Introduction
Hepatocellular carcinoma (HCC) is a major cause of
cancer-related death worldwide with extremely poor prognosis
(1). Liver transplantation and
surgical resection are potentially curative options for HCC
patients at an early stage of cancer progression. However, when the
disease develops to an advanced stage, these treatments are not
useful. Moreover, the most challenging problem for HCC clinical
treatment is a high incidence of tumor recurrence and metastasis
(2). Currently, sorafenib, an oral
multikinase inhibitor targeting RAF kinases, is the only approved
targeted therapy to prolong the overall survival of patients with
advanced HCC (3). Molecular
mechanisms of malignant transformation of hepatocytes, and
development and progression of HCC still remain largely elusive.
Therefore, identification of key genes that serve as early
diagnosis and novel therapeutic targets is urgently needed.
AP-2α extensively functions as a tumor suppressor in
various types of human solid tumors, including melanoma, breast
cancer, prostate cancer and colorectal cancer. Clinical studies
demonstrated that loss of AP-2α expression associates with cancer
progression and predicts poor prognosis (4–9).
Exogenous AP-2α in cancer cell lines inhibited cell growth in
vitro (10), and transfection
of AP-2α reduced the tumorigenicity and metastatic potential in
nude mice, whereas inactivation of AP-2α enhanced tumorigenicity
(7,11,12).
However, some data showed that low cytoplasmic and high nuclear
AP-2α expression were related to increased tumor malignancy and
poor survival (13–17), suggesting that AP-2α acts as an
oncogene in certain histological subtypes, such as acute myeloid
leukemia (18) squamous cell
carcinomas (19) and head and neck
squamous cell carcinoma (HNSCC) (20).
AP-2α was able to inhibit cancer cell growth and
motility, by modulating many downstream pathways including the
HIF-1α-mediated VEGF/PEDF signaling pathway, β-catenin/TCF/LEF
signaling, bax/cytochrome c/Apaf1/caspase 9-dependent mitochondrial
pathway, CdK-inhibitor p21WAF in p53-dependent and -independent
pathways (7,21–24),
thus suppressing angiogenesis, cell cycle progression, cell
proliferation and invasion. AP-2α expression sensitized cancer
cells to chemotherapy and loss of AP-2α led to chemoresistance
(25,26). Adriamycin, cisplatin, taxol,
etoposide and carboplatin could synergistically combine with AP-2α
expression to enhance tumor killing (26). Alteration or post-translational
modification in AP-2α protein can affect molecular pathogenesis of
human cancers (27,28). Although AP-2α is reported to
function in hepatocellular carcinogenesis (10), the molecular mechanisms are still
elusive. To further study the role of AP-2α in human
carcinogenesis, we compared the expression of AP-2α between human
hepatocellular carcinoma tissues and live cancer cell lines.
Moreover, the effects of AP-2α on cellular proliferation,
migration, invasion and stem cell sphere-forming were detected not
only in vitro but also in vivo assays. Furthermore,
whether AP-2α increased the sensitivity of hepatocellular cancer
cells to cisplatin and the molecular mechanisms of AP-2α in
hepatocellular cancer cells were investigated in this study.
Materials and methods
Immunohistochemistry
Ninety hepatocellular carcinoma samples and 18
adjacent normal tissues were examined. The experiment was approved
by Hunan Normal University Human Ethics Committee and informed
consent was obtained from all patients. The immunohistochemical
staining was performed on paraffin-embedded samples. Sections (5-μm
thick) were deparaffinized and rehydrated. Endogenous peroxidase
was quenched with 3% hydrogen peroxide (H2O2)
for 10 min. For antigen retrieval, sections were immersed in 0.01 M
citrate buffer (pH 6.0) and boiled for 10 min in microwave oven.
Non-specific binding was blocked by 5% normal goat serum in PBS for
30 min. The mouse monoclonal anti-AP-2α antibody (1:100 dilution,
Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added at 4ºC
overnight in a moist chamber. The sections were then sequentially
incubated with HRP-conjugated goat anti-mouse IgG (1:500 dilution,
Sigma-Aldrich Corp., St. Louis, MO, USA) for 45 min and
3,3-diaminobenzidine (DAB)/H2O2 for 10 min at
room temperature. The sections were counterstained with
hematoxylin, mounted and photographed using an optical microscope
(Olympus CX41, Tokyo, Japan). The percentage of tumor cells stained
was scored as: 0 (no cell staining), 1 (≤30%), 2 (31–60%) and 3
(61–100%). The staining between two score values was given 0.5.
Cell culture
SMMC-7721, Hep3B, HepG2, MHHC 97-H and HeLa cells
were cultured in DMEM medium (Gibco BRL, Grand Island, NY, USA)
supplemented with 10% fetal calf serum (FBS, Hyclone, Australia),
100 U/ml penicillin and 100 μg/ml streptomycin. All the cells were
maintained in a humidified atmosphere with 5% CO2 at
37ºC.
AP-2α overexpression lentivirus
generation
The full-length AP-2α cDNA was subcloned to
pGC-FU-3xFlag-IRES-Puromycin vector, and pGCFU-GFP-3xFlag
-IRES-Puromycin vector served as a negative control (NC).
Lentiviral particles were prepared as described in our previous
work (18). Briefly, the
lentivirus expression plasmid and packaging plasmids (pHelper 1.0
and pHelper 2.0) were cotransfected into 293T cells, supernatants
were harvested 48 h after transfection and filtered through a
0.45-μm pore size filter (Millipore, Billerica, MA, USA) and
concentrated by ultracentrifugation. The infectious titer was
determined using hole-by-dilution titer assay. SMMC-7721 and Hep3B
cells were infected with AP-2α-Flag-lentivirus or
GFP-Flag-lentivirus at the multiplicity of infection (MOI) of 10 in
the presence of 5 μg/ml polybrene (Sigma) and detected on the 4th
day by the invert fluorescence microscope followed by the screening
of 1.5 μg/ml of puromycine for stable cell lines.
Cell survival assay
100,000 cells stably expressing AP-2α-Flag or
GFP-Flag and parental cells were plated in triplicate in 6-well
plates in complete medium. After 1–4 days, cell numbers were
counted with a hemocytometer after trypan blue staining of viable
cells.
MTT assay
To detect the cell growth rate, 3,000 cells per well
were plated in 96-well plates untreated or treated with 30 μM of
cisplatin (DDP) or 0.9% NaCl (NS) for 48 h. On day 1–7, cells were
analyzed with 1 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
Sigma) at 37ºC for 4 h. Then 100 μl dimethylsulfoxide (DMSO) per
well was added to dissolve the formazan crystals. The absorbency at
490 nm was measured with a spectrophotometer (UV-2102C, Changsha,
China). Triplicate independent assays were performed in
octuplicate.
Liquid colony formation was performed as
previously described (29)
Briefly, 2,000 cells were seeded in duplicate in
6-well plates and grown for over 10 days. Colonies were fixed with
methanol, stained with Giemsa (BBI International, Cardiff, UK) and
photographed with the digital camera (Olympus). Only colonies
containing over 30 cells were counted. All experiments were carried
out for at least three times.
Tumor xenograft mouse model
Approximately 2×107 of
lentivirus-infected AP-2α-SMMC-7721 or GFP-SMMC-7721 cells in 0.2
ml of sterile PBS were injected subcutaneously into the left and
right dorsal regions of 4- to 5-week-old female nude mice (n=6
mice), respectively. Mice were checked every 2 days and the formed
tumors were measured as previously described (30). After 41 days, mice were sacrificed,
and tumors were excised, weighed and photographed. The mouse
experiments were carried out according to the ethical guidelines
for laboratory animal use and approved by the Ethics Committee of
Hunan Normal University.
Wound-healing assay
SMMC-7721 cells were cultured in 6-well plates until
over 95% confluence. A 100-μl pipette tip was used to generate
wounds. After wound creation, the medium was changed to remove
cellular debris. Three wounded areas in each well were marked on
the bottom of plates and photographed at 1 and 3 days with an
invert microscope (Zeiss Axioskop 2, LLC, Thornwood, NY, USA).
Cell migration and invasion assay
The Transwell cell migration and invasion assays
were performed in polyethylene terephthalate (PET)-based migration
chambers and BD BioCoat Matrigel Invasion Chambers (BD Biosciences,
Bedford, MA, USA) with 8 μm porosity according to the
manufacturer's instructions. Tumor cells (4×104) in
serum-free DMEM/F12 were seeded onto uncoated or Matrigel-coated
filters in the upper chambers. DMEM/F12 containing 15% FBS was
added to the lower chambers. After 48–72 h of incubation, cells on
the upper surface of the filters were removed with a cotton swab,
and the filters were fixed with 100% methanol and stained with
Giemsa. The migration and invasive ability of the tumor cells was
expressed as the mean number of cells in all fields. The
experiments were performed three times independently.
Sphere-forming and self-renewal
assay
Tumor cells were seeded at a density of 10,000
cells/ml in low-attachment culture bottles and grown in serum-free
DMEM/F12 supplemented with 20 ng/ml epidermal growth factor, 10
ng/ml basic fibroblast growth factor (PeproTech, Inc., Rocky Hill,
NJ, USA), 0.4 μg/ml insulin and B27 (1:50; Gibco, Karlsruhe,
Germany). The number of spheres was counted after 5 or 10 days,
respectively. After sphere-forming culture, photos were observed
with the invert microscope every 2 days to observe cell size and
morphology. The sphere-forming cells were separated into single
cells using 1× Trypsin-EDTA and analyzed by cell survival and MTT
assays.
To investigate self-renewal capacity of liver cancer
spheres, single cell suspension from the sphere forming cells was
prepared and plated in 96-well ultra-low plates containing 200 μl
serum-free medium per well. Single cell in these wells was
monitored daily under the invert microscope for 9 days.
To analyze the effects of cisplatin on sphere
forming of AP-2α-overexpressing SMMC-7721 cells, suspension cells
from the sphere forming cells were plated at a density of 10,000
cells/well in 24-well ultra-low plate. Cisplatin (DDP) (30 μM) or
0.9% NaCl (NS) was added to medium on day 2. After further
culturing for 3 days, the spheres were analyzed by optical
microscopy, MTT assays and cell counting.
Western blotting
Cells were lysed in RIPA buffer as previously
described (31). Briefly, the
lysates were denatured and heated to 95ºC for 5 min. Samples were
then separated on 10–15% SDS-PAGE gels and transferred to
polyvinylidene difluoride (PVDF) membranes. The blots were detected
with rabbit polyclonal antibodies against GFP and MMP9, mouse
monoclonal antibodies against ERK and phosphorylated ERK, cyclin D1
(CCND1), Flag, c-Myc, p53, CD133, E-cadherin, Vimentin, β-catenin
and GAPDH (Santa Cruz Biotechnology). HRP-conjugated goat
anti-rabbit and goat anti-mouse secondary antibodies (Sigma) were
used. The signal was detected with SuperSignal West Pico
chemiluminescent Substrate (Thermo Scientific Pierce, Rockford, IL,
USA) and visualized with tanon-5200 system (Bio-Tanon, Shanghai,
China). The data are presented as mean ± SD (n=3).
Luciferase reporter assays
Hep3B cells were cultured in 12-well plates and
transfected with reporter plasmid pTOP-Flash and the indicated
plasmids using Lipofectamine 2000 as previously described (32).
Statistical analysis
All statistical analyses were performed using the
SPSS 11.0 software (SPSS Inc., Chicago, IL, USA). Data are shown as
mean ± SD from at least 3 independent experiments. Statistical
significance was determined using Student's t-test at P-value
<0.05.
Results
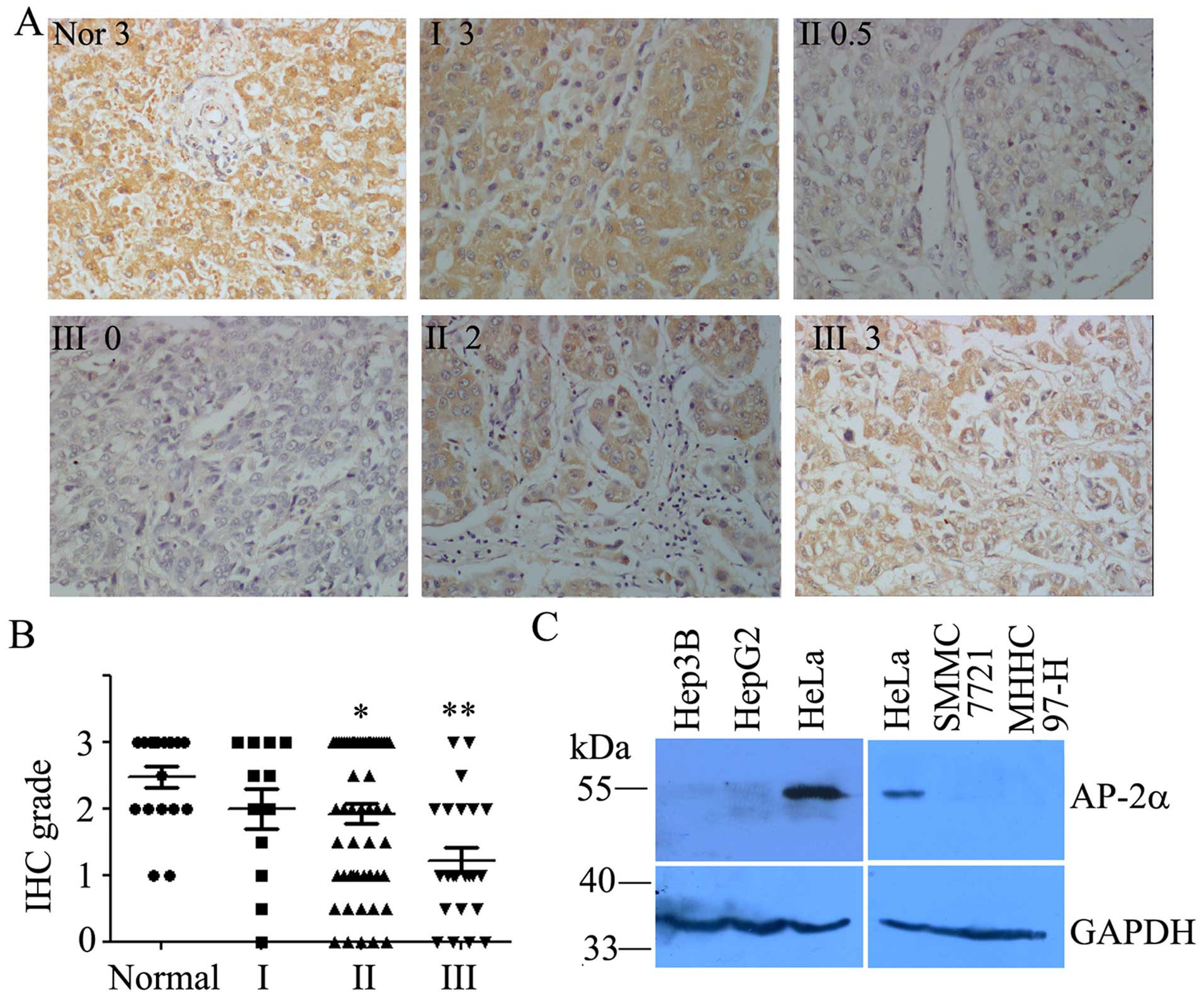
AP-2α is lowly expressed in high-grade
hepatocellular cancer tissues and high-invasive cell lines
The expression level of AP-2α was examined in 12 WHO
grade I, 56 WHO grade II, 22 WHO grade III hepatocellular cancers
and 18 adjacent normal tissues by immunohistochemistry analysis
using mouse monoclonal anti-AP-2α antibody. We found that AP-2α was
completely localized in the nucleus (Fig. 1A). AP-2α expression was detected in
34 (37.8%) of the 90 hepatocellular cancers with strong staining
(3+), 20 (22.2%) of the 90 hepatocellular cancers were moderately
stained (2+), and 36 (40%) were weakly stained or negative for
AP-2α expression (+/0) (Fig. 1B),
which indicated AP-2α was lowly expressed in hepatocellular cancers
(P<0.05) according to Student's t-test. A high expression of
AP-2α was observed in human normal liver and hepatocellular cancers
(I) (Fig. 1A and B). Therefore,
AP-2α expression was significantly decreased in human high-grade
hepatocellular cancer tissues (II and III) than normal liver and
hepatocellular cancer tissues (I). Patient characteristics are
summarized in Table I.
 | Table IPatient clinicopathological
characteristics in 90 cases of hepatocellular carcinoma. |
Table I
Patient clinicopathological
characteristics in 90 cases of hepatocellular carcinoma.
| Clinical
features | Number (%) |
|---|
| Gender |
| Female | 13 (14.3) |
| Male | 77 (85.7) |
| Age (median, 48
years) |
| <48 years | 33 (36.7) |
| ≥48 years | 57 (63.3) |
| Tumor size, cm |
| ≤5 | 61 (67.8) |
| >5 | 29 (32.2) |
| Cell
differentiation |
| Well (I–II) | 47 (52.2) |
| Moderately
(III) | 43 (47.8) |
| Tumor stage |
| Stage III | 22 (24.5) |
| Stage II | 56 (62.2) |
| Stage I | 12 (13.3) |
| Primary tumor |
| Tx | 2 (2.2) |
| T1 | 5 (5.6) |
| T2 | 56 (62.2) |
| T3 | 24 (26.7) |
| T4 | 3 (3.3) |
| Normal tissue | 18 |
We next analyzed the expression of AP-2α proteins in
human hepatocellular cancer cell lines. Low expression or loss of
AP-2α proteins was evident in Hep3B, HepG2, SMMC-7721 and MHHC 97-H
cells compared to HeLa cells with high expression of AP-2α
(Fig. 1C). Thus, the level of
AP-2α expression appears low in hepatocellular cancer cell lines.
These cell lines were further selected to overexpress AP-2α
proteins, respectively.
AP-2α overexpression inhibits the
proliferation of human hepatocellular cancer cell lines in vitro
and in vivo
To further investigate the role of AP-2α in human
hepatocellular cancer cells, we overexpressed AP-2α and asked
whether AP-2α inhibits the proliferation of hepatocellular cancer
cells. Then, lentivirus-based vector
Ubi-gene-3xFlag-IRES-puromycin-AP-2α containing the full-length
AP-2α and packaging plasmids pHelper 1.0 and pHelper 2.0 were
cotransfected to 293T cells. pGCFU-GFP-3xFlag-IRES-Puromycin served
as negative control. Lentiviral particles were prepared to infect
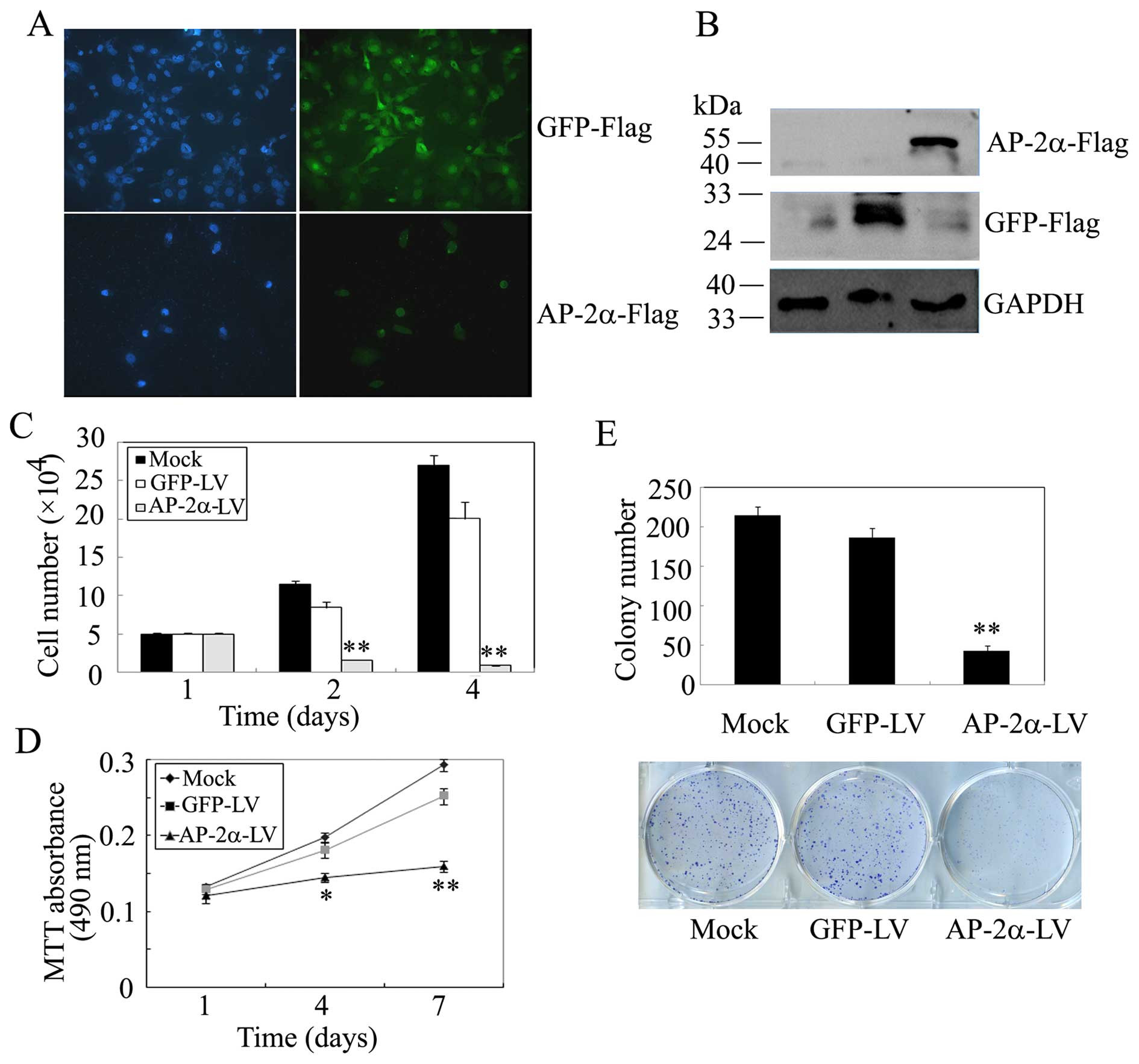
SMMC-7721 cells. The fluorescence intensity was markedly increased
4 days after infection and the infection efficiency was ~95% in
SMMC-7721 followed by the screening of 1.5 μg/ml of puromycin
(Fig. 2A). Western blot analysis
demonstrated overexpression of AP-2α and GFP (Fig. 2B).
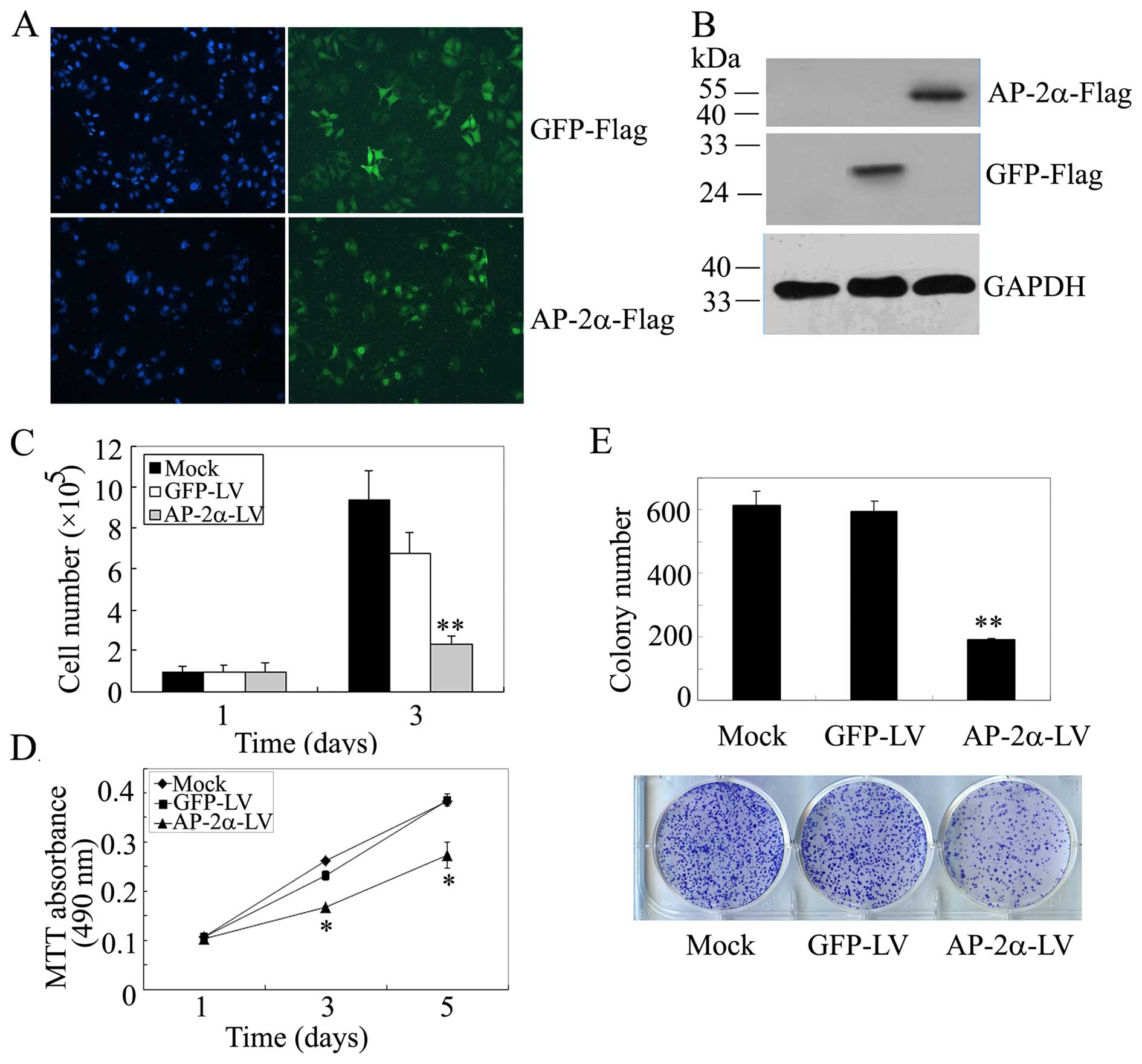 | Figure 2Effects of AP-2α overexpression on
SMMC-7721 hepatocellular cancer cell proliferation. (A)
Immunofluorescence staining of AP-2α and GFP expression in
SMMC-7721 cells stably expressing AP-2α and GFP. (B) Western blot
of GFP/Flag and AP-2α/Flag expression in SMMC-7721 cells using
anti-Flag antibodies. GAPDH served as a loading control. (C) Cell
survival assays of AP-2α-infected SMMC-7721 cells, GFP-infected
SMMC-7721 cells and parental SMMC-7721 cells. Cells (100,000) were
plated into 6-well plates in triplicate, grown in DMEM with 10% FBS
for 3 days and stained with trypan blue in PBS, viable cells were
counted. (D) MTT assays of mock or infected SMMC-7721 cells. Cells
(3,000) were plated in octuplicate in 96-well plates and grown in
DMEM with 10% FBS. The absorbance was analyzed for 1, 3 and 5 days.
(E) Liquid colony formation analysis of mock or infected SMMC-7721
cells. Cells (1,000) were seeded in triplicate in 6-well plates,
and grown for 10 days. Colonies were fixed with methanol, stained
with Giemsa, images were taken (lower panel) and counted (upper
panel). The data represent at least three independent experiments
with similar results. **P<0.01, compared with
parental and control cells. |
Next, we examined whether AP-2α is a critical
regulator of hepatocellular cancer cell proliferation and detected
the effect of AP-2α overexpression on SMMC-7721 cell growth. The
same number of cells was plated in triplicate in 6-well plates, and
cell number was counted on day 1 and 3. We found that the
overexpression of AP-2α in SMMC-7721 cells showed a decreased
cellular growth compared with controls (Fig. 2C). Likely, we examined cell
viability using MTT assays, AP-2α overexpression resulted in a
remarkable decrease in viable cells (Fig. 2D). Further, the liquid colony
formation assay indicated a great decrease in colony number and
size (Fig. 2E). These results
indicated that AP-2α over-expression inhibits hepatocellular cancer
cell SMMC-7721 proliferation in vitro.
To further demonstrate the effects of AP-2α on cell
proliferation, we performed the above experiments in Hep3B cells.
From Fig. 3A–D, we observed that
AP-2α overexpression significantly inhibits the survival and
proliferation of Hep3B cells, whereas GFP has no effect compared
with parental cells. The liquid colony formation assays showed that
exogenous AP-2α displays considerably fewer and smaller colonies in
Hep3B cells, while control GFP had no effect compared with
uninfected parental cells (Fig.
3E). Therefore, AP-2α over expression suppresses the
proliferation of human hepatocellular cancer Hep3B cells in
vitro.
The inhibitory effect of AP-2α on hepatocellular
cancer cell proliferation in vitro suggested that AP-2α
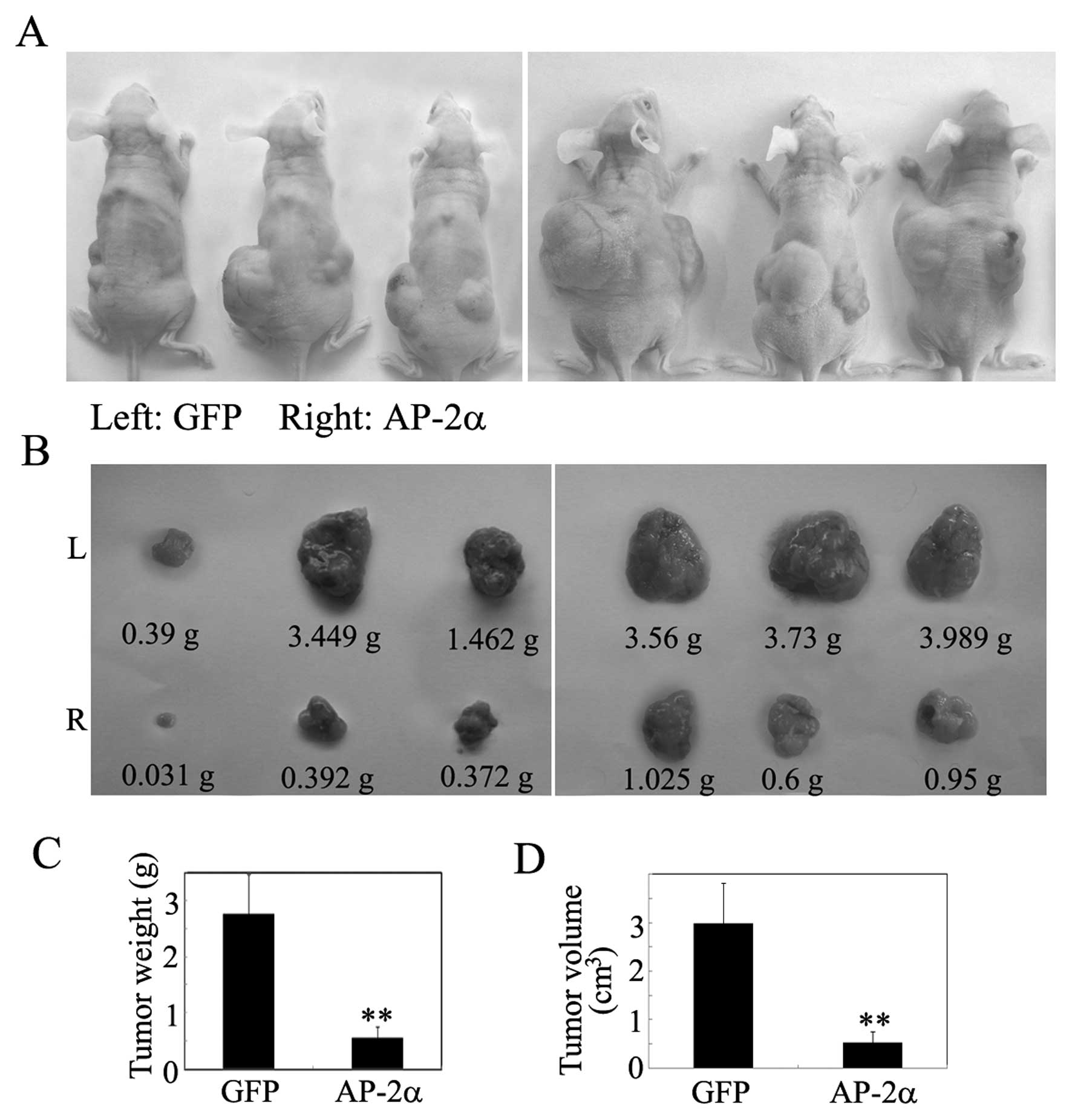
might suppress tumor growth in vivo. Tumorigenicity assay
was performed by subcutaneous injection of AP-2α cells into nude
mice, whereas GFP was used as controls. Within 41 days, solid
tumors were readily visible in left and right dorsal regions of all
mice (Fig. 4A and B), but the
average tumor weight and volume of the AP-2α group was reduced by
>70% compared with the negative controls (Fig. 4C and D).
AP-2α overexpression suppresses
hepatocellular cancer cell migration and increases the sensitivity
of hepatocellular cancer cells to cisplatin
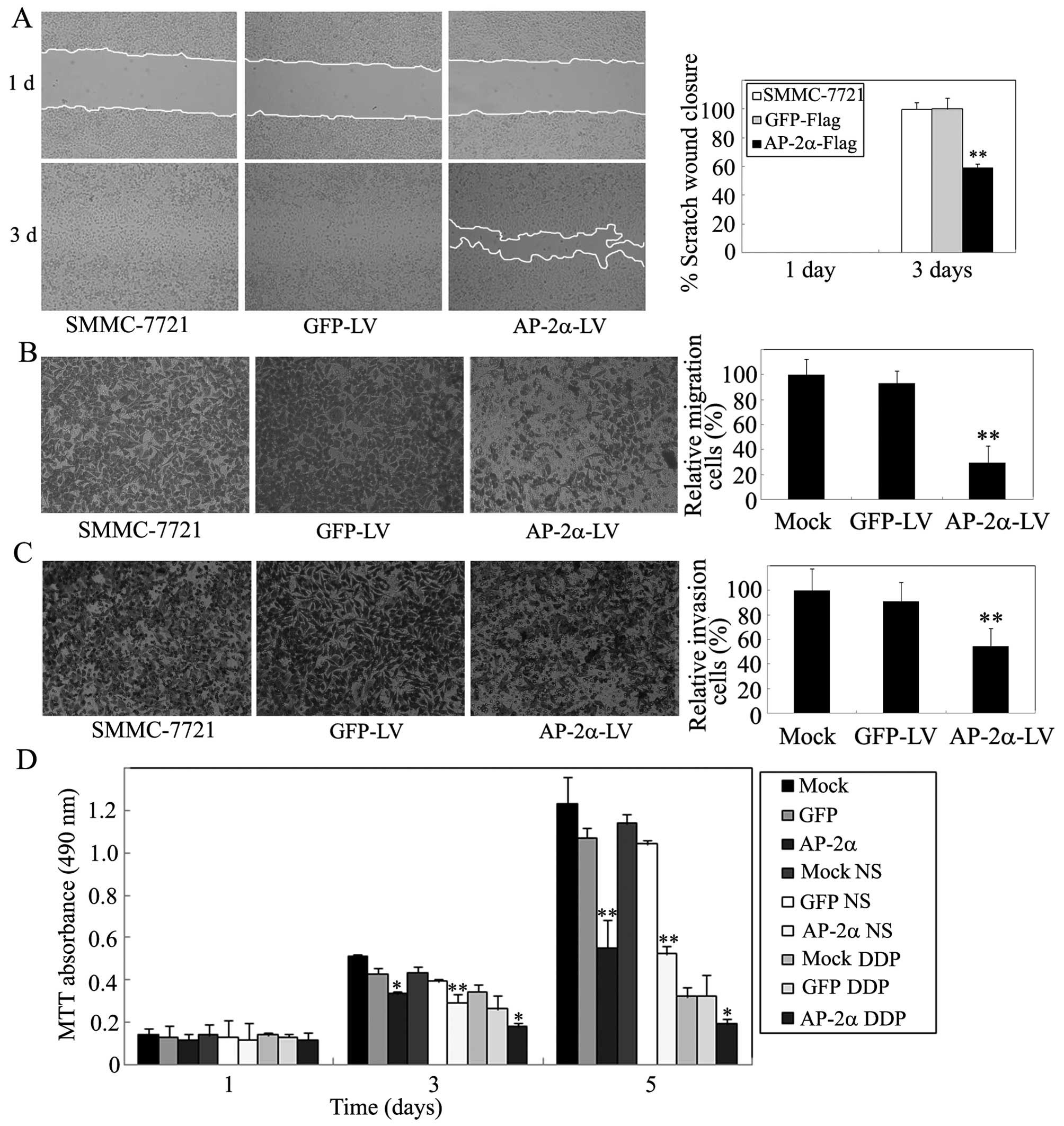
Since cell motility is an important factor
regulating cancer invasion and metastasis, the effect of AP-2α on
cell motility was characterized by woundhealing, Transwell
migration, and Matrigel invasion assays. As shown in Fig. 5A, the wound-healing assay showed
that AP-2α overexpression produces 62% inhibition of cell migration
at the edge of exposed regions in SMMC-7721 cells. In contrast,
control groups significantly increased cell migration.
Subsequently, the Transwell migration assay showed that
overexpression of AP-2α led to a marked decrease in cell motility,
whereas more cells were observed to migrate through the 8-μm pores
in control GFP compared with AP-2α (Fig. 5B). Similarly, the invasion assay
showed that AP-2α cells obtained a significantly slower rate of
cell invasion than that of control cells (Fig. 5C). Thus, these data demonstrated
that AP-2α is able to significantly decrease cell motility.
We next investigated the implication of AP-2α in
chemosensitivity of hepatocellular cancer cells. SMMC-7721 cells
were treated with cisplatin, chemotherapeutic agents. Reduction of
MTT absorbance (63%) was detected in AP-2α overexpressing cells
compared with 48% inhibition in their control cells (Fig. 5D). Therefore, AP-2α could sensitize
SMMC-7721 cells to cytotoxicity of cisplatin as reported in
endometrial cancer cells (25).
AP-2α overexpression attenuates the
self-renewal ability of liver TICs
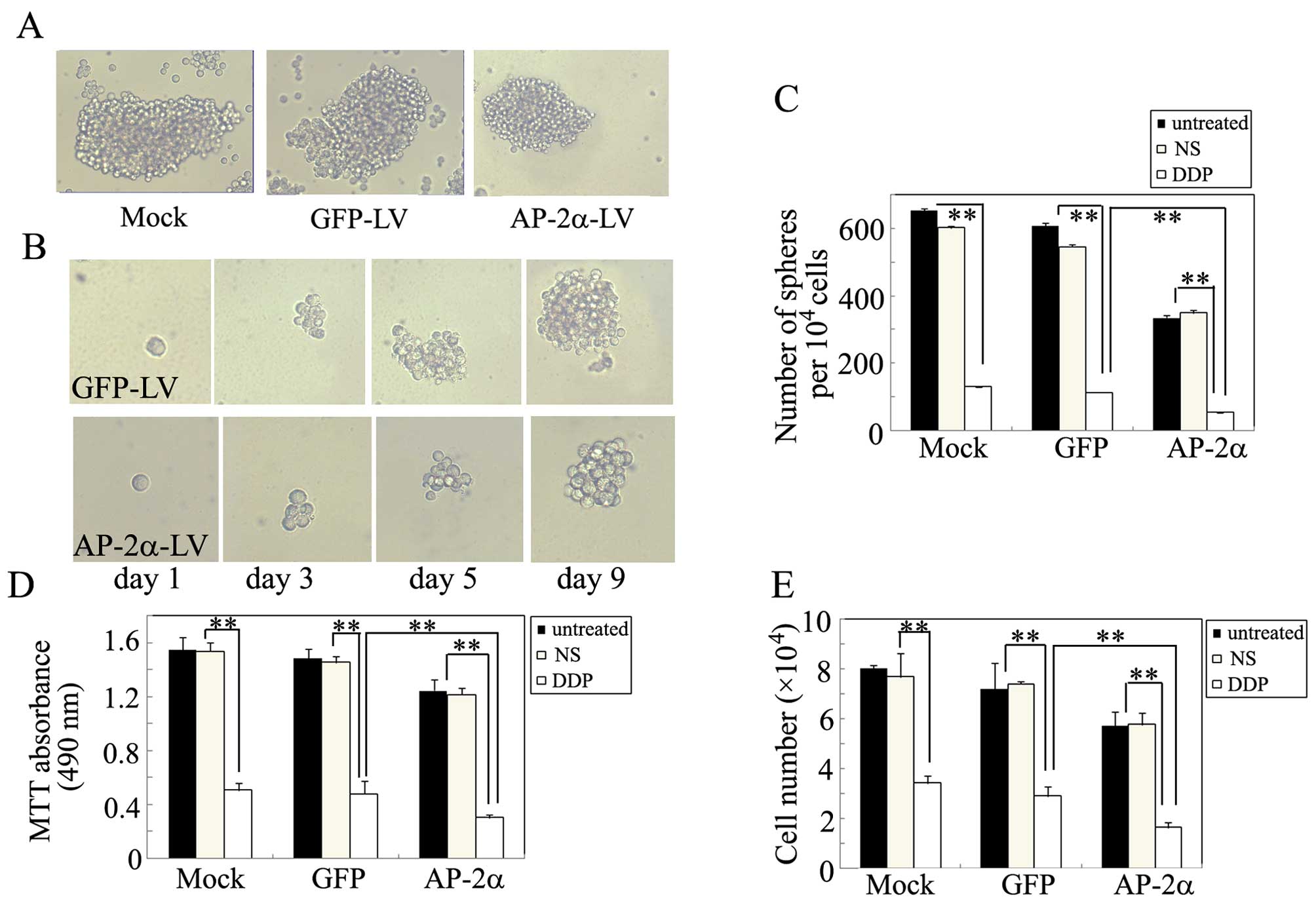
We next decided to examine whether AP-2α can
regulate the self-renewal ability of liver TICs. As shown in
Fig. 6A and C, AP-2α
overexpression significantly suppressed the spheroid formation
ability of these cells when grown in non-adherent serum-free
conditions in vitro. Moreover, we evaluated whether AP-2α
could suppress the self-renewal of TICs in vitro, single
cell suspension from the primary tumorspheres was used to form the
secondary sphere. AP-2α overexpression significantly decreased the
size of the secondary tumorsphere, suggesting a decreased
self-renewal ability of these TICs (Fig. 6B). Furthermore, we also
investigated the effects of AP-2α overexpression combined with
cisplatin on TICs. As shown in Fig.
6C–E, AP-2α overexpression displayed a markedly lower growth
and proliferation, when cells were treated with cisplatin for 3
days, cisplatin synergistically inhibited the survival and growth
of TICs.
AP-2α affects the expression of
phosphorylated ERK, β-catenin, p53 and EMT markers
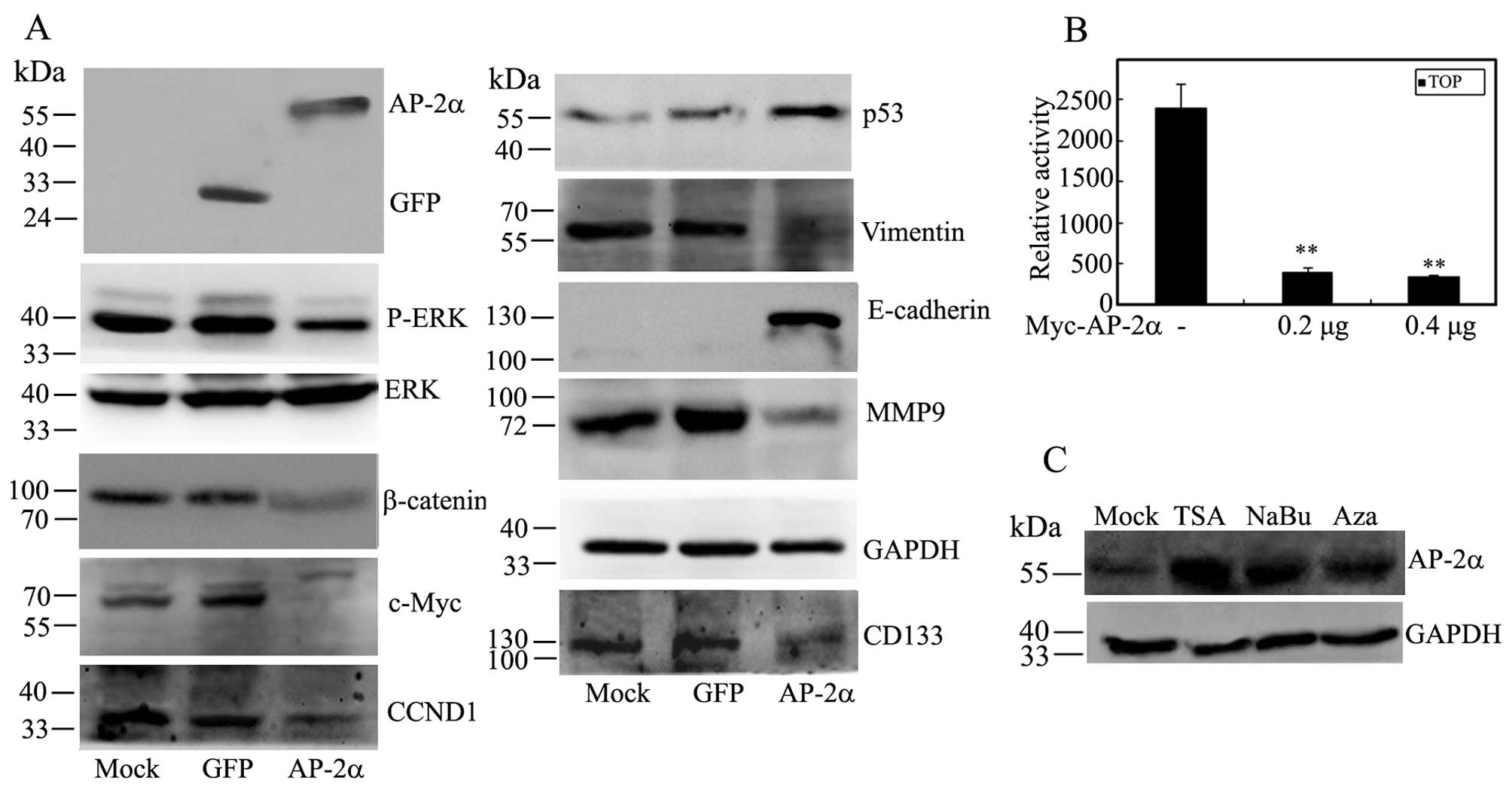
Since AP-2α influences many genes involved in the
development and progression of human cancers, we decided to
investigate whether AP-2α regulates these target genes in
hepatocellular cancer cells. As shown in Fig. 7A, AP-2α overexpression had no
effect on ERK protein, but it decreased the expression of β-catenin
and the phosphorylated level of ERK expression, and downregulated
the expression of c-Myc and cyclin D1 as a downstream target of ERK
signaling and Wnt signaling. Moreover, AP-2α markedly suppressed
the TOPFlash reporter activity and gave an inhibitory effect on Wnt
signaling pathway (Fig. 7A and B).
AP-2α overexpression increased the expression of tumor suppressor
p53. Furthermore, the level of EMT epithelial marker E-cadherin was
concomitantly increased in AP-2α-infected cells while AP-2α
overexpression decreased the levels of mysenchymal marker vimentin
and MMP9 (Fig. 7A). Taken
together, these data showed that AP-2α regulates cell proliferation
and migration, at least in part, by affecting ERK signaling and
EMT-related cell movement.
We wished to clarify whether the epigenetic
modification might regulate low expression of AP-2α in
hepatocellular carcinoma, thus, we determined the effect of the
histone deacetylase inhibitor, trichostatin A (TSA) or sodium
butyrate (NaBu) and/or the DNA methyltransferase inhibitor,
5-aza-2'deoxycytidine (5-aza-dC) on AP-2α expression in SMMC-7721
cells. We found that TSA, NaBu and 5-aza-dC significantly increased
the expression of AP-2α protein (Fig.
7C), suggesting the possible modification influences of AP-2α
function in hepatocellular carcinoma.
Discussion
Hepatocellular carcinoma (HCC) is a common malignant
tumor. Carcinogenesis of HCC is a multi-factor, multi-step and
complex process, which is associated with genetic alterations that
involve the gain-of-function mutation, amplification, and/or
overexpression of key oncogenes together with the loss-of-function
mutation, deletion, and/or epigenetic silencing of critical tumor
suppressors. Previously, loss of AP-2α has been associated with
tumor malignancy in various cancers, including melanoma (12), breast cancer (27), and glioma (14). It has been well reported that DNA
methylation of CpG islands near the promoter and exon 1 affects the
transcription of AP-2α in breast cancer (27), and acetylation of AP-2α upregulates
Toll-like receptor 2 gene expression in THP-1 cells, a human
monocytic cell line (28). In the
present study, that loss or low expression of AP-2α was detected in
40% of HCC samples, indicating a tumor suppressive role of AP-2α in
HCC. Moreover, the levels of demethylated and acetylated AP-2α were
increased in SMMC-7721 cells after 5-aza-dC or TSA treatment.
Therefore, in a future study we will investigate whether the
downregulation of AP-2α might associate with DNA hypermethylation
and deacetylation in hepatocellular carcinoma.
The in vitro and in vivo functional
assays were performed to characterize the role of AP-2α in
regulating cell proliferation, migration and invasion of
hepatocellular cancer, and the results showed that AP-2α has strong
tumor-suppressive ability. We found that AP-2α-infected cells were
able to form smaller and fewer colonies in liquid colony formation
and inhibited tumor formation in nude mice. Furthermore, AP-2α
decreased HCC cell migration and invasion by wound healing and a
serial of Transwell assays. More intriguingly, we showed that AP-2α
overexpression significantly enhanced cisplatin-induced death of
HCC cells, supporting that AP-2α increased the sensitivity of liver
cancer cells to cisplatin. Consistent with our previous data that
miR-200b/200c/429 induced cisplatin resistance by repressing AP-2α
expression in endometrial cancer cells (25), all these findings strongly
supported that AP-2α plays an important role in HCC growth and
motility.
Recent studies in a variety of solid tumors and
leukemia show that there is significant heterogeneity with respect
to tumor-forming ability within a given population of tumor cells,
suggesting that the capability to maintain tumorigenesis is found
only in a small population of cells called cancer stem cells (CSCs)
or tumor-initiating cells (TI Cs) (33). CSCs have been shown to be resistant
to conventional therapies and are thus thought to drive disease
relapse (34). Hepatic
stem/progenitor cell markers including CD44 and EpCAM with the
transmembrane cell-surface glycoprotein CD133 can mark liver TICs
(35). Our results showed that
AP-2α overexpression significantly suppressed the growth and
self-renewal of liver CSCs by downregulating CD133 expression.
Many pathways including Ras/MAPK, PI3K/AKT, and
Wnt/β-catenin signaling, have been shown to be of significance in
HCC. The Ras-Raf-MEK-ERK signaling cascade plays a crucial role in
regulating cellular processes including differentiation,
proliferation, survival and apoptosis (36). We demonstrated that AP-2α inhibits
the expression of phosphorylated ERK and downstream genes c-Myc and
CCND1 in cellular growth of liver cancer. Moreover, c-myc is a
major driver of tumor angiogenesis and its expression has been
associated with HCC recurrence (37). In addition, c-Myc and CCND1 also
serve as canonical Wnt pathway downstream target genes. β-catenin
is the central effector of the canonical Wnt signaling, which is a
highly conserved pathway regulating critical cellular processes
such as proliferation, differentiation, survival and self-renewal
(38,39). The Wnt/β-catenin pathway has been
implicated in a subset of HCC where activating mutations in the
β-catenin gene have been reported in 20–40% of patients (40). Knockdown of β-catenin in hepatoma
cells leads to decreased growth and survival, and oncogene
β-catenin is considered a valuable therapeutic target (41). AP-2α has been identified to
associate with adenomatous polyposis coli/β-catenin and inhibits
β-catenin/T-cell factor transcriptional activity in colorectal
cancer cells (23). However, no
evidence indicates that AP-2α regulates Wnt/β-catenin pathway in
liver cancer. Here, we found that AP-2α overexpression can inhibit
the Wnt/β-catenin pathway in HCC cells through downregulating
β-catenin expression. In addition, the tumor suppressor gene p53
inhibits the development and growth of the majority of human tumors
(42). Moreover, the tumor
suppressor activity of AP-2α is mediated through a direct
interaction with p53 (43), we
also found that AP-2α augments p53 activation in HCC cells.
Epithelial-mesenchymal transition (EMT) is a
critical process for tumor invasion and metastasis in which
epithelial cells lose their junctions and apical-basal polarity,
remodel their cytoskeleton, undergo a change in cell shape and
reprogramme gene expression to increase cell motility and develop
an invasive phenotype, while MET describes the reverse process
(44,45). In this study we found that AP-2α
over-expression gives a significant effect on EMT by increasing the
expression of epithelial markers (E-cadherin) and decreasing
mesenchymal markers (vimentin), and might achieve lower motility
and invasiveness. Thus, these data suggested that AP-2α might
partly inhibit EMT-like transition and invasion in HCC. Moreover,
the invasiveness in tumor cells was enhanced by upregulation of
MMPs such as MMP9 for extracellular matrix remodeling (46). We provided evidence that the levels
of MMP9 are decreased in AP-2α overexpressed HCC cells.
In conclusion, our data suggest the pivotal role of
AP-2α in hepatocellular cancer progression. Epigenetically modified
AP-2α is lowly expressed in hepatocellular cancer tissues and cell
lines. AP-2α regulates the proliferation, migration, invasion and
self-renewal ability of hepatocellular cancer cells, at least in
part, by affecting the expression of phosphorylated ERK, β-catenin,
p53 and EMT markers. Preliminary data showed that AP-2α increases
the cytotoxicity of chemo-therapeutic drugs in hepatocellular
cancer cells. Taken together, the tumor suppressor AP-2α might have
an important role as molecular therapeutic and/or prognostic
markers in human hepatocellular cancers.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81272318 to J.Z., no. 81272190 to
X.D.), Excellent Youth Foundation of Hunan Provincial Education
Department (no. 13B068 to X.D.), Distinguished Youth Foundation of
Hunan Province (no. 2015JJ1011 to X.D.), Excellent Young Talents of
Hunan Normal University (no. ET14104 to X.D.), Youth innovation and
entrepreneurship platform of Hunan Province (no. 1 to X.D.) and the
postgraduate Research Innovation Fund of Hunan Province (no.
CX2015B170 to W.H.).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Portolani N, Coniglio A, Ghidoni S,
Giovanelli M, Benetti A, Tiberio GA and Giulini SM: Early and late
recurrence after liver resection for hepatocellular carcinoma:
Prognostic and therapeutic implications. Ann Surg. 243:229–235.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forner A, Hessheimer AJ, Isabel Real M and
Bruix J: Treatment of hepatocellular carcinoma. Crit Rev Oncol
Hematol. 60:89–98. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pellikainen J, Naukkarinen A, Ropponen K,
Rummukainen J, Kataja V, Kellokoski J, Eskelinen M and Kosma VM:
Expression of HER2 and its association with AP-2 in breast cancer.
Eur J Cancer. 40:1485–1495. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pellikainen JM and Kosma VM: Activator
protein-2 in carcinogenesis with a special reference to breast
cancer - a mini review. Int J Cancer. 120:2061–2067. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ropponen KM, Kellokoski JK, Lipponen PK,
Pietiläinen T, Eskelinen MJ, Alhava EM and Kosma VM: p22/WAF1
expression in human colorectal carcinoma: Association with p53,
transcription factor AP-2 and prognosis. Br J Cancer. 81:133–140.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ruiz M, Pettaway C, Song R, Stoeltzing O,
Ellis L and Bar-Eli M: Activator protein 2alpha inhibits
tumorigenicity and represses vascular endothelial growth factor
transcription in prostate cancer cells. Cancer Res. 64:631–638.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schwartz B, Melnikova VO, Tellez C,
Mourad-Zeidan A, Blehm K, Zhao YJ, McCarty M, Adam L and Bar-Eli M:
Loss of AP-2alpha results in deregulation of E-cadherin and MMP-9
and an increase in tumorigenicity of colon cancer cells in vivo.
Oncogene. 26:4049–4058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tellez C, McCarty M, Ruiz M and Bar-Eli M:
Loss of activator protein-2alpha results in overexpression of
protease-activated receptor-1 and correlates with the malignant
phenotype of human melanoma. J Biol Chem. 278:46632–46642. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng YX, Somasundaram K and el-Deiry WS:
AP2 inhibits cancer cell growth and activates p21WAF1/CIP1
expression. Nat Genet. 15:78–82. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bar-Eli M: Gene regulation in melanoma
progression by the AP-2 transcription factor. Pigment Cell Res.
14:78–85. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang S, Jean D, Luca M, Tainsky MA and
Bar-Eli M: Loss of AP-2 results in downregulation of c-KIT and
enhancement of melanoma tumorigenicity and metastasis. EMBO J.
17:4358–4369. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anttila MA, Kellokoski JK, Moisio KI,
Mitchell PJ, Saarikoski S, Syrjänen K and Kosma VM: Expression of
transcription factor AP-2alpha predicts survival in epithelial
ovarian cancer. Br J Cancer. 82:1974–1983. 2000.PubMed/NCBI
|
|
14
|
Heimberger AB, McGary EC, Suki D, Ruiz M,
Wang H, Fuller GN and Bar-Eli M: Loss of the AP-2alpha
transcription factor is associated with the grade of human gliomas.
Clin Cancer Res. 11:267–272. 2005.PubMed/NCBI
|
|
15
|
Karjalainen JM, Kellokoski JK, Eskelinen
MJ, Alhava EM and Kosma VM: Downregulation of transcription factor
AP-2 predicts poor survival in stage I cutaneous malignant
melanoma. J Clin Oncol. 16:3584–3591. 1998.PubMed/NCBI
|
|
16
|
Lipponen P, Aaltomaa S, Kellokoski J,
Ala-Opas M and Kosma V: Expression of activator protein 2 in
prostate cancer is related to tumor differentiation and cell
proliferation. Eur Urol. 37:573–578. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ropponen KM, Kellokoski JK, Pirinen RT,
Moisio KI, Eskelinen MJ, Alhava EM and Kosma VM: Expression of
transcription factor AP-2 in colorectal adenomas and
adenocarcinomas; comparison of immunohistochemistry and in situ
hybridisation. J Clin Pathol. 54:533–538. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding X, Yang Z, Zhou F, Wang F, Li X, Chen
C, Li X, Hu X, Xiang S and Zhang J: Transcription factor AP-2α
regulates acute myeloid leukemia cell proliferation by influencing
Hoxa gene expression. Int J Biochem Cell Biol. 45:1647–1656. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oyama N, Takahashi H, Tojo M, Iwatsuki K,
Iizuka H, Nakamura K, Homma Y and Kaneko F: Different properties of
three isoforms (alpha, beta, and gamma) of transcription factor
AP-2 in the expression of human keratinocyte genes. Arch Dermatol
Res. 294:273–280. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bennett KL, Romigh T and Eng C: AP-2alpha
induces epigenetic silencing of tumor suppressive genes and
microsatellite instability in head and neck squamous cell
carcinoma. PLoS One. 4:e69312009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stabach PR, Thiyagarajan MM, Woodfield GW
and Weigel RJ: AP2alpha alters the transcriptional activity and
stability of p53. Oncogene. 25:2148–2159. 2006. View Article : Google Scholar
|
|
22
|
Shi D, Xie F, Zhang Y, Tian Y, Chen W, Fu
L, Wang J, Guo W, Kang T, Huang W, et al: TFAP2A regulates
nasopharyngeal carcinoma growth and survival by targeting HIF-1α
signaling pathway. Cancer Prev Res (Phila). 7:266–277. 2014.
View Article : Google Scholar
|
|
23
|
Li Q and Dashwood RH: Activator protein
2alpha associates with adenomatous polyposis coli/beta-catenin and
Inhibits beta-catenin/T-cell factor transcriptional activity in
colorectal cancer cells. J Biol Chem. 279:45669–45675. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wajapeyee N, Britto R, Ravishankar HM and
Somasundaram K: Apoptosis induction by activator protein 2alpha
involves transcriptional repression of Bcl-2. J Biol Chem.
281:16207–16219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu Y, Xiao Y, Ding X, Zhuo Y, Ren P, Zhou
C and Zhou J: A miR-200b/200c/429-binding site polymorphism in the
3′ untranslated region of the AP-2α gene is associated with
cisplatin resistance. PLoS One. 6:e290432011. View Article : Google Scholar
|
|
26
|
Wajapeyee N, Raut CG and Somasundaram K:
Activator protein 2alpha status determines the chemosensitivity of
cancer cells: Implications in cancer chemotherapy. Cancer Res.
65:8628–8634. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Douglas DB, Akiyama Y, Carraway H,
Belinsky SA, Esteller M, Gabrielson E, Weitzman S, Williams T,
Herman JG and Baylin SB: Hypermethylation of a small CpGuanine-rich
region correlates with loss of activator protein-2alpha expression
during progression of breast cancer. Cancer Res. 64:1611–1620.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li M, Li X, Wang E and Luo E: Upregulation
of Toll-like receptor 2 gene expression by acetylation of AP-2
alpha in THP-1 cells, a human monocytic cell line. Int J Biochem
Cell Biol. 45:1594–1599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding X, Zhou F, Wang F, Yang Z, Zhou C,
Zhou J, Zhang B, Yang J, Wang G, Wei Z, et al: Eps8 promotes
cellular growth of human malignant gliomas. Oncol Rep. 29:697–703.
2013.
|
|
30
|
Chen C, Liang Z, Huang W, Li X, Zhou F, Hu
X, Han M, Ding X and Xiang S: Eps8 regulates cellular proliferation
and migration of breast cancer. Int J Oncol. 46:205–214. 2015.
|
|
31
|
Ding X, Fan C, Zhou J, Zhong Y, Liu R, Ren
K, Hu X, Luo C, Xiao S, Wang Y, et al: GAS41 interacts with
transcription factor AP-2beta and stimulates AP-2beta-mediated
transactivation. Nucleic Acids Res. 34:2570–2578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li X, Chen C, Wang F, Huang W, Liang Z,
Xiao Y, Wei K, Wan Z, Hu X, Xiang S, et al: KCTD1 suppresses
canonical Wnt signaling pathway by enhancing β-catenin degradation.
PLoS One. 9:e943432014. View Article : Google Scholar
|
|
33
|
Shekhani MT, Jayanthy AS, Maddodi N and
Setaluri V: Cancer stem cells and tumor transdifferentiation:
Implications for novel therapeutic strategies. Am J Stem Cells.
2:52–61. 2013.PubMed/NCBI
|
|
34
|
Lobo NA, Shimono Y, Qian D and Clarke MF:
The biology of cancer stem cells. Annu Rev Cell Dev Biol.
23:675–699. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang KH, Ma S, Lee TK, Chan YP, Kwan PS,
Tong CM, Ng IO, Man K, To KF, Lai PB, et al: CD133(+) liver
tumor-initiating cells promote tumor angiogenesis, growth, and
self-renewal through neurotensin/interleukin-8/CXCL1 signaling.
Hepatology. 55:807–820. 2012. View Article : Google Scholar
|
|
36
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cui J, Dong BW, Liang P, Yu XL and Yu DJ:
Effect of c-myc, Ki-67, MMP-2 and VEGF expression on prognosis of
hepatocellular carcinoma patients undergoing tumor resection. World
J Gastroenterol. 10:1533–1536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lustig B and Behrens J: The Wnt signaling
pathway and its role in tumor development. J Cancer Res Clin Oncol.
129:199–221. 2003.PubMed/NCBI
|
|
39
|
Willert K, Brown JD, Danenberg E, Duncan
AW, Weissman IL, Reya T, Yates JR III and Nusse R: Wnt proteins are
lipid-modified and can act as stem cell growth factors. Nature.
423:448–452. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wong CM, Fan ST and Ng IO: beta-Catenin
mutation and over-expression in hepatocellular carcinoma:
Clinicopathologic and prognostic significance. Cancer. 92:136–145.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zeng G, Apte U, Cieply B, Singh S and
Monga SP: siRNA-mediated beta-catenin knockdown in human hepatoma
cells results in decreased growth and survival. Neoplasia.
9:951–959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Soussi T and Béroud C: Assessing TP53
status in human tumours to evaluate clinical outcome. Nat Rev
Cancer. 1:233–240. 2001. View Article : Google Scholar
|
|
43
|
McPherson LA, Loktev AV and Weigel RJ:
Tumor suppressor activity of AP2alpha mediated through a direct
interaction with p53. J Biol Chem. 277:45028–45033. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thomas GJ, Lewis MP, Hart IR, Marshall JF
and Speight PM: AlphaVbeta6 integrin promotes invasion of squamous
carcinoma cells through up-regulation of matrix
metalloproteinase-9. Int J Cancer. 92:641–650. 2001. View Article : Google Scholar : PubMed/NCBI
|