the transcription factor E2F1 suppresses
expression of specific S phase genes. The regulation of E2F1 DNA
binding and transactivation has been proposed to represent an S
phase checkpoint which when disrupted due to decreased cyclin
A/cdk2 activity results in S phase delay/arrest followed by
regrowth or apoptosis (16).
Enhanced DNA binding of hypophosphorylated E2F1 has been correlated
with increased sensitivity of human tumor cells to cytotoxic drug
and ionizing radiation treatments (17,18).
Two metastatic variants of the human cancer cell
line (MDA-MB-435) isolated from lung and brain metastasis in nude
mice (19) demonstrate markedly
different levels of sensitivity to antimetabolite chemotherapeutic
drugs. In this study, we investigated the involvement of cell cycle
regulatory proteins in eliciting differential response in these two
variants to the antimetabolite drug PALA. PALA is a potent
inhibitor of aspartate transcarbamoyl transferase, the enzyme
catalyzing the second step of de novo pyrimidine
biosynthesis.
We observed elevation of cyclin A expression and
activation of its catalytic subunit kinase in the drug sensitive
L-2 cells undergoing apoptosis but not in the resistant Brl cells.
Further, we demonstrated that induced ectopic expression of cyclin
A was sufficient to cause apoptosis in the resistant Br1 cells when
exposed to PALA. In cells undergoing apoptosis, elevated cyclin A
expression and kinase activities also correlated with increased
E2Fl DNA binding activity. Therefore, this study provides evidence
that apoptotic response in antimetabolite drug-treated tumor cells
involves enhanced cyclin A/cdk2 activity concomitant with increased
E2Fl DNA binding activity. Taken together these results suggest
that cyclin A and associated kinase activity are regulators of a
checkpoint response that is activated in drug-treated cells leading
to induction of apoptosis.
Materials and methods
Cell lines and drug selection
Br-l and L-2 cell lines established from metastasis
in nude mouse injected with the human tumor cell line MDA-MB-435
were provided by Dr Janet E. Price of the Department of Cancer
Biology, The University of Texas M.D. Anderson Cancer Center.
MDA-MB-435, isolated from plural effusion of a 31-year-old breast
cancer patient, was later reported to show similarity with
melanocyte/melanoma cells based on gene expression profiling data.
The Br-l cell line was established from a brain metastasis while
L-2 cells were selected by two cycles of growth and metastasis to
lung in nude mice (19). Cell
lines were maintained in Dulbecco’s modified Eagle’s medium
supplemented with 10% dialyzed FBS (Gibco, Grand Island, NY, USA).
The cells were grown on plastic and incubated in 5% CO2
in air at 37°C in a humidified incubator. Three independent clones
isolated from the L-2 (L-2, L-2-1, L-2-2) and Br-1 (Brl-3prl,
Brl-3pr2, Brl3pr3) were utilized in this study. Population doubling
time, for each of these cell lines were estimated to be ~24 h. Drug
resistance levels and proliferative response to drug treatment
among the clonal isolates from each cell type variant were very
similar.
Cell lines were tested for their potential to
acquire resistance against the DNA antimetabolite drug PALA.
Frequency of drug resistant cells developing at 5xLD50
concentration of the drug were <10−5 for L-2 and
<10−3 for Br-1 cells. PALA was obtained from Drug
synthesis Branch (Division of Cancer Treatment, National Cancer
Institute). At 20xLD50, Br-1 cells gave rise to
resistant colonies but L-2 cells did not. Further experiments to
study early proliferative response and cell cycle regulatory
protein expression were done with cells exposed to
20xLD50 of PALA (300 μM).
Drug treatment
L-2 and Brl-3prl cells were plated at a density of
1-2×106 and 48 h later 300 μM of PALA was added. Cells
were initially harvested after 12, 24 and 48 h of PALA treatment
for flow cytometry analysis and oligonucleosomal DNA analyses. In
another set of experiments, cells treated for 48 h were washed,
re-plated in drug-free medium and harvested at 0, 4, 10, 24 and 48
h for flow cytometry, oligonucleosomal DNA analysis and protein
analysis. Cells harvested immediately after 48 h of PALA at 0 h
were considered as those representing the control time point.
Growth rate analysis
Exponentially growing L-2 and Brl-3prl were plated
in 60-mm dishes at a cell density of 3×105. After 48 h,
regular medium was replaced with medium containing 300 μM of PALA
(20xLD50). PALA was washed off after 48 h and regular
medium was added. Cells were counted from day 0 through day 5 for
every 24 h with trypan blue staining.
Flow cytometry analysis
Approximately 1×106 cells were washed
with phosphate buffered saline (PBS), fixed overnight in 70%
ethanol, stained with propidium iodide (final concentration was
0.01μg/ml in PBS and analyzed on a FACScan cytometer (Becton
Dickinson). Resolution of G1, S and G2/M phases was done with LYSIS
II analysis software (Becton Dickinson).
Effect of PALA on cellular
nucleotides
To determine the PALA-mediated perturbation in the
ribonucleotide pools, each cell line was incubated with 300 μM PALA
for 48 h. After treatment cells were washed with phosphate-buffered
saline, enumerated with Coulter counter analyzer, and mean cell
volume was determined with the Coulter channelyzer (Coulter
Electronics Inc., Hialeah, FL, USA). Nucleotides were extracted
from cells by standard procedures using HC104 before (control) and
48 h after incubation with 300 μM PALA. Ribonucleotides were
separated using an anion-exchange Partisil-10 SAX (Waters Corp.,
Milford, MA, USA) column by high-pressure liquid chromatography
(45). The intracellular
concentration was calculated and expressed as the quantity of
nucleotides contained in the extract from a given number of cells
of a determined mean volume. This calculation assumes that
nucleotides are uniformly distributed in total cell water. In
general, the lower limit of sensitivity of this assay was 50 pmol
in an extract of 1×107 cells, corresponding to a
cellular concentration of ~2.5 μM.
Oligonucleosomal DNA analysis
For DNA fragmentation assay, both adherent and
non-adherent cells were washed in PBS and lysed in 100 μl ice cold
PBS and 400 μl lysis buffer (Tris-EDTA, 0.1% Triton X-100) on ice
for 20 min. The supernatant containing fragmented DNA was isolated
by centrifuging and precipitating with NaCl and isopropanol at
−20°C overnight. The pellets were resuspended in 150 μl of buffer
containing 10 mM Tris pH 7.8, 25 mM EDTA, 0.5% SDS and 100 μg of
proteinase K at 37°C overnight. The samples were extracted twice
with phenol:chloroform (1:1 by volume). The low molecular weight
DNA was recovered by ethanol precipitation, resuspended in 25 μl
Tris-EDTA, and treated with RNAse A for 2 h at 37°C prior to
electrophoresis on 1.8% agarose gel.
Western blots
Total cell lysates were prepared by lysing cells in
lysis buffer (50 mM Tris pH 7.4, 5 mM EDTA, 250 mM NaCl, 50 mM NaF,
1 μg/ml leupeptin and aprotinin) at 4°C for 20 min. The protein
concentration of supernatant was determined by Bio-Rad protein
assay. Equal amount of protein was resolved on 8–10% SDS-PAGE gels
and transferred to Immobilon P (Millipore). Membranes were blocked
with 5% non-fat milk and probed with antibodies against p53, Rb,
p21, myc, cyclin A, cyclin E, cdc2, bcl-2 (Oncogene Science), cdk2
(Upstate Biotechnology). After incubation of blot with horse-radish
peroxidase-conjugated secondary antibody, reactive polypetides were
detected by the enhanced chemiluminescence (ECL) system
(Amersham).
Hl kinase activity
Cyclin A, cyclin E, cdc2 and cdk2 associated kinase
activities were measured in vitro after immunoprecipitation,
using histone (Boehringer Mannheim) as a substrate. Total cell
lysates were made from 300 μM PALA treated cells at indicated time
points. For each immunoprecipitation 50 μg of protein saturated
with proper antibody was incubated with protein G+A agarose beads
(Oncogene Science) overnight at 4°C. After extensive washing in
lysis and kinase buffer the beads were assayed for 30 min at 37°C
in kinase buffer (50 mM Tris pH 7.4, 10 mM MgCl2, 1 mM
DTT, 50 μM ATP, 1 μCi of (γ32P-ATP) containing l mg/ml
substrate. Kinase reactions were analyzed by autoradiography
following 12% SDS-PAGE and quantitation with the help of
phosphorimager.
E2F1 DNA binding activity assay
Cells were washed in PBS containing 1 mM PMSF, and
then harvested to prepare nuclear proteins for assessing E2Fl DNA
binding activity by gel retardation assay (46). Harvested cells were washed in wash
buffer (20 mM HEPES, 2 mM DTT, 2 mM MgCl2, 1 mM PMSF, 2
μg/ml leupeptin, 1 μg/ml aprotinin) and lysed in wash buffer
containing 0.2% Triton X-100 by incubation on ice for 10 min.
Nuclei were then collected by centrifugation at 13,000 rpm for 10
sec at 4°C. The nuclei were resuspended in 50 μl of nuclear
extraction buffer (20 mM HEPES, 2 mM MgCl2, 2 mM DTT,
420 mM KCl, 25% glycerol, 1 mM EDTA and protease inhibitors) and
incubated on ice for 20 min. Cellular debris was removed by
centrifugation at 13,000 rpm for 5 min at 4°C. The protein/DNA
interaction assay was performed by preincu-bating 5 μg of nuclear
proteins in a binding buffer (4% Ficoll 400, 20 mM HEPES pH 7–9, 2
mM MgCl2, 0.5 μg of salmon sperm DNA and 100 mM KCl) for
10 min at room temperature. γ32P-ATP labeled E2
oligonucleotide probe (10 pmol) was then added and incubated at
room temperature for 30 min. The reaction products were resolved on
a 4% polyacrylamide gel in 0.5 × TBE run at 160 V at 4°C for 2–3 h.
Gels were dried and exposed to X-ray film.
Brl transfection with pMTCyc A
Cells bearing a Zn2+ responsive cyclin A
construct were generated by transfecting the pMTCyc A plasmid into
Brl-3pr1 cells (Br-l pMTCyc A). pMTCyc A contains a 2.3 kb
EcoR1 fragment of human cyclin A cDNA downstream of a sheep
metallothionein promoter and a neomycin-resistant marker gene.
Thirty randomly selected clones were expanded and screened by
southern blot analysis of total genomic DNA with cyclin A probe.
Four positive clones were analyzed further to see inducibility of
the cyclin A transgene. Two of these clones showed
distinct-induction of cyclin A expression in the presence of 100 μM
Zn2+. Expression of induced cyclin A in the clones was
determined after induction with 100 μM of Zn2+ for
different time intervals. Total cell lysates were prepared and
Western blot analysis was done as described above. Oligonucleosomal
DNA fragmentation analysis of clones was done as described above
after treatment with 300 μM PALA for 48 h and simultaneous
induction of cyclin A with 100 μM Zn2+ for 9 h.
Results
Growth rate analysis in L-2 and
Brl-3prl
Effect of 48 h PALA treatment at 20xLD50
on the growth rate of the two cell types, was assessed over a 5 day
period. L-2 and Brl-3prl showed clear difference in their growth
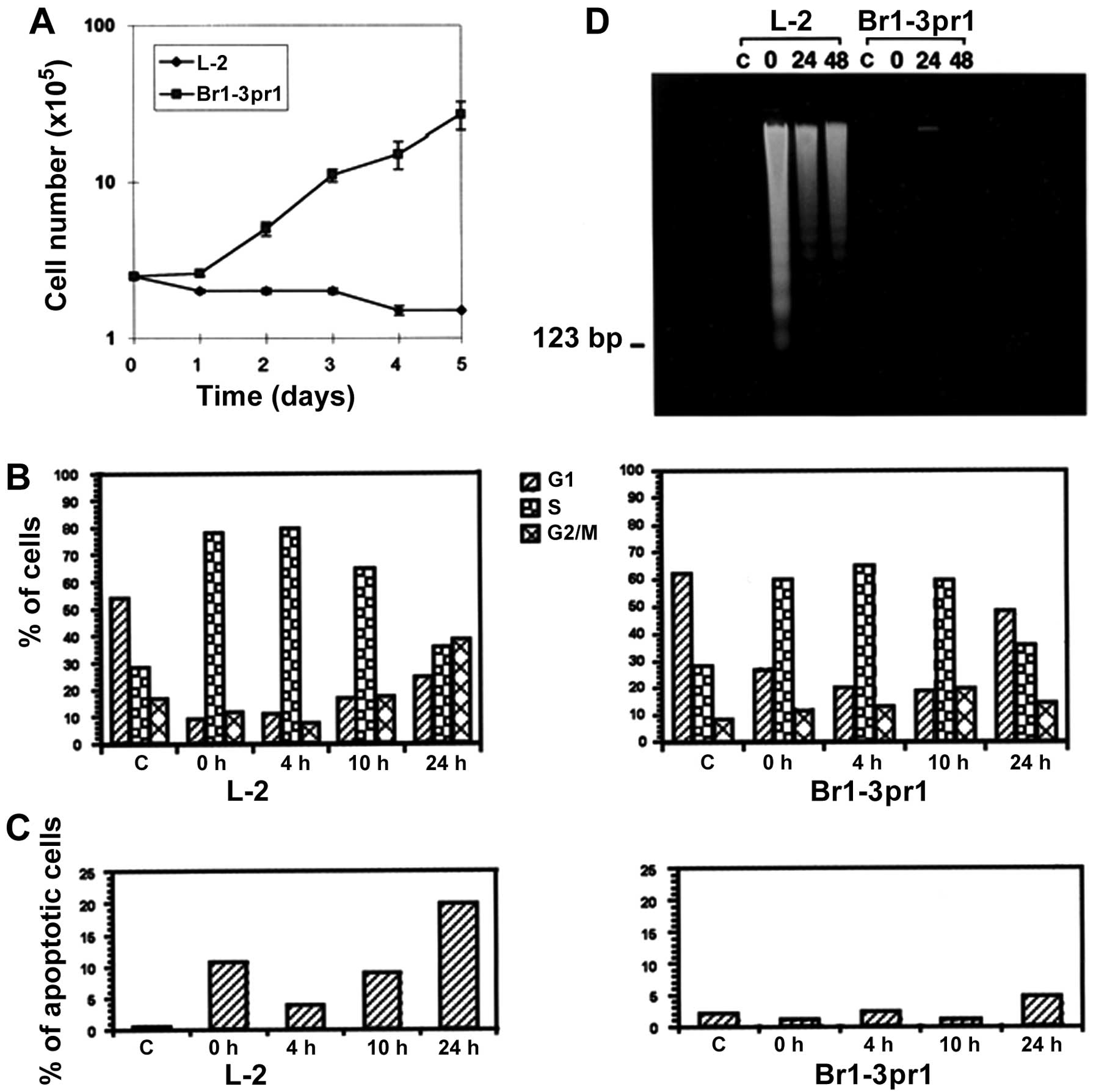
potential following drug treatment (Fig. 1A). After an initial growth arrest
in both cell lines, Brl-3prl recovered and continued proliferation
whereas L-2 cells failed to grow. These results demonstrated that
the differential levels of drug sensitivity in the two cell types
were manifested in their markedly different proliferation pattern
within the first 24 h after exposure to the drug.
Cell cycle distribution pattern following
PALA administration
We investigated how PALA exposure affected
proliferative pattern through the cell cycle in the two cell types.
Untreated cells of both variants showed a similar cell cycle
distribution pattern (C in Fig.
1B). It was noted that both cell lines treated for 48 h showed
progressive accumulation of cells in S phase. However, the
difference in cell cycle stage specific distribution pattern of the
cells became evident following release from PALA treatment of L-2
and Brl-3prl cells. At the end of 48 h treatment (or same as 0 h,
Fig. 1B), cells were predominantly
in S phase in both the cell lines, although slightly higher
proportion of cells in S phase were noted in L-2 than Brl-3prl
cells. At 4 h post-treatment S phase cell population showed further
increase in the two cell types but at 10 h, there was a decline in
the percentage of cells in S phase in both cases with increase in
G2/M phase cells. At 24 h post-treatment cell cycle distribution
profile of L-2 cells showed more cells in G2/M phase than in G1 or
S phases. Brl-3prl cells, however, revealed a pattern similar to
the one seen in untreated controls. In view of our findings that
there was significant loss in proliferative capacity as well as
decrease in the number of surviving L-2 cells after PALA treatment,
we compared the percentage of apoptotic cells using the sub-G1 FACS
assay in both cell types.
There was significant cell death detected in L-2
cells immediately after 48 h (11%, 0 h, Fig. 1C) PALA treatment. Following a lower
incidence (4%) at 4 h, there was a second wave of apoptosis
induction detected in the L-2 cells at 10 h (9%) and 24 h (21%).
Brl-3prl cells on the other hand did not undergo significant
apoptosis up to 10 h post-treatment but showed slight increase (5%)
at 24 h post-treatment. These results suggested that both cell
types underwent an initial S phase delay/arrest following PALA
treatment. Except for a small percentage, most Brl-3pr1 cells
recovered and proliferated but increasing number of L-2 cells
progressively underwent apoptosis.
DNA fragmentation assay
Apoptosis in response to PALA treatment was assayed
by the presence of oligonucleosomal DNA fragments in the two cell
types. No significant DNA fragmentation was observed in L-2 cells
treated with PALA for less than 48 h. At the end of 48 h (time 0 h,
Fig. 1D) significant DNA
fragmentation indicative of apoptosis was observed in agreement
with the flow cytometry data reported earlier. Brl-3pr1 cells did
not reveal any significant DNA fragmentation following PALA
treatment at all the time points checked. These results reveal that
L-2 cells treated for 48 h with PALA show progressive accumulation
of cells in S phase and activation of an apoptotic pathway leading
to cell death. Brl-3pr1 cells on the other hand showed a transient
S phase arrest and recovered to proliferate without significant
evidence of apoptosis in these cells. L-2 cells from several dishes
had to be pooled for all subsequent protein expression and kinase
activity studies since massive apoptosis was induced in these cells
following drug treatment.
Effect of PALA on cellular
nucleotides
To determine if the apoptotic response of these cell
lines to PALA treatment was due to differential effect on the
nucleotide pools, cellular NTPs were quantitated in control
(untreated cells) and in cells incubated with PALA for 48 h. As
presented in Table I, there was a
significant reduction in the pyrimidine nucleotide pools in both
cell lines after PALA treatment. CTP pool size was lowered to
<8% in both cases (p=0.024 for L-2 and p=0.002 for Brl-3prl
cells). There was >95% decrease in the concentration of UTP
(p=0.018 and 0.0002 for L-2 and Brl-3prl cells, respectively). In
contrast to pyrimidine nucleotides, there was 30–60% increase in
the purine nucleotide concentration. These data clearly
demonstrated that the action of PALA in both these cell lines was
comparable resulting in similar reduction in the pyrimidine
NTPs.
 | Table IEffect of PALA on ribonucleotides in
L-2 and Br1-3pr1 cells. |
Table I
Effect of PALA on ribonucleotides in
L-2 and Br1-3pr1 cells.
| % of Control NTP,
Mean ± SD |
|---|
|
|
|---|
| NTP | L-2 | Br1-3pr1 |
|---|
| CTP | 7.1±2.7 | 7.5±2.2 |
| UTP | 3.3±0.2 | 4.7±1.2 |
| ATP | 141.7±18.8 | 161.6±28.5 |
| GTP | 123.0±8.0 | 159.2±31.5 |
Expression of cell cycle regulatory
protein
The expression profile of cell cycle and apoptosis
regulatory proteins after PALA treatment were assessed to evaluate
if they correlated with induction of apoptosis process. In order to
assess the pattern of expression of cell cycle regulatory proteins
in L-2 cells undergoing apoptosis and Brl-3prl cells recovering to
proliferate following S phase arrest, western blot analysis of p53,
p21, Rb, c-myc, cyclin A, cyclin E, cdk2, cdc2 and bcl-2 was
performed at different time intervals after release from 48 h
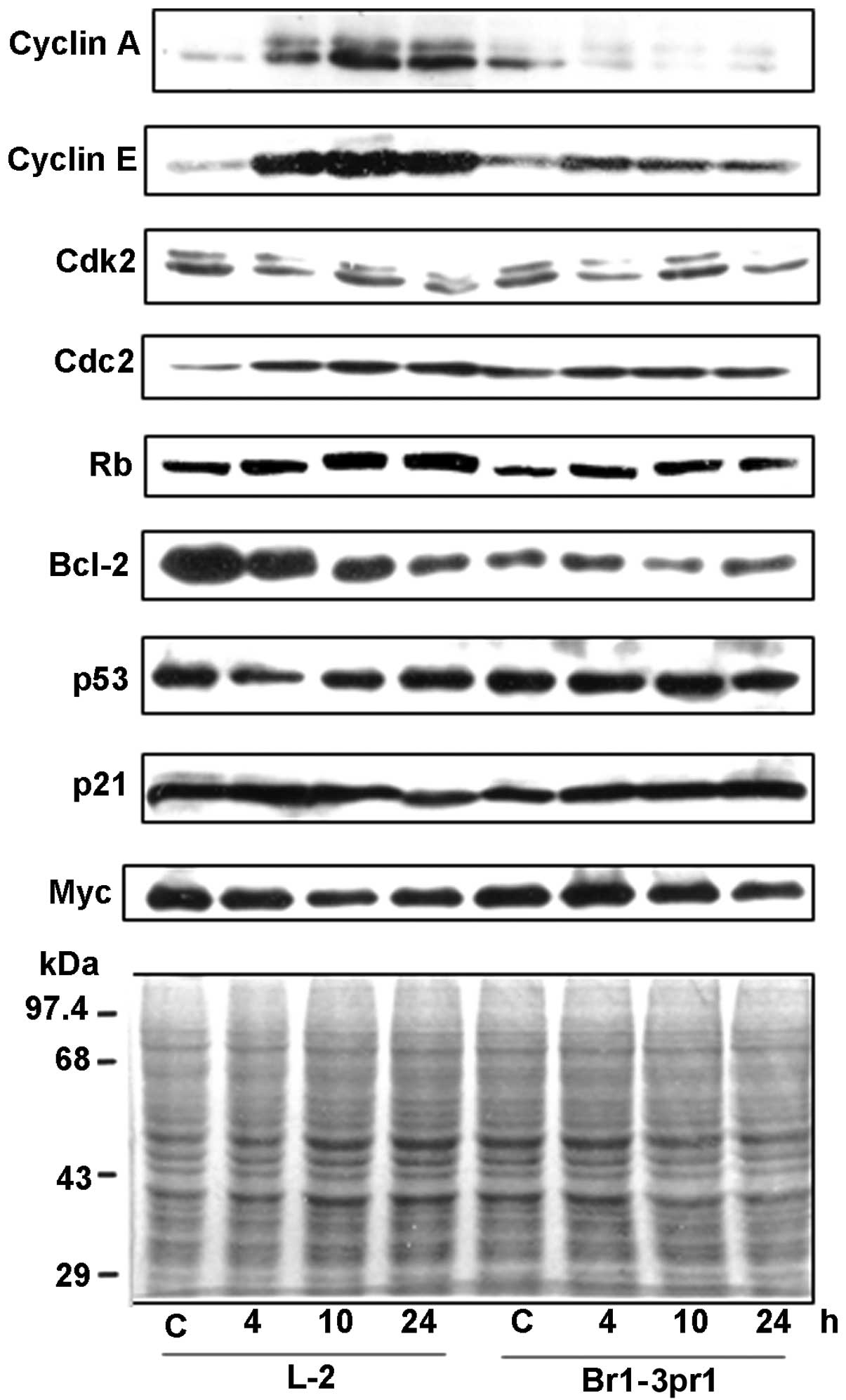
treatment of PALA (Fig. 2).
Compared to the untreated cells (C), no apparent effect in the
levels of p53, p21 and myc proteins were observed in the two cell
types. There was moderate difference in the level of phosphorylated
Rb proteins seen in the two cell types. L-2 cells showed increased
accumulation of phosphorylated Rb compared with Brl-3pr1 cells.
Marked increase in the amount of cyclin A protein was detected in
the L-2 cells undergoing apoptosis with the highest level detected
at 10 h post-drug treatment. In contrast, there was no increase in
the level of cyclin A seen in the Brl-3prl cells. Cyclin E protein
was found elevated in the L-2 cells and Brl-3prl cells compared to
their respective controls (lanes C). The increase in cyclin E
protein in L-2 cells despite being more than that seen in Brl-3prl
cells was not as significant as that seen for cyclin A protein, at
similar time intervals.
Expression of both cdk2 and cdc2 proteins were also
monitored. Cdc2 protein amount was elevated in drug-treated L-2
cells compared to the control at all the time points checked. There
was no distinct alteration in the level of cdk2 protein detected in
the cell types. L-2 demonstrated a gradual decrease in the
expression of bcl-2 from 4 through 24 h post-treatment; whereas
bcl-2 expression levels remained virtually unchanged in Brl-3pr1
cells. Taken together these data document that in contrast to
proliferating Br1-3pr1 cells, apoptosis in PALA treated L-2 cells
is accompanied with discrete changes in the levels of some
proteins. These include increase in the cyclin A and cdc2 along
with decline in the bcl-2 proteins.
Kinase activity assay
In order to determine if the levels of cyclins and
kinases correlated with their associated kinase activities,
immunoprecipitates of cyclin A, cyclin E, cdc2 and cdk2 were
subjected to HI kinase assays. Quantitation of these data indicated
a significant elevation in cyclin A, cyclin E associated kinase
activities in L-2 cells at 4 and 10 h after PALA removal as
compared to the control and there was relatively moderate increase
in these kinase activities of Br1-3pr1 cells (Table II). Cyclin A and E are known to
bind both cdK2 and cdc2, the catalytic subunits (20,21).
Cdk2 and cdc2 kinase activities were markedly elevated in L-2 cells
while in Brl-3pr1 cells, only cdk2 kinase was 3× higher post-PALA
treatment. In L-2, cdk2 kinase activity was elevated ~9-fold at 4 h
and 7.5-fold at 10 h while cdc2 kinase activity was ~1.6- and
2.1-fold elevated at these time points. The results indicate that
higher cyclin A, cdc2 associated kinase activities correlated with
higher expression levels of these proteins. Cdk2 kinase activity on
the other hand was significantly elevated with no apparent increase
in the amount of the protein in these cells.
| Table IIKinase activities in L-2 and Br1-3prl
cells. |
Table II
Kinase activities in L-2 and Br1-3prl
cells.
| L-2 | Br1-3pr1 |
|---|
|
|
|
|---|
| Kinase | Control | 4 h | 10 h | 24 h | Control | 4 h | 10 h | 24 h |
|---|
| Cyclin A | 1 | 3.1 | 2.2 | 1.2 | 1 | 1.4 | 2.0 | 1.8 |
| Cyclin E | 1 | 3.4 | 2.2 | 1.8 | 1 | 1.5 | 1.5 | 1.7 |
| Cdk2 | 1 | 9.0 | 7.5 | 5.0 | 1 | 3.0 | 2.0 | 1.7 |
| Cdc2 | 1 | 1.6 | 2.1 | 1.5 | 1 | 1.0 | 1.1 | 1.2 |
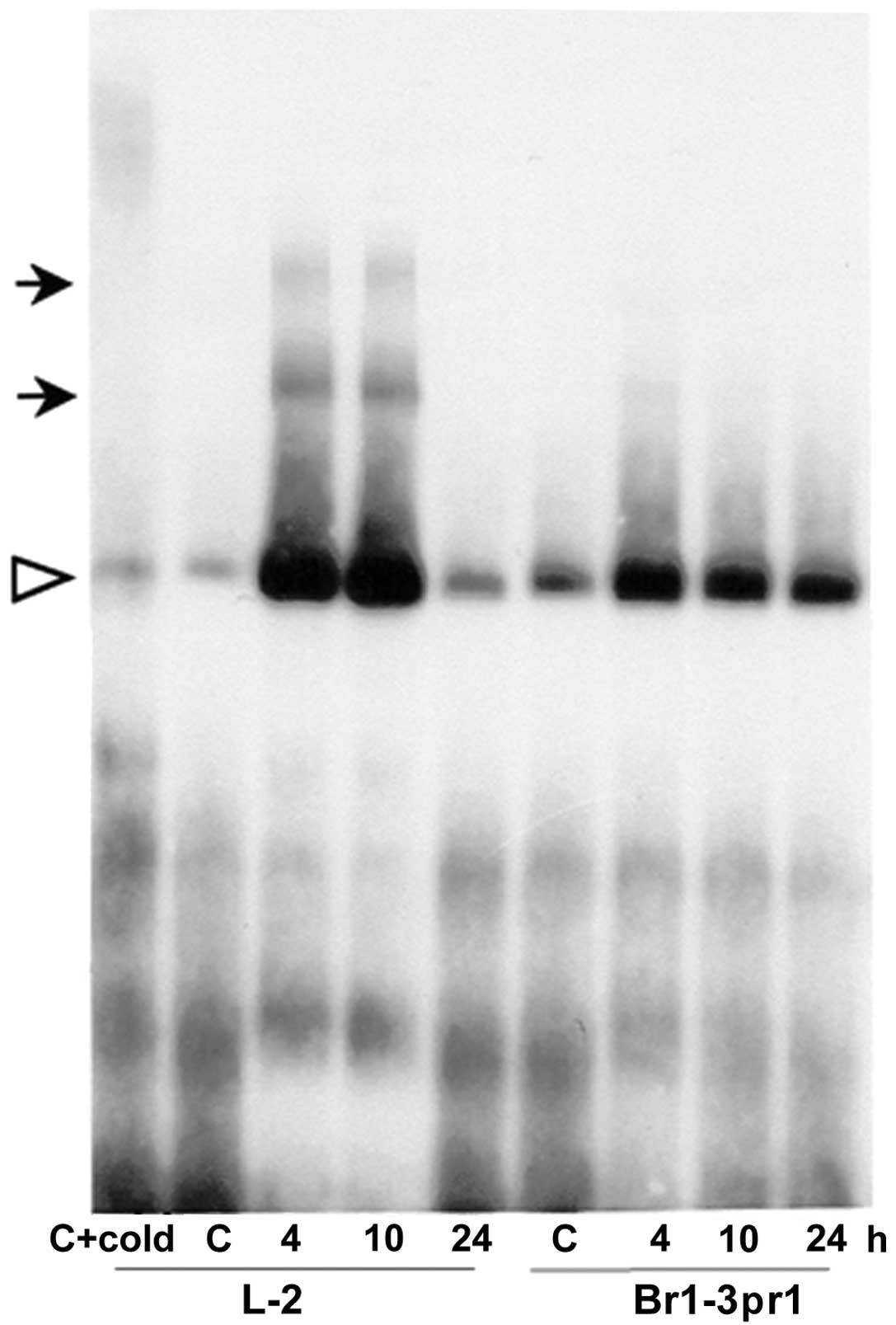
E2F1 DNA binding assay
Cyclin A kinase regulated E2F1 transactivation
function has been proposed to represent an S-phase checkpoint that
is activated in cells exposed to cytotoxic effects of drugs and
ionizing radiation. We assayed for E2F expression and DNA binding
activity in untreated and PALA treated cells by gel retardation
assay. There was no difference in the level of expression of E2F
protein detected in the treated and untreated cells (data not
shown). As shown in Fig. 3, E2F
complexes (bands shown with solid head arrows) were detected in L-2
cell extracts at 4 and 10 h post-treatment but were not seen in the
control untreated or in the PALA treated Brl-3prl cells. It is
important to mention that these L-2 cells also revealed
progressively increasing incidence of apoptosis with enhanced
cyclin A expression and associated kinase activities. At 24 h
post-treatment sample, however, the gel retardation assay did not
show discrete E2F complexes and only the nonspecific band (shown
with open arrowhead) similar to untreated control cell extracts was
seen. This might be explained by the fact that at this time,
extensive apoptosis in treated cells may have eliminated most of
the cells containing E2F with binding affinity for the cognate
promoter sequence.
Induction of apoptosis and E2F1-DNA
binding in pMTCyc A transfected Brl-3prl cells after PALA and
Zn2+ treatment
In order to confirm whether elevated expression of
cyclin A in DNA antimetabolite treated cells can mediate or
accelerate apoptotic response, stably transfected and conditionally
over-expressing cyclin A cell lines were generated by tranfecting
Brl-3prl cells with pMTCyc A (Br-lpMTCyc A). Cyclin A expression
was induced in Br-lpMTCyc A cells by 100 μM Zn2+.
Highest level of cyclin A protein expression was consistently
achieved after 9 h induction with 100 μM Zn2+ in the
clones tested. Fig. 4A
demonstrates the inducibility of cyclin A protein in two different
clones of Br-lpMTCyc A cells by 100 μM Zn2+ for 9 h.
Cell viability was checked after treatment of cells with 300 μM
PALA for 48 h and simultaneous induction of cyclin A with 100 μM
Zn2+ for 9 h in the two clones (Br-lpMTCyc A1,
Br-lpMTCyc A-2). Cell death was seen at all the time points, with
the percentage of apoptotic cells being maximum at 24 h
post-treatment (Fig. 4B).
Apoptotic death was further confirmed by DNA fragmentation analysis
(Fig. 4C). A time-dependent
increase in the DNA fragmentation was noted in both clones from
0–24 h; with maximum fragmentation seen at 24 h. In contrast
neither controls nor Zn2+ treated cells displayed
presence of oligonucleosomal DNA fragments. Transfected cells,
after 48 h PALA treatment, undergoing apoptosis were analyzed for
E2F1-DNA binding activity also by gel retardation assay. As shown
in Fig. 4D, E2F-DNA complex was
clearly detected in cells induced to express cyclin A in parallel
with PALA treatment. Low level E2F binding was also seen in cells
treated with PALA in the absence of Zn2+. The data
suggested that, even in the absence of Zn2+ induction,
slightly elevated cyclin A expression due to leaky regulation of
the promoter in the transfected expression vector may have caused
low level E2F DNA binding in these cells.
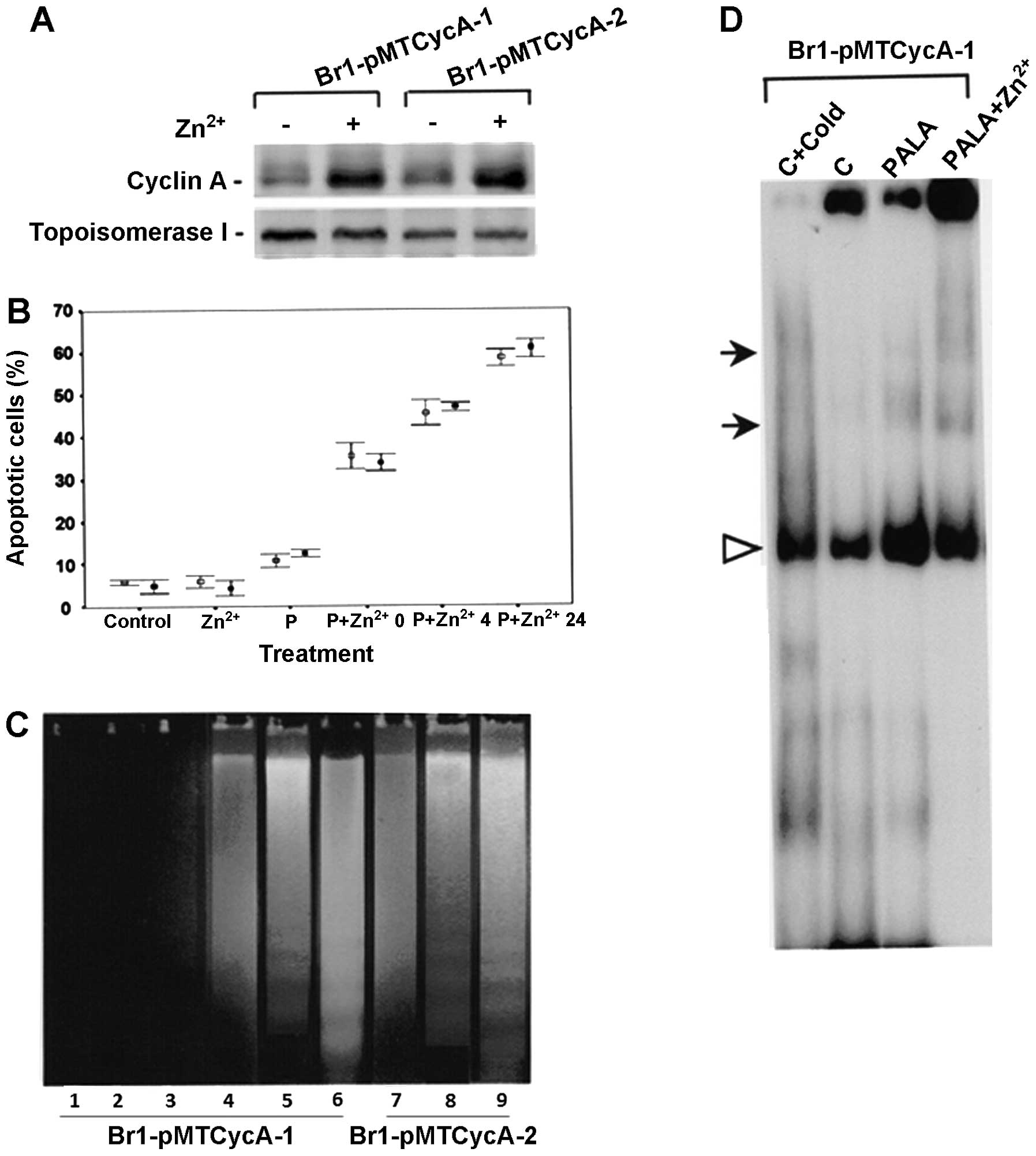 | Figure 4Characterization of inducible cyclin
A expressing cells. (A) Cyclin A expression analysis. Brl-pMT Cyc
A-1 and Br1-pMT Cyc A-2 cells were treated with 300 μM PALA for 48
h with simultaneous induction of cyclin A with 100 μM
Zn2+ for 9 h. (B) Percentage of Br1-pMT CycA-l (open
squares) and Br1-pMT CycA-2 (solid squares) cells undergoing
apoptosis in controls, Zn2+ alone, PALA alone and PALA +
Zn2+ at different time intervals. (C) DNA fragmentation
in Br-l pMTCyc A-I and Brl-pMTCyc A-2 cells: Lane 1, Control; 2,
Zn2+ treated; 3, PALA treated; 4–9, PALA +
Zn2+ treated and harvested at 0 h, lanes 4 and 7; 4 h,
lanes 5 and 8; and 24 h, lanes 6 and 9. (D) E2F1 DNA binding assay
in BR 1-pMTCyc A-1 cells: Nuclear extracts from control, 300 μM
PALA and 300 μM PALA + Zn2+ treated cells. |
Discussion
Resistance to chemotherapeutic drugs remains largely
unresolved and a germane issue of cancer biology. Molecular events
which influence cancer cell sensitivity to chemo therapeutic drugs
may include components involved in apoptosis and DNA repair
pathways. It has been hypothesized that loss of apoptotic response
often leads to chemotherapy resistance in cancer cells. Distinct
induction of apoptosis in the L-2 cells and the absence of similar
response in the Br1 cells demonstrated that the varying sensitivity
to antimetabolite PALA observed in these two cell types was due to
the difference in the drug-induced apoptotic response elicited.
It has been established that PALA cytotoxicity is
mediated through its inhibitory action on the pyrimidine
biosynthetic pathway and TAp73-dependent expression of Noxa and Bim
(22–24). Hence differential action of PALA on
the pyrimidine nucleotides in these cell lines could result in the
observed contrast in apoptotic response. To determine if the action
of PALA varied in these cell lines, intracellular ribonucleotides
were analyzed after incubating cells for 48 h with
(20xLD50) 300 μM concentration of the drug. This
treatment resulted in a >90% decrease in pyrimidine pools, yet
the extent of decline was similar in both cell lines. These data
and the fact that the isogenic cell lines (Brl cells and cyclin A
transfected Brl cells) also varied in their apoptotic response to
PALA suggest that this difference is not due to the PALA-mediated
alteration in the pyrimidine pools.
Expression of genes such as p53, bcl-2, c-myc and
cyclin A are considered critical determinants of apoptotic response
in cells (24–27). Cyclin A associated kinase activity
has also been reported to be essential for paclitaxel sensitivity
of human breast and ovarian cancer cells (28). It has been reported earlier that
loss of p53 function in cells leads to resistance to cytotoxic
treatments (29,30). Presence of an identical homozygous
mutation in p53 in the two cell types (unpublished data) and lack
of any perceptible alterations in the expression pattern of the
mutant protein following drug treatment suggested that the
apoptotic pathway, preferentially activated in the sensitive L-2
cells, was p53 independent.
A number of antitumor agents preferentially induce
apoptosis at specific phases of the cell cycle (31–33).
Cells undergoing apoptosis frequently do so in late G1 or early S
in response to a wide range of stimuli (34–36)
indicating that the gene products expressed at these stages may be
involved in the activation of apoptotic pathway. Many of the gene
products which appear to control apoptotic pathway are regulators
of cell cycle progression; thus, cell cycle control and cell death
appear to be tightly linked processes (37). Inappropriate activation of p34cdc2
kinase has been reported to lead to apoptosis (38). More recently dominant negative
mutants of cdc2, cdk2 and cdk3 were found to suppress apoptosis,
induced by diverse agents (39),
known to be mediated by the active cyclin A kinase complex
(40). Elevation in cdk2 and cdc2
kinase activities in L-2 cells undergoing apoptosis therefore
reflected their involvement in activation of the apoptotic pathway.
Activation of these kinases following induced cyclin A expression
in Br-1pMTcyc A cells also suggested that failure to activate
cyclin A kinase checkpoint in PALA treated cells allowed Br-l cells
to escape apoptosis and proliferate. Induction of apoptosis has
earlier been associated with activation of cyclin A dependent
kinases but not activity associated with cyclin E or B (40).
It has been reported that overexpression of cyclin A
alone in BHK cells cause ‘mitotic catastrophes’ with chromosomal
fragmentation that resembles apoptosis (41). Cyclin A protein has also been
implicated in myc-induced apoptosis (27). While our study provides strong
evidence in support of cyclin A being a critical determinant of
drug-induced apoptosis response, lack of correlation between cyclin
A and myc expression suggests that cyclin A mediated apoptotic
pathway may be myc independent in these tumor cells. It is however
noteworthy that cyclin A kinase activation correlated with higher
activity of cyclin E kinase in the L-2 cells. This result may be
explained in view of the earlier observation that cyclin E kinase
activity precedes and mediates upregulation of cyclin A kinase
(42).
Enhanced cyclin A kinase induced apoptotic response,
described in this paper, presents an apparent paradox to the
concept that cyclin A underlies an S phase checkpoint that acts
through phosphorylation of E2F1, to eliminate E2FI-DNA binding.
Phosphorylation of E2Fl regulated by cyclin A/cdk2 in S phase is
known to reduce the ability of the E2F1-DPl heterodimer to bind to
DNA and mediate transcriptional transactivation. Mutant E2FI
lacking cyclin A/cdk2 binding domain and not phosphorylated by
cyclin A/cdk2, prolongs S phase causing apoptosis and increased
sensitivity to the cytotoxic effects of S phase specific agents
(43). Inhibition of cyclin A/cdk2
activity causing reduced E2F1 phosphorylation was also shown to
increase sensitivity of human tumor cells lacking pRb to various
classes of anticancer drugs (18).
Induction of apoptotic response in the
antimetabolite treated cells displaying high cyclin A in parallel
with augmented E2F1-DNA binding detected in this study provide
contradiction to the above findings and suggests that cyclin A
kinase effect on E2F1-DNA binding involves a complex regulatory
process. Since cyclin A/cdk2 activation as well as E2Fl
transactivation are both required during Gl to S transition, it is
likely that additional factor(s) such as those required for
initiation and/or elongation of DNA-replication modulate the effect
of cyclin A kinase on E2Fl and vice versa. It is relevant here to
mention that in vitro studies have convincingly demonstrated
that addition of cyclin A alone reconstitutes both kinase activity
as well as DNA replication in mammalian S phase cell extracts and
does so by acting at a stage prior to elongation of nascent DNA
(44). Although targets of
cyclin-dependent kinases at the replication origin remain to be
established, the activation pathway may either involve
phosphorylation of replication proteins or phosphorylation of
downstream kinases. Enhanced expression of cyclin A in drug treated
L-2 and Brl-pMTcyc A cells may therefore have activated such
downstream effectors of DNA replication to stimulate the elongation
process leading to an abortive S phase.
Our results further suggest that such enhanced
cyclin A driven replication may also induce increased E2Fl-DNA
binding in the cells leading to apoptosis. Low cyclin A activity in
drug treated cells, on the contrary, may slow down the replication
machinery due to reduced activity of the downstream effectors
allowing the cells to complete DNA synthesis without activating an
apoptotic response. It is conceivable that inappropriate activation
of both E2F1 as well as cyclin A in drug treated cells may induce
apoptotic response. Cyclin A kinase, however, may enhance or reduce
E2F1-DNA binding based upon the status of effector proteins
involved in DNA synthesis. We do not know if loss of apoptotic
response seen in drug treated Br1 cells occurs due to
downregulation of cyclin A gene expression at the transcriptional
or translational level. Further elucidation of this mechanism for
cyclin A and other cell cycle regulatory proteins involved in
activation of drug-induced apoptotic pathways will help identify
useful therapeutic targets to overcome drug resistance in tumor
cells.
Acknowledgements
This study was supported by grants from the National
Institutes of Health (RO1CA089716 and NCI/EDRN UO1CA111302);
University Cancer Foundation, UTMDACC to S.S.
Abbreviations:
|
PALA
|
N(phosphonoacetyl)-L-aspartate
|
|
Mtx
|
methotrexate
|
|
BHK
|
baby hamster kidney
|
|
Rb
|
retinoblastoma protein
|
|
PAGE
|
polyacrylamide gel electrophoresis
|
References
|
1
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: The significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J, Chan JY, Fong CC, Tzang CH, Fung
KP and Yang M: Transcriptional analysis of doxorubicin-induced
cytotoxicity and resistance in human hepatocellular carcinoma cell
lines. Liver Int. 29:1338–1347. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sen S and D’Incalci M: Apoptosis.
Biochemical events and relevance to cancer chemotherapy. FEBS Lett.
307:122–127. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Colombel M, Olsson CA, Ng PY and Buttyan
R: Hormone-regulated apoptosis results from reentry of
differentiated prostate cells onto a defective cell cycle. Cancer
Res. 52:4313–4319. 1992.PubMed/NCBI
|
|
5
|
Ucker DS: Death by suicide: One way to go
in mammalian cellular development? New Biol. 3:103–109.
1991.PubMed/NCBI
|
|
6
|
Katayama H, Wang J, Treekitkarnmongkol W,
Kawai H, Sasai K, Zhang H, Wang H, Adams HP, Jiang S, Chakraborty
SN, et al: Aurora kinase-A inactivates DNA damage-induced apoptosis
and spindle assembly checkpoint response functions of p73. Cancer
Cell. 21:196–211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elledge SJ and Spottswood MR: A new human
p34 protein kinase, CDK2, identified by complementation of a cdc28
mutation in Saccharomyces cerevisiae, is a homolog of Xenopus Eg1.
EMBO J. 10:2653–2659. 1991.PubMed/NCBI
|
|
8
|
Zhu J, Sen S, Wei C and Frazier ML: Cyclin
D1b represses breast cancer cell growth by antagonizing the action
of cyclin D1a on estrogen receptor α-mediated transcription. Int J
Oncol. 36:39–48. 2010.
|
|
9
|
Nurse P: Ordering S phase and M phase in
the cell cycle. Cell. 79:547–550. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
King RW, Jackson PK and Kirschner MW:
Mitosis in transition. Cell. 79:563–571. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Forsburg SL and Nurse P: Cell cycle
regulation in the yeasts Saccharomyces cerevisiae and
Schizosaccharomyces pombe. Annu Rev Cell Biol. 7:227–256. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeGregori J, Leone G, Ohtani K, Miron A
and Nevins JR: E2F-1 accumulation bypasses a G1 arrest resulting
from the inhibition of G1 cyclin-dependent kinase activity. Genes
Dev. 9:2873–2887. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikeda MA, Jakoi L and Nevins JR: A unique
role for the Rb protein in controlling E2F accumulation during cell
growth and differentiation. Proc Natl Acad Sci USA. 93:3215–3220.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krek W, Xu G and Livingston DM: Cyclin
A-kinase regulation of E2F-1 DNA binding function underlies
suppression of an S phase checkpoint. Cell. 83:1149–1158. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang Y, Ishiko T, Nakada S, Utsugisawa T,
Kato T and Yuan ZM: Role for E2F in DNA damage-induced entry of
cells into S phase. Cancer Res. 57:3640–3643. 1997.PubMed/NCBI
|
|
18
|
Li WW, Fan J, Hochhauser D and Bertino JR:
Overexpression of p21waf1 leads to increased inhibition of E2F-1
phosphorylation and sensitivity to anticancer drugs in
retinoblastoma-negative human sarcoma cells. Cancer Res.
57:2193–2199. 1997.PubMed/NCBI
|
|
19
|
Price J, Fabra A, Zhang R, Radinsky R and
Pathak S: Characterization of variants of a human breast-cancer
cell-line isolated from metastases in different organs of
nude-mice. Int J Oncol. 5:459–467. 1994.PubMed/NCBI
|
|
20
|
Graña X and Reddy EP: Cell cycle control
in mammalian cells: Role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
21
|
Elledge SJ, Richman R, Hall FL, Williams
RT, Lodgson N and Harper JW: CDK2 encodes a 33-kDa cyclin
A-associated protein kinase and is expressed before CDC2 in the
cell cycle. Proc Natl Acad Sci USA. 89:2907–2911. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moyer JD, Smith PA, Levy EJ and
Handschumacher RE: Kinetics of N-(phosphonacetyl)-L-aspartate and
pyrazofurin depletion of pyrimidine ribonucleotide and
deoxyribonucleotide pools and their relationship to nucleic acid
synthesis in intact and permeabilized cells. Cancer Res.
42:4525–4531. 1982.PubMed/NCBI
|
|
23
|
Grem JL, King SA, O’Dwyer PJ and
Leyland-Jones B: Biochemistry and clinical activity of
N-(phosphonacetyl)-L-aspartate: A review. Cancer Res. 48:4441–4454.
1988.PubMed/NCBI
|
|
24
|
Ruhul Amin AR, Thakur VS, Gupta K, Agarwal
MK, Wald DN, Shin DM and Agarwal ML: N-(phosphonacetyl)-L-aspartate
induces TAp73-dependent apoptosis by modulating multiple Bcl-2
proteins: Potential for cancer therapy. Oncogene. 32:920–929. 2013.
View Article : Google Scholar
|
|
25
|
Katayama H, Sasai K, Kawai H, Yuan ZM,
Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA and Sen
S: Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lotem J and Sachs L: Regulation by bcl-2,
c-myc, and p53 of susceptibility to induction of apoptosis by heat
shock and cancer chemotherapy compounds in
differentiation-competent and -defective myeloid leukemic cells.
Cell Growth Differ. 4:41–47. 1993.PubMed/NCBI
|
|
27
|
Hoang AT, Cohen KJ, Barrett JF, Bergstrom
DA and Dang CV: Participation of cyclin A in Myc-induced apoptosis.
Proc Natl Acad Sci USA. 91:6875–6879. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takahashi T, Yamasaki F, Sudo T, Itamochi
H, Adachi S, Tamamori-Adachi M and Ueno NT: Cyclin A-associated
kinase activity is needed for paclitaxel sensitivity. Mol Cancer
Ther. 4:1039–1046. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Debbas M and White E: Wild-type p53
mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev.
7:546–554. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lowe SW, Ruley HE, Jacks T and Housman DE:
p53-dependent apoptosis modulates the cytotoxicity of anticancer
agents. Cell. 74:957–967. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Evans DL and Dive C: Effects of cisplatin
on the induction of apoptosis in proliferating hepatoma cells and
nonproliferating immature thymocytes. Cancer Res. 53:2133–2139.
1993.PubMed/NCBI
|
|
32
|
Gorczyca W, Gong J, Ardelt B, Traganos F
and Darzynkiewicz Z: The cell cycle related differences in
susceptibility of HL-60 cells to apoptosis induced by various
antitumor agents. Cancer Res. 53:3186–3192. 1993.PubMed/NCBI
|
|
33
|
Wang J, Fong CC, Tzang CH, Xiao P, Han R
and Yang M: Gene expression analysis of human promyelocytic
leukemia HL-60 cell differentiation and cytotoxicity induced by
natural and synthetic retinoids. Life Sci. 84:576–583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ashwell JD, Cunningham RE, Noguchi PD and
Hernandez D: Cell growth cycle block of T cell hybridomas upon
activation with antigen. J Exp Med. 165:173–194. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Howard MK, Burke LC, Mailhos C, Pizzey A,
Gilbert CS, Lawson WD, Collins MK, Thomas NS and Latchman DS: Cell
cycle arrest of proliferating neuronal cells by serum deprivation
can result in either apoptosis or differentiation. J Neurochem.
60:1783–1791. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ryan JJ, Danish R, Gottlieb CA and Clarke
MF: Cell cycle analysis of p53-induced cell death in murine
erythroleukemia cells. Mol Cell Biol. 13:711–719. 1993.PubMed/NCBI
|
|
37
|
Kastan MB, Canman CE and Leonard CJ: P53,
cell cycle control and apoptosis: Implications for cancer. Cancer
Metastasis Rev. 14:3–15. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi L, Nishioka WK, Th’ng J, Bradbury EM,
Litchfield DW and Greenberg AH: Premature p34cdc2 activation
required for apoptosis. Science. 263:1143–1145. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Meikrantz W and Schlegel R: Apoptosis and
the cell cycle. J Cell Biochem. 58:160–174. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Meikrantz W, Gisselbrecht S, Tam SW and
Schlegel R: Activation of cyclin A-dependent protein kinases during
apoptosis. Proc Natl Acad Sci USA. 91:3754–3758. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Heald R, McLoughlin M and McKeon F: Human
wee1 maintains mitotic timing by protecting the nucleus from
cytoplasmically activated Cdc2 kinase. Cell. 74:463–474. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rudolph B, Saffrich R, Zwicker J, Henglein
B, Müller R, Ansorge W and Eilers M: Activation of cyclin-dependent
kinases by Myc mediates induction of cyclin A, but not apoptosis.
EMBO J. 15:3065–3076. 1996.PubMed/NCBI
|
|
43
|
Logan TJ, Evans DL, Mercer WE, Bjornsti MA
and Hall DJ: Expression of a deletion mutant of the E2F1
transcription factor in fibroblasts lengthens S phase and increases
sensitivity to S phase-specific toxins. Cancer Res. 55:2883–2891.
1995.PubMed/NCBI
|
|
44
|
Fotedar A, Cannella D, Fitzgerald P,
Rousselle T, Gupta S, Dorée M and Fotedar R: Role for cyclin
A-dependent kinase in DNA replication in human S phase cell
extracts. J Biol Chem. 271:31627–31637. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gandhi V, Danhauser L and Plunkett W:
Separation of 1-beta-D-arabinofuranosylcytosine 5′-triphosphate and
9-beta-D-arabinofuranosyl-2-fluoroadenine 5′-triphosphate in human
leukemia cells by high-performance liquid chromatography. J
Chromatogr A. 413:293–299. 1987. View Article : Google Scholar
|
|
46
|
Shirodkar S, Ewen M, DeCaprio JA, Morgan
J, Livingston DM and Chittenden T: The transcription factor E2F
interacts with the retinoblastoma product and a p107-cyclin A
complex in a cell cycle-regulated manner. Cell. 68:157–166. 1992.
View Article : Google Scholar : PubMed/NCBI
|