Introduction
Lung cancer is still the leading cause of
cancer-related death throughout the world (1). In recent genome studies, oncogenic
driver mutations including EGFR gene mutation and ALK fusion gene
have been discovered in non-small cell lung cancer (NSCLC) patients
(2–4). First-line treatments with
EGFR-tyrosine kinase inhibitors (TKIs) including gefitinib,
erlotinib and afatinib and ALK-inhibitor crizotinib showed higher
response rate and superior progression-free survival (PFS) compared
with standard cytotoxic chemotherapy and are recognized as the
standard therapies for the treatment of advanced NSCLC with driver
mutations (5–8). Based on these findings, another
small-molecular-targeted drug has recently been developed and its
efficacy and safety have been evaluated in NSCLC patients.
Nintedanib (BIBF1120) is a potent, oral angiokinase
inhibitor that targets VEGFR-1-3, PDGFR-α and β, and FGFR-1-3
signaling (9). Nintedanib has been
evaluated for the treatment of idiopathic pulmonary fibrosis (IPF)
(10,11). Recent phase III studies
demonstrated that nintedanib reduced the decline in forced vital
capacity, resulting in decreased frequency of acute exacerbations
in patients with IPF with tolerable adverse events (10,11).
The US Food and Drug Administration (FDA) approved nintedanib as a
new drug for IPF in 2014 based on these trials. IPF treatment has
entered a new era by the development of nintedanib. Nintedanib has
also been evaluated for the treatment of various solid tumors
including advanced NSCLC (12).
The LUME-Lung-1 phase III trial showed that nintedanib in
combination with docetaxel is an effective second-line therapy for
advanced NSCLC patients, especially for lung adenocarcinoma
patients (12). This was the first
evidence for a molecular targeted agent showing an effect in
combination therapy with cytotoxic agents after failure of platinum
doublet chemotherapy in NSCLC patients. Therefore, identification
of predictive biomarkers for the response to nintedanib and patient
selection based on the biomarker may have a clinical benefit for
NSCLC patients treated with nintedanib.
MicroRNAs (miRNAs) are single-stranded,
18–24-nucleotide, non-coding molecules that post-transcriptionally
modulate gene expression through binding to 3′UTRs of target mRNAs.
miRNAs, which usually induce gene silencing, can function as either
tumor suppressors or oncogenes (13,14).
Previous studies revealed that miRNAs were diagnostic, prognostic
and therapeutic biomarkers in lung cancer (15,16).
Our previous study demonstrated that inhibition of miR-21 and
miR-134/487b/655 cluster expression could be used as a therapeutic
strategy in connection with EGFR-tyrosine kinase inhibitor
(EGFR-TKI) treatment (17,18). These findings suggest that miRNAs
may be promising predictive biomarkers and therapeutic targets in
NSCLC.
In the present study, we examined the miRNA profile
in order to clarify which miRNA is associated with sensitivity to
nintedanib using 16 NSCLC cell lines. We demonstrate that miR-200b
and miR-141 associated with epithelial-mesenchymal transition (EMT)
are predictive biomarkers and therapeutic targets of nintedanib in
NSCLC cells. Furthermore, we found that nintedanib inhibited EMT
and reversed the resistance to EGFR-TKI with TGF-β-induced EMT
through miR-200 family induction in NSCLC cells.
Materials and methods
Cell lines
We used the following 16 NSCLC cell lines in the
present study: A549, PC-3, PC-9, PC-14, NCI-HCC827, NCI-H1650,
NCI-H1975, LC-2/ad and RERF-LC-KJ adeno-carcinoma (AC) cell lines
and PC-1, PC-10, LK-2, SQ5, QG56, EBC-1 and LC-1/sq squamous-cell
carcinoma (SQ) cell lines. A549, NCI-H1650 and NCI-H1975 were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA); RERF-LC-KJ, SQ5, LC-2/ad and LC-1/sq were
obtained from Riken Cell Bank (Ibaraki, Japan); PC-1, PC-3, PC-9,
PC-10 and PC-14 were obtained from Immuno-Biological Laboratories
(Gunma, Japan); and EBC-1, LK-2, and QG56 were purchased from
Health Science Research Resources Bank (Osaka, Japan). Lung cancer
cell lines were maintained in RPMI-1640 medium (Gibco, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS). These cell
lines were obtained from 2008 to 2009, amplified and frozen, and
one aliquot of each was thawed for this project. All cells were
routinely screened for the absence of mycoplasma.
Drugs and growth-inhibition assay
Nintedanib and gefitinib were purchased from Selleck
Chemicals (Houston, TX, USA). Growth inhibition was assessed by the
MTS assay to examine the effect of nintedanib on the 16 NSCLC cell
lines. Cell suspensions (5,000 cells/well) were seeded into 96-well
plates and increasing concentrations of nintedanib (0, 0.01. 0.1,
1.0 and 10 μM) were added. After incubation at 37ºC for 72 h, MTS
was added to each well and incubated at 37ºC for 2 h, after which
absorbance was measured using a microplate reader with a test
wavelength of 450 nm. The IC50 value was defined as the
concentration needed for 50% reduction of the growth by treatment
with nintedanib by SigmaPlot12 (Hulinks, Inc., Tokyo, Japan). A549
and PC-1R cells (5,000 cells/well) were seeded into 96-well plates
for 24 h, and then incubated in the various concentrations of
nintedanib at 37ºC for 72 h after exposure to miR-200 mimic,
miR-141 mimic or miR-mimic-control (Cont-mimic) at a final
concentration of 40 nM for 24 h.
RNA extraction and miRNA microarray
analysis
Total RNA was extracted from four lines with TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) as previously described
(19,20). Total RNA (5 μg) was employed for
hybridization on miRNA micro-array chips containing 667 probes with
the TaqMan® Array Human MicroRNA A + B Cards Set v2.0
(Life Technologies, Carlsbad, CA, USA) on a 7900 Real-Time PCR
System (Applied Biosystems, Foster City, CA, USA). Processed slides
were scanned with a PerkinElmer ScanArray XL5K scanner.
Experimental data were analyzed by DataAssist™ software (Life
Technologies) using RNU44 and RNU48 as endogenous controls. Ct
values were provided from all miRNAs represented on the cards and
fold changes in expression were calculated using the delta Ct
(ΔΔCt) method. Expression levels of MammU6 on the array card were
defined as positive controls for the purpose of ΔΔCt
calculation.
Real-time quantitative reverse
transcription-PCR
The expression levels of miR-200 family members were
measured by quantitative reverse transcription-PCR (qRT-PCR) using
TaqMan® MicroRNA assay system (Applied Biosystems). The
RNU66 expression level was determined by qRT-PCR as an internal
control (Applied Biosystems). miRNA expression was quantified and
reported as 2−ΔΔCT value (21).
Western blot analysis and receptor
tyrosine kinase phos-pholylation array
Cells were dissolved in buffer containing 50 mM
Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1%
Nonidet P-40 and 0.5% sodium-deoxycholate. The lysates were cooled
with ice for 30 min, and then centrifuged at 13,000 × g for 30 min.
Proteins (10 μg) in the collected supernatant were separated by
SDS-PAGE on 12% gels and then transferred to nitrocellulose
membranes. The membrane, after a block with 5% skim milk, was
incubated with antibodies to E-cadherin, vimentin, ZEB1, β-actin
(Cell Signaling Technology, Beverley, MA, USA) and ZEB2
(Sigma-Aldrich, St. Louis, MO, USA). Each protein was detected by
immunoblotting with ECL-Plus reagents (GE Healthcare Bio-Science
Corp., Piscataway, NJ, USA). We also performed human receptor
tyrosine kinase (RTK) phosphorylation antibody arrays including 71
antibodies (RayBiotech, Inc., Norcross, GA, USA) as previously
described (22).
Oligonucleotide transfection
miR-200b and miR-141 mimics, and their negative
control (Cont-mimic), were synthesized by Ambion (Austin, TX, USA).
All mimics were treated with Lipofectamine 2000 transfection
reagent 24 h after seeding, as per the manufacturer's instructions
(Life Technologies). The mimic complexes were transfected into
cells at a final concentration of 40 nM. The transfection medium
was replaced 6 h later and cells were then incubated at 37ºC for 48
h. TGF-β1 was purchased from R&D Systems, Inc. (Minneapolis,
MN, USA). Cells were exposed to 5 ng/ml TGF-β1 for the indicated
period of time.
Statistical analysis
Data were expressed as the mean (SD) of three
independent experiments and evaluated with the Student's t-test.
P<0.05 were defined as statistically significant.
Results
Effect of nintedanib on the growth of 16
NSCLC cell lines
Growth-inhibitory effects of nintedanib on 16 NSCLC
cell lines were assessed by MTS assay. Gene status of EGFR
mutation, ALK rearrangement, KRAS mutation and MET amplification
were evaluated by LCI Medience Corp. (Tokyo, Japan). Table I shows the sensitivity to
nintedanib of 16 NSCLC cell lines and their genetic status.
Nintedanib had IC50 values of >1 μM, which is above
the pharmacologically achievable concentration in mammals (0.45
μM), in the 16 NSCLC cell lines (9). However, PC-1 cells without genetic
alterations were relatively sensitive to nintedanib (1.0 μM). There
were no correlations between nintedanib IC50 and
pathological type or genetic status.
 | Table ISensitivity to nintedanib of 16 NSCLC
cell lines and their genetic status. |
Table I
Sensitivity to nintedanib of 16 NSCLC
cell lines and their genetic status.
| Cell lines | Pathology | Nintedanib
IC50 | EGFR mut | ALK fusion | KRAS mut | MET amp |
|---|
| PC-1 | SQ | 1.0 | - | - | − | − |
| QG56 | SQ | 1.9 | - | - | − | − |
| LK-2 | SQ | 2.5 | - | - | − | − |
| EBC-1 | SQ | 3.2 | - | - | − | + |
| PC-9 | AC | 3.2 | Ex19 del | - | − | − |
| NCI-HCC827 | AC | 3.9 | Ex19 del | - | − | − |
| NCI-H1975 | AC | 5.9 | Ex19 del,
T790M | - | − | − |
| RERF-LC-KJ | AC | 7.3 | - | - | − | − |
| PC-10 | SQ | 7.7 | - | - | + | − |
| PC-14 | AC | 7.7 | - | - | − | − |
| NCI-H1650 | AC | 20 | L858R | - | − | − |
| A549 | AC | 25 | - | - | + | − |
| SQ5 | SQ | 27 | - | - | − | − |
| PC-3 | AC | 71 | - | - | − | − |
| LC-1/sq | SQ | 78 | - | - | − | − |
| LC-2/ad | AC | >100 | - | - | + | − |
Correlation between FGFR/PDGFR/VEGFR
expression and drug sensitivity
Based on the IC50, we placed NSCLC cell
lines into two groups, namely, nintedanib-sensitive and
nintedanib-resistant cell lines. Five cell lines (PC-1, QG56, LK-2,
EBC-1 and PC-9) were sensitive (IC50 of ≤3.2 μM), and 5
other cell lines were resistant to nintedanib (A549, SQ5, PC-3,
LC-1/sq and LC-2/ad) (IC50 of >25 μM) (Table I). First, RTK phosphorylation
profiles including FGFR, PDGFR and VEGFR family kinases were
investigated in the 5 nintedanib-sensitive and 5
nintedanib-resistant cell lines to clarify whether the kinases were
associated with sensitivity to nintedanib. The relationship between
RTKs phosphorylation and nintedanib sensitivity is shown in
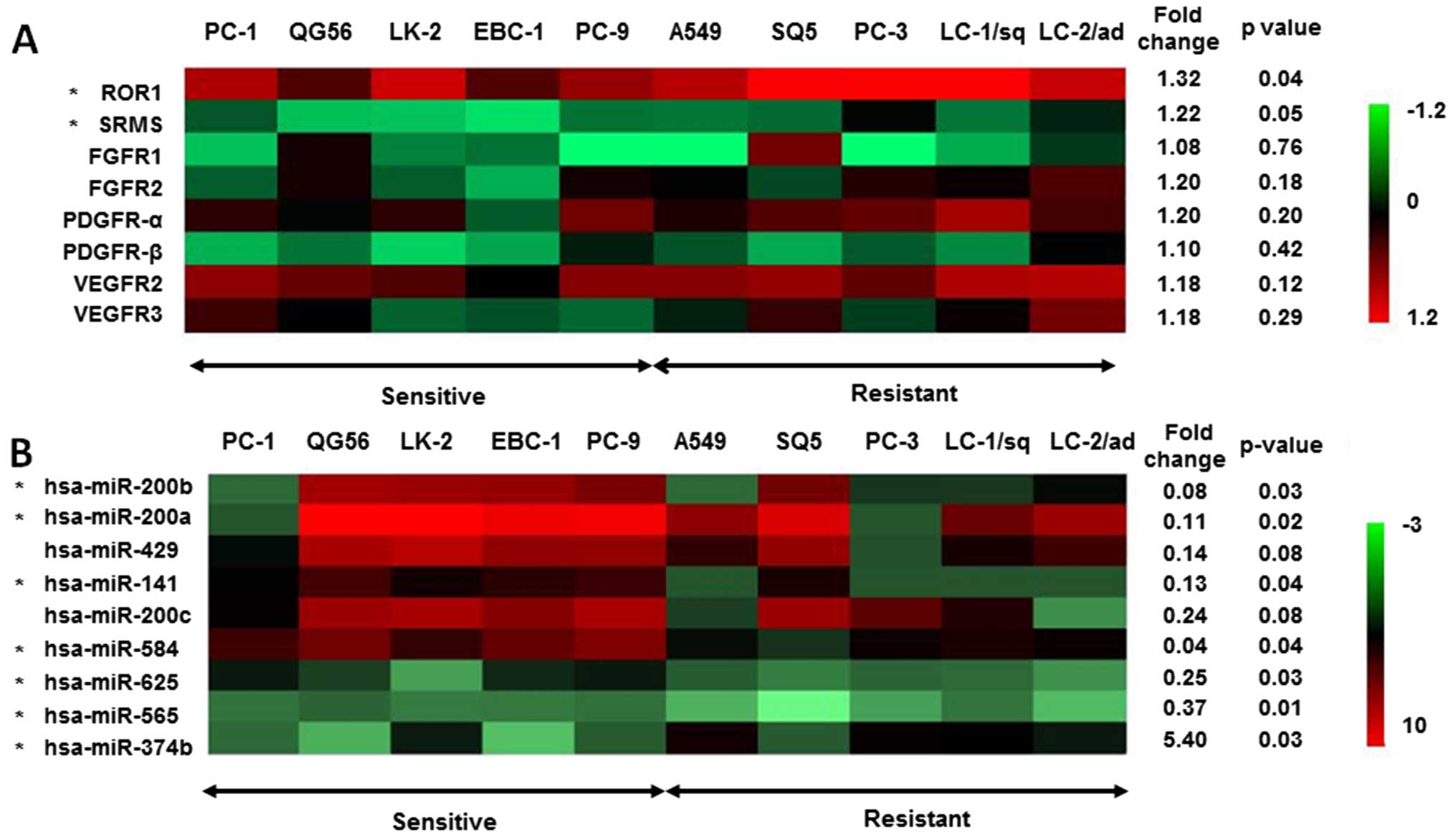
Fig. 1A. Phosphorylation of ROR1
and SRMS was significantly increased in nintedanib-resistant cells.
However, the phosphorylation status of FGFR1/2, PDGFRα/β and
VEGFR2/3 showed no significant differences between the sensitive
and resistant cell lines (Fig.
1A). This result suggested that activation of targets of
nintedanib, FGFR, PDGFR and VEGFR kinases, does not affect the
sensitivity of NSCLC cells to nintedanib.
Correlation between miRNA expression and
drug sensitivity
In order to identify miRNAs that contribute to the
sensitivity to nintedanib, we compared miRNA expression profiles
between the 5 nintedanib-sensitive and 5 nintedanib-resistant cell
lines by miRNA array. The expression of 7 miRNAs was significantly
altered in the nintedanib-resistant cell group compared to the
nintedanib-sensitive cell group (fold changes of >2.0 or
<0.5) (Fig. 1B). Among them,
expression of three miR-200 family members, miR-200b, miR-200a and
miR-141, were significantly downregulated in the
nintedanib-resistant cell lines (Fig.
1B). The miR-200 family contains five miRNAs: miR-200a/200b/429
on chromosome 1 and miR-200c/141 on chromosome 12. The levels of
miR-200c and miR-429 expression tended to be decreased in the
nintedanib-resistant cell group (Fig.
1B). The expression levels of miR-200 family members were
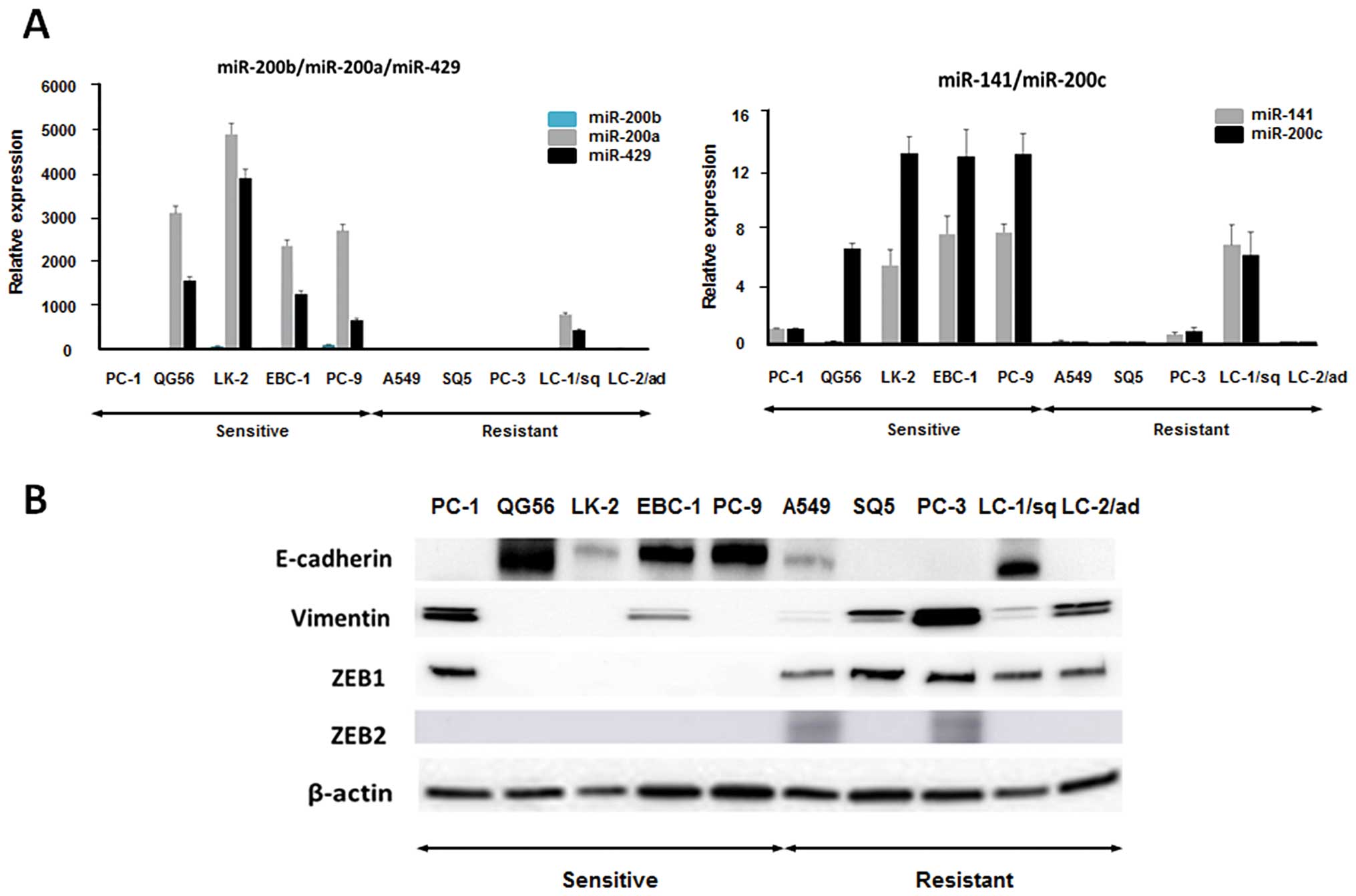
validated by qRT-PCR (Fig. 2A).
The expression levels of miR-200 family members were higher in the
5 nintedanib-sensitive cell lines than in the resistant cell lines
except for LC-1/sq cells (Fig.
2A). The miR-200 family members have been described as a main
suppressor of EMT by targeting ZEB1 and ZEB2 (23). Therefore, we evaluated the protein
expression of EMT markers in the 10 NSCLC cell lines. Epithelial
marker E-cadherin expression was lower, and mesenchymal marker
vimentin expression was higher in the 5 nintedanib-resistant cell
lines (Fig. 2B). Expression of
ZEB1 which is well known as a direct target of the miR-200 family
was also increased in the 5 nintedanib-resistant cell lines
(Fig. 2B). ZEB2 expression was
found in 2 of the 5 nintedanib-resistant cell lines (Fig. 2B). These results suggested that the
mesenchymal phenotype may be involved in the resistance to
nintedanib in NSCLC cells.
Establishment of nintedanib-resistant PC1
cells and evaluation of miR-200 family expression and EMT
markers
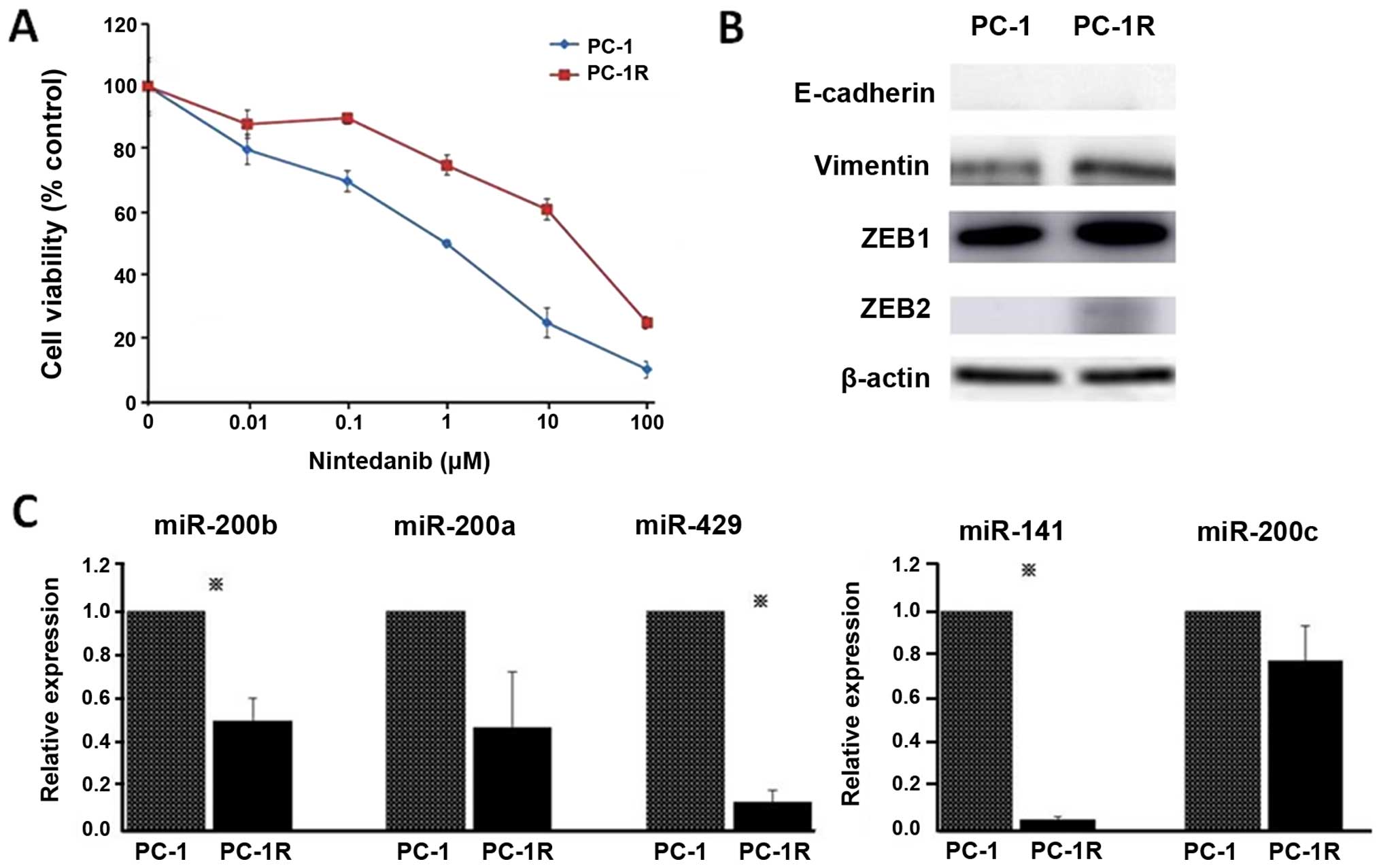
To further clarify the mechanism of resistance to
nintedanib, we tried to establish nintedanib-resistant PC-1 cells
by continuous exposure to increasing concentrations of nintedanib
in a stepwise manner. After two months, we established
nintedanib-resistant PC-1 cell lines; PC-1R survived upon
incubation with 25 μM nintedanib and we used these PC1-R cells in
further investigations (Fig. 3A).
The expression levels of vimentin, ZEB1 and ZEB2 were increased in
PC1-R cells compared to PC-1 cells (Fig. 3B). Furthermore, miR-200b, miR-141
and miR-429 expression levels were significantly decreased in PC1-R
cells compared to PC-1 cells (Fig.
3C). Based on these results, we focused on the miR-200 family
and ZEB, especially miR-200b and miR-141 and their target ZEB1, as
candidate miRNAs and their target associated with sensitivity to
nintedanib in NSCLC cells.
miR-200 family regulated EMT and
nintedanib sensitivity in NSCLC cells
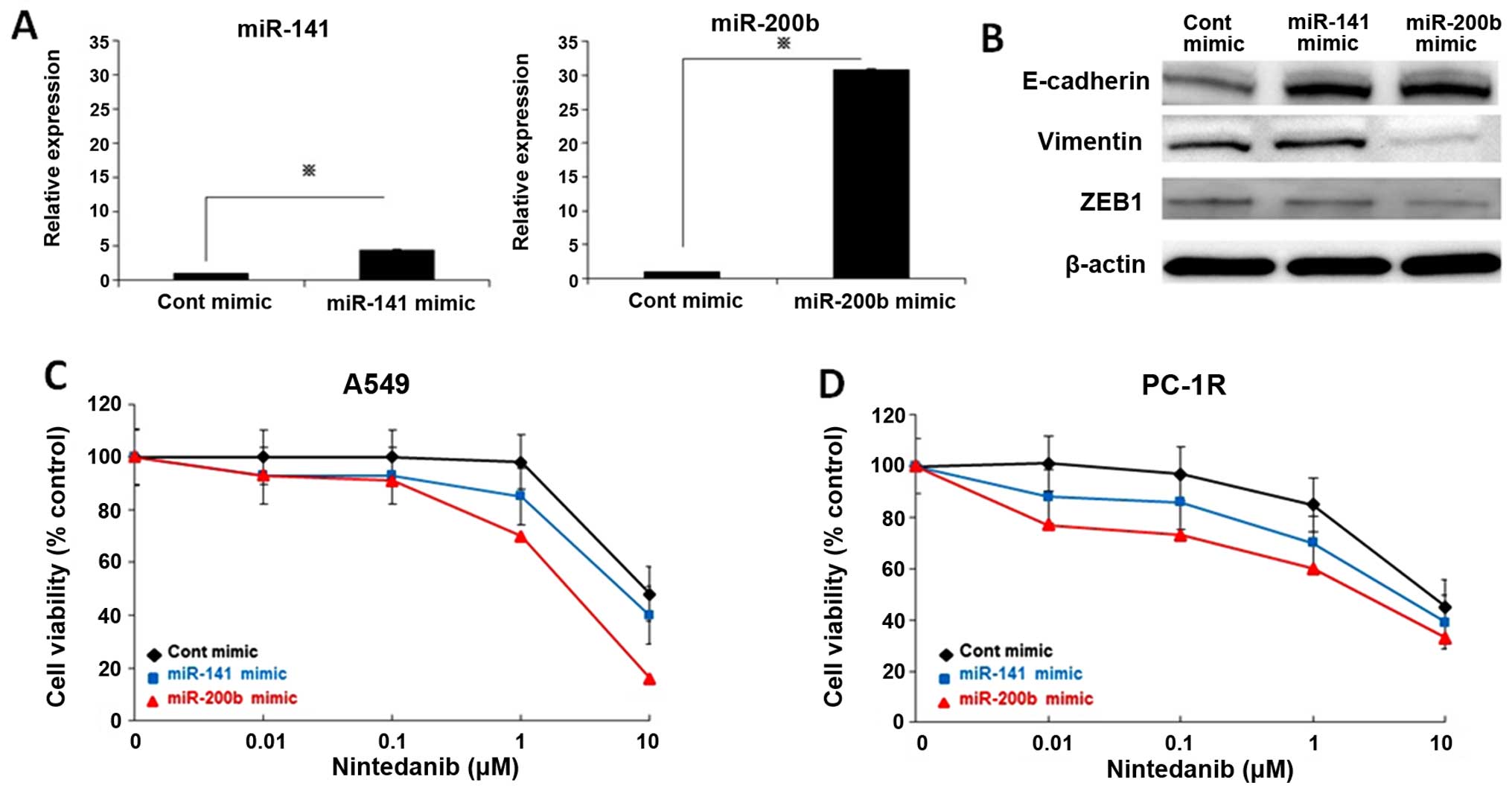
To further examine whether the miR-200 family
regulates the response to nintedanib, nintedanib-resistant A549
cells were transfected with a miR-200b or miR-141 mimic and control
mimic for 72 h. We confirmed that miR-200b and miR-141 were
overexpressed after the miR-200b or miR-141 mimic was incubated
with A549 cells (Fig. 4A). ZEB1
expression was diminished resulting in increased E-cadherin and
decreased vimentin expression in A549 cells treated with miR-200b
mimic (Fig. 4B). Overexpression of
miR-141 decreased ZEB1 and increased E-cadherin expression
(Fig. 4B). Next, we investigated
the effect of overexpression of the two miRNAs on the sensitivity
of A549 cells to nintedanib. Induction of miR-200b or miR-141
mimics enhanced the sensitivity of A549 cells to nintedanib
(Fig. 4C). Furthermore, miR-200b
or miR-141 overexpression reversed the resistance to nintedanib of
PC1-R cells (Fig. 4D). These
results suggested that suppression of the miR-200 family is
involved in the resistance to nintedanib in NSCLC cells.
Nintedanib restored drug resistance to
gefitinib by activating the miR-200 family
EMT has been recognized as one of the mechanisms of
resistance to EGFR-TKI in NSCLC with EGFR mutation (24,25).
In addition, a recent study demonstrated that nintedanib induced
mesenchymal-epithelial transition (MET) in A549 lung cancer cells
(26). Therefore, we evaluated
whether nintedanib overcomes EMT and the resistance to the EGFR-TKI
gefitinib in NSCLC cells. A549 cells were exposed to TGF-β1 and
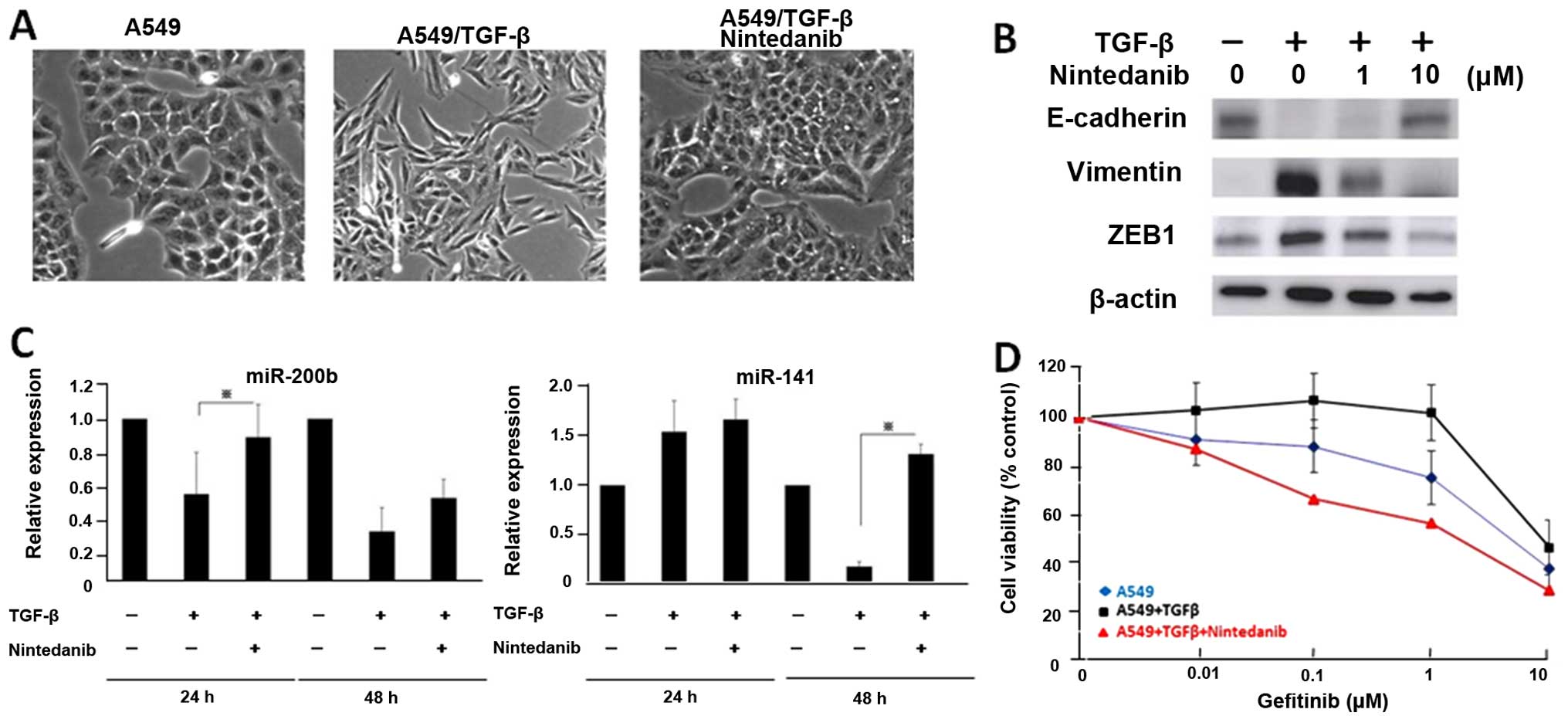
then treated with nintedanib for 72 h. Fig. 5A shows that the conversion from
epithelial phenotype to mesenchymal phenotype induced by TGF-β1 was
more inhibited in A549 cells treated with nintedanib (Fig. 5A). We observed that the increases
in vimentin and ZEB1 expression and decrease in E-cadherin
expression activated by TGF-β1 were more reduced in A549 cells
treated with nintedanib (Fig. 5B).
Furthermore, decreased miR-200b and miR-141 expression caused by
TGF-β1 was restored after treatment with nintedanib (Fig. 5C). We also measured the response to
gefitinib in A549 cells after treatment with nintedanib in
combination with TGF-β1. Nintedanib restored the resistance to
gefitinib caused by TGF-β1 in naturally resistant A549 cells
(Fig. 5D). These results suggest
that nintedanib combined with EGFR-TKI might be a novel therapeutic
strategy for NSCLC cells with EMT phenotype.
Discussion
Although the effect of nintedanib monotherapy on
NSCLC patients was limited, PFS was significantly improved in
advanced NSCLC patients treated with nintedanib combined with
docetaxel in the LUME-Lung-1 phase III study (12). This evidence suggests that
nintedanib combined with anticancer agents might be a promising
therapy in previously treated NSCLC patients. Therefore,
identification of mechanisms of resistance to nintedanib is
required for developing a therapeutic strategy in advanced NSCLC
patients.
In the present study, low expression of miR-200b and
miR-141, resulting in high level of ZEB1 and low level of
E-cadherin, was associated with the resistance to nintedanib in
NSCLC cells. The miR-200 family has been recognized as a main
suppressor of EMT by targeting ZEB1 (23). EMT has been implicated in
prevention of apoptosis and resistance to anticancer agents
including EGFR-TKI in NSCLC cells. However, a method of overcoming
EMT-associated resistance to anticancer drugs has not been
established yet. In functional analysis in a recent report,
nintedanib could reverse the EMT phenotype in lung cancer and
pancreatic cancer cells (26). Our
results showed that retention of the epithelial phenotype with high
levels of miR-200 family ensured good sensitivity to nintedanib in
NSCLC cells. In addition, induction of miR-200b and miR-141 showed
reversal of EMT and increased sensitivity for nintedanib in both
naturally resistant A549 and acquired resistant PC1-R cells.
Expression of the miR-200 family and ZEB1 may be used as a
predictive marker and therapeutic target for nintedanib therapy in
NSCLC cells. Nintedanib therapy may be effective in NSCLC patients
with the epithelial phenotype. A recent study showed that
inhibition of miR-200 family targeting ZEB1 significantly enhanced
the chemosensitivity to docetaxel in vitro and in
vivo in lung adenocarcinoma cells with the EMT phenotype
(27). This result may provide
supporting evidence of the effectiveness of nintedanib combined
with docetaxel in the LUME-Lung-1 study (12). The miR-200 family and its target
ZEB1 may be common attractive targets of nintedanib and docetaxel
therapies.
We also found that treatment with nintedanib caused
reversal of TGF-β1-induced EMT and resistance to gefitinib through
upregulation of miR-200b and miR-141 in A549 lung cancer cells.
These effects may be due to the multitargeted function of
nintedanib, which inhibits FGFR as well as VEGFR and PDGFR.
Reversal of EMT by nintedanib might be attributed to inhibition of
fibroblast function. A recent report showed decreased levels of
α-SMA and S-100A4 in fibroblasts in pancreatic cancer xenografts
after treatment with nintedanib (26). Alternatively, FGF pathway
activation could provide an escape mechanism from anti-molecular
targeted therapy in various cancers (28). FGFR promotes metastasis through EMT
in breast tumors (29). FGFR1
inhibitor also restored EMT in head and neck squamous cell
carcinoma (30). Taken together
with our results, the reversal of EMT might be mainly regulated by
FGFR inhibition of nintedanib. These findings demonstrated a novel
role of nintedanib as a potential therapeutic strategy for
resistance to EGFR-TKI associated with TGF-β1-induced EMT in NSCLC
cells. Overcoming EMT-associated resistance to EGFR-TKI would have
extremely great benefit for EGFR-mutant NSCLC patients.
Nintedanib is also one of the promising drugs for
IPF patients (10,11). FDA approved nintedanib for the
treatment of IPF. In addition, EMT is considered to contribute to
IPF (31,32). The mesenchymal markers collagen I,
vimentin and α-SMA were expressed in the bleomycin IPF model
(31). A human IPF study has shown
co-localization of epithelial and mesenchymal markers (32). It has been recognized that
nintedanib has the potential to reduce disease progression, slowing
the decline of lung function by blocking signaling pathways that
are involved in fibrotic processes (31,32).
The miR-200 family also inhibited fibrogenic activity of pulmonary
fibroblasts obtained from mice with experimental pulmonary fibrosis
and from IPF patients (33). IPF
is one of the most common complications in patients with lung
cancer. Optimal treatments for lung cancer with IPF have not been
established because of acute exacerbation of IPF caused by
anticancer treatment in lung cancer patients with IPF (34). Our findings suggest that nintedanib
can be used for the treatment of NSCLC patients with IPF as well as
IPF patients.
In conclusion, the miR-200 family and ZEB1 could be
used as predictive markers for sensitivity to nintedanib in NSCLC
cells. Selection of patients for nintedanib therapy based on
miR-200 family or ZEB1 expression may be useful in NSCLC patients.
Nintedanib combined with EGFR-TKI might be a new therapeutic
strategy for NSCLC patients with acquired resistance to EGFR-TKI by
EMT. Further studies should be performed to clarify the effect of
nintedanib on EMT and EGFR-TKI therapy in NSCLC.
Acknowledgements
We would like to thank Ms. Junko Murase and Mr.
Hiroshi Terasaki of LSI Medience Corporation for analyzing genetic
alterations.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soda M, Choi YL, Enomoto M, Takada S,
Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K,
Hatanaka H, et al: Identification of the transforming EML4-ALK
fusion gene in non-small-cell lung cancer. Nature. 448:561–566.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al; North-East Japan Study Group. Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang
Y, Li W, Hou M, Shi JH, Lee KY, et al: Afatinib versus cisplatin
plus gemcitabine for first-line treatment of Asian patients with
advanced non-small-cell lung cancer harbouring EGFR mutations
(LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet
Oncol. 15:213–222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó
L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al:
Crizotinib versus chemotherapy in advanced ALK-positive lung
cancer. N Engl J Med. 20;368:2385–2394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hilberg F, Roth GJ, Krssak M, Kautschitsch
S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel
A, Quant J, et al: BIBF 1120: triple angiokinase inhibitor with
sustained receptor blockade and good antitumor efficacy. Cancer
Res. 15;68:4774–4782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Richeldi L, Costabel U, Selman M, Kim DS,
Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G,
et al: Efficacy of a tyrosine kinase inhibitor in idiopathic
pulmonary fibrosis. N Engl J Med. 22;365:1079–1087. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Richeldi L, du Bois RM, Raghu G, Azuma A,
Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y,
et al: Efficacy and safety of nintedanib in idiopathic pulmonary
fibrosis. N Engl J Med. 370:2071–2082. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reck M, Kaiser R, Mellemgaard A, Douillard
JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I,
Liao M, et al; LUME-Lung 1 Study Group. Docetaxel plus nintedanib
versus docetaxel plus placebo in patients with previously treated
non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind,
randomised controlled trial. Lancet Oncol. 15:143–155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yanaihara N, Caplen N, Bowman E, Seike M,
Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, et
al: Unique microRNA molecular profiles in lung cancer diagnosis and
prognosis. Cancer Cell. 9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seike M, Goto A, Okano T, Bowman ED,
Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H, et
al: MiR-21 is an EGFR-regulated anti-apoptotic factor in lung
cancer in never-smokers. Proc Natl Acad Sci USA. 106:12085–12090.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kitamura K, Seike M, Okano T, Matsuda K,
Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K and Gemma A:
MiR-134/487b/655 cluster regulates TGF-β-induced
epithelial-mesenchymal transition and drug resistance to gefitinib
by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther.
13:444–453. 2014. View Article : Google Scholar
|
|
19
|
Seike M, Yanaihara N, Bowman ED, Zanetti
KA, Budhu A, Kumamoto K, Mechanic LE, Matsumoto S, Yokota J,
Shibata T, et al: Use of a cytokine gene expression signature in
lung adeno-carcinoma and the surrounding tissue as a prognostic
classifier. J Natl Cancer Inst. 99:1257–1269. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimokawa T, Seike M, Soeno C, Uesaka H,
Miyanaga A, Mizutani H, Kitamura K, Minegishi Y, Noro R, Okano T,
et al: Enzastaurin has anti-tumour effects in lung cancers with
over-expressed JAK pathway molecules. Br J Cancer. 106:867–875.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bustin SA: Absolute quantification of mRNA
using real-time reverse transcription polymerase chain reaction
assays. J Mol Endocrinol. 25:169–193. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sugano T, Seike M, Noro R, Soeno C, Chiba
M, Zou F, Nakamichi S, Nishijima N, Matsumoto M, Miyanaga A, et al:
Inhibition of ABCB1 overcomes cancer stem cell-like properties and
acquired resistance to MET inhibition in non-small lung cancer. Mol
Cancer Ther. 14:2433–2440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yauch RL, Januario T, Eberhard DA, Cavet
G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al:
Epithelial versus mesenchymal phenotype determines in vitro
sensitivity and predicts clinical activity of erlotinib in lung
cancer patients. Clin Cancer Res. 11:8686–8698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Suda K, Tomizawa K, Fujii M, Murakami H,
Osada H, Maehara Y, Yatabe Y, Sekido Y and Mitsudomi T: Epithelial
to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kutluk Cenik B, Ostapoff KT, Gerber DE and
Brekken RA: BIBF 1120 (nintedanib), a triple angiokinase inhibitor,
induces hypoxia but not EMT and blocks progression of preclinical
models of lung and pancreatic cancer. Mol Cancer Ther. 12:992–1001.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ren J, Chen Y, Song H, Chen L and Wang R:
Inhibition of ZEB1 reverses EMT and chemoresistance in
docetaxel-resistant human lung adenocarcinoma cell line. J Cell
Biochem. 114:1395–1403. 2013. View Article : Google Scholar
|
|
28
|
Casanovas O, Hicklin DJ, Bergers G and
Hanahan D: Drug resistance by evasion of antiangiogenic targeting
of VEGF signaling in late-stage pancreatic islet tumors. Cancer
Cell. 8:299–309. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qian X, Anzovino A, Kim S, Suyama K, Yao
J, Hulit J, Agiostratidou G, Chandiramani N, McDaid HM, Nagi C, et
al: N-cadherin/FGFR promotes metastasis through
epithelial-to-mesenchymal transition and stem/progenitor cell-like
properties. Oncogene. 33:3411–3421. 2014. View Article : Google Scholar :
|
|
30
|
Nguyen PT, Tsunematsu T, Yanagisawa S,
Kudo Y, Miyauchi M, Kamata N and Takata T: The FGFR1 inhibitor
PD173074 induces mesenchymal-epithelial transition through the
transcription factor AP-1. Br J Cancer. 109:2248–2258. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DeMaio L, Buckley ST, Krishnaveni MS,
Flodby P, Dubourd M, Banfalvi A, Xing Y, Ehrhardt C, Minoo P, Zhou
B, et al: Ligand-independent transforming growth factor-β type I
receptor signalling mediates type I collagen-induced
epithelial-mesenchymal transition. J Pathol. 226:633–644. 2012.
View Article : Google Scholar
|
|
32
|
Kage H and Borok Z: EMT and interstitial
lung disease: A mysterious relationship. Curr Opin Pulm Med.
18:517–523. 2012.PubMed/NCBI
|
|
33
|
Yang S, Banerjee S, de Freitas A, Sanders
YY, Ding Q, Matalon S, Thannickal VJ, Abraham E and Liu G:
Participation of miR-200 in pulmonary fibrosis. Am J Pathol.
180:484–493. 2012. View Article : Google Scholar :
|
|
34
|
Minegishi Y, Sudoh J, Kuribayasi H,
Mizutani H, Seike M, Azuma A, Yoshimura A, Kudoh S and Gemma A: The
safety and efficacy of weekly paclitaxel in combination with
carboplatin for advanced non-small cell lung cancer with idiopathic
interstitial pneumonias. Lung Cancer. 71:70–74. 2011. View Article : Google Scholar
|