Introduction
The phenomenon of multidrug resistance (MDR) still
represents a major obstacle for successful chemotherapy of cancer
(1–4). The emergence of the MDR phenotype is
mainly caused by the increase in expression of MDR-associated
genes, such as ATP-binding cassette, subfamily B 1 gene (ABCB1)
(MDR1), ABCC1 (MRP1) and ABCG2 (BCRP1). Numerous efforts have been
made to develop strategies for modulation of MDR-associated genes.
The classical type of MDR is mediated by overexpression of the
P-glycoprotein (PgP), the gene product of the ATP-binding cassette
(ABC) transporter ABCB1. Therefore reversal approaches target the
expression and function of ABCB1/PgP (4). Among these approaches the use of
cytokines has shown promise for downregulation of ABCB1 in
association with increased drug uptake and chemosensitization of
tumor cells (5). Several studies
have shown, that cytokines, such as tumor necrosis factor α
(TNF-α), interferon γ (IFN-γ), leukoregulin and interleukin-2
(IL-2) are capable of reducing ABCB1 gene expression and increasing
chemosensitivity of different cancer cell lines in vitro,
and more recently also for endothelial cells in the blood-brain
barrier (6–12). It has been shown, that particularly
persistent treatment with these cytokines is of importance to
achieve significant reduction in ABCB1 gene expression (6,7,9,13).
Consistent with the pre-clinical findings of cytokine-mediated
chemosensitization, clinical trials have demonstrated synergism of
combinations of cytostatic drugs with TNF-α or interferones in
cancer patients (14,15). Among all cytokines used for ABCB1
expression modulation, TNF-α has shown the highest efficiency in
vitro, in vivo and in clinical approaches (9,11,13,16).
Although the TNF-α mediated effects on ABCB1 expression were
repeatedly shown at functional level, it is still not fully
understood, what mechanism in cell signalling is reponsible for
these effects.
TNF-α exerts its intracellular activities via
TNF-receptor 1 (TNFR1, p55) and TNF-receptor 2 (TNFR2, p75). Among
other factors, the receptor-mediated intracellular signalling
cascade requires nuclear factor κ light chain enhancer (NF-κB) as
one key mediator and TNF-α is one of the most potent activators of
NF-κB signaling (17).
Activation of NF-κB, which is sequestered in the
cytoplasm as an inactive factor, is mediated via the nuclear factor
of κ light polypeptide gene enhancer in B-cells inhibitor (IκB)
kinase (IKK) complex, which is triggered by proinflammatory
cytokines including TNF-α or cytostatic drugs (18). IKK phosphorylates the NF-κB
inhibitor IκBα, which is then degraded by the 26S proteasome
complex. NF-κB is released, unmasking its nuclear localization
signal and enters the nucleus to activate transcription of specific
target genes, including of its own inhibitor IκBα. Within this
signaling cascade IκBα mediates both, rapid activation of NF-κB and
strong negative feedback (19).
The link between NF-κB and ABCB1 gene expression
regulation has been analyzed in several studies (20). These studies demonstrate that
transient induction of NF-κB is associated with increase in ABCB1
gene expression and inversely, inhibition of NF-κB leads to
downregulation of ABCB1 expression (20–23).
Promoter analyses revealed that the human ABCB1 promoter harbors
NF-κB responsive elements, which bind NF-κB to mediate regulation
of ABCB1 gene expression (24).
However, other studies revealed a more complex picture of TNF-α
triggered NF-κB activation and its target gene regulation. They
provide detailed insights into the diverging nature of TNF-α/NF-κB
signaling. This is strongly dependent on duration of pathway
activation, showing rather contrary effects of early transient and
of late persistent phase effects, mediated either by IκBα or IκBβ
(25). Interestingly, such
differential action can lead to negative feedback mechanisms of
TNF-α/NF-κB signaling which dampen the initial early transient
phase effects, in which IκBα and IκBβ are important regulators
(26–28).
In this context our study analyzed both, the
transient and more importantly persistent TNF-α mediated effects on
ABCB1 expression in human colorectal cancer cells as diverging
action of this cytokine. We show that NF-κB/p65 exerts signal
transduction via TNFR1, which is strictly time-dependent and
tightly associated with the modulation of ABCB1 expression. These
findings extend the picture of previously described diverging
nature of TNF-α action. They provide a potential link to chronic
inflammation, persistent TNF-α release and drug sensitivity in
TNF-α exposed cells or tissues, as is clinically observed in e.g.
inflammatory intestine diseases (29–32).
More importantly, this study might open new insights for targeted
interventions of MDR reversal in the treatment of colon cancer.
Materials and methods
Cell culture
The human colorectal carcinoma cell line HCT15 was
cultured at 37ºC, 5% CO2 in RPMI-1640 medium (Gibco BRL,
Gaithersburg, MD, USA), containing 10% FCS. This cell line
endogenously expresses high-level ABCB1 and possesses a strong MDR
phenotype (33). Authentification
of the cell line was performed by STR DNA typing (DSMZ,
Braunschweig, Germany).
Treatment with TNF-α
Briefly, 5×104 cells were seeded into
24-well plates and grown for 24 h. Then, cells were treated with 30
ng TNF-α/ml (Invitrogen, Carlsbad, CA, USA) for 5–120 min in
short-term incubations and 24–72 h in long-term incubations. The
cells were harvested for further analyses at indicated time points
(5, 10, 15, 30, 60 and 120 min; 24, 48 and 72 h). In the long-term
incubations in addition cells were harvested shortly after TNF-α
application at the respective days, indicated as 24, 48 and 72
h+.
Blocking of TNF-receptors
The blocking of TNF-receptors was performed in cells
treated for 72 h with TNF-α. For this, 5×104 cells were
seeded into 24-well plates and grown for 24 h. One hour before each
treatment with 30 ng TNF-α/ml (Invitrogen) cells were incubated
with 8 or 20 μg mouse anti-TNFR1 monoclonal antibody
(R&D Systems, Inc., Minneapolis, MN, USA) and 4 or 12 μg
mouse anti-TNFR2 monoclonal antibody (R&D Systems, Inc.)
respectively, to specifically block the receptor (as recommended by
manufacturer). The control cells were treated with 30 ng TNF-α/ml
only, or remained untreated.
Real-time quantitative RT-PCR
(qRT-PCR)
Total RNA from cells was isolated using the TRIzol™
method (Invitrogen). Reverse transcriptase (RT) reaction was
performed with 50 ng of total RNA (MuLV reverse transcriptase;
Perkin-Elmer, Weiterstadt, Germany). Each quantitative real-time
PCR (95ºC for 10 sec, 45 cycles of 95ºC 10 sec, 60ºC 30 sec and
72ºC 1 sec) was done using the LightCycler (LightCycler Fast Start
DNA master hybridization probes kit; Roche Diagnostics GmbH,
Mannheim, Germany). Expression of human ABCB1, NF-κB/p65, IκBα,
IκBβ and of the housekeeping gene glucose-6-phosphate dehydrogenase
(G6PDH) was determined in parallel from the same RT-reaction, each
done in duplicate per sample. For ABCB1 a 167 bp amplicon (forward,
5′-CCCATCATTGCAATAGCAGG-3′; FITC-labeled probe forward,
5′-CACTGAAAGATAAGAAAGAACTAGAAGGTGCT-3′; LCRed640-labeled probe
forward, 5′-GGAAGATCGCTACTGAAGCAATAGAAAACT-3′ and reverse,
5′-GTTCAAACTTCTGCTCCTGA-3′); for NF-κB/p65 a 365 bp amplicon
(forward, 5′-AGATCAATGGCTACACAGGA-3′ and reverse,
5′-GATGGGATGAGAAAGGACA-3′); for IκBα a 354 bp amlicon (forward,
5′-CCGAGACTTTCGAGGAAAT-3′ and reverse, 5′-GTGAGCTGGTAGGGAGAATA-3′);
for IκBβ a 417 bp amplicon (forward, 5′-AGTACATGGACCTGCAGAAT-3′ and
reverse, 5′-GGACCATCTCCACATCTTTg-3′), and for G6PDH a 123 bp
amplicon was produced, which were detected by gene-specific
fluorescein- and LCRed640-labeled hybridization probes [(primers
for ABCB1, NF-κB, IκBα, IκBβ; BioTeZ, Berlin, Germany); (probes for
ABCB1; TIB Molbiol, Berlin, Germany); (primers and probes for
G6PDH; Roche Diagnostics GmbH)]. The calibrator cDNA, derived from
the human ABCB1 expressing cell line HCT15, was employed in serial
dilutions (in duplicate) simultaneously in each run.
Western blotting
Cells were lysed in lysis buffer (50 mM Tris-HCl, pH
7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1X complete
protease inhibitor cocktail (Roche Diagnostics GmbH). Cytoplasmic
and nuclear extracts were prepared from cells by using the NE-PER
extraction kit according to manufacturer's instructions (Pierce
Biotechnology, Inc., Rockford, IL, USA). The protein content was
quantified by using the Coomassie Plus protein assay according to
manufacturer's instructions (Pierce Biotechnology, Inc.). Precast
NuPAGE 4–12% gradient polyacrylamide gels (Invitrogen) were loaded
with 50 μg protein of either total cell lysates, cytoplasmic
extracts, or 10 μg of nuclear extracts and electrophorezed
at 200 V, 60 min. The gels were blotted onto nitrocellulose filter
(Hybond-C Extra; Amersham, Freiburg, Germany). The filters were
blocked 1 h at room temperature in TBS blocking buffer (50 mM Tris,
150 mM NaCl, pH 7.5, 5% fat-free dry milk) and washed in TBST
(0.05% Tween-20 in TBS buffer) at RT.
For detection anti-human-TNFR1 mouse IgG monoclonal
antibody (1:100), anti-human-TNFR2 mouse IgG monoclonal antibody
(1:250) (both from Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA), anti-human PgP monoclonal mouse IgG-antibody C219 (1:100;
Calbiochem, San Diego, CA, USA), and anti-human NF-κB/p65 mouse
monoclonal IgG-antibody (1:500), anti-human IκBα mouse monoclonal
IgG-antibody (1:500), anti-human IκBβ mouse monoclonal IgG-antibody
(1:100) (all from Santa Cruz Biotechnology, Inc.), anti-human
β-tubulin mouse monoclonal IgM antibody (1:500; BD Pharmingen,
Heidelberg, Germany), anti-human nuclear matrix protein (NMP) p84
mouse monoclonal IgG antibody (1:2,000; Abcam, Cambridge, UK) and
anti-human GAPDH goat polyclonal antibody (1:1,000; Santa Cruz
Biotechnology, Inc.) were used. As secondary antibodies HRP-labeled
goat anti-mouse IgG-antibody (1:6,000; Pierce Biotechnology, Inc.),
goat anti-mouse IgM-antibody (1:5,000; Sigma-Aldrich, St. Louis,
MO, USA) or mouse anti-goat antibody (1:5,000; Santa Cruz
Biotechnology, Inc.) were used. All antibodies were diluted in TBST
containing 5% BSA. After incubation with the respective primary and
secondary antibodies, the filters were washed in TBST. After
washing, the respective protein was detected using ECL-solution
(Amersham) and exposed to Kodak X-Omat AR film.
Electrophoretic mobility shift assay
(EMSA)
The nuclear extracts were prepared from cells by
using the NE-PER extraction kit according to manufacturer's
instructions (Pierce Biotechnology, Inc.). The EMSA was performed
with 3 μg of nuclear extract using biotin end-labeled
double-stranded oligonucleotides harboring the NF-κB/p65-binding
site of the wild-type human ABCB1 promoter (sense,
5′-GCTGCTCTGGCCGCGATGGGCACTGCAGGGGCTTTCCTGTGCGCGGGGTCTCCAGCATCT-3′
and antisense,
5′-AGATGCTGGAGACCCCGCGCACAGGAAAGCCCCTGCAGTGCCCATCGCGGCCAGAGCAGC-3′);
or mutant (sense,
5′-GCTGCTCTGGCCGCGATGGGCACTGCACTCGCTTTCCTGTGCGCGGGGTCTCCAGCATCT-3′
and antisense,
5′-AGATGCTGGAGACCCCGCGCACAGGAAAGCGAGTGCAGTGCCCATCGCGGCCAGAGCAGC-3′).
In competition experiments unlabeled double-stranded
oligonucleotides were used to control binding specificity of
NF-κB/p65. For the supershift experiments, the NF-κB/p65 mouse
monoclonal IgG-antibody (0.5 or 1.0 μg; Santa Cruz
Biotechnology, Inc.) was used. For the EMSA 6% TBS pre-cast gels
(Invitrogen) were used. Gels were blotted onto nitrocellulose
filters (Amersham). After UV cross-linking shifted and supershifted
bands were detected by the LightShift kit (Pierce Biotechnology,
Inc.) according to manufacturer's instructions and exposed to Kodak
X-Omat AR film.
Drug uptake assay
After the pretreatment with 30 ng TNF-α/ml
(Invitrogen) for 2 or 72 h, respectively, cells were incubated with
doxorubicin (50 μM; Sigma, Taufkirchen, Germany) in phenol
red-free RPMI-1640 medium for 3 h at 37ºC, and were washed with
phenol red-free medium and kept on ice. Fluorescence intensity of
1×104 cells was measured in duplicate per sample by
using the FACSCalibur (Cell Quest program; Becton-Dickinson, San
Diego, CA, USA). The drug uptake is expressed as fold-increase
compared to untreated cells, which did not receive TNF-α.
Cytotoxicity assay
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT)
For the cytotoxicity assay, 5×103 cells
were seeded into 96-well plates. The short- or long-term TNF-α
effects on drug sensitivity of cells was determined by 2 or 72 h
pretreatment with 30 ng TNF-α/ml. Thereafter cells were washed and
treated with 50, 100, 200, 400, 1,000, 1,250 and 1,500 ng
doxorubicin/ml (Sigma) for 72 h at 37ºC. Then, MTT (5 mg/ml; Sigma)
was added and absorbance was measured in triplicates at 560 nm in a
microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
Values are expressed as percent of untreated controls. Cells
treated 2 or 72 h with TNF-α only served as additional control.
Cell signalling reporter assay
To determine TNF-α mediated short- and long-term
induction via NF-κB/p65, we used the Cignal NF-κB/p65
Renilla/firefly dual luciferase reporter assay kit
(SABiosciences, Frederick, MD, USA). For each well of a 96-well
plate 100 ng Cignal NF-κB reporter or Cignal negative control
plasmid-DNA, respectively, were reverse transfected with FuGENE HD
(Roche Diagnostics GmbH) into 1×104 cells and seeded in
serum-free conditions, as recommended by the manufacturer. Then, 24
h after transfection medium was replaced with RPMI-1640 medium +10%
FBS and treatment with 30 ng TNF-α/ml (Invitrogen) was started. For
short-term TNF-α affected reporter expression cells were incubated
2, 4, 6, 8, 10 and 12 h, washed with PBS and lysed in passive lysis
buffer (Promega). For evaluation of TNF-α affected reporter
expression in long-term treatment, transfected cells were incubated
for 8, 24, 48 and 72 h with 30 ng TNF-α/ml. In addition, time
points of 8 h after the respective TNF-α treatment were included,
indicated as 24, 48 and 72 h+. Cells were washed with PBS and lysed
in passive lysis buffer. The luciferase assay was performed in
triplicates with Dual-Luciferase Reporter Assay system (Promega)
using 3 μl cell lysate in triplicates and using the Centro
LB 960 luminometer (Berthold Technologies GmbH & Co., Bad
Wildbad, Germany).
ABCB1 promoter driven luciferase reporter
assay
To analyze the short- and long-term effects of TNF-α
on the human ABCB1 promoter activity, the NF-κB/p65 consensus
harboring 990 bp promoter sequence was PCR-cloned (24). This sequence spans the first exon
and intron, and was used to drive the pGL3 plasmid luciferase
reporter expression (Promega).
For the assay 150 ng ABCB1 promoter luciferase or
promotorless pGL3 plasmid-DNA as negative control respectively were
reverse transfected in 1×104 cells in each well of a
96-well plate with LTX-RG (Invitrogen) in serum-free conditions.
Medium was replaced with RPMI-1640 medium and 10% FBS 24 h after
transfection and TNF-α treatment was started. For determination of
short-term TNF-α affected reporter expression cells were incubated
with 30 ng TNF-α/ml (Biosource) for 2, 4, 6, 8, 10 and 12 h, washed
with PBS and lysed passive lysis buffer (Promega). For evaluation
of TNF-α-affected reporter expression in long-term treatment,
transfected cells were incubated for 8, 24, 48 and 72 h with TNF-α,
washed with PBS and lysed in passive lysis buffer. In addition,
cells were harvested 8 h after the respective TNF-α application in
the long-term treatment, indicated as 24, 48 and 72 h+. The
luciferase activity was quantified by luciferase reporter assay in
triplicates by the Centro LB 960 luminometer.
Statistical analysis
Analyses for statistical significance were performed
with GraphPad Prism version 5 (GraphPad Software Inc., La Jolla,
CA, USA). Comparison of several groups was done by one-way analysis
of variance (ANOVA) and Bonferroni post hoc multiple comparison.
Statistical significance was set at P-values <0.05.
Results
TNF-α mediated regulation of ABCB1
expression, drug uptake and cytotoxicity
The TNF-α mediated effects on ABCB1 expression were
analyzed in HCT15 human colon carcinoma cells, which is an
intrinsically high expressor of ABCB1. First, we evaluated the
effects of human TNF-α on ABCB1 expression and ABCB1-mediated
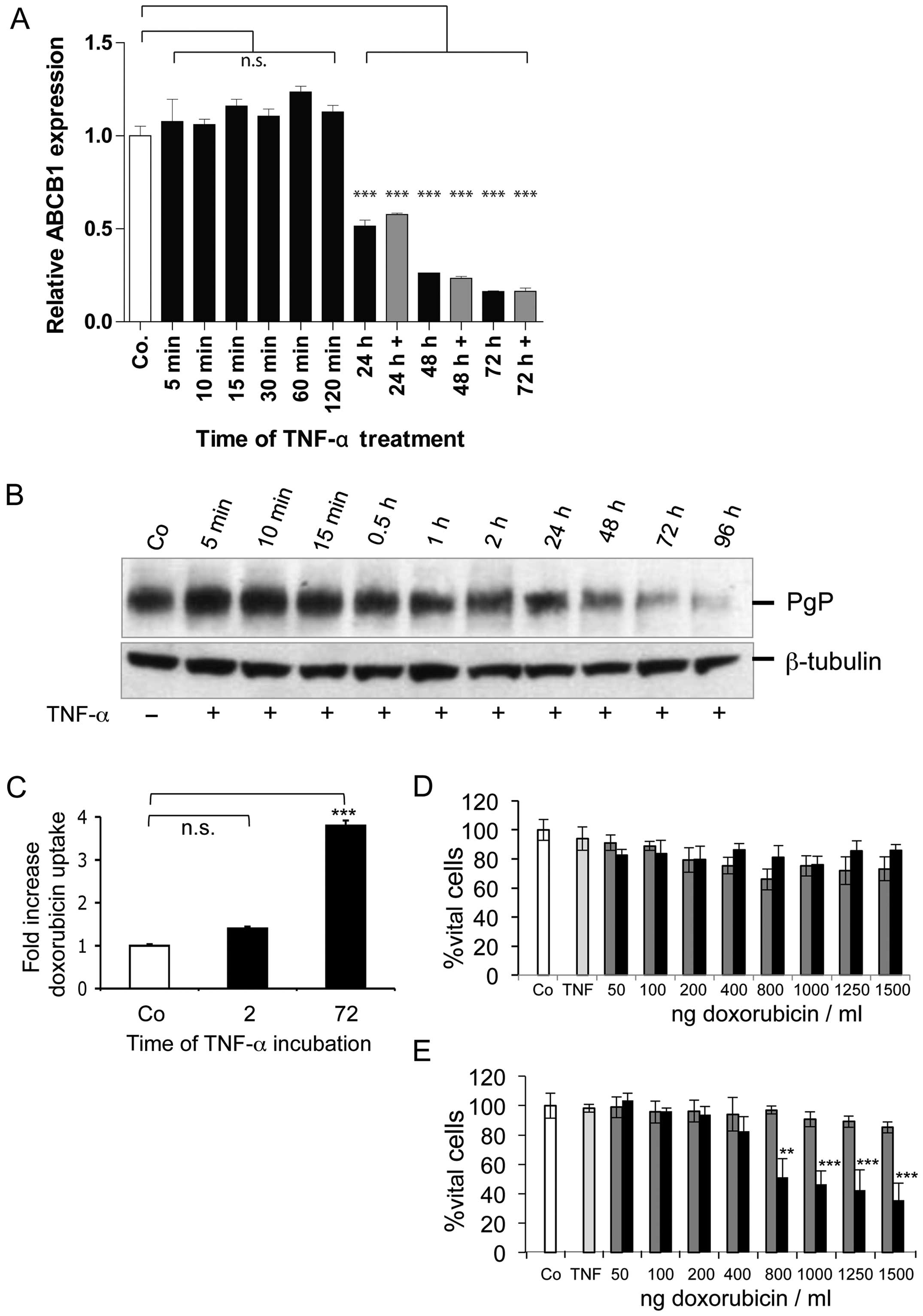
resistance. The treatment with 30 ng TNF-α/ml significantly reduced
ABCB1 expression at mRNA level starting from 24–72 h (Fig. 1A). This long-term treatment caused
1.9-fold reduction in mRNA expression after 24 h, 3.8-fold
reduction after 48 h and 6-fold reduction after 72 h, whereas
short-term treatment (0.5–2 h) did not reduce ABCB1 expression.
Similarly, western blot analysis revealed reduced PgP-expression at
24 h up to 96 h of TNF-α treatment (Fig. 1B). The short-term treatment (5 min
to 2 h) however, did not alter PgP-expression. In a next step we
determined possible functional effects of TNF-α mediated ABCB1
downregulation. For this, the uptake of the fluorescent drug
doxorubicin as PgP substrate was determined (Fig. 1C). TNF-α treatment for 72 h leads
to significant up to 3.8-fold increase in doxorubicin uptake,
whereas 2 h TNF-α treatment caused only 1.4-fold increase in drug
uptake. We next asked if the increased drug uptake after long-term
TNF-α treatment has impact on doxorubicin cytotoxicity. Long-term
treatment did increase cytotoxicity leading to 64% growth
inhibition at highest dose of 1,500 ng/ml (Fig. 1E). By comparison, 2 h TNF-α
treatment did not exert any effect on doxorubicin cytotoxicity even
at highest drug doses (Fig. 1D).
Taken together, we demonstrated in our model that long-term TNF-α
treatment downregulated ABCB1 expression at mRNA and protein levels
in colorectal cancer cells. This leads to high drug accumulation
and sensitization towards doxorubicin, reflected by increased
cytotoxicity.
TNFR signaling and PgP expression
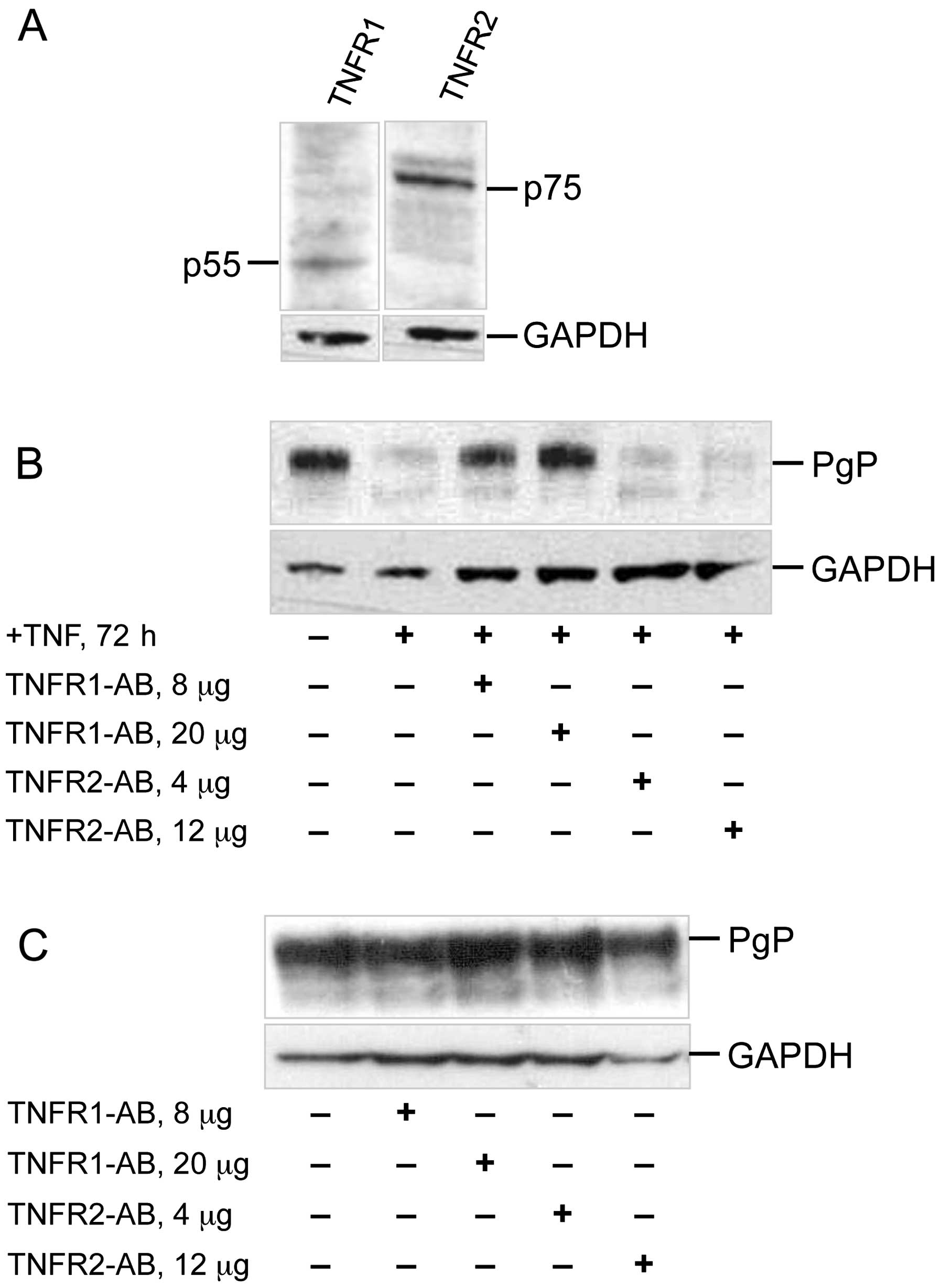
Next we determined, whether TNFR1/p55 or TNFR2/p75
is essential in mediating the observed TNF-α effects on ABCB1
expression. Western blot analysis revealed, that both receptors are
present in the colorectal cancer cells (Fig. 2A). For blocking the TNF-α binding
to either TNFR1 or TNFR2 we pre-treated the cells with anti-TNFR1
or anti-TNFR2 antibody (Fig. 2B).
As shown, addition of anti-TNFR1 antibody prevented PgP
downregulation of the long-term 72 h TNF-α treatment, whereas
addition of the anti-TNFR2 antibody could not prevent
downregulation of PgP. In control experiments sole addition of the
antibodies, however, did not alter PgP expression. This excluded
any antibody mediated unspecific effect on PgP expression (Fig. 2C). Thus, binding of TNF-α to TNFR1
is essential for TNF-α mediated downregulation of ABCB1 expression
after long-term TNF-α treatment.
Effects of short- and long-term TNF-α
mediated NF-κB/p65 signaling on ABCB1
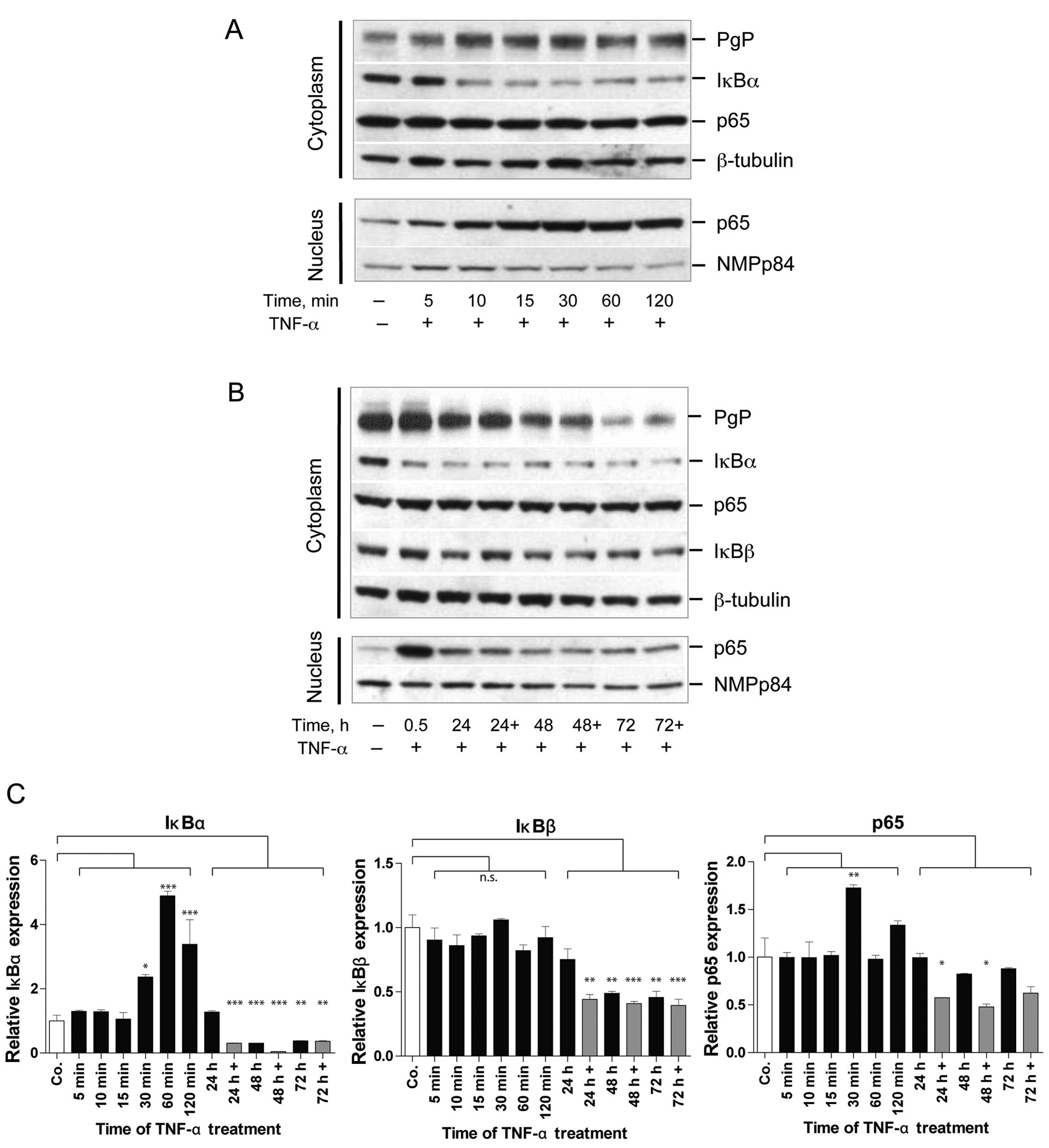
TNFR1 mediates signaling via the NF-κB pathway.
Therefore, we determined the effect of short- and long-term TNF-α
on NF-κB/p65 and on IκBα, IκBβ as regulators of NF-κB/p65 (Fig. 3A and B). The short-term (5–120 min)
TNF-α mediated effects show strong cytoplasmatic reduction in IκBα
levels after 10 min of TNF-α treatment, whereas, no detectable
alteration for cytoplasmic NF-κB level was observed (Fig. 3A). As early as 10 min of TNF-α
treatment the NF-κB/p65 protein starts to accumulate in the cell
nuclei. This differs from what is observed for long-term (24–72 h)
TNF-α mediated effects. Here IκBα levels remain at very low level
and NF-κB shuttling to the nucleus is reduced. This is paralleled
by reduction in PgP levels in the same experiment (Fig. 3B). Therefore, long-term TNF-α
treatment reduces shuttling of NF-κB/p65 to the nucleus and
prevents re-appearance of IκBα to levels of control cells in the
cytoplasm. The analysis of IκBα mRNA expression revealed that this
reduction in IκBα for long-term TNF-α treatment is the result of
significant up to 3-fold reduced transcription (Fig. 3C). The cytoplasmic NF-κB level,
however, seems to be unaffected by long-term TNF-α action. Even if
analyzing the time point of 30 min after re-application of TNF-α
(indicated as 24, 48 and 72 h+), no increase in nuclear
accumulation of NF-κB/p65 is seen. For NF-κB/p65 shows only slight
and insignificant reduction in mRNA expression by the long-term
TNF-α treatment as seen in Fig.
3C.
Furthermore, we analyzed the fate of IκBβ in
long-term TNF-α treatment where IκBβ protein persists in the
cytoplasm, however, for 24–72 h TNF-α treatment at slightly lower
level (Fig. 3C). The expression
analysis also revealed an up to 2.5-fold reduction of IκBβ mRNA for
the long-term TNF-α treatment, which correlates with the reduction
at protein level (Fig. 3B and
C).
NF-κB/p65 binding to its consensus
sequence in the ABCB1 promoter after long-term TNF-α treatment
Since long-term TNF-α reduced nuclear NF-κB/p65 we
determined if this is associated with reduced binding of NF-κB/p65
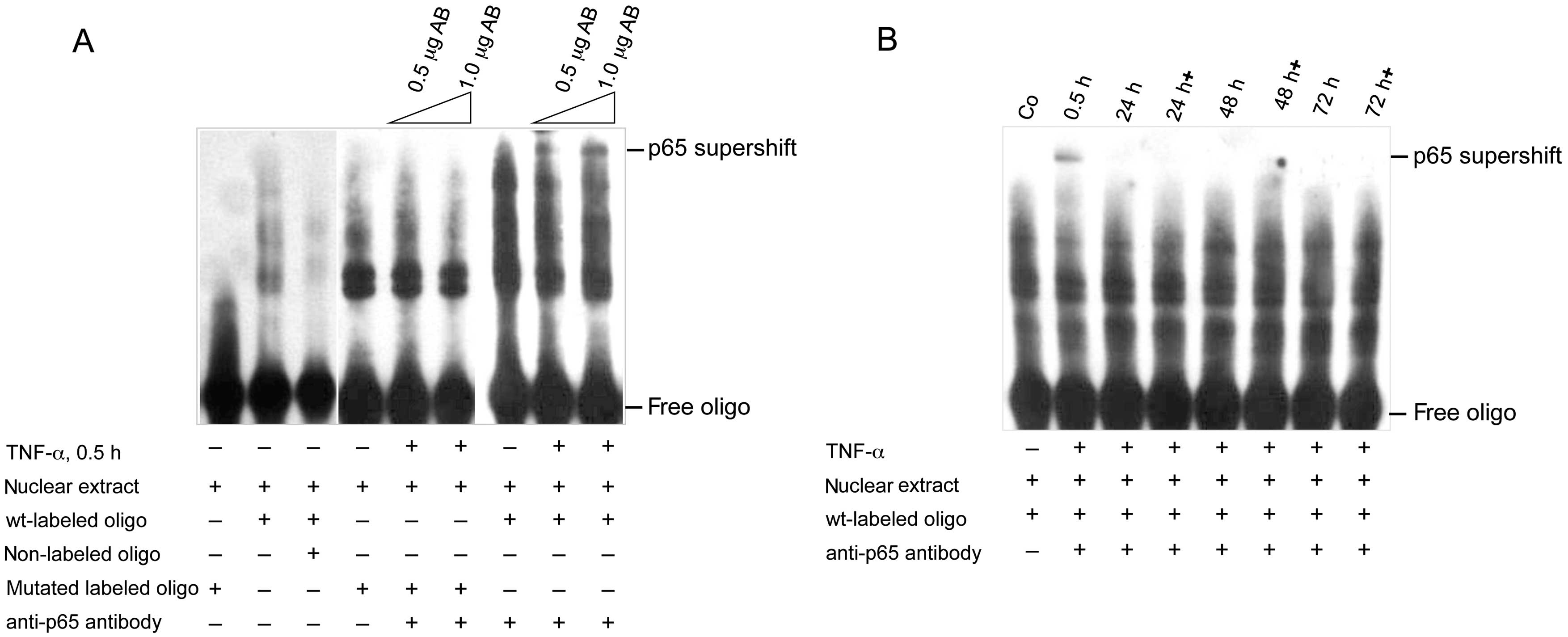
to its consensus sequence in the human ABCB1 promoter. In the EMSA
we first analyzed, if binding to the NF-κB consensus sequence of
the ABCB1 promoter is specific (Fig.
4A). Under non-stimulated conditions there is binding of the
nuclear extract, which by addition of excess nonlabeled oligo
disappers due to competition. The mutated oligo however shows no
binding. After treatment with TNF-α for 30 min the mutated oligo
shows binding of nuclear extract. However, addition of
anti-NF-κB/p65 antibody does not lead to supershift. By contrast,
use of the wild-type oligo shows binding of the nuclear extract,
and supershift bands after addition of the anti-p65 antibody. This
indicates, that the short-term TNF-α treatment induces NF-κB
binding to the consensus sequence. Next we analyzed the impact of
long-term TNF-α action on NF-κB binding to its consensus sequence
within the human ABCB1 promoter by supershift experiment using the
anti-p65 antibody. As positive control, the 30 min TNF-α
stimulation again induced NF-κB binding, verified by appearace of a
supershift band (Fig. 4B). This
supershift disappeared when TNF-α was added for 24, 48 and 72 h,
respectively. Interestingly, this is also seen for the samples,
collected 30 min after each re-application of TNF-α, indicated as
24, 48 and 72 h+. This suggests that long-term TNF-α treatment
desensitizes the colorectal cancer cells to TNF-α mediated NF-κB
signaling and prevents NF-κB from binding to its consensus sequence
within the ABCB1 promoter. This in turn leads to downregulation of
ABCB1 expression.
Diverging effect of TNF-α mediated
regulation on ABCB1 promoter activity
Since long-term TNF-α action prevents nuclear
translocation and binding to consensus sequence of NF-κB,
determination of functional consequences of this is important.
First, we analyzed the effects of short- and long-term TNF-α
treatment using the Cignal NF-κB/p65 Renilla/firefly dual
luciferase reporter assay, which harbors tandem repeats of NF-κB
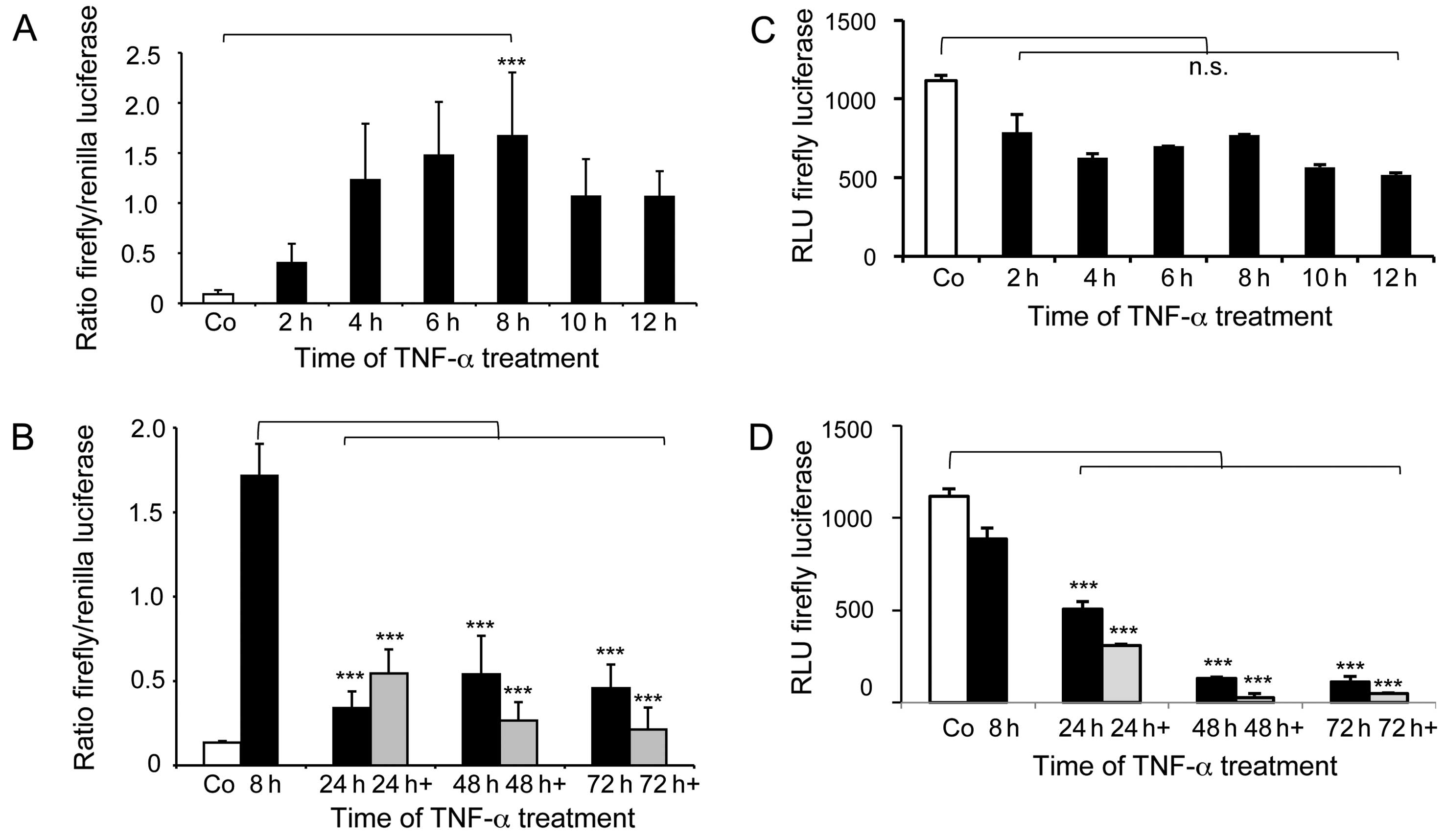
transcriptional response element (Fig.
5). In this assay short-term application of TNF-α induces
reporter expression peaking at 8 h with a 1.7-fold increase
compared to the control (Fig. 5A).
The long-term effects of TNF-α dramatically differ from this. Here,
persistent TNF-α application leads to significant downregulation of
reporter expression compared to the short-term TNF-α effects
(Fig. 5B). This is reflected by
the 3- to 5-fold decreases in promoter activity at 24–72 h of TNF-α
application. Even at the time points of 8 h after each respective
re-application of TNF-α (indicated as 24, 48 and 72 h+) the
reporter system remains unresponsive towards TNF-α mediated
induction. To further validate this observation, we used the NF-κB
consensus sequence containing human ABCB1 promoter for luciferase
expression. These assays revealed that short-term TNF-α application
slightly reduced basal promoter activity in the colorectal cancer
cells (Fig. 5C). The picture again
drastically changes for long-term TNF-α treatment. The results show
significant 2- to 9-fold decreases in ABCB1 promoter activity
(Fig. 5D). At 8 h after respective
TNF-α re-application (indicated as 24, 48 and 72 h+) this reporter
system remains also unresponsive for TNF-α mediated induction and
the reporter expression is even further reduced. Taken together,
these analyses support that long-term TNF-α inhibits ABCB1 promoter
activity. This results in reduced ABCB1 transcription and PgP
expression (Fig. 1) leading to MDR
reversal in the resistant cells.
Discussion
MDR still represents a leading obstacle for
successful chemotherapy of cancer. Many different approaches are
used to overcome MDR including the use of MDR reversing drugs or
cytokines (1,5,34).
One long known molecule, which mediates MDR in cancer is the ATP
binding cassette ABCB1/PgP transporter. This membrane protein is
responsible for the drug extrusion from cancer cells causing drug
resistance. Therefore great efforts are aimed at downregulation of
ABCB1 expression to sensitize tumor cells towards chemotherapy. One
such approach is the use of pro-inflammatory cytokines, such as
TNF-α or INF-γ for chemosensitization of resistant tumor cells
(7,35). From numerous studies it is well
accepted that particularly TNF-α is able to modulate the ABCB1
expression. In this regard several studies report, that treatment
with TNF-α mediates ABCB1 downregulation, which in turn leads to
improved drug accumulation in association with increased
cytotoxicity. This has been found in different tumor models,
including glioblastoma, colon, breast and hepatocellular cancer,
and more recently also for endothelial cells in the blood-brain
barrier (5,8–12).
By contrast there are reports with the opposite observations,
stating that treatment with TNF-α or other factors, which lead to
activation of the NF-κB pathway rather act as inducers of ABCB1
expression (20,36–38),
thus, interference in NF-κB signaling by inhibitors has been shown
to downregulate ABCB1 expression, which then sensitizes tumor cells
towards chemotherapy (20).
Due to the complex and partially contradictory
picture on TNF-α mediated effects we were interested to determine
the interplay between short- and long-term TNF-α treatment, NF-κB
signaling and the resulting modulation in this signaling, which
leads to ABCB1 repression in colon cancer cells.
In this study we determined in more detail the
impact of short-term and of long-term action of TNF-α on the
expression regulation of ABCB1 in intrinsically resistant human
HCT15 colon carcinoma cells. We have shown, that persistent
treatment with TNF-α for 24–72 h leads to significant
downregulation of ABCB1 in association with sensitization of these
cells towards drug treatment. This is in line with previous
publications, in which TNF-α leads to MDR reversal (5,9).
Apart from these studies, the question remained,
what mechanism is responsible for the TNF-α mediated downregulation
of ABCB1, which is mostly observed for persistent treatment. We
therefore evaluated by which TNFR-signaling such modulation of
ABCB1 expression is permitted. This analysis revealed, that TNFR1
mediated signaling is important for this process. Since TNF-α
triggers NF-κB activation and nuclear translocation via TNFR1, we
focused on this pathway under conditions of short- and of long-term
TNF-α activation. The human ABCB1 promoter harbors NF-κB binding
sites linking NF-κB signaling and ABCB1 expression regulation
(24). Our study shows short-term
TNF-α treatment triggers NF-κB activation. This is demonstrated by
nuclear accumulation of NF-κB, accompanied by disappearance of IκBα
from the cytoplasm, and binding of the transcription factor to its
consensus sequence in the EMSA. By contrast, long-term TNF-α
treatment does the opposite: NF-κB is not shuttled to the nucleus,
even at time points shortly after TNF-α re-stimulation, although
NF-κB is still present in the cytoplasm. This is associated with
lack of IκBα re-appearance in the cytoplasm. This re-appearance
would be expected after TNF-α treatment to rescue NF-κB from the
nucleus and to reduce its DNA-binding ability leading to the known
termination of NF-κB signaling (39). Interestingly, similar observation
regarding lack of IκBα re-appearance was made in FS-4 fibroblasts
treated for up to 15 h with TNF-α. In this study long-term TNF-α
prevents re-appearance of IκBα due to persistent
proteasome-mediated degradation of the protein (28,40).
It is known that IκBα is not only important for the termination of
NF-κB signaling but is also essential for re-initiation of the
signaling after its resynthesis (27). The absence of IκBα leads to delayed
and reduced cytokine-induced NF-κB activation (27). Here we show, that the persistent
TNF-α action prevents re-appearance of IκBα due to reduced IκBα
transcription, which is then unavailable for maintenance of ABCB1
expression. In addition, we did also observe significant reduction
in IκBβ transcription and reduced protein level in the long-term
TNF-α treated HCT15 cells. This reduction however, does not reach
the extent observed for IκBα. It is suggested that the remaining
IκBβ level might be sufficient for dumping the long-term
oscillations in NF-κB signaling, as described by others (26). In this context almost unaltered
presence and action of IκBβ might overrule the remaining small IκBα
activity and hinder NF-κB activation, which finally results in
ABCB1 downregulation.
As we observed, persistent TNF-α treatment not only
reduces nuclear translocation of NF-κB, but also DNA binding to its
consensus sequence in the ABCB1 promoter, explaining why ABCB1
transcription is reduced. This is further supported by the reporter
assays. They clearly show, that long-term TNF-α treatment has
inhibitory effects, whereas for short-term TNF-α treatment
induction in reporter gene expression was seen. Interestingly, even
re-application of TNF-α in the long-term treatment was unable to
restimulate reporter gene expression, from either the Cignal
NF-κB/p65 Renilla/firefly dual luciferase reporter system or
the authentic ABCB1 promoter construct.
In conclusion, our study provides new perspectives
on the mechanism how long-term treatment of TNF-α is reducing NF-κB
signaling, which results in the downregulation of ABCB1. This
strongly supports the MDR reversing potential of the
pro-inflammatory cytokine, which in turn mediates effective
chemosensitization of colon cancer cells. Furthermore, this
mechanism might also apply to the phenomena observed in situations
of chronic inflammation of the gut associated with persistent
presence of TNF-α, such as ulcerative colitis. These conditions are
reported to be associated with reduced expression of ABC
transporters, including ABCB1 (29,30).
Interestingly, apart from colon cancer such observation has
recently also been made in an inflammation model for microglia
parenchymal cells, where ABCB1 expression is reduced pointing to a
potential more general property of TNF-α on regulation of ABCB1
(41).
Acknowledgements
We thank Liselotte Malcherek for excellent technical
assistance.
Abbreviations:
|
ABCB1
|
ATP-binding cassette, subfamily B 1
gene
|
|
EMSA
|
electromobility shift assay
|
|
MDR
|
multidrug resistance
|
|
NF-κB
|
nuclear factor κ light chain
enhancer
|
|
IκBα/IκBβ
|
nuclear factor of κ light polypeptide
gene enhancer in B-cells inhibitor, α/β
|
|
PgP
|
P-glycoprotein
|
|
TNFR
|
TNF-receptor
|
|
TNF-α
|
tumor necrosis factor α
|
References
|
1
|
Binkhathlan Z and Lavasanifar A:
P-glycoprotein inhibition as a therapeutic approach for overcoming
multidrug resistance in cancer: Current status and future
perspectives. Curr Cancer Drug Targets. 13:326–346. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Breier A, Gibalova L, Seres M, Barancik M
and Sulova Z: New insight into P-glycoprotein as a drug target.
Anticancer Agents Med Chem. 13:159–170. 2013. View Article : Google Scholar
|
|
3
|
Nooter K and Stoter G: Molecular
mechanisms of multidrug resistance in cancer chemotherapy. Pathol
Res Pract. 192:768–780. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Teodori E, Dei S, Martelli C, Scapecchi S
and Gualtieri F: The functions and structure of ABC transporters:
Implications for the design of new inhibitors of Pgp and MRP1 to
control multidrug resistance (MDR). Curr Drug Targets. 7:893–909.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stein U and Walther W: Cytokine-mediated
reversal of multidrug resistance. Cytotechnology. 27:271–282. 1998.
View Article : Google Scholar
|
|
6
|
Walther W and Stein U: Influence of
cytokines on mdr1 expression in human colon carcinoma cell lines:
Increased cytotoxicity of MDR relevant drugs. J Cancer Res Clin
Oncol. 120:471–478. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stein U, Walther W and Shoemaker RH:
Modulation of mdr1 expression by cytokines in human colon carcinoma
cells: An approach for reversal of multidrug resistance. Br J
Cancer. 74:1384–1391. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stein U, Walther W and Shoemaker RH:
Reversal of multidrug resistance by transduction of cytokine genes
into human colon carcinoma cells. J Natl Cancer Inst. 88:1383–1392.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding L, Chen XP, Zhang ZW, Guan J, Zhang
WG, Wang HP, Wang ZH and Li CL: Synergistic effect of bromocriptine
and tumor necrosis factor-alpha on reversing hepatocellular
carcinoma multidrug resistance in nude mouse MDR1 model of liver
neoplasm. World J Gastroenterol. 11:5621–5626. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee NY and Kang YS: The decrease of
paclitaxel efflux by pretreatment of interferon-γ and tumor
necrosis factor-α after intracerebral microinjection. Brain Res.
1499:158–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee G and Piquette-Miller M: Cytokines
alter the expression and activity of the multidrug resistance
transporters in human hepatoma cell lines; analysis using RT-PCR
and cDNA micro-arrays. J Pharm Sci. 92:2152–2163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iqbal M, Ho HL, Petropoulos S, Moisiadis
VG, Gibb W and Matthews SG: Pro-inflammatory cytokine regulation of
P-glycoprotein in the developing blood-brain barrier. PLoS One.
7:e430222012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Belliard AM, Lacour B, Farinotti R and
Leroy C: Effect of tumor necrosis factor-alpha and interferon-gamma
on intestinal P-glycoprotein expression, activity, and localization
in Caco-2 cells. J Pharm Sci. 93:1524–1536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kreuser ED, Wadler S and Thiel E:
Biochemical modulation of cytotoxic drugs by cytokines: Molecular
mechanisms in experimental oncology. Recent Results Cancer Res.
139:371–382. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vacchelli E, Galluzzi L, Eggermont A,
Galon J, Tartour E, Zitvogel L and Kroemer G: Trial Watch:
Immunostimulatory cytokines. OncoImmunology. 1:493–506. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lejeune FJ and Rüegg C: Recombinant human
tumor necrosis factor: An efficient agent for cancer treatment.
Bull Cancer. 93:E90–E100. 2006.PubMed/NCBI
|
|
17
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biol.
11:372–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Karin M: How NF-kappaB is activated: The
role of the IkappaB kinase (IKK) complex. Oncogene. 18:6867–6874.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang TT, Kudo N, Yoshida M and Miyamoto
S: A nuclear export signal in the N-terminal regulatory domain of
IkappaBalpha controls cytoplasmic localization of inactive
NF-kappaB/IkappaBalpha complexes. Proc Natl Acad Sci USA.
97:1014–1019. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bentires-Alj M, Barbu V, Fillet M, Chariot
A, Relic B, Jacobs N, Gielen J, Merville MP and Bours V: NF-kappaB
transcription factor induces drug resistance through MDR1
expression in cancer cells. Oncogene. 22:90–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takada Y, Kobayashi Y and Aggarwal BB:
Evodiamine abolishes constitutive and inducible NF-kappaB
activation by inhibiting IkappaBalpha kinase activation, thereby
suppressing NF-kappaB-regulated antiapoptotic and metastatic gene
expression, up-regulating apoptosis, and inhibiting invasion. J
Biol Chem. 280:17203–17212. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang W, McLeod HL and Cassidy J:
Disulfiram-mediated inhibition of NF-kappaB activity enhances
cytotoxicity of 5-fluorouracil in human colorectal cancer cell
lines. Int J Cancer. 104:504–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakanishi C and Toi M: Nuclear
factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat
Rev Cancer. 5:297–309. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ogretmen B and Safa AR: Negative
regulation of MDR1 promoter activity in MCF-7, but not in multidrug
resistant MCF-7/Adr, cells by cross-coupled NF-κ B/p65 and c-Fos
transcription factors and their interaction with the CAAT region.
Biochemistry. 38:2189–2199. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thompson JE, Phillips RJ,
Erdjument-Bromage H, Tempst P and Ghosh S: I kappa B-beta regulates
the persistent response in a biphasic activation of NF-kappa B.
Cell. 80:573–582. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoffmann A, Levchenko A, Scott ML and
Baltimore D: The IkappaB-NF-kappaB signaling module: Temporal
control and selective gene activation. Science. 298:1241–1245.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schmidt C, Peng B, Li Z, Sclabas GM,
Fujioka S, Niu J, Schmidt-Supprian M, Evans DB, Abbruzzese JL and
Chiao PJ: Mechanisms of proinflammatory cytokine-induced biphasic
NF-kappaB activation. Mol Cell. 12:1287–1300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ladner KJ, Caligiuri MA and Guttridge DC:
Tumor necrosis factor-regulated biphasic activation of NF-kappa B
is required for cytokine-induced loss of skeletal muscle gene
products. J Biol Chem. 278:2294–2303. 2003. View Article : Google Scholar
|
|
29
|
Gibson PR: Increased gut permeability in
Crohn's disease: Is TNF the link? Gut. 53:1724–1725. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Englund G, Jacobson A, Rorsman F,
Artursson P, Kindmark A and Rönnblom A: Efflux transporters in
ulcerative colitis: Decreased expression of BCRP (ABCG2) and Pgp
(ABCB1). Inflamm Bowel Dis. 13:291–297. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Petrovic V, Teng S and Piquette-Miller M:
Regulation of drug transporters during infection and inflammation.
Mol Interv. 7:99–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blokzijl H, Vander Borght S, Bok LI,
Libbrecht L, Geuken M, van den Heuvel FA, Dijkstra G, Roskams TA,
Moshage H, Jansen PL, et al: Decreased P-glycoprotein (P-gp/MDR1)
expression in inflamed human intestinal epithelium is independent
of PXR protein levels. Inflamm Bowel Dis. 13:710–720. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu L, Smythe AM, Stinson SF, Mullendore
LA, Monks A, Scudiero DA, Paull KD, Koutsoukos AD, Rubinstein LV,
Boyd MR, et al: Multidrug-resistant phenotype of disease-oriented
panels of human tumor cell lines used for anticancer drug
screening. Cancer Res. 52:3029–3034. 1992.PubMed/NCBI
|
|
34
|
Wu CP, Calcagno AM and Ambudkar SV:
Reversal of ABC drug transporter-mediated multidrug resistance in
cancer cells: Evaluation of current strategies. Curr Mol Pharmacol.
1:93–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ho EA and Piquette-Miller M: Regulation of
multidrug resistance by pro-inflammatory cytokines. Curr Cancer
Drug Targets. 6:295–311. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ros JE, Schuetz JD, Geuken M, Streetz K,
Moshage H, Kuipers F, Manns MP, Jansen PL, Trautwein C and Müller
M: Induction of Mdr1b expression by tumor necrosis factor-alpha in
rat liver cells is independent of p53 but requires NF-kappaB
signaling. Hepatology. 33:1425–1431. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Um JH, Kang CD, Lee BG, Kim DW, Chung BS
and Kim SH: Increased and correlated nuclear factor-kappa B and Ku
auto-antigen activities are associated with development of
multidrug resistance. Oncogene. 20:6048–6056. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe
S, Mills GB and Unate H: Induction of human MDR1 gene expression by
2-acetyl aminofluorene is mediated by effectors of the
phosphoinositide 3-kinase pathway that activate NF-kappaB
signaling. Oncogene. 21:1945–1954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Arenzana-Seisdedos F, Thompson J,
Rodriguez MS, Bachelerie F, Thomas D and Hay RT: Inducible nuclear
expression of newly synthesized I kappa B alpha negatively
regulates DNA-binding and transcriptional activities of NF-kappa B.
Mol Cell Biol. 15:2689–2696. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Poppers DM, Schwenger P and Vilcek J:
Persistent tumor necrosis factor signaling in normal human
fibroblasts prevents the complete resynthesis of I kappa B-alpha. J
Biol Chem. 275:29587–29593. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gibson CJ, Hossain MM, Richardson JR and
Aleksunes LM: Inflammatory regulation of ATP binding cassette
efflux transporter expression and function in microglia. J
Pharmacol Exp Ther. 343:650–660. 2012. View Article : Google Scholar : PubMed/NCBI
|