Introduction
Head-and-neck squamous cell carcinoma (HNSCC) is the
most common cancer in the head and neck region, and affects
~550,000 patients worldwide (1).
The overall 5-year survival rate of HNSCC including oral squamous
cell carcinoma (OSCC) is ~50%, which has not improved markedly
during the last decade (2,3). Thus, new effective therapeutic
modalities are needed to improve survival of HNSCC patients.
The epidermal growth factor receptor (EGFR) is a
member of the receptor tyrosine kinase (TK) or HER family, which
consists of EGFR (HER1/ErbB1), HER2/Neu (ErbB2), HER3 (ErbB3), and
HER4 (ErbB4). Stimulation of the receptors through ligand binding
activates the receptor tyrosine kinase and promotes its
homodimerization or heterodimerization with another HER. EGFR
activation stimulates a number of downstream signaling cascades,
such as the RAS/RAF/ERK/MAPK, phosphatidylinositol 3-kinase
(PI3K)/AKT pathway, and the phospholipase C-γ/protein kinase C
(PLCγ/PKC) pathway, and including the Src family kinases (SFKs),
and the signal transducers and activators of transcription (STATs).
These pathways affect various cellular responses, including
proliferation, survival, migration, angiogenesis and metastasis
(4–10).
EGFR is constitutively distributed in normal
epithelial cells, but it is highly expressed in various cancers,
including those of the breast, prostate, and lung cancers, as well
as gliomas (11). EGFR is
expressed at higher levels in >95% of HNSCCs compared to normal
mucosa (12). Furthermore,
upregulation of EGFR expression in HNSCC has been reported to be
associated with an unfavorable prognosis and is a useful prognostic
biomarker of low survival rate (13,14).
Therefore, EGFR is considered to be one of the most promising
molecular targets in oncology, and EGFR-targeted therapies have
been developed using monoclonal antibodies (mAbs) to the
extra-cellular ligand-binding domain of the EGFR.
Cetuximab is a chimeric mAb consisting of a Fv
region of mouse anti EGFR antibody and human IgG1 heavy and κ
light-chain constant regions, which binds with high affinity to the
extracellular domain of EGFR (15)
and subsequently blocks EGFR activation by preventing TK-mediated
phosphorylation of the protein (16). As a result, cetuximab promotes
apoptosis, and inhibits cell cycle progression, tumor cell invasion
and angiogenesis.
Cetuximab has already been used in the clinic,
however, intrinsic or acquired resistance to EGFR therapy remains a
major obstacle to achieving positive clinical outcomes with
cetuximab (17,18). In order to resolve these problems,
it is important to understand both the mechanisms of action and
resistance to cetuximab.
The OSCC cell line SAS, exhibits proliferation that
is not sensitive to cetuximab treatment, although SAS cells do
express EGFR and also undergo phosphorylation. However, SAS growth
has been reported to be inhibited by cetuximab under aggregation
culture conditions (19). In this
study, we investigated the molecular basis of changes in cetuximab
sensitivity facilitated by growth signals provided by culture
conditions in SAS cells.
Materials and methods
Cell culture and reagents
Three OSCC cell lines, HSC3, HSC4, and SAS, were
purchased from RIKEN Bioresource Center (Ibaraki, Japan). Cells
were cultured in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% (v/v) fetal bovine serum (FBS) at 37ºC in a
humidified atmosphere of 5% CO2. DMEM and FBS were
purchased from Gibco (Life Technologies, Tokyo, Japan). Antibodies
used included anti-EGFR (Cell Signaling Technology, Tokyo, Japan),
anti-phospho-EGFR (Tyr1068, Cell Signaling Technology), anti-ERK1
(Santa Cruz Biotechnology, CA, USA), anti-phospho-ERK (Santa Cruz
Biotechnology), anti-AKT (Cell Signaling Technology),
anti-phospho-AKT (Ser473, Cell Signaling Technology),
anti-caveolin-1 (Santa Cruz Biotechnology), anti-phospho-caveolin-1
(Tyr14, Cell Signaling Technology), and anti-α-tubulin
(Sigma-Aldrich, Tokyo, Japan). Cetuximab (Erbitux®) was
purchased from Merck Serono (Tokyo, Japan). AG1478, TAPI-2,
LY294002 and MK2206 were from Calbiochem (Merk Millipore, Tokyo,
Japan).
Cell proliferation assay
Human OSCC cells (2×103/well) were plated
in 96-well plates. After 24 h of growth, various reagents were
added at the indicated concentrations and growth continued for an
additional 2, 4, or 6 days. All experiments were performed in
triplicate. Cell proliferation was assessed using the CellTiter
96® Non-Radioactive Cell Proliferation assay (Promega,
Tokyo, Japan).
Aggregation cultures
When aggregation culture conditions were utilized,
1–5×103 cells were seeded into each well of low-adhesive
96-well plates (Sumitomo Bakelite, Tokyo, Japan) and cultured in
DMEM supplemented with 10% (v/v) FBS at 37 ºC under 5%
CO2.
Western blotting
Cells were washed with phosphate-buffered saline
(PBS) and then lysed with RIPA buffer consisting of 150 mM NaCl, 10
mM Tris-HCl, pH 8.0, 1% (V/V) Nonidate P-40, 0.5% (W/V) deoxycholic
acid, 0.1% (W/V) SDS, 5 mM EDTA, 1X Halt™ protease inhibitor
cocktail (Thermo Fisher Scientific, Yokohama, Japan), and 1X Halt™
Protein phosphatase inhibitor (Thermo Fisher Scientific). The
protein concentration of the lysates was determined using a
BCA™ Protein assay kit (Thermo Fisher Scientific) and
equal amounts of protein were subjected to SDS-polyacrylamide gel
electrophoresis. The separated proteins were electrophoretically
transferred onto PVDF membranes (GE Healthcare, UK). Non-specific
binding was blocked by incubation in 5% (W/V) bovine serum albumin
(BSA) in TBS/Tween-20 (TBS-T) for 1 h at room temperature.
Membranes were probed with antibodies in TBS-T overnight at 4ºC and
then incubated with HRP-conjugated secondary antibody.
Antibody-antigen complexes were detected by ECL plus western
blotting detection reagent (GE Healthcare).
Scratch wound healing assay
Cell migration was determined by a scratch wound
healing assay as described (20),
with slight modifications. Briefly, cells at a semi-confluence in
12-well plates were treated with 10 μg/ml of mitomycin C for 4 h to
block proliferation and subsequently wounded with a sterile 200-μl
pipette tip to generate a cell-free gap ~1 mm in width. Cells were
then washed with PBS and photographed to record the wound width at
0 h. Next, one group of cells was cultured in DMEM with 10% FBS for
24 h as a control. Other groups were treated with various
concentrations of cetuximab. Twenty-four hours later, photographs
were taken to evaluate migration.
Immunofluorescence staining
Cultured cells were fixed in 3.5% (w/v)
formaldehyde, permeabilized in 0.2% (v/v) Triton X-100, and blocked
in 2% (w/v) BSA. The primary antibodies were incubated at 4ºC
overnight. Alexa fluor 488-conjugated IgG (Life Technologies) was
used as the secondary antibody. After incubation with the
antibodies, SlowFade Gold Antifade reagent with
4′,6-diamidino-2-phenylindole (DAPI; Invitrogen/Life Technologies)
was added. The specimens were observed using fluorescence
microscopy.
Results
Cetuximab inhibits EGFR phosphorylation,
resulting in reduced cell migratory activity, but not growth, of
SAS, an OSCC cell line
We previously reported that SAS cell growth is
poorly sensitive to cetuximab or AG1478 treatment in a monolayer
culture, even though EGFR is expressed and phosphorylated (19), indicating that EGFR phosphorylation
is minimally involved in SAS cell proliferation.
To evaluate the concentration and time-dependency of
cetuximab or AG1478 on oral squamous cell carcinoma (OSCC) cell
proliferation, we exposed HSC3, HSC4, and SAS cells to various
concentrations of cetuximab or AG1478 for 2–6 days and measured
cell activity by MTT assays. The growth rate of SAS cells was not
affected by each concentration of cetuximab in 6 days of culture;
however, proliferation of HSC3 and HSC4 cells were reduced after
2–4 days in culture (Fig. 1A).
Proliferation of HSC3 and HSC4 cells ceased after 2 days in culture
in the presence of >5 μM AG1478; however, growth of SAS cells
was maintained until day 4 of culture and then inhibited after 6
days in 10 μM AG1478 (Fig.
1A).
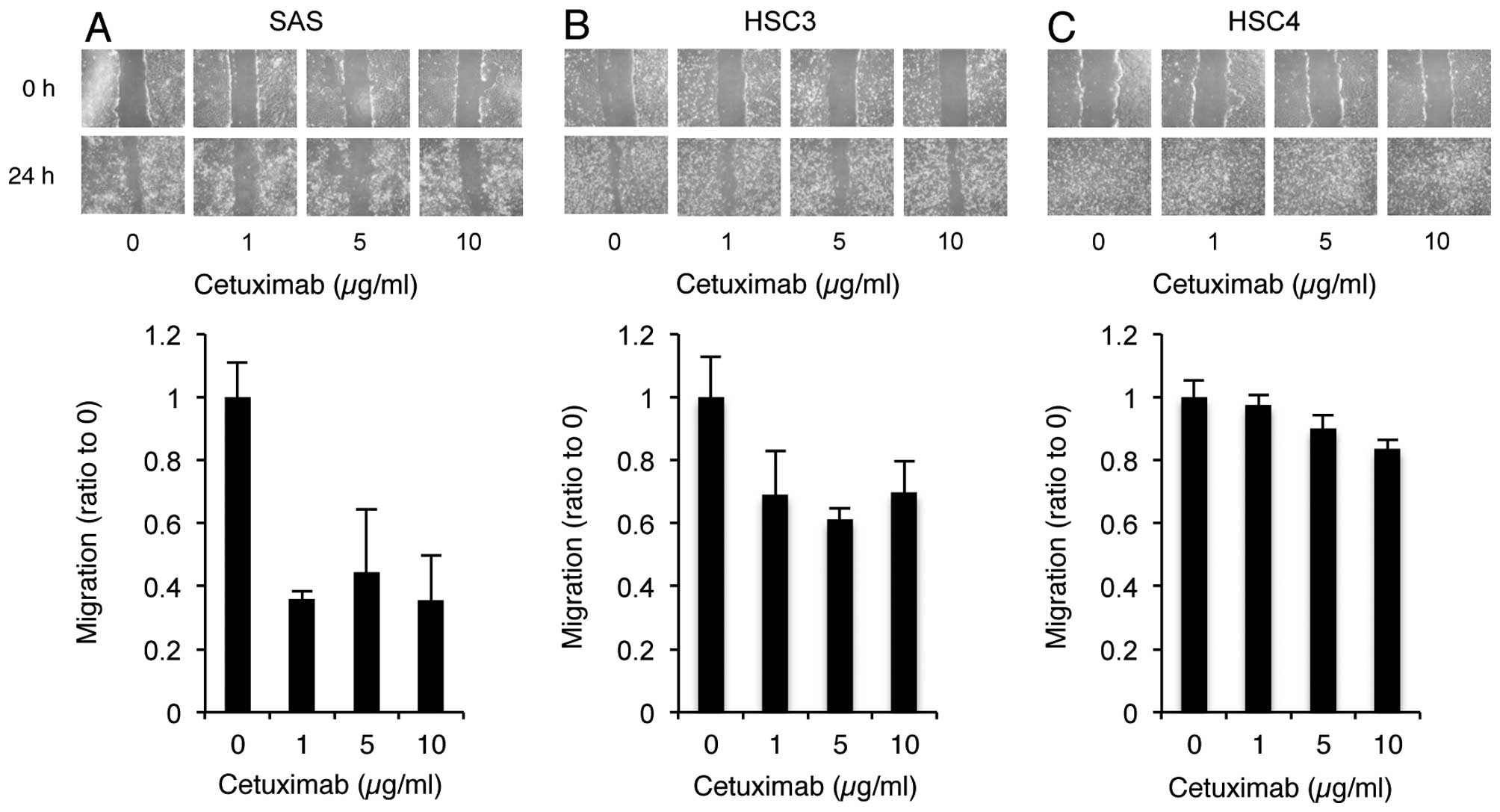
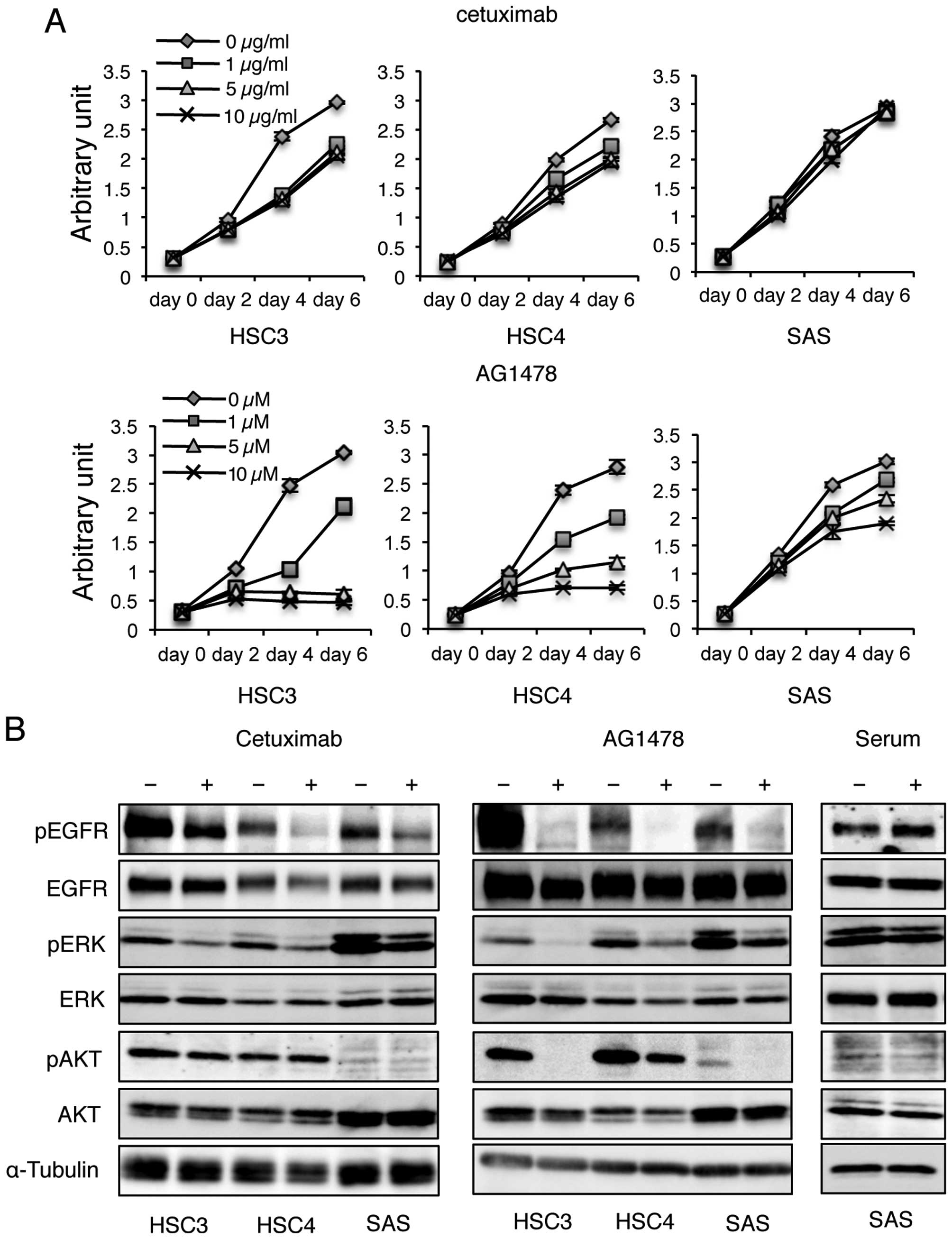 | Figure 1Cetuximab reduces the phosphorylation
levels of EGFR, but does not affect the growth of SAS cells. (A)
Each cell line was cultured in cetuximab or AG1478 at the indicated
concentration. MTT activity was measured on each day of culture.
(B) Cells were treated with cetuximab, AG1478, or serum and the
phosphorylation levels of EGFR, ERK, and AKT were determined by
immunoblotting for EGFR, pEGFR Y1068, ERK, pERK Y209, AKT and pAkt
S473, respectively. α-tubulin was used as loading control. |
Next, we examined whether cetuximab inhibits
phosphorylation of EGFR in SAS monolayer cultures by performing
western blotting using an anti-phospho-EGFR antibody. Cetuximab
treatment clearly reduced the phosphorylation level of EGFR,
although the inhibition by cetuximab was less effective than that
observed by AG1478 (Fig. 1B).
AG1478 treatment almost eliminated EGFR phosphorylation in all
cells examined (Fig. 1B).
Furthermore, EGFR phosphorylation levels were increased by serum
stimulation (Fig. 1B), although
SAS cells actively proliferate in serum-free culture conditions as
described previously (19). These
results indicate that the inhibitory effects of cetuximab on EGFR
phosphorylation are weaker than those of AG1478, and EGFR
phosphorylation signals are not the main factor inducing
proliferation of SAS cells in monolayer cultures.
To investigate the effects of cetuximab treatment on
downstream EGFR signaling events, western blotting was performed
using anti-phospho-ERK and -AKT antibodies. ERK and AKT protein
levels were not altered in any of the tested cell lines following
cetuximab treatment. However, phosphorylated ERK levels were
reduced in all cells, but phosphorylated AKT levels were unchanged
(Fig. 1B), even though
phosphorylated AKT was faintly detected in SAS cells. In response
to AG1478 treatment, ERK phosphorylation levels were further
reduced compared to cetuximab treatment, and AKT phosphorylation
levels were markedly suppressed by AG1478 treatment. These results
suggest that cetuximab and AG1478 treatments exert different
inhibitory effects on EGFR signaling. Inhibition of EGFR
phosphorylation by cetuximab affects the ERK pathway, while
AG1478-induced inhibition of EGFR phosphorylation affects both the
ERK and AKT pathways.
In addition to cell proliferation, EGFR signaling
impacts other important physiological properties, including
migration, differentiation, and apoptosis (21,22).
To determine if cell migration was inhibited in response to the
cetuximab-induced reduction in EGFR phosphorylation, a wound
healing assay in the presence of cetuximab was performed. Cultured
cells that formed a confluent sheet were treated with mitomycin C,
and scratched with a plastic tip. After 24-h culture, the distance
of migration of non-treated and treated cells was measured.
Cetuximab treatment markedly inhibited the migratory activity of
SAS cells (Fig. 2A). Inhibitory
effects of cetuximab were moderate in HSC3 cells, and almost absent
in HSC4 cells (Figs. 2B and C).
These results suggest that alterations in EGFR signaling induced by
cetuximab play an important role in SAS cell migration.
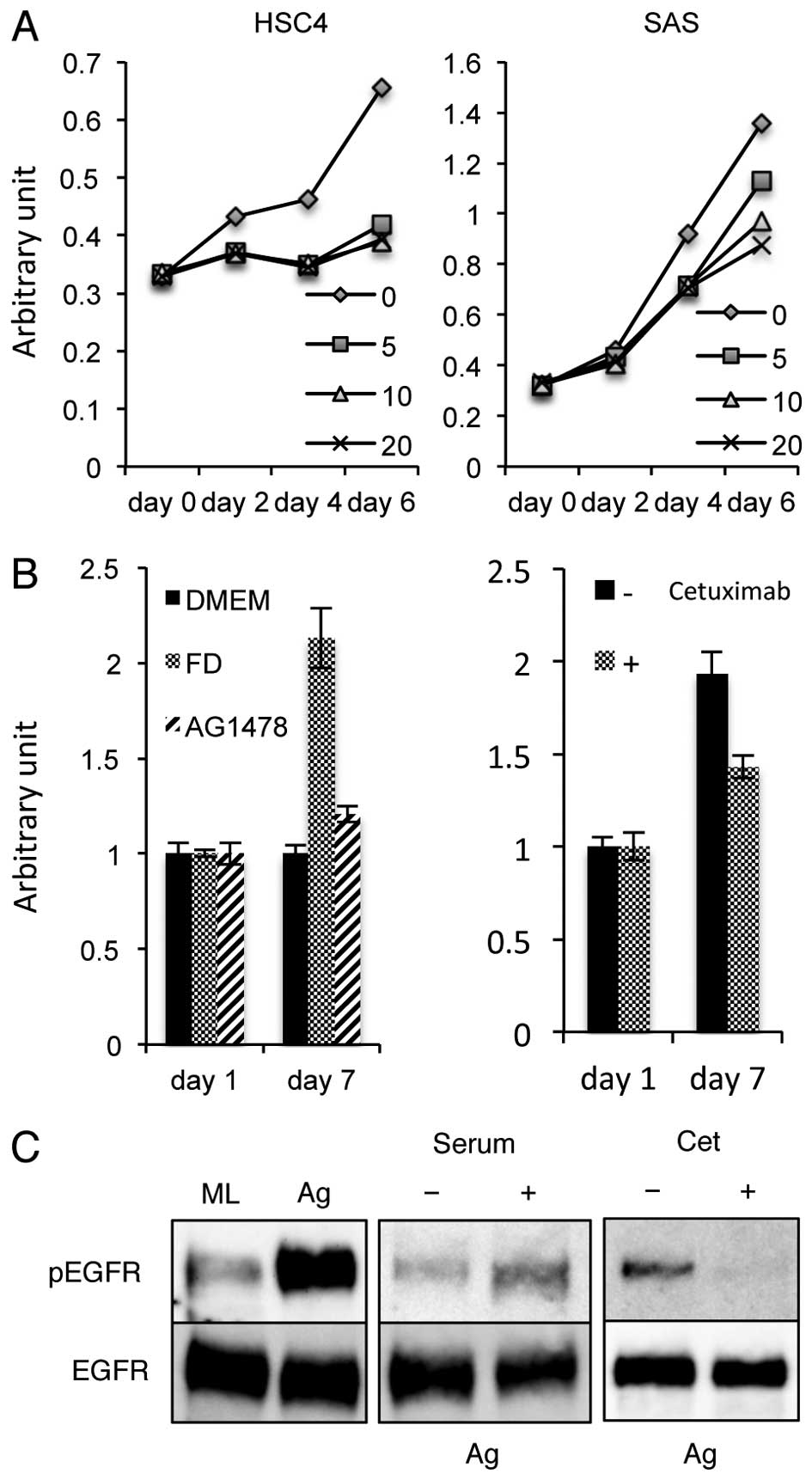
Growth of SAS aggregates is induced by
EGFR signaling through serum stimulation and cetuximab
sensitivity
SAS cell growth was actively maintained in
serum-free culture conditions (19). In order to determine whether the
serum-independent growth of SAS cells is induced by
autocrine/paracrine regulation, the effects of the sheddase
inhibitor TAPI-2 on SAS cell proliferation were examined. HSC4
cells, which proliferate weakly in serum-free culture conditions,
ceased to undergo proliferation in response to TAPI-2 treatment,
but SAS cell growth was maintained throughout 6 days of culture
with TAPI-2 (Fig. 3A), indicating
that the serum-independent growth of SAS cells is not induced
through the release of factors by ADAM17, as observed in HSC4.
These data suggest that SAS cell growth in monolayer culture
conditions is independent of ligand stimulation.
SAS cells become aggregates in floating culture in
low-adhesive U-shaped 96-well plates. Growth of SAS aggregates
ceased in serum-free culture conditions, and when treated with
cetuximab or AG1478 treatment (Fig.
3B), consistent with a previous report (19). These data indicate that EGFR
stimulation is involved in the growth of SAS aggregates. To
investigate the level of EGFR phosphorylation in aggregates treated
with cetuximab, we performed western blotting using phospho-EGFR
specific antibodies. Phosphorylation levels of EGFR were increased
in aggregates compared to cells cultured in a monolayer and were
slightly upregulated by the addition of serum (Fig. 3C). Furthermore, cetuximab treatment
of SAS aggregates almost inhibited the EGFR phosphorylation
(Fig. 3C). These results suggest
that EGFR phosphorylation of SAS aggregates is induced in response
to serum stimulation, and facilitates the transition of SAS
aggregate growth to become sensitive to cetuximab.
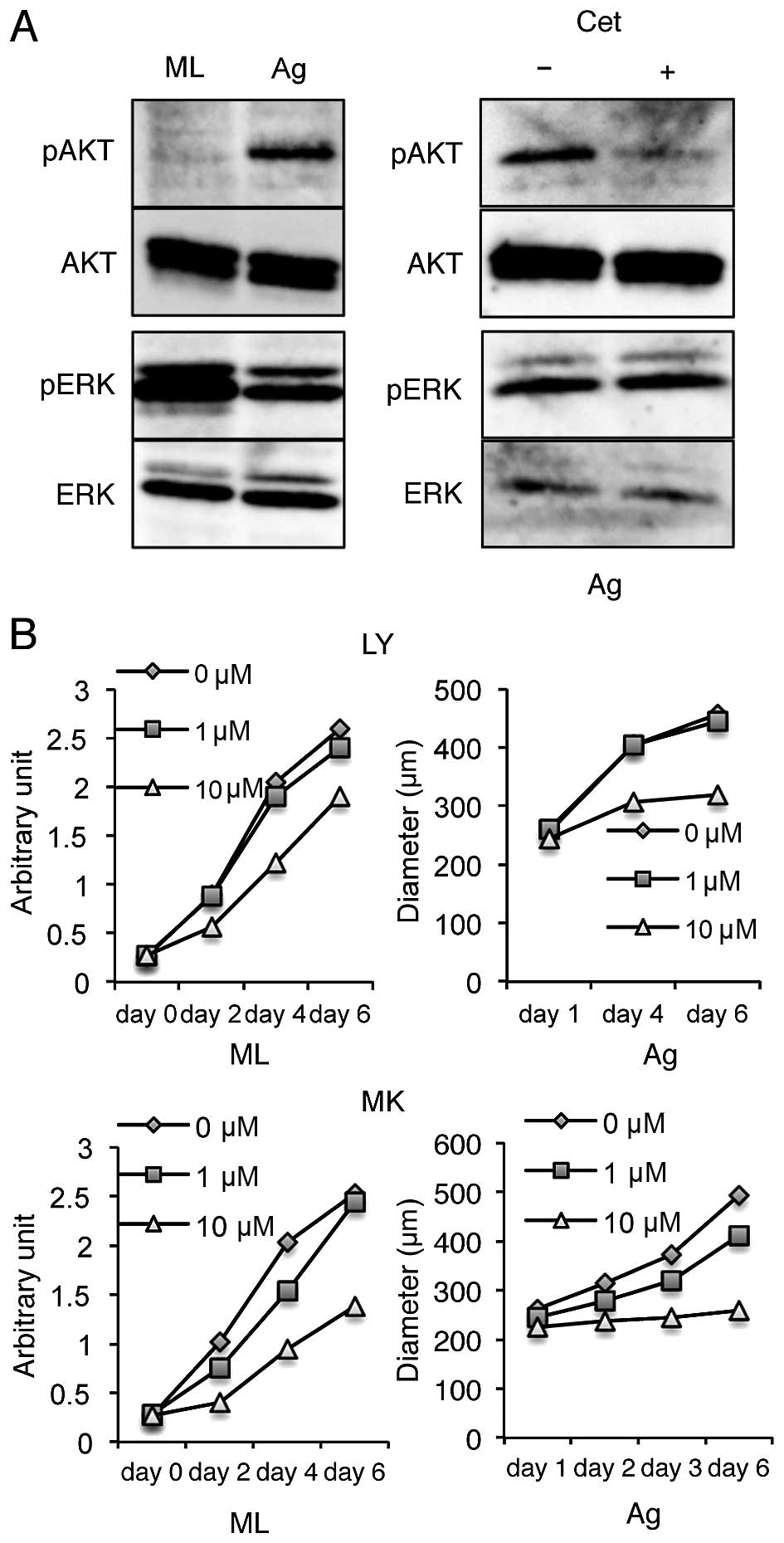
Cetuximab sensitivity of SAS aggregate
growth is EGFR-PI3K-AKT pathway-dependent
Signaling through the EGFR is transmitted to the
nucleus through various routes. In the present study, we
investigated the involvement of the MAPK/ERK and the PI3K-AKT
pathways downstream of EGFR activation in SAS aggregates. ERK is
phosphorylated in SAS monolayer cultures, whereas, AKT is weakly
phosphorylated (Fig. 4A). The
phosphorylation of Akt increased, but the phosphorylation of ERK
decreased in SAS aggregates (Fig.
4A) compared to monolayer cultures. Moreover, AKT
phosphorylation was suppressed by cetuximab treatment in
aggregates, and the phosphorylation of ERK was not affected by
cetuximab treatment in aggregates (Fig. 4A). These results indicate that AKT
phosphorylation in SAS aggregates was suppressed by the inhibition
of EGFR phosphorylation.
To assess whether the PI3K-AKT pathway is required
for the growth of SAS aggregates, the effect of the PI3K inhibitor
LY294002 or the AKT inhibitor MK2206 on SAS aggregate proliferation
was investigated. Proliferation of SAS cells was not inhibited by
either agent at 10 μM in a monolayer culture (Fig. 4B). However, proliferation of SAS
aggregates nearly ceased following addition of 10 μM of both
inhibitors (Fig. 4B). These
results suggest that the EGFR-PI3K-AKT pathway plays a cruicial
role in SAS growth under anchorage-independent conditions.
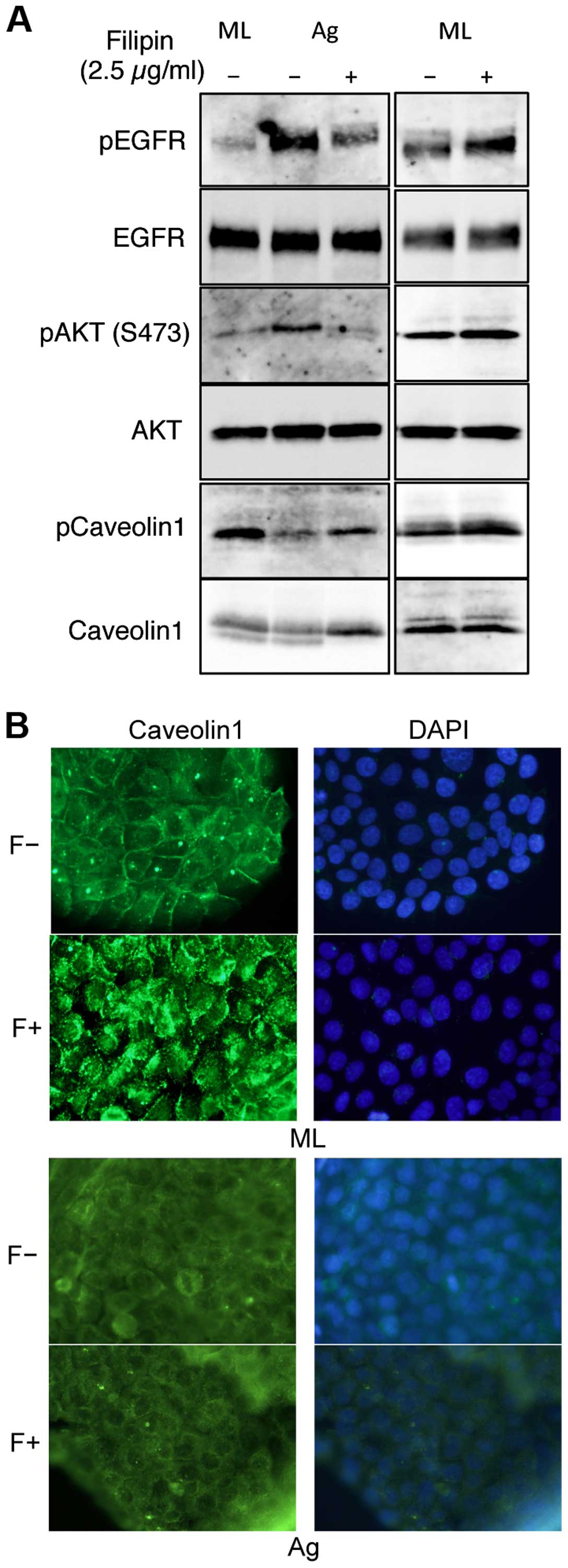
EGFR is stimulated in lipid rafts in SAS
aggregates
The finding that phosphorylation of EGFR and AKT was
upregulated by aggregation in floating cultures of SAS cells
indicates that distinct EGFR signaling pathways are used in
different culture conditions. EGFR is localized mainly at the
plasma membrane and is activated by signals from the environment.
The plasma membrane contains discrete heterogeneous micro-domains
(23), including lipid rafts that
act as platforms for cellular signaling (24). EGFR has been reported to be
localized in the lipid rafts (25). To explore whether lipid rafts may
also provide such a platform of EGFR phosphorylation in SAS
aggregates, we sought to assess the effects of lipid raft
disruption. We used the filipin III, which preferentially removes
cholesterol from plasma membranes, to perturb the lipid rafts
(26–28). Filipin III treatment inhibited the
phophorylation of EGFR and AKT in SAS aggregates, but not in
monolayer cultures (Fig. 5A),
indicating that lipid rafts are involved in EGFR transactivation in
SAS aggregates.
A previous report showed that caveolin-1 (cav1)
phosphorylation is required for EGFR and AKT activation in a
specific environment (29). Thus,
we next examined cav1 expression and its phosphorylation in SAS
aggregates. Cav1 was detected in equal amounts in both SAS cell
aggregates and in monolayers. However, its phosphorylation levels
were only downregulated in aggregates (Fig. 5A). Immunocytological staining
demonstrated that in SAS monolayer cultures, cav1 was localized on
the cell surface and transloated into the cytoplasm in response to
filipin III treatment (Fig. 5B,
ML). However, cav1 was localized in the cytoplasm in SAS aggregates
and its localization was not affected by filipin III treatment
(Fig. 5B, Ag). These results
indicate that in SAS aggregates, non-phosphorylated cav1 was
localized in the cytoplasm and not involved with lipid rafts.
Discussion
The mechanism of EGFR activation has been described
as the ligand binding inducing the dimerization and activation of
the cytoplasmic kinase domains, resulting in the phosphorylation of
tyrosine residues in the C-terminus and the subsequent recruitment
of downstream effectors (6,30).
In addition, several reports have shown that EGFR can be activated
without its ligand (29,31,32).
Cetuximab binds to the EGFR with a high affinity comparable to that
of its ligands (34), and prevents
ligand binding and receptor activation. Thus, the effects of
cetuximab are restricted to cellular physiology regulated by
ligand-dependent EGFR activation. Growth of SAS cells in monolayer
cultures persists in serum-free medium and was inhibited by AG1478
treatment, but not by cetuximab treatment (Fig. 1). Wound repair by SAS cells was
inhibited by cetuximab treatment (Fig.
2). These data show that dual systems of EGFR activation,
including ligand-independent and ligand-dependent EGFR activation,
play distinct roles in SAS monolayer cultures, in cell growth and
in wound repair, respectively.
Cetuximab treatment reduced phosphorylation levels
of EGFR and ERK in all cell lines examined, whereas phosphorylation
of Akt was generally not affected by cetuximab treatment (Fig. 1). AG1478 treatment suppressed
growth of SAS cells and the phosphorylation levels of ERK and Akt
in monolayer culture conditions (Fig.
1). However, the PI3K inhibitor LY294002 and the Akt inhibitor
MK2204 did not prevent SAS cell growth in a monolayer culture
(Fig. 3). Taken together, these
data suggest that the EGFR-ERK pathway may play a role in the
growth of SAS cells in monolayer cultures. In contrast, the
phosphorylation levels of EGFR and Akt were upregulated in SAS
aggregates (Fig. 3), growth of
which is serum-dependent. Furthermore, growth was inhibited by
cetuximab, AG1478, LY294002, and MK2206 (Figs. 3 and 4), indicating the growth of SAS
aggregates was promoted by ligand-dependent EGFR activation through
the PI3K-Akt pathway.
Anchorage-mediating structures on the extracellular
matrix of epithelial cells serve a mechanical function and provide
important survival signals to the cell. Detachment from the
substrate, loss of cell anchorage, and concomitant loss of such
survival signals leads to the induction of apoptosis, which is
termed anoikis, in the majority of adherent cells (34–36).
Acquisition of anoikis resistance of cancer cells constitutes an
essential prerequisite for tumor progression and metastases in most
cancers of epithelial origin (37,38).
In addition, anchorage-independent growth is a hallmark of cell
transformation and is connected to elevated tumorigenic potential
(39). Herein, we demonstrated
that activation of the ligand-dependent EGFR/PI3K/Akt pathway,
other than anoikis resistance, is necessary for
anchorage-independent growth during metastasis, consistent with a
recent report (40).
Lipid rafts, a plasma membrane subdomain, facilitate
the organization of the specific molecular distribution and
regulate the activity of receptors and proximal effectors of
signaling (41). Our results
demonstrated that the high levels of EGFR and Akt phosphorylation
observed in suspension cultures of SAS cells were decreased when
lipid rafts were disrupted with filipin III (Fig. 5). Thus, we speculate that lipid
rafts serve as a platform in which EGFR and PI3K co-localize in the
plasma membrane of SAS aggregates, thereby transmitting growth
signals to the PI3K-Akt pathway through EGFR activation by ligand
binding.
Cav1, a major component of caveolae and the cav1
scaffold in the plasma membrane subdomain (41), is involved in both tumor
suppression and oncogenesis, depending on the tumor type and stage
of progression (42,43). Cav1 interacts with EGFR and
negatively regulates EGFR activity (44). In the present study, Cav1 protein
was detected in the cytoplasm of SAS aggregates by immunostaining
(Fig. 5), indicating that Cav1 was
not involved with EGFR activity in the plasma membrane of
aggregated cells.
Wound repair of non-dividing SAS cells was markedly
inhibited by cetuximab treatment (Fig.
2), suggesting that ligand-dependent EGFR activation is
associated with migration of SAS cells in monolayers, as cell
migration has been shown to be a fundamental step in wound repair
(45–49). Tyrosine 14 phosphorylation of Cav1
regulates its interaction with integrins, various signaling
adaptors, and protein tyrosine phosphatases (50). A galectin-3/phosphorylated Cav1/Rho
A signaling module that mediates integrin signaling downstream of
growth factor activation, leading to actin and matrix remodeling
and tumor-cell migration in metastatic cancer cells (51). Thus, phosphorylated Cav1 may
promote cell migration through EGFR activation in SAS monolayer
cultures and de-phosphorylated Cav1 moves into the cytoplasm in SAS
aggregates. We demonstrate the possibility that cetuximab inhibits
metastasis and cell growth, because cell migration is essential for
cancer metastasis.
Cetuximab treatment in combination with chemotherapy
and radiotherapy has shown a survival benefit (52,53).
However, resistance to cetuximab caused by mutations in EGFR or
downstream effectors have been reported (54–58).
This study provides evidence that environmental stimuli alter the
direction of EGFR signal transduction, resulting in a change in
cetuximab sensitivity.
Acknowledgements
This manuscript has been edited for English language
by Textcheck English consultants. Funding for this study was
provided by Osaka University (M.N.) and Osaka Dental University
(K.K.).
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
|
OSCC
|
oral squamous cell carcinoma
|
|
DMEM
|
Dulbeco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen EE, Lingen MW and Vokes EE: The
expanding role of systemic therapy in head and neck cancer. J Clin
Oncol. 22:1743–1752. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cooper JS, Pajak TF, Forastiere AA, Jacobs
J, Campbell BH, Saxman SB, Kish JA, Kim HE, Cmelak AJ, Rotman M, et
al; Radiation Therapy Oncology Group 9501/Intergroup. Postoperative
concurrent radiotherapy and chemotherapy for high-risk
squamous-cell carcinoma of the head and neck. N Engl J Med.
350:1937–1944. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biscardi JS, Tice DA and Parsons SJ:
c-Src, receptor tyrosine kinases, and human cancer. Adv Cancer Res.
76:61–119. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abram CL and Courtneidge SA: Src family
tyrosine kinases and growth factor signaling. Exp Cell Res.
254:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schlessinger J: Cell signaling by receptor
tyrosine kinases. Cell. 103:211–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Blume-Jensen P and Hunter T: Oncogenic
kinase signalling. Nature. 411:355–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Prenzel N, Fischer OM, Streit S, Hart S
and Ullrich A: The epidermal growth factor receptor family as a
central element for cellular signal transduction and
diversification. Endocr Relat Cancer. 8:11–31. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marmor MD, Skaria KB and Yarden Y: Signal
transduction and oncogenesis by ErbB/HER receptors. Int J Radiat
Oncol Biol Phys. 58:903–913. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37(Suppl 4): S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grandis JR and Tweardy DJ: Elevated levels
of transforming growth factor α and epidermal growth factor
receptor messenger RNA are early markers of carcinogenesis in head
and neck cancer. Cancer Res. 53:3579–3584. 1993.PubMed/NCBI
|
|
13
|
Rubin Grandis J, Melhem MF, Gooding WE,
Day R, Holst VA, Wagener MM, Drenning SD and Tweardy DJ: Levels of
TGF-α and EGFR protein in head and neck squamous cell carcinoma and
patient survival. J Natl Cancer Inst. 90:824–832. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ang KK, Berkey BA, Tu X, Zhang H-Z, Katz
R, Hammond EH, Fu KK and Milas L: Impact of epidermal growth factor
receptor expression on survival and pattern of relapse in patients
with advanced head and neck carcinoma. Cancer Res. 62:7350–7356.
2002.PubMed/NCBI
|
|
15
|
Galizia G, Lieto E, De Vita F, Orditura M,
Castellano P, Troiani T, Imperatore V and Ciardiello F: Cetuximab,
a chimeric human mouse anti-epidermal growth factor receptor
monoclonal antibody, in the treatment of human colorectal cancer.
Oncogene. 26:3654–3660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ,
Kussie P and Ferguson KM: Structural basis for inhibition of the
epidermal growth factor receptor by cetuximab. Cancer Cell.
7:301–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boeckx C, Baay M, Wouters A, Specenier P,
Vermorken JB, Peeters M and Lardon F: Anti-epidermal growth factor
receptor therapy in head and neck squamous cell carcinoma: Focus on
potential molecular mechanisms of drug resistance. Oncologist.
18:850–864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rebucci M, Peixoto P, Dewitte A, Wattez N,
De Nuncques MA, Rezvoy N, Vautravers-Dewas C, Buisine MP, Guerin E,
Peyrat JP, et al: Mechanisms underlying resistance to cetuximab in
the HNSCC cell line: Role of AKT inhibition in bypassing this
resistance. Int J Oncol. 38:189–200. 2011.
|
|
19
|
Ohnishi Y, Minamino Y, Kakudo K and Nozaki
M: Resistance of oral squamous cell carcinoma cells to cetuximab is
associated with EGFR insensitivity and enhanced stem cell-like
potency. Oncol Rep. 32:780–786. 2014.PubMed/NCBI
|
|
20
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFbeta-mediated fibroblastic transdifferentiation and cell
migration. J Cell Sci. 115:3193–3206. 2002.PubMed/NCBI
|
|
21
|
Pal HC, Sharma S, Strickland LR, Agarwal
J, Athar M, Elmets CA and Afaq F: Delphinidin reduces cell
proliferation and induces apoptosis of non-small-cell lung cancer
cells by targeting EGFR/VEGFR2 signaling pathways. PLoS One.
8:e772702013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Morozevich GE, Kozlova NI, Ushakova NA,
Preobrazhenskaya ME and Berman AE: Integrin α5β1 simultaneously
controls EGFR-dependent proliferation and Akt-dependent
pro-survival signaling in epidermoid carcinoma cells. Aging
(Albany, NY). 4:368–374. 2012.
|
|
23
|
Maa MC, Leu TH, McCarley DJ, Schatzman RC
and Parsons SJ: Potentiation of epidermal growth factor
receptor-mediated oncogenesis by c-Src: Implications for the
etiology of multiple human cancers. Proc Natl Acad Sci USA.
92:6981–6985. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Simons K and Ikonen E: Functional rafts in
cell membranes. Nature. 387:569–572. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu W, Graves LM, Gill GN, Parsons SJ and
Samet JM: Src-dependent phosphorylation of the epidermal growth
factor receptor on tyrosine 845 is required for zinc-induced Ras
activation. J Biol Chem. 277:24252–24257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bolard J: How do the polyene macrolide
antibiotics affect the cellular membrane properties? Biochim
Biophys Acta. 864:257–304. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Montesano R, Vassalli P and Orci L:
Structural heterogeneity of endocytic membranes in macrophages as
revealed by the cholesterol probe, filipin. J Cell Sci. 51:95–107.
1981.PubMed/NCBI
|
|
28
|
Santos NC, Ter-Ovanesyan E, Zasadzinski
JA, Prieto M and Castanho MA: Filipin-induced lesions in planar
phospholipid bilayers imaged by atomic force microscopy. Biophys J.
75:1869–1873. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang B, Peng F, Wu D, Ingram AJ, Gao B
and Krepinsky JC: Caveolin-1 phosphorylation is required for
stretch-induced EGFR and Akt activation in mesangial cells. Cell
Signal. 19:1690–1700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen X and Kramer RH: Adhesion-mediated
squamous cell carcinoma survival through ligand-independent
activation of epidermal growth factor receptor. Am J Pathol.
165:1315–1329. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lambert S, Vind-Kezunovic D, Karvinen S
and Gniadecki R: Ligand-independent activation of the EGFR by lipid
raft disruption. J Invest Dermatol. 126:954–962. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goldstein NI, Prewett M, Zuklys K,
Rockwell P and Mendelsohn J: Biological efficacy of a chimeric
antibody to the epidermal growth factor receptor in a human tumor
xenograft model. Clin Cancer Res. 1:1311–1318. 1995.PubMed/NCBI
|
|
34
|
Frisch SM and Francis H: Disruption of
epithelial cell-matrix interactions induces apoptosis. J Cell Biol.
124:619–626. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frisch SM, Vuori K, Ruoslahti E and
Chan-Hui PY: Control of adhesion-dependent cell survival by focal
adhesion kinase. J Cell Biol. 134:793–799. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Meredith JE Jr, Fazeli B and Schwartz MA:
The extracellular matrix as a cell survival factor. Mol Biol Cell.
4:953–961. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu Z, Li H, Wu X, Yoo BH, Yan SR, Stadnyk
AW, Sasazuki T, Shirasawa S, LaCasse EC, Korneluk RG, et al:
Detachment-induced upregulation of XIAP and cIAP2 delays anoikis of
intestinal epithelial cells. Oncogene. 25:7680–7690. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rosen K, Coll ML, Li A and Filmus J:
Transforming growth factor-alpha prevents detachment-induced
inhibition of c-Src kinase activity, Bcl-XL down-regulation, and
apoptosis of intestinal epithelial cells. J Biol Chem.
276:37273–37279. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu P, Begley M, Michowski W, Inuzuka H,
Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al:
Cell-cycle-regulated activation of Akt kinase by phosphorylation at
its carboxyl terminus. Nature. 508:541–545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lajoie P, Goetz JG, Dennis JW and Nabi IR:
Lattices, rafts, and scaffolds: Domain regulation of receptor
signaling at the plasma membrane. J Cell Biol. 185:381–385. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Williams TM, Hassan GS, Li J, Cohen AW,
Medina F, Frank PG, Pestell RG, Di Vizio D, Loda M and Lisanti MP:
Caveolin-1 promotes tumor progression in an autochthonous mouse
model of prostate cancer: Genetic ablation of Cav-1 delays advanced
prostate tumor development in tramp mice. J Biol Chem.
280:25134–25145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Quann K, Gonzales DM, Mercier I, Wang C,
Sotgia F, Pestell RG, Lisanti MP and Jasmin JF: Caveolin-1 is a
negative regulator of tumor growth in glioblastoma and modulates
chemosensitivity to temozolomide. Cell Cycle. 12:1510–1520. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Couet J, Sargiacomo M and Lisanti MP:
Interaction of a receptor tyrosine kinase, EGF-R, with caveolins.
Caveolin binding negatively regulates tyrosine and serine/threonine
kinase activities. J Biol Chem. 272:30429–30438. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Blay J and Brown KD: Epidermal growth
factor promotes the chemotactic migration of cultured rat
intestinal epithelial cells. J Cell Physiol. 124:107–112. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen JD, Kim JP, Zhang K, Sarret Y, Wynn
KC, Kramer RH and Woodley DT: Epidermal growth factor (EGF)
promotes human keratinocyte locomotion on collagen by increasing
the alpha 2 integrin subunit. Exp Cell Res. 209:216–223. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fujii K, Dousaka-Nakajima N and Imamura S:
Epidermal growth factor enhancement of HSC-1 human cutaneous
squamous carcinoma cell adhesion and migration on type I collagen
involves selective up-regulation of alpha 2 beta 1 integrin
expression. Exp Cell Res. 216:261–272. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matthay MA, Thiery JP, Lafont F, Stampfer
F and Boyer B: Transient effect of epidermal growth factor on the
motility of an immortalized mammary epithelial cell line. J Cell
Sci. 106:869–878. 1993.PubMed/NCBI
|
|
49
|
Basson MD, Modlin IM and Madri JA: Human
enterocyte (Caco-2) migration is modulated in vitro by
extracellular matrix composition and epidermal growth factor. J
Clin Invest. 90:15–23. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Goetz JG, Lajoie P, Wiseman SM and Nabi
IR: Caveolin-1 in tumor progression: The good, the bad and the
ugly. Cancer Metastasis Rev. 27:715–735. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Boscher C and Nabi IR: Galectin-3- and
phospho-caveolin-1-dependent outside-in integrin signaling mediates
the EGF motogenic response in mammary cancer cells. Mol Biol Cell.
24:2134–2145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, et al: Platinum-based chemotherapy plus cetuximab in head and
neck cancer. N Engl J Med. 359:1116–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sartore-Bianchi A, Martini M, Molinari F,
Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P,
De Dosso S, Mazzucchelli L, et al: PIK3CA mutations in colorectal
cancer are associated with clinical resistance to EGFR-targeted
monoclonal antibodies. Cancer Res. 69:1851–1857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sartore-Bianchi A, Di Nicolantonio F,
Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani
T, Marrapese G, Mazzucchelli L, et al: Multi-determinants analysis
of molecular alterations for predicting clinical benefit to
EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One.
4:e72872009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Laurent-Puig P, Lievre A and Blons H:
Mutations and response to epidermal growth factor receptor
inhibitors. Clin Cancer Res. 15:1133–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
De Rock W, Lambrechts D and Tejpar S:
K-ras mutations and cetuximab in colorectal cancer. N Engl J Med.
360:8342009.
|
|
58
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|