Introduction
IR treatment leads to alteration of multiple
signaling pathways that control cell survival or cell death
(1–4). It is well recognized that ionizing
radiation induces DNA damage in human cells and triggers expression
of a number of genes (5–7). However, radiation can also alter
expression of many genes that are not typically involved in DNA
damage and repair pathways (8,9). For
example, it has been shown that IR can modulate expression of
important immunogenic genes, making tumor cells more susceptible to
immune responses (9–12).
Tumor cells escape immune responses by
downregulating genes that are essential for effective antitumor
immunity (13) such as genes
involved in presentation of antigens to T cells (14), stimulation of T cells (15), and susceptibility to apoptotic
signals (16,17). Members of the tumor necrosis factor
(TNF) receptor superfamily 4-1BBL/CD137L/TNFSF9 and OX-40L/
CD134L/TNFSF4 are expressed on antigen presenting cells and have
been reported to be expressed on tumor cells (9). Interaction between these ligands with
their cognate receptors, 4-1BB and OX-40 promotes T cell division,
survival, cytokine production and effector cell activity (18–20).
Following proper stimulation, cytotoxic T lymphocytes (CTLs)
commonly use death receptors such as DR4 (TRAIL-R1), DR5
(TRAIL-R2), and Fas (CD95/Apo-1) to kill tumor cells (21). Interaction between these death
receptors with their ligands on antitumor immune cells is essential
for driving apoptosis in many types of tumor cells (22). Thus, modulation of these molecules
is a promising approach for improving the activity of
tumor-specific T cells against resistant cancer cells and for
enhancing the efficacy of cancer immunotherapies (23).
A tumor cell undergoes heritable changes in gene
function to acquire specific growth advantages (24). Oncogenes may undergo DNA
hypomethylation and histone hyperacetylation to promote cell growth
and survival (25,26). Conversely, tumor suppressor genes
commonly undergo DNA hypermethylation and histone deacetylation to
silence these genes and to prevent inhibition of cell growth and
survival (27–31). DNA accessibility during
transcription is affected by differential packaging of DNA with
histone and non-histone proteins into chromatin (32). Histone tails are subjected to
several modifications that influence the ability of nucleosomes to
form stable higher chromatin structures (33). For example, histone acetylation
facilitates gene expression whereas histone deacetylases (HDACs)
return DNA to a less accessible conformation by removing acetyl
groups (34). Cancer cells are
known to increase deacetylation and silence genes that would
prevent tumor development. Deacetylation is mediated by HDACs which
are highly expressed in many cancers, including CRC (35). Class I histone deacetylases (HDAC1,
HDAC2 and HDAC3) are recruited into specific transcriptional
repression complexes resulting in chromatin condensation and
transcriptional silencing (36).
In addition to altered acetylation of histones, cancer cells also
increase DNA methylation to silence genes whose loss of expression
contributes to cancer progression. DNA hypermethylation of CpG
dinucleotides frequently contributes to loss of tumor suppressor
genes by accumulating methylation in their promoter regions
(37). DNA methylation is carried
out by DNA methyltransferases (DNMTs) via catalyzing transfer of
methyl groups to cytosine residues in DNA (38). In mammalian cells, three active
DNMTs have been identified; DNMT3a and DNMT3b facilitate formation
of de novo DNA methylation patterns while DNMT1 is mainly
required for maintenance of established patterns of DNA methylation
(39). We reported that
pharmacological inhibition of HDACs and DNMTs alter expression of
OX-40L and 4-1BBL (9). Others
showed that expression of death receptors can be influenced by DNA
methylation and histone acetylation (40–42);
however the molecular details at specific gene promoters remain
unknown, as well as how to manipulate their expression in
tumors.
Ionizing radiation (IR) is a pivotal treatment
modality for several cancers and has been shown to downregulate the
expression of class I HDACs (43)
and to decrease the activity of DNA methyltransferases (1,44).
We have recently shown that sublethal radiation enhances expression
of multiple death receptors (10),
as well as co-stimulatory molecules (9), in colorectal cancer (CRC) cells. We
have also observed increased histone acetylation at the promoter
for 4-1BBL (9). These findings
suggest that tumor cells surviving radiation treatment could be
upregulating transcript expression of immunogenic genes, through
modulation of epigenetic enzymes. However, the activity of DNA
methylating and histone deacetylating enzymes in the expression of
death receptors and co-stimulatory molecules in response to
radiation have not been investigated.
These are important questions because epigenetically
altered expression of genes can result in changes that are
sustained within a tumor cell population, and knowledge about such
changes could be further exploited to improve combination cancer
immunotherapy strategies. In the present study, we hypothesize that
CRC cells exposed to radiation, at a dose simulating a
hypofractionated dose (5 Gy), enhance expression of co-stimulatory
molecules and death receptors through modulation of epigenetic
enzyme activity at specific genes. We show that enhanced gene
expression of several immune-relevant genes following radiation
treatment is due, in part, to changes in DNA methylation and
histone acetylation. Further, we show that IR increases expression
of co-stimulatory molecules and death receptors by decreasing
DNMT1, HDAC2 and HDAC3 levels at the promoter regions of these
genes.
Materials and methods
Reagents and cell lines
5-Aza-2′-deoxycytidine (5-Aza-dC) and Trichostatin A
(TSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Antibodies recognizing histone H3 and acetylated histone H3 were
from Millipore (Lake Placid, NY, USA). Antibody recognizing HDAC1
were from Santa Cruz. Antibodies recognizing HDAC2, HDAC3, DNMT1,
DNMT3a and DNMT3b were from Abcam. Human colorectal carcinoma cell
line HCT116 and SW620 cells were purchased from ATCC. All cells
were cultured in media designated by ATCC for propagation and
maintenance. Cells were incubated at 37°C incubator with 5%
CO2 and tested to ensure absence of Mycoplasma.
Irradiation
Tumor cells were irradiated by using an RS-2000
Biological X-ray Irradiator (Rad Source Technology, Suwanee, GA,
USA). Cells were irradiated at a dose rate of 2 Gy/min for 2.5 min
by setting irradiator voltage and current at 160 kV and 25 mA.
During irradiation, the cells were maintained in recommended media
and kept on ice. Following irradiation, the culture media was
replaced with the fresh media.
Cell surface staining and flow cytometry
analysis
HCT116 cells (p53 WT) (45) and SW620 cells (p53 mutant)
(46) were irradiated (5 Gy), TSA
(125 nM) or 5-AZA-dC (20 μM)- treated. Cell surface staining of
tumor and normal cells were performed using the following primary
labeled antibodies; 4-1BBL-PE, OX-40L-PE, Fas-PE, DR4-PE, and
DR5-APC and the appropriate isotype matched controls (BioLegend.
San Diego, CA, USA). Surface staining was performed in cell
staining buffer for 1 h on ice. Stained cells were acquired on a BD
Fortessa flow cytometer (BD Pharmingen, San Diego, CA, USA). Dead
cells were excluded from the analysis based on scatter profile.
Isotype control staining was <5% for all samples analyzed.
Western blot analyses
Cell lysates were electrophoresed through
polyacrylamide gels and transferred onto nitrocellulose membranes.
Membranes were incubated with antibodies specific for histone H3
(Cell Signaling Technology, Danvers, TX, USA), acetylated histone
H3 (Millipore, Lake Placid, NY, USA), HDAC1 (Santa Cruz
Biotechnology), HDAC2, HDAC3, DNMT1, DNMT3a and DNMT3b (Abcam,
Cambridge, MA, USA), then revealed with secondary horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG (Santa Cruz
Biotechnology) and HRP-conjugated anti-mouse IgG (Promega, Madison,
WI, USA).
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed as previously described
in Cacan et al (30).
Briefly, HCT116 cells were irradiated (5 Gy) or TSA (125 nM)-
treated. Cells were then cross-linked with 1% formaldehyde and cell
nuclei were isolated and concentrated. Cell nuclei were sonicated
using a Bioruptor to generate an average of 500–700 bp of sheared
DNA. Sonicated lysates were then precleared with
salmon-sperm/agarose beads (Upstate) and 5% of the total lysate was
stored as input for normalization. Half of the remaining lysate was
immunoprecipitated with control antibody, and the other half was
immunoprecipitated with 5 μg of indicated antibody overnight at
4°C. Following an additional 2-h immunoprecipitation with
salmon-sperm/ agarose beads, all samples were washed and DNA was
isolated. Isolated DNA was quantified by real-time PCR on an ABI
PRISM 7900 (Applied Biosystems, Foster City, CA, USA) using the
following primers and probe for CD95/Fas: forward, 5′-TCG AGG TCC
TCA CCT GAA G-3′, reverse, 5′-TGC ACA AAT GGG CAT TCC T-3′ and
probe, 5′-CCA GCC ACT GCA GGA ACG CC-3′; for DR5: forward, 5′-CCC
AAG TGC CTC CCT CAA-3′, reverse, 5′-CCG GGC TGT GGT TTG TTT C-3′
and probe, 5′-CCC CAA GTT TCG GTG CCT GTC CT-3′; for 4-1BBL:
forward, 5′-CAG CAA GAG ACC CAC AGA GAT-3′, reverse, 5′-CTC CCT GCT
GTC TCT CCA T-3′ and probe, 5′-AGA GCG GCA GAG AGG GAG AAC C-3′;
for OX-40L: forward, 5′-GGG TTC AAT GTT TAG TTA CA-3′, reverse,
5′-GGG TCA CTT GGT AAA GAT A-3′ and probe, 5′-GCT GCT ACA TTA ACG
GAT GAT GAT T-3′; and for GAPDH: forward, 5′-AAT GAA TGG GCA GCC
GTT A-3′, reverse, 5′-TAG CCT CGC TCC ACC TGA CT-3′ and probe,
5′-CCT GCC GGT GAC TAA CCC TGC GCT CCT-3′. Values generated from
real-time PCR reactions were calculated based on standard curves
generated, were run in triplicate reactions and were analyzed using
the SDS 2.0 program.
Methylation-specific polymerase chain
reaction
HCT116 and SW620 cells were plated or irradiated
(5Gy) and then incubated for 48 h. Genomic DNA was extracted using
the E.Z.N.A. kit according to the manufacturer's protocol. Using
the EZ-DNA Methylation-Direct ™ Kit (Zymo Research, Irvine, CA,
USA), 750 ng of DNA was bisulfite converted per the manufacturer's
instructions. The primers for detection of unmethylated Fas gene
were 5′-ATA GGA ATG TTT ATT TGT GTA ATG A-3′ (forward) and 5′-CAA
AAT CAA AAA CAA ACT CAC AAA-3′ (reverse) to generate a 186-bp
product. The cycling conditions were 2 min at 94°C, followed by 34
cycles of 30sec at 94°C, 30 sec at 48°C and 30 sec at 72°C. To
detect the methylated Fas gene, 5′-GGG ATA GGA ATG TTT ATT TGT GTA
AC-3′ (forward) and 5′-GAA ATC AAA AAC GAA CTC ACG A-3′ (reverse)
were used to generate a 188-bp product. The cycling conditions were
2 min at 94°C, followed by 34 cycles of 30 sec at 94°C, 30 sec at
52°C and 30 sec at 72°C. The primers for detection of methylated
β-actin gene were 5′-TTT TGC AGG GTT CAC CCT CCT G-3′ (forward) and
5′-CGA GCA TCC CCC AAA GTT CAC AA-3′ (reverse). The cycling
conditions were 2 min at 94°C, followed by 34 cycles of 30 sec at
94°C, 30 sec at 58°C and 30 sec at 72°C.
Statistical analyses
Statistical differences between groups were
calculated using Student's t-test and calculated at 95% confidence.
Values represent mean ± SEM of three independent experiments. The
p-values <0.05 are indicated by one asterisk (*). The p-values
<0.005 are indicated by two asterisks (**). The p-values
<0.0005 are indicated by three asterisks (***).
Results
Surface expression of death receptors and
co-stimulatory molecules following treatment with a DNMT inhibitor,
HDAC inhibitor or IR in colorectal tumor cells
Epigenetic regulation and suppression of death
receptors have been reported in lung, medulloblastoma, and melanoma
tumor cell lines (17,47,48),
and we have recently reported that expression of OX-40L and 4-1BBL
co-stimulatory molecules appear to be sensitive to epigenetic
modifications in CRC cells (9).
Collectively, these proteins are important in influencing the
sensitivity of tumor cells to antitumor immune cells as well as
providing positive signals to these responding cells.
Interestingly, the expression of these molecules has also been
reported to be modulated following irradiation of tumor cells
(9,10), however, whether radiation is
altering expression of these molecules through specific chromatin
remodeling enzymes remains unknown. We first sought to determine if
pharmacologic inhibitors of histone deacetylation and DNA
methylation could alter the expression of multiple death receptors
in colorectal cancer cells in a manner similar to previous
observations of co-stimulatory molecules (9). The HDAC inhibitor, TSA, and DNMT
inhibitor, 5-Aza-dC, were used to inhibit HDACs and DNMTs activity,
respectively. HCT116 cells (p53 WT) treated with 125 nM TSA for 2
days, or with 20 μM 5-Aza-dC for 3 days, were compared to cells
irradiated with 5 Gy and cell surface protein expression of DR4,
DR5, Fas, 4-1BBL and OX-40L was quantified by flow cytometry. Both
IR and 5-Aza-dC treatment significantly increased cell surface
protein expression of DR4 in HCT116 colorectal carcinoma cells
while TSA resulted in a less robust increase that was not
significant (Fig. 1A). HCT116
cells uniformly express DR5 (100% positive) and Fas (>98%
positive) on their surface (Fig. 1B
and C) and there was no significant decrease in the frequency
of cells expressing DR5 and Fas. However, changes in the median
fluorescence intensity (MFI) values show a significant change in
the expression (density) levels of both DR5 and Fas following
radiation, TSA or 5-Aza-dC treatment (Fig. 1, insets B and C). Interestingly,
radiation increased both Fas and DR5 MFI more robustly than TSA or
5Aza-dC in these cells. Inhibition of DNA methylation robustly
increased expression of all three death receptors with a maximum
increase of 3-fold seen in Fas (Fig.
1C). We previously reported that inhibition of HDACs by TSA and
treatment with 10 Gy IR could influence expression of
co-stimulatory molecules on HCT116 tumor cells. We further
evaluated HCT116 cells to determine how significant these
differences were and if they occurred at a dose of radiation <10
Gy. In addition, having previously observed increased expression of
co-stimulatory molecule mRNA following inhibition of DNMT using
5-Aza-dC in these cells, we wanted to determine if this treatment
could also increase surface expression of the protein. Inhibition
of DNMT, HDACs and 5 Gy irradiation significantly increased 41BBL
surface expression (Fig. 1D),
while only IR significantly increased OX-40L surface expression.
While treatment with TSA or 5-Aza-dC appeared to mildly increase
OX-40L expression in HCT116 cells, the difference was not
significant (Fig. 1E). These data
suggest that the contribution of DNA methylation and histone
acetylation on the regulation of these two co-stimulatory genes is
different.
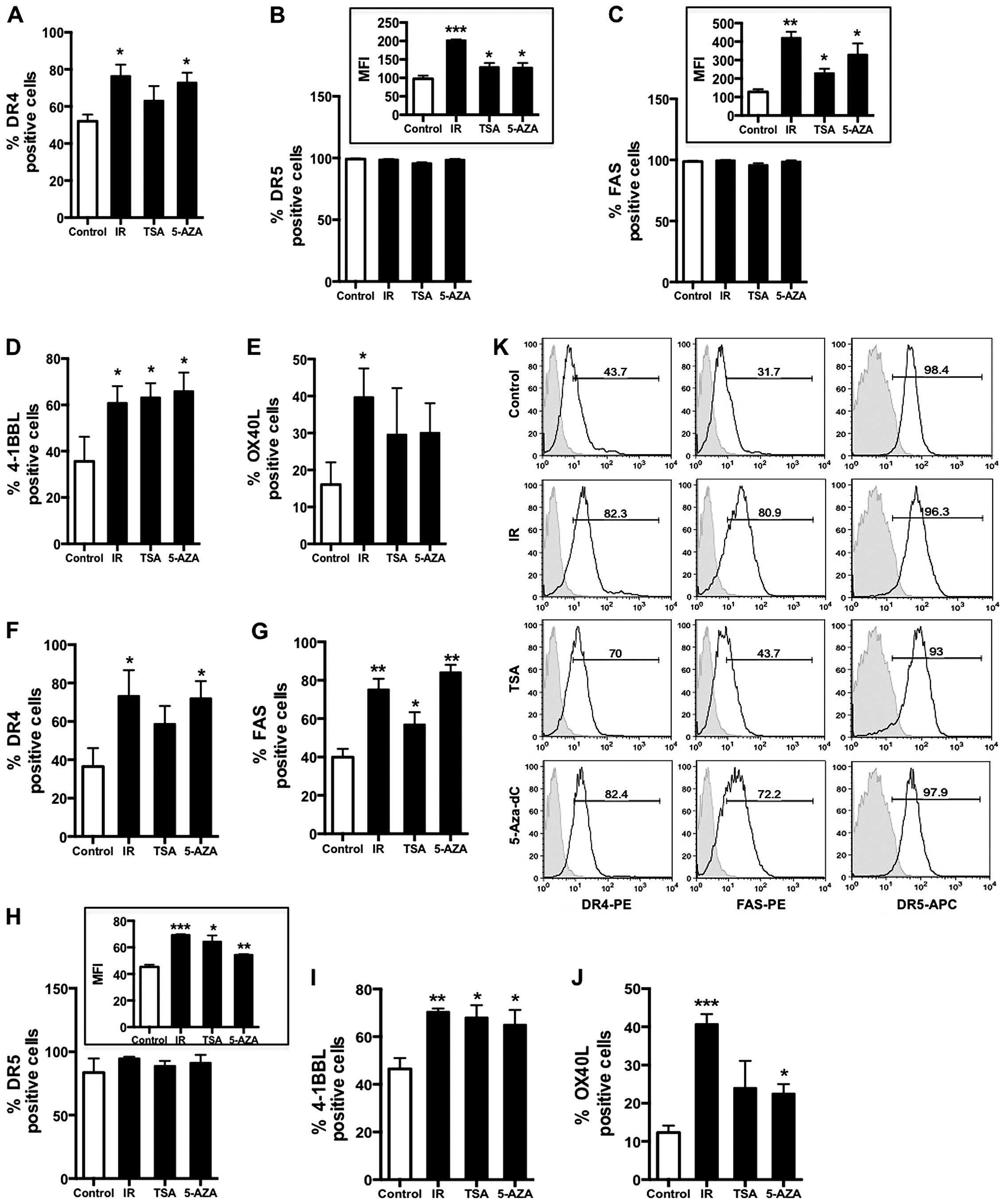 | Figure 1Cell surface expression of death
receptors and co-stimulatory molecules are altered by inhibition of
chromatin remodeling enzymes in colorectal cancer cells. Surface
expression of death receptors and co-stimulatory molecules were
evaluated using flow cytometry. Cells were plated and treated with
5-Aza-dC (20 μM), TSA (125 nM) or ionizing radiation (5Gy).
Adherent cells were harvested and stained with PE-labeled antibody
to human DR4, Fas, 4-1BBL and OX-40L or APC-labeled antibody to
human DR5 48 h after IR or TSA and 72 h post-5-Aza-dC. Isotype
control stained cells were ≤5% positive and analyzed for each
treatment group individually. The mean of data is graphed with
error bars representing SEM and the p-values are based on three
experimental replicates. Experiments were repeated at least three
times with similar results. Percent positive values were compared
to the level of gene expression see in untreated control samples.
*P<0.05, **P<0.005 and
***P<0.0005. A) DR4, B) DR5, C) Fas, D) 4-1BBL and E)
OX-40L surface expression in HCT116 cells. Cells surface expression
of F) DR4, G) Fas, H) DR5, I) 4-1BBL and J) OX-40L in SW620 cells.
K) Representative FACS plots showing DR4, Fas and DR5 expression
following IR, TSA and 5-Aza-dC treatments in SW620 cells. |
To confirm that these changes are not limited to
HCT116 cells and could occur similarly in another CRC cell line, we
evaluated surface expression in treated SW620 (p53 mutated)
colorectal cancer cells. SW620 cells were irradiated, treated with
TSA or 5-Aza-dC and surface protein expression was measured by flow
cytometry. Consistent with observations in HCT116 cells, the
treatments upregulated the surface expression of DR5, Fas and
4-1BBL most robustly in SW620 cells (Fig. 1G–I). Interestingly, while TSA
treatment did appear to increase expression of DR4 and OX-40L the
difference was not significant (Fig.
1F and J). Furthermore, we observed a much more robust increase
in the expression of OX-40L 48 h post-IR than previously observed
at 24 h post-IR using a higher dose. In contrast to the significant
increase in surface expression of 4-1BBL seen with all three
treatments, only IR and 5-Aza-dC induced a significant increase in
the expression of OX40L. While the magnitude of 4-1BBL increase was
similar between IR-treated cells and cells treated with the
epigenetic drugs, the increase in OX-40L was much lower in
drug-treated cells than observed in irradiated cells. Overall,
these data indicate that expression of these genes collectively is
most consistently increased following exposure to radiation in CRC
cell lines. Moreover, we observed that TSA and 5-Aza-dC treatments
were also able to significantly enhance expression of DR5, Fas and
4-1BBL across the cell lines.
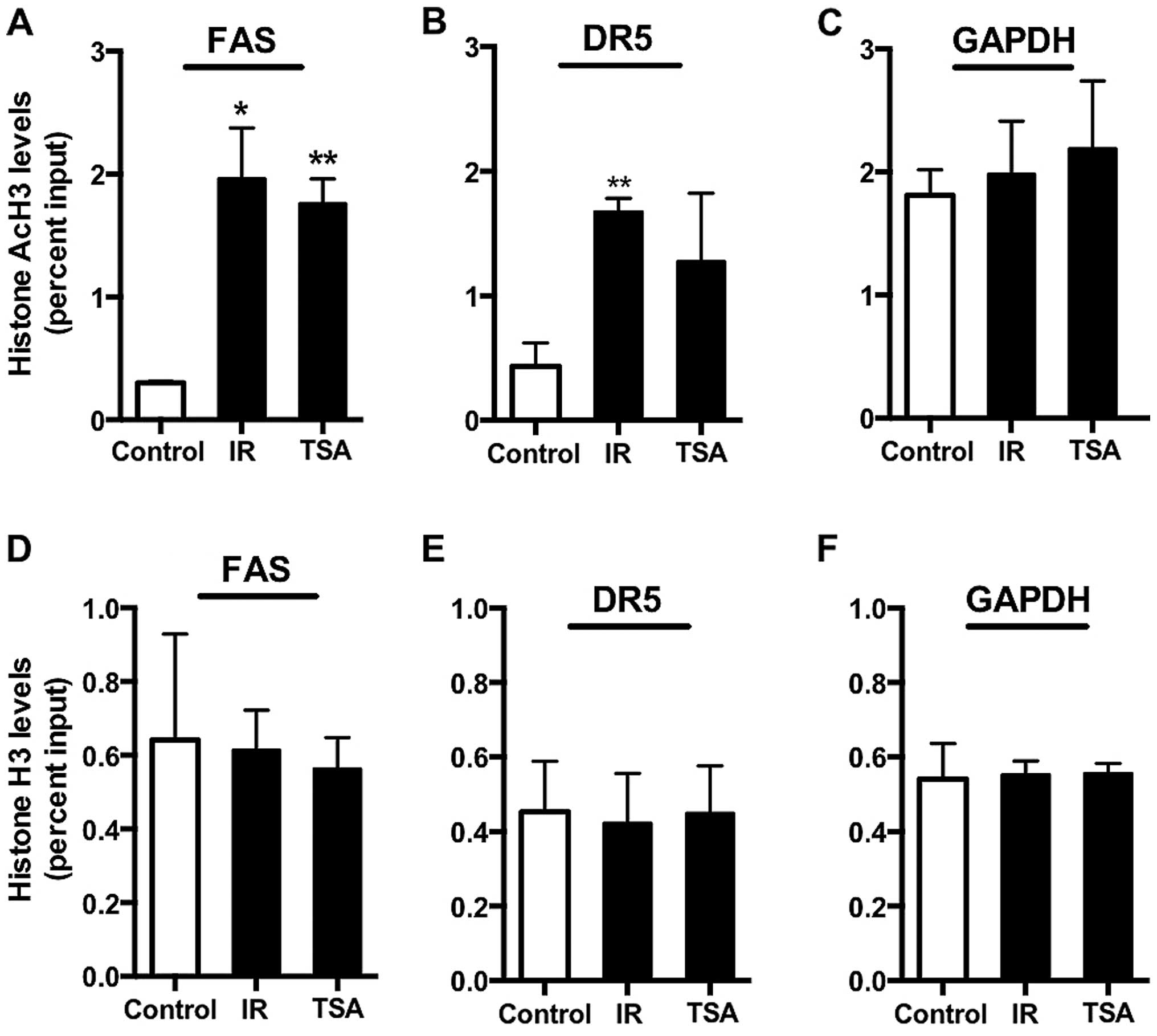
Histone acetylation at Fas, DR5, 4-1BBL
and OX-40L promoters following IR
Our data indicate that expression of death receptors
and co-stimulatory molecules can be influenced by drugs that
inhibit epigenetic activities in the cell, and that radiation also
increases expression of these genes in CRC cell lines. We observed
a significant increase in the expression of DR5, Fas, 4-1BBL and
OX-40L following IR in both colorectal cancer cells. We previously
reported that 10 Gy IR increases histone acetylation at the 4-1BBL
promoter (9). To explore whether
histone modifications also regulate death receptor expression
post-IR, we first assessed the levels of histone acetylation at the
promoters of DR5 and Fas by ChIP assays in both untreated and
irradiated HCT116 cells. TSA inhibits HDACs activity and
TSA-treated HCT116 cells were used as a positive control for
promoter acetylation. As expected, we observed a robust increase in
histone acetylation status following TSA treatment. Histone H3
acetylation levels significantly increased at both the Fas and DR5
promoters following 5 Gy radiation as compared to untreated control
cells (Fig. 2A and B), while we
observed similar amounts of total H3 histone binding at Fas, DR5
promoters (Fig. 2D and E). GAPDH
was evaluated in parallel since this gene is stably and
constitutively expressed at high levels in most tissues and cells.
In contrast, to the increased acetylation we saw at the death
receptors promoters, we observed no change in the levels of
acetylated histone H3 at the GAPDH promoter between untreated and
irradiated HCT116 cells (Fig. 2C),
nor any change in the total H3 histone binding (Fig. 2F). As GAPDH is gene that is
constitutively on and thus in an open, or acetylated, conformation;
we also evaluated a gene that would be expected to be closed and
remain closed in tumor cells. For this purpose, we looked at the
global histone H3 and acetylated histone H3 levels at the promoter
of CD4 and did not observe any alteration in acetylated histone H3
levels at this promoter following radiation treatment (data not
shown). These data suggest that radiation is specifically causing
promoter acetylation of Fas and DR5 and not uniformly across all
genes.
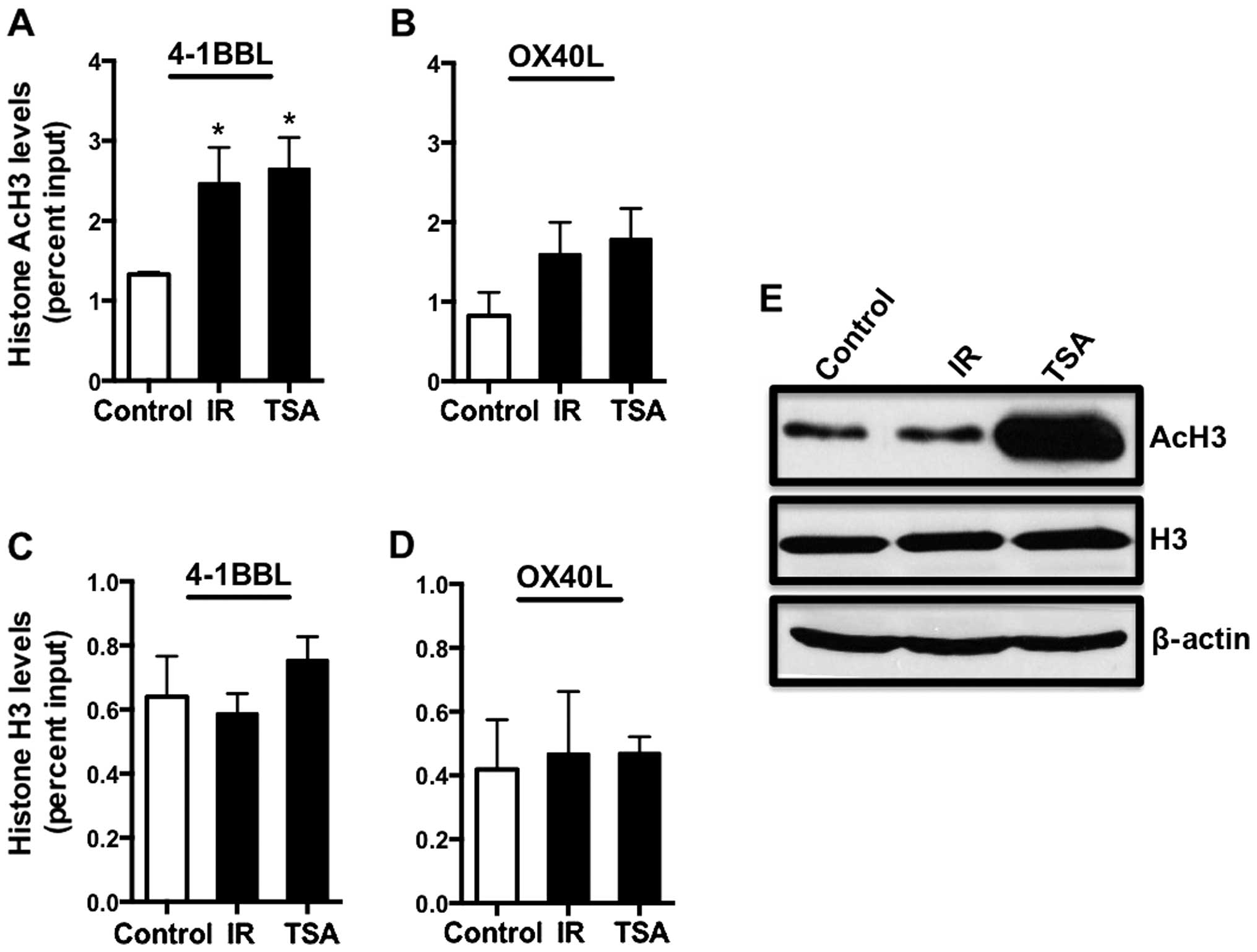
We previously reported that 10 Gy IR significantly
increased H3 acetylation at the 4-1BBL promoter in CRC cells and
wanted to determine if the same occurred at the promoter of another
effector T cell co-stimulatory molecule. For this we assessed the
levels of histone acetylation at the promoter of OX-40L by ChIP
assay. Again, TSA-treated HCT116 cells were used as a positive
control for promoter acetylation. Since we have already reported
enhanced histone acetylation at 4-1BBL promoter following 10 Gy IR,
here we used 4-1BBL promoter acetylation as a positive control but
also to determine if 5 Gy IR was sufficient to alter histone
acetylation status of this promoter. We found that histone H3
acetylation levels significantly increased at the 4-1BBL promoter
following 5 Gy radiation as compared to untreated control cells
(Fig. 3A). Interestingly, we
observed a 2-fold increase in histone acetylation level at the
OX-40L promoter following 5 Gy IR, however the difference was not
significant (Fig. 3B). Thus, we
saw no significant change in OX-40L surface expression following
TSA treatment in either HCT116 nor SW620 cells (Fig. 1E and J), and no significant
increase in acetylation at the promoter for this gene. These data
suggest that, in contrast to 4-1BBL expression, OX-40L expression
is not strongly regulated by histone acetylation. As before, we
observed no change in the overall levels of H3 and total H3 histone
binding was similar at 4-1BBL and OX-40L promoters (Fig. 3C and D), suggesting that the
increased acetylation at these promoters is not simply due to
increased overall histone H3 levels in the cells. Overall, our data
suggest that radiation increases the expression of some
co-stimulatory molecules and death receptors by increasing promoter
histone acetylation. This would facilitate transcription initiation
by loosening interactions between the histones and DNA; however,
the regulation of these genes by histone acetylation shows
variability across the genes. We further determined the total
cellular histone H3 and acetylated histone H3 protein levels in
HCT116 cells. TSA-treated cells were used as positive control for
histone acetylation and we found that 5 Gy IR did not alter total
cellular H3 or AcH3 levels in HCT116 cells (Fig. 3G). These data suggest that IR
induces histone acetylation in a gene specific manner.
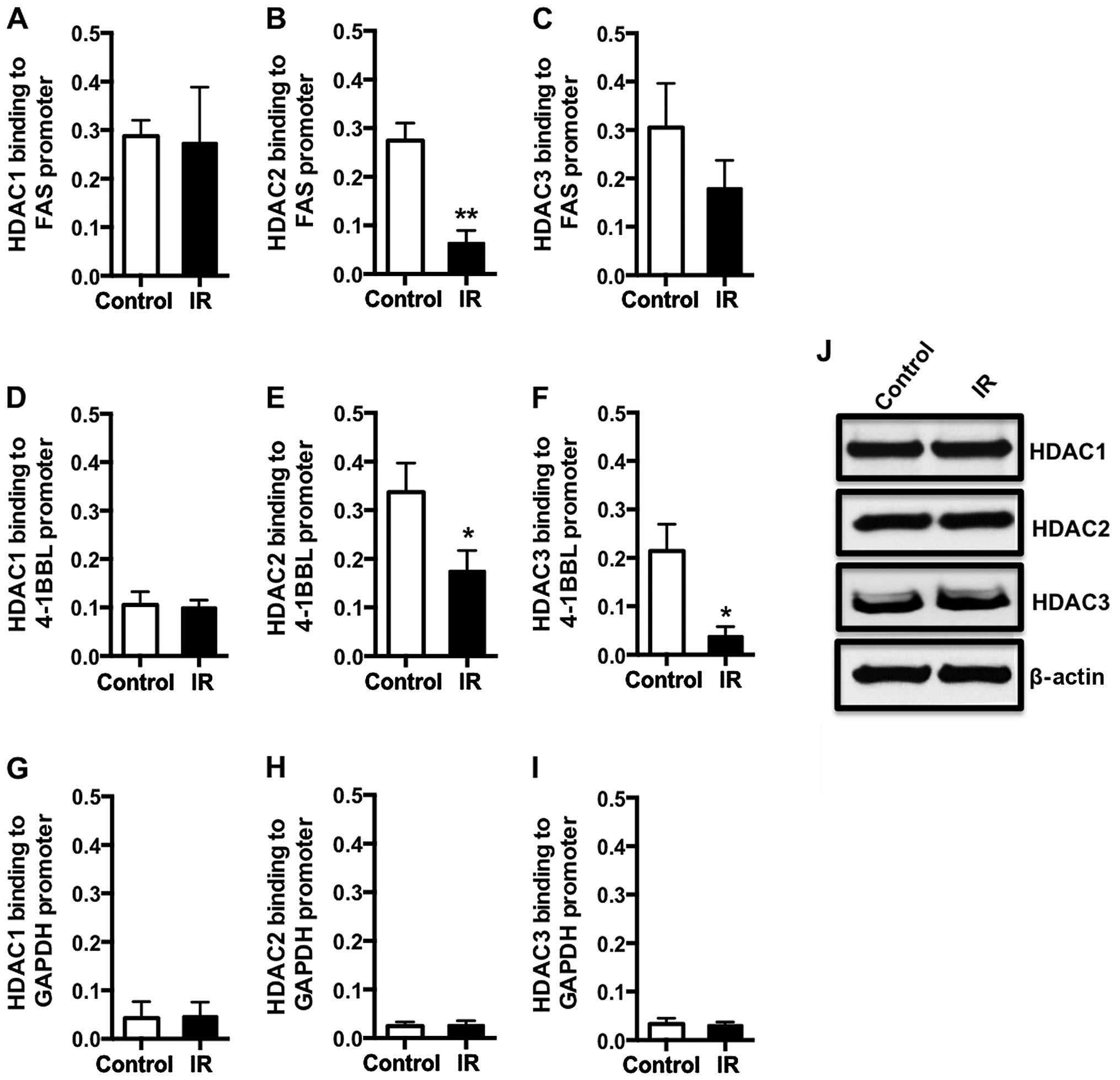
Radiation treatment influences binding of
HDAC2 and HDAC3 but not HDAC1 to specific promoters
Histone acetylation is dynamically regulated in
cells by the opposing actions of histone acetyltransferases (that
add the acetyl functional group to histones), and HDACs (that
remove them). We have observed an increase in histone acetylation
levels at promoters of co-stimulatory genes and death receptors
following radiation treatment of cancer cells. To determine the
potential mechanism that could be responsible for the enhancement
of histone acetylation following radiation treatment, we sought to
evaluate the binding of HDACs to the promoter region of Fas and
4-1BBL since we observed a robust and significant increase in
histone acetylation levels at the promoter region of both of these
genes. HCT116 cells were irradiated (5 Gy) and ChIP assays were
performed to determine binding of class I HDACs to the promoter
regions of Fas, 4-1BBL and control gene, GAPDH. We observed that
HDAC2 and HDAC3 proteins bind with decreased frequency to the Fas
promoter in radiation-treated cells compared to untreated cells
(Fig. 4B and C), and the change in
HDAC2 binding was more significant than that observed with HDAC3.
We did not observe any alterations in the binding of HDAC1 to the
Fas promoter between control and irradiated cells (Fig. 4A). Similarly, we observed a
significant decrease in binding of both HDAC2 and HDAC3 to the
4-1BBL promoter, and no change in binding of HDAC1 to the promoter.
These data suggest that HDAC2 and HDAC3 play a role in regulating
Fas and 4-1BBL transcription following radiation of tumor cells.
Moreover, radiation mediated decrease in HDAC2 and HDAC3 binding to
the Fas and 4-1BBL promoters is gene specific, as there was no
significant change in the level of HDAC association with the GAPDH
promoter between irradiated and control cells (Fig. 4G–I). To determine if radiation was
simply reducing the overall cellular levels of HDAC2 and HDAC3, we
performed western blot analysis. HDAC1, HDAC2 and HDAC3 expression
levels were similar in both radiation-treated and untreated cells
(Fig. 4J). Together, these data
indicate that radiation increases expression of Fas and 4-1BBL
molecules, in part, by decreasing HDAC2 and HDAC3 accumulation at
the Fas and 4-1BBL promoters in colorectal cancer cells.
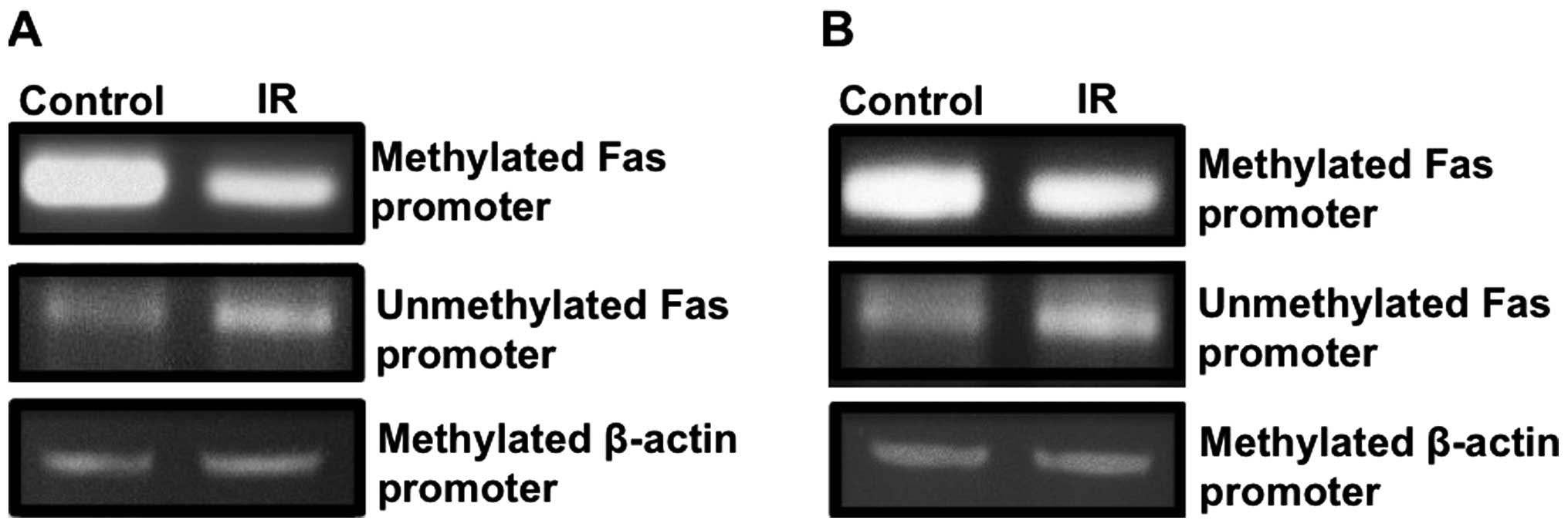
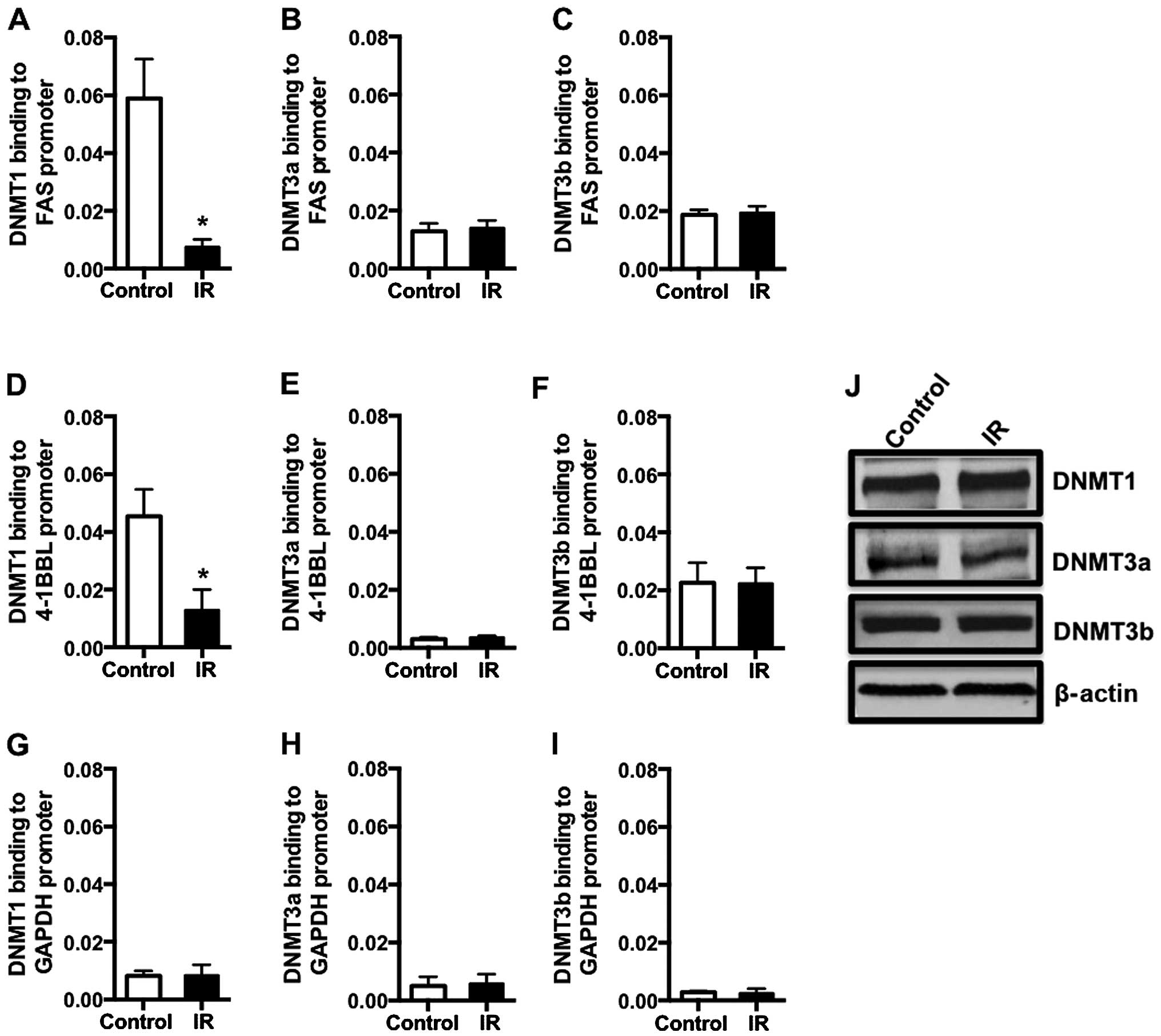
Radiation treatment decreases DNMT1
binding to Fas and 4-1BBL promoters
DNMTs are responsible for methylating DNA and for
silencing genes. Recent studies suggest that radiation treatment
alters expression of some target genes by changing DNA methylation
status of these genes (49). We
observed that treatment of CRC cells with 5-Aza-dC or IR increased
surface expression of Fas and 4-1BBL in both cell lines evaluated
(Fig. 1), suggesting that
expression of these molecules is regulated, in part, by DNMTs.
However, the role of methylating enzymes in expression of
co-stimulatory molecules and death receptors in response to
radiation has not been investigated. To determine whether the Fas
promoter is methylated in colorectal cancer cells, we utilized
methylation specific PCR. We observed that Fas promoter is
methylated in HCT116 and SW620 colorectal cancer cells and 5 Gy
radiation decreased DNA methylation at Fas promoter in both cell
lines (Fig. 5A and B). To
determine if radiation treatment causes changes in DNA methylating
enzyme status at co-stimulatory and death receptor genes, the human
colorectal cell line HCT116 was irradiated and DNMTs binding to the
Fas and 4-1BBL promoters was assessed by ChIP assays. We observed a
similar binding of DNMT3a and DNMT3b to the Fas promoter following
radiation treatment (Fig. 6B and
C). However, we detected a significant decrease in DNMT1
binding to the promoter region of Fas (Fig. 6A). Radiation induced similar
changes at the 4-1BBL promoter, and there was no change in the
binding of either DNMT3a and DNMT3b (Fig. 6E and F), and a decrease in the
binding of DNMT1 (Fig. 6D)
following radiation treatment. Conversely, 5 Gy radiation did not
cause any alteration in the binding of DNMTs to the promoter of the
housekeeping gene, GAPDH (Fig.
6G–I). Further, we determined that overall cellular DNMT1,
DNMT3a and DNMT3b levels were unchanged by 5 Gy radiation (Fig. 6J), suggesting that radiation was
specifically reducing the binding of DNMT1 to these promoters.
These data suggest that DNMT1 also contributes to regulation of Fas
and 4-1BBL expression post-IR.
Discussion
Radiation is clinically used for the treatment
various cancers. A better understanding of how radiation modulates
the expression of co-stimulatory molecules and death receptors will
extend the use of radiation and will be useful in improving cancer
immunotherapy strategies used in combination with radiation. We
investigated the molecular mechanisms of epigenetic regulation of
gene expression in CRC cells in response to IR. We identified that
radiation treatment specifically and significantly alters histone
acetylation at the promoters of two death receptor genes, Fas and
DR5. Further we determined that IR decreases HDAC2, HDAC3 and DNMT1
binding to the promoters of the death receptor Fas and the
co-stimulatory molecule 4-1BBL.
DNA methylation and histone acetylation play
important roles in chromatin organization and gene regulation in
eukaryotic cells. Multiple epigenetic modifications are commonly
disrupted during carcinogenesis. Oncogenes undergo hypomethylation
on DNA and acetylation and hypermethylation on histones in order to
drive enhanced expression (25,26).
Conversely, DNA hypermethylation and histone deacetylation commonly
occur on tumor suppressor genes to silence expression (27–29).
HDACs and DNMT inhibitors induce a potent anticancer response by
inhibiting histone deacetylation and DNA hypermethylation to induce
expression of pro-apoptotic genes (50–52).
Our data indicate that inhibition of DNMTs and HDACs by 5-Aza-dC
and TSA induce expression of co-stimulatory molecules and death
receptors in colorectal cancer cells. The data presented here
further indicate accumulation of DNMT1 and HDAC2/3 at the promoters
of such genes suppressing their expression in colorectal cancer
cells. Interestingly, not all of the genes analyzed are regulated
the same way. For example, expression of DR4 and Fas appear to be
dominantly regulated by DNA methylation, based on expression
following treatment with epigenetic drugs (Fig. 1), while 4-1BBL seems to be equally
regulated by both DNA methylation and histone deacetylation.
Several studies have reported that radiation can
alter histone modifications (53–55),
however, the direct effect of radiation on histone acetylation and
HDACs remains unclear. It has been reported that DR4 expression was
downregulated in medulloblastoma tumor samples and increased
histone H3 and H4 acetylation at the DR4 promoter sensitized
medulloblastoma cell lines to TRAIL-induced apoptosis (47). To our knowledge, this is the first
report showing the effect of IR on promoter acetylation of multiple
death receptors (Fig. 2) and
co-stimulatory molecules (Fig. 3)
and alterations in direct binding of class I HDACs to the promoter
region of these genes. Our data demonstrate that radiation
treatment enhances histone acetylation by decreasing binding of
HDAC2 and HDAC3 to the Fas and 4-1BBL promoters in HCT116
colorectal cancer cell line (Fig.
4). Interestingly, HDAC1 was found to bind to the Fas promoter
in T cells (56), which suggests
that different HDACs may play role in regulation of Fas in
different cells or tissues. Pollack et al observed an
increase at the level of histone H3 acetylation following UVR
exposure at several gene promoters, such as ATF3, COX2 and IL-8,
but no increase at the promoter region of the MHC class II gene in
HaCaT cells (54). Consistent with
their data, which suggest gene promoter specificity following
exposure to radiation, we did not observe changes at the GAPDH
promoter acetylation following IR in our current study (Fig. 3). This is further supported by our
previous report that 10 Gy radiation did not alter acetylation of
the CIITA promoter in HCT116 cells (9). Yu et al observed that UV
irradiation triggers genome-wide histone hyperacetylation at both
histone H3 and H4 in yeast (53),
however we did not observe a global histone hyperacetylation
following IR (Fig. 3), which could
be due to differences between cell responses to UV radiation versus
ionizing radiation or due to differences between yeast and human
tumor cells.
Promoter deacetylation and aberrant DNA methylation
are two major mechanisms associated with inactivation of
tumor-suppressor genes in cancer. Suzuki et al screened over
10,000 genes that are epigenetically silenced in human colorectal
cancer (57). They reported that
many of the hypermethylated genes have high potential for roles in
tumorigenesis. These data further suggest that gene silencing by
DNA hypermethylation and histone deacetylation commonly occur on
tumor suppressor and proapoptotic genes. Fas downregulation by
DNMT1 accumulation has been observed in osteosarcoma and lung
carcinoma cell lines (58), and
aberrant methylation of Fas promoter was observed in bladder
carcinoma (48,59). It has also been reported that DNMT1
silencing upregulates DR5 expression and sensitizes human hepatoma
cells to TRAIL-mediated apoptosis (60) and hypermethylation of the DR4
promoter was noted in invasive gastric carcinoma (61). Furthermore, Fas and DR4 suppression
was reported in small cell lung carcinoma cell lines and 5-Aza-dC
treatment induced expression of these genes (17). Some studies have suggested that
radiation can induce global DNA hypomethylation (62), while others observe both DNA
hypomethylation and hypermethylation (63). Different biological responses to
radiation could be influenced by different doses or tissue types
evaluated. We began our investigation by treating cells with
5-Aza-dC to inhibit DNA methylation in order to determine if this
would alter expression of some death receptors and co-stimulatory
molecules in human CRC cells. We observed increased expression of
five of the genes of interest in at least one of the CRC cell lines
evaluated (Fig 1). In our studies,
we observed a decrease in the binding of DNMT1 to the Fas and
4-1BBL promoters, but no change in global DNA methylation in HCT116
colorectal cancer cells (Fig 6).
DNMT1 is required for maintenance of the established patterns of
DNA methylation and is highly associated with tumor progression.
DNMT1 is recruited to the promoter region of target genes via
multiple steps and associates with methyl-CpG binding protein
(MeCP2) in order to perform maintenance methylation (64). A recent report indicates that
radiation treatment decreases level of proteins that function in
DNA methylation such as MeCP2 (49). These data suggest that radiation
treatment may not directly regulate DNMT1 expression, but may alter
DNMT1 recruitment to target gene promoters. Indeed, we observed
that IR did not alter global DNMT1 level, but did decrease DNMT1
binding to Fas promoters. If this is a consequence of MeCP2
activity is the subject of future investigation. Interestingly, it
has been reported that immunoprecipitated MeCP2 complexes show DNA
methyltransferase activity in hemimethylated DNA (64). In contrast to DNMT1 activity on
hemimethylated DNA, DNMT3a and DNMT3b are responsible for de
novo methylation of DNA. Thus this could be a reason why we did
not observe any alterations in the binding of DNMT3a and DNMT3b to
the promoter regions of Fas and 4-1BBL.
DNA methylation and histone deacetylation are often
associated with transcriptional repression of gene expression
(65,66) and with decreased responsiveness to
chemotherapy (67,68). DNA hypermethylation and histone
deacetylation contribute to cancer progression and chemoresistance
through amplification of master regulators of multiple cell
survival pathways (69–73). Clinical trials have demonstrated
HDAC inhibitors to be an effective anti-tumor drug therapy
(74) and such epigenetic
inhibitors have recently shown great therapeutic promise against
colorectal cancer (75). Thus, it
seems clear that altered gene expression via manipulation of
epigenetic control of intrinsic cell death and survival genes in
tumors contributes to increased sensitivity to apoptotic signals
from subsequent drugs. Here, we report findings suggesting that
manipulation of epigenetic regulation of extrinsic cell death genes
(i.e. death receptors), as well as immune co-stimulatory molecules,
by IR, results in increased sensitivity to attack by immune cells.
Indeed, we and others have reported increased killing of tumor
cells by T cells post-IR (11,12,76).
While the ability of radiation to modulate
expression of immune relevant genes in tumor cells has been
reported by many, detailed investigation into how radiation is
altering expression of these genes in tumor cells surviving
radiation has been limited. It has recently been reported that
micro-RNAs may regulate expression of some immune relevant genes in
non-malignant cells (77), and the
data presented here demonstrate that IR can also modulate the
binding of epigenetic enzymes to specific promoters. Thus,
radiation may alter expression of immune relevant genes in cells
that survive treatment via multiple mechanisms. Epigenetic changes
could greatly enhance our ability and approach of using IR to
enhance immunotherapy strategies as they are retained for quite
some time in tumor cell populations (78). Indeed we have observed sustained
changes in death receptor expression post-IR (10) and co-stimulatory molecule
expression post-IR (unpublished observations). Moreover, because
radiation therapy is already a standard of care for many diverse
cancers, it may be more amenable than pharmacological epigenetic
drugs for use in combination with novel immune-based therapies.
Overall, our study suggests that sub-lethal irradiation can
immunomodulate tumor cells through epigenetic regulatory mechanisms
of specific death receptors and co-stimulatory molecules. Whether
radiation treatment will alter expression of other immune relevant
genes through these same mechanisms is the subject of further
investigation as well as if this regulation occurs similarly in
other tissue and cell types.
References
|
1
|
Szumiel I: Ionising radiation-induced
oxidative stress, epigenetic changes and genomic instability: The
pivotal role of mitochondria. Int J Radiat Biol. 91:1–12. 2015.
View Article : Google Scholar
|
|
2
|
Agassi AM, Myslicki FA, Shulman JM,
Rotterman Y, Dosoretz DE, Fernandez E, Mantz CA and Finkelstein SE:
The promise of combining radiation therapy and immunotherapy:
Morbidity and toxicity. Future Oncol. 10:2319–2328. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Finkelstein SE, Salenius S, Mantz CA,
Shore ND, Fernandez EB, Shulman J, Myslicki FA, Agassi AM,
Rotterman Y, DeVries T, et al: Combining immunotherapy and
radiation for prostate cancer. Clin Genitourin Cancer. 13:1–9.
2015. View Article : Google Scholar
|
|
4
|
Garnett-Benson C, Hodge JW and Gameiro SR:
Combination regimens of radiation therapy and therapeutic cancer
vaccines: Mechanisms and opportunities. Semin Radiat Oncol.
25:46–53. 2015. View Article : Google Scholar
|
|
5
|
Di Leonardo A, Linke SP, Clarkin K and
Wahl GM: DNA damage triggers a prolonged p53-dependent G1 arrest
and long-term induction of Cip1 in normal human fibroblasts. Genes
Dev. 8:2540–2551. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
d'Adda di Fagagna F, Reaper PM,
Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G,
Carter NP and Jackson SP: A DNA damage checkpoint response in
telomere-initiated senescence. Nature. 426:194–198. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ostling O and Johanson KJ:
Microelectrophoretic study of radiation-induced DNA damages in
individual mammalian cells. Biochem Biophys Res Commun.
123:291–298. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kumari A, Cacan E, Greer SF and
Garnett-Benson C: Turning T cells on: Epigenetically enhanced
expression of effector T-cell costimulatory molecules on irradiated
human tumor cells. J Immunother Cancer. 1:172013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ifeadi V and Garnett-Benson C: Sublethal
irradiation of human colorectal tumor cells imparts enhanced and
sustained susceptibility to multiple death receptor signaling
pathways. PLoS One. 7:e317622012. View Article : Google Scholar
|
|
11
|
Gameiro SR, Jammeh ML, Wattenberg MM,
Tsang KY, Ferrone S and Hodge JW: Radiation-induced immunogenic
modulation of tumor enhances antigen processing and calreticulin
exposure, resulting in enhanced T-cell killing. Oncotarget.
5:403–416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garnett CT, Palena C, Chakraborty M, Tsang
KY, Schlom J and Hodge JW: Sublethal irradiation of human tumor
cells modulates phenotype resulting in enhanced killing by
cytotoxic T lymphocytes. Cancer Res. 64:7985–7994. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim R, Emi M and Tanabe K: Cancer
immunoediting from immune surveillance to immune escape.
Immunology. 121:1–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bubeník J: MHC class I down-regulation:
Tumour escape from immune surveillance (Review)? Int J Oncol.
25:487–491. 2004.
|
|
15
|
Lee H, Kim JH, Yang SY, Kong J, Oh M,
Jeong DH, Chung JI, Bae KB, Shin JY, Hong KH, et al: Peripheral
blood gene expression of B7 and CD28 family members associated with
tumor progression and microscopic lymphovascular invasion in colon
cancer patients. J Cancer Res Clin Oncol. 136:1445–1452. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
French LE and Tschopp J: Defective death
receptor signaling as a cause of tumor immune escape. Semin Cancer
Biol. 12:51–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hopkins-Donaldson S, Ziegler A, Kurtz S,
Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U and Stahel
R: Silencing of death receptor and caspase-8 expression in small
cell lung carcinoma cell lines and tumors by DNA methylation. Cell
Death Differ. 10:356–364. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Croft M, So T, Duan W and Soroosh P: The
significance of OX40 and OX40L to T-cell biology and immune
disease. Immunol Rev. 229:173–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tu TH, Kim CS, Nam-Goong IS, Nam CW, Kim
YI, Goto T, Kawada T, Park T, Yoon Park JH, Ryoo ZY, et al: 4-1BBL
signaling promotes cell proliferation through reprogramming of
glucose metabolism in monocytes/macrophages. FEBS J. 282:1468–1480.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eun SY, Lee SW, Xu Y and Croft M: 4-1BB
ligand signaling to T cells limits T cell activation. J Immunol.
194:134–141. 2015. View Article : Google Scholar
|
|
21
|
Guicciardi ME and Gores GJ: Life and death
by death receptors. FASEB J. 23:1625–1637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koornstra JJ, Kleibeuker JH, van Geelen
CM, Rijcken FE, Hollema H, de Vries EG and de Jong S: Expression of
TRAIL (TNF-related apoptosis-inducing ligand) and its receptors in
normal colonic mucosa, adenomas, and carcinomas. J Pathol.
200:327–335. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Driscoll PC: Structural studies of death
receptors. Methods Enzymol. 545:201–242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gronbaek K, Hother C and Jones PA:
Epigenetic changes in cancer. APMIS. 115:1039–1059. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin B, Yao B, Li JL, Fields CR, Delmas AL,
Liu C and Robertson KD: DNMT1 and DNMT3B modulate distinct
polycomb-mediated histone modifications in colon cancer. Cancer
Res. 69:7412–7421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Müller BM, Jana L, Kasajima A, Lehmann A,
Prinzler J, Budczies J, Winzer KJ, Dietel M, Weichert W and Denkert
C: Differential expression of histone deacetylases HDAC1, 2 and 3
in human breast cancer--overexpression of HDAC2 and HDAC3 is
associated with clinicopathological indicators of disease
progression. BMC Cancer. 13:2152013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Esteller M: Cancer epigenomics: DNA
methylomes and histone-modification maps. Nat Rev Genet. 8:286–298.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nguyen CT, Gonzales FA and Jones PA:
Altered chromatin structure associated with methylation-induced
gene silencing in cancer cells: Correlation of accessibility,
methylation, MeCP2 binding and acetylation. Nucleic Acids Res.
29:4598–4606. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cacan E, Ali MW, Boyd NH, Hooks SB and
Greer SF: Inhibition of HDAC1 and DNMT1 modulate RGS10 expression
and decrease ovarian cancer chemoresistance. PLoS One.
9:e874552014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ali MW, Cacan E, Liu Y, Pierce JY,
Creasman WT, Murph MM, Govindarajan R, Eblen ST, Greer SF and Hooks
SB: Transcriptional suppression, DNA methylation, and histone
deacetylation of the regulator of G-protein signaling 10 (RGS10)
gene in ovarian cancer cells. PLoS One. 8:e601852013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ramakrishnan V: Histone structure and the
organization of the nucleosome. Annu Rev Biophys Biomol Struct.
26:83–112. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kuo MH and Allis CD: Roles of histone
acetyltransferases and deacetylases in gene regulation. BioEssays.
20:615–626. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stypula-Cyrus Y, Damania D, Kunte DP, Cruz
MD, Subramanian H, Roy HK and Backman V: HDAC up-regulation in
early colon field carcinogenesis is involved in cell tumorigenicity
through regulation of chromatin structure. PLoS One. 8:e646002013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Millard CJ, Watson PJ, Celardo I,
Gordiyenko Y, Cowley SM, Robinson CV, Fairall L and Schwabe JW:
Class I HDACs share a common mechanism of regulation by inositol
phosphates. Mol Cell. 51:57–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Esteller M: CpG island hypermethylation
and tumor suppressor genes: A booming present, a brighter future.
Oncogene. 21:5427–5440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rhee I, Jair KW, Yen RW, Lengauer C,
Herman JG, Kinzler KW, Vogelstein B, Baylin SB and Schuebel KE: CpG
methylation is maintained in human cancer cells lacking DNMT1.
Nature. 404:1003–1007. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin B, Li Y and Robertson KD: DNA
methylation: Superior or subordinate in the epigenetic hierarchy?
Genes Cancer. 2:607–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Venza M, Visalli M, Catalano T, Fortunato
C, Oteri R, Teti D and Venza I: Impact of DNA methyltransferases on
the epigenetic regulation of tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) receptor expression in malignant
melanoma. Biochem Biophys Res Commun. 441:743–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Maecker HL, Yun Z, Maecker HT and Giaccia
AJ: Epigenetic changes in tumor Fas levels determine immune escape
and response to therapy. Cancer Cell. 2:139–148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jazirehi AR and Arle D: Epigenetic
regulation of the TRAIL/ Apo2L apoptotic pathway by histone
deacetylase inhibitors: An attractive approach to bypass melanoma
immunotherapy resistance. Am J Clin Exp Immunol. 2:55–74. 2013.
|
|
43
|
Mueller S, Yang X, Sottero TL, Gragg A,
Prasad G, Polley MY, Weiss WA, Matthay KK, Davidoff AM, DuBois SG,
et al: Cooperation of the HDAC inhibitor vorinostat and radiation
in metastatic neuroblastoma: Efficacy and underlying mechanisms.
Cancer Lett. 306:223–229. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Antwih DA, Gabbara KM, Lancaster WD, Ruden
DM and Zielske SP: Radiation-induced epigenetic DNA methylation
modification of radiation-response pathways. Epigenetics.
8:839–848. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kaeser MD, Pebernard S and Iggo RD:
Regulation of p53 stability and function in HCT116 colon cancer
cells. J Biol Chem. 279:7598–7605. 2004. View Article : Google Scholar
|
|
46
|
Rodrigues NR, Rowan A, Smith ME, Kerr IB,
Bodmer WF, Gannon JV and Lane DP: p53 mutations in colorectal
cancer. Proc Natl Acad Sci USA. 87:7555–7559. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Aguilera DG, Das CM, Sinnappah-Kang ND,
Joyce C, Taylor PH, Wen S, Hasselblatt M, Paulus W, Fuller G, Wolff
JE, et al: Reactivation of death receptor 4 (DR4) expression
sensitizes medulloblastoma cell lines to TRAIL. J Neurooncol.
93:303–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Watson CJ, O'Kane H, Maxwell P, Sharaf O,
Petak I, Hyland PL, O'Rouke D, McKnight J, Canning P and Williamson
K: Identification of a methylation hotspot in the death receptor
Fas/ CD95 in bladder cancer. Int J Oncol. 40:645–654. 2012.
|
|
49
|
Bae JH, Kim JG, Heo K, Yang K, Kim TO and
Yi JM: Identification of radiation-induced aberrant hypomethylation
in colon cancer. BMC Genomics. 16:562015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Johnstone RW: Histone-deacetylase
inhibitors: Novel drugs for the treatment of cancer. Nat Rev Drug
Discov. 1:287–299. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Takai N, Desmond JC, Kumagai T, Gui D,
Said JW, Whittaker S, Miyakawa I and Koeffler HP: Histone
deacetylase inhibitors have a profound antigrowth activity in
endometrial cancer cells. Clin Cancer Res. 10:1141–1149. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lyko F and Brown R: DNA methyltransferase
inhibitors and the development of epigenetic cancer therapies. J
Natl Cancer Inst. 97:1498–1506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yu Y, Teng Y, Liu H, Reed SH and Waters R:
UV irradiation stimulates histone acetylation and chromatin
remodeling at a repressed yeast locus. Proc Natl Acad Sci USA.
102:8650–8655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pollack BP, Sapkota B and Boss JM:
Ultraviolet radiation-induced transcription is associated with
gene-specific histone acetylation. Photochem Photobiol. 85:652–662.
2009. View Article : Google Scholar
|
|
55
|
Ji S, Tian Y, Lu Y, Sun R, Ji J, Zhang L
and Duan S: Irradiation-induced hippocampal neurogenesis impairment
is associated with epigenetic regulation of bdnf gene
transcription. Brain Res. 1577:77–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zimmerman MA, Singh N, Martin PM,
Thangaraju M, Ganapathy V, Waller JL, Shi H, Robertson KD, Munn DH
and Liu K: Butyrate suppresses colonic inflammation through
HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T
cells. Am J Physiol Gastrointest Liver Physiol. 302:G1405–G1415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Suzuki H, Gabrielson E, Chen W, Anbazhagan
R, van Engeland M, Weijenberg MP, Herman JG and Baylin SB: A
genomic screen for genes upregulated by demethylation and histone
deacetylase inhibition in human colorectal cancer. Nature genetics.
31:141–149. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
58
|
Thaler R, Spitzer S, Karlic H, Berger C,
Klaushofer K and Varga F: Ibandronate increases the expression of
the pro-apoptotic gene FAS by epigenetic mechanisms in tumor cells.
Biochem Pharmacol. 85:173–185. 2013. View Article : Google Scholar :
|
|
59
|
Li W, Xia D, Wang Y, Li Y, Xue Y, Wu X and
Ye Z: Relationship between aberrant methylation of FAS promoter and
biological behavior of bladder urothelial carcinoma. J Huazhong
Univ Sci Technol Med Sci. 31:794–798. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kurita S, Higuchi H, Saito Y, Nakamoto N,
Takaishi H, Tada S, Saito H, Gores GJ and Hibi T: DNMT1 and DNMT3b
silencing sensitizes human hepatoma cells to TRAIL-mediated
apoptosis via up-regulation of TRAIL-R2/DR5 and caspase-8. Cancer
Sci. 101:1431–1439. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lee KH, Lim SW, Kim HG, Kim DY, Ryu SY,
Joo JK, Kim JC and Lee JH: Lack of death receptor 4 (DR4)
expression through gene promoter methylation in gastric carcinoma.
Langenbecks Arch Surg. 394:661–670. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chaudhry MA and Omaruddin RA: Differential
DNA methylation alterations in radiation-sensitive and -resistant
cells. DNA Cell Biol. 31:908–916. 2012. View Article : Google Scholar
|
|
63
|
Aypar U, Morgan WF and Baulch JE:
Radiation-induced epigenetic alterations after low and high LET
irradiations. Mutat Res. 707:24–33. 2011. View Article : Google Scholar
|
|
64
|
Kimura H and Shiota K: Methyl-CpG-binding
protein, MeCP2, is a target molecule for maintenance DNA
methyltransferase, Dnmt1. J Biol Chem. 278:4806–4812. 2003.
View Article : Google Scholar
|
|
65
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fuks F: DNA methylation and histone
modifications: Teaming up to silence genes. Curr Opin Genet Dev.
15:490–495. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Esteller M, Garcia-Foncillas J, Andion E,
Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB and Herman JG:
Inactivation of the DNA-repair gene MGMT and the clinical response
of gliomas to alkylating agents. N Engl J Med. 343:1350–1354. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Giacinti L, Vici P and Lopez M: Epigenome:
A new target in cancer therapy. Clin Ter. 159:347–360.
2008.PubMed/NCBI
|
|
69
|
Ng JM and Yu J: Promoter hypermethylation
of tumour suppressor genes as potential biomarkers in colorectal
cancer. Int J Mol Sci. 16:2472–2496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lin PC, Lin JK, Lin CH, Lin HH, Yang SH,
Jiang JK, Chen WS, Chou CC, Tsai SF and Chang SC: Clinical
relevance of plasma DNA methylation in colorectal cancer patients
identified by using a genome-wide high-resolution array. Ann Surg
Oncol. Dec 4–2014.(Epub ahead of print). View Article : Google Scholar
|
|
71
|
Gan L, Chen S, Zhong J, Wang X, Lam EK,
Liu X, Zhang J, Zhou T, Yu J, Si J, et al: ZIC1 is downregulated
through promoter hypermethylation, and functions as a tumor
suppressor gene in colorectal cancer. PLoS One. 6:e169162011.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Vaish V, Khare T, Verma M and Khare S:
Epigenetic therapy for colorectal cancer. Methods Mol Biol.
1238:771–782. 2015. View Article : Google Scholar
|
|
73
|
Lee SC, Cheong HJ, Kim SJ, Yoon J, Kim HJ,
Kim KH, Kim SH, Kim HJ, Bae SB, Kim CK, et al: Low-dose
combinations of LBH589 and TRAIL can overcome TRAIL-resistance in
colon cancer cell lines. Anticancer Res. 31:3385–3394.
2011.PubMed/NCBI
|
|
74
|
Marks PA, Miller T and Richon VM: Histone
deacetylases. Curr Opin Pharmacol. 3:344–351. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tampakis A, Tampaki EC, Nebiker CA and
Kouraklis G: Histone deacetylase inhibitors and colorectal cancer:
What is new? Anticancer Agents Med Chem. 14:1220–1227. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Chakraborty M1, Wansley EK, Carrasquillo
JA, Yu S, Paik CH, Camphausen K, Becker MD, Goeckeler WF, Schlom J
and Hodge JW: The use of chelated radionuclide
(samarium-153-ethylenediaminetetramethylenephosphonate) to modulate
phenotype of tumor cells and enhance T cell-mediated killing. Clin
Cancer Res. 14:4241–4249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Palayoor ST, John-Aryankalayil M, Makinde
AY, Falduto MT, Magnuson SR and Coleman CN: Differential expression
of stress and immune response pathway transcripts and miRNAs in
normal human endothelial cells subjected to fractionated or
single-dose radiation. Mol Cancer Res. 12:1002–1015. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Khare S and Verma M: Epigenetics of colon
cancer. Methods Mol Biol. 863:177–185. 2012. View Article : Google Scholar : PubMed/NCBI
|