Introduction
Glioblastoma (glioblastoma multiforme, GBM) is a
grade IV astrocytic tumour, the most aggressive form of astrocytic
malignancy and the most common brain tumour in adults (1). The disease can arise de novo,
which accounts for 90% of cases, or as a secondary GBM, which is
more common in younger people and progresses from a lower grade
glial tumour. GBM is notoriously resistant to therapy, surgery
cannot target the diffuse margins of the tumour, while cells have
limited susceptibility to conventional radiation and temo-zolomide
chemotherapy. With standard therapy, the median survival is 19
months post diagnosis (2). This
dismal prognosis is in part due to the presence of cancer stem
cells (CSC) in GBM. While the definition and characteristics of GBM
CSC are controversial (3–6), these cells can generally considered
to be intrinsically resistant to therapy (7), have self-renewal activity so can
re-establish tumours after treatment (8) and are highly migratory and invasive
(9), meaning they are likely to be
present in the infiltrating edges of the tumour left behind after
surgery. Therapies that target CSC and the characteristics of
survival and self-renewal should dramatically improve the outcome
for people with GBM.
Normal cellular processes of proliferation and
survival are tightly regulated through a number of signalling
pathways, including through the phosphatoinositide 3-kinase (PI3K)
family proteins. The genes PIK3CA, PIK3CB, PIK3CD encode the
catalytic subunit of class IA kinases p110α, p110β and
p110δ, respectively, while the gene PIK3CG codes for a separate
subunit class IB kinase p110γ. Mutation and
dysregulation of the PI3K/AKT/mTOR signalling axis is a major
contributor to tumorigenic behaviour in cells, with key roles in
proliferation, migration and epigenetic silencing of developmental
pathways. Hyper-phosphorylation of AKT is frequent in cancers,
particularly where PI3K activity is decoupled from EGF signal
transduction through the loss of PTEN function (10). This increases proliferation and
migration of cells (11,12) particularly in glioblastoma, where
85% of tumours have activating mutations in the RTK/PI3K/AKT
pathway (13). This has made the
PI3K pathway a potential pharmacological target in glioblastoma
treatment (14). Inhibition of
various kinases within these pathways is an effective treatment for
GBM in vitro, however, clinical translation of these
findings remains to be clarified. This is due in part to cross-talk
and plasticity between cell signalling pathways (15) and to the intrinsic resistance of
GBM cells to apoptosis (16,17).
To overcome the adaptive response to PI3K/AKT/mTOR signalling, a
complete understanding of the role of each individual effector is
required.
The PI3K enzyme family mediate signal transduction
via the phosphorylation of the 3′-OH of phosphatidylinositols
localised to the internal surface of the cell membrane. Once
activated, these messengers are responsible for downstream
transduction through phosphorylation of AKT and the associated
cellular responses. While class-I PI3K p110 isoforms share obvious
structural similarities, a growing body of evidence describes
discrete physiological roles for each isoform (18–24),
with marked alterations in dominant isoform across malignancies
(25), especially in PTEN
deficient solid tumours (26).
Activating point mutations are reported in the class IA
PI3K p110α isoform in glioblastoma (27,28),
along with gene copy amplifications of both PI3K p110α and PI3K
p110δ (29,30). Isoform specific inhibition may
provide significant benefits if used in the appropriate genetic
background (31). A number of
strategies using isoform specific PI3K inhibitors, alone or in
combination with additional compounds are currently in clinical
trial (26,32).
Most investigation has focused on PI3K activity in
the proliferative and migratory phenotypes of differentiated
glioblastoma cells (14,25, 33–36),
however, little is known regarding the activity of PI3K isoforms in
GBM CSC. In the present study, we examined expression and
signalling of class IA PI3K isoforms in two models of
GBM CSC. The cell line 08/04 has high expression of embryonic and
neural stem progenitor genes including SOX2, OCT4 and MSI1 and
recapitulates a GBM phenotype following intracranial implantation
(37). These cells were selected
to model the effect of PI3K inhibition in maintenance of an
established cancer stem cell phenotype. To determine how PI3K
inhibition affects the acquisition of stem-like properties, an LN18
neurosphere model was utilised (37). PI3K isoforms p110α, p110β and p110δ
were selectively inhibited and effects on proliferation and
migration assessed. This revealed a different dominant isoform in
each model. Furthermore, changes in gene expression were evaluated
following inhibition of the dominant isoform, to explore effects on
stem-like and differentiation phenotypes. Inhibition increased
transcription of cancer stem cell genes in both models, suggesting
de-differentiation in response to blockade of the PI3K/AKT/mTOR
signalling axis.
Materials and methods
Cell lines and tissue culture
The human GBM cell line LN18 was obtained from the
American Type Culture Collection and was maintained in RPMI-1640
growth medium supplemented with 10% (v/v) fetal bovine serum (Life
Technologies, Auckland, New Zealand). Cells were discarded within
20 passages of initial receipt and replaced with cryopreserved
stock cultures. Primary human glioblastoma stem cells 08/04
(37) were maintained as an
adherent monolayer in serum-free stem cell medium (SCM)
supplemented with heparin, hEGF and bFGF (NeuroCult NS-A
proliferation kit; Stem Cell Technologies, Tullamarine, VIC,
Australia) as recommended by the manufacturer. All cultures were
maintained at 37°C in a humidified 5% CO2 atmosphere and
subject to regular screening for mycoplasma contamination (e-Myco™
Mycoplasma PCR detection kit; Intron Biotechnology,
Sangdaewon-dong, Korea).
PI3K inhibitors
A66 (38), TGX221
(39), IC87114 (40), BEZ235 (41), targeting p110α, p110β, p110δ and
pan-specific PI3K, respectively, were dissolved in sterile DMSO.
The starting dose for each inhibitor was chosen based on the
IC50 in a cell-free kinase assay, then titrated down
from 100× IC50 to find the maximal tolerated dose. A66
was used at 537.5 nM (12.5× IC50), TGX221 at 850 nM
(100× IC50), IC87114 at 6 μM (100× IC50) and
BEZ235 at 112.5 nM (1.5× IC50).
RT-qPCR
RNA was extracted using the ZR Quick-RNA MiniPrep
kit (Zymo Research, Irvine, CA, USA) in accordance with the
manufacturer's protocol. cDNA was synthesised from 250 ng of total
RNA via the iScript cDNA synthesis kit (Bio-Rad Laboratories,
Auckland, New Zealand). The reverse transcription was run with a
Bio-Rad iCycle PCR machine with the following parameters: 25°C 5
min, 42°C 30 min, 85°C 5 min and the sample was then held at 4°C.
QuantiTect Primer Assays (Qiagen, Melbourne, VIC, Australia)
targeted to 18S (QT00199367), HPRT1 (QT00059066), SOX-2
(QT00237601), MSI-1 (QT00025389), OCT-4 (QT00210840), GFAP
(QT00081151), PIK3CA (QT00014861), PIK3CB (QT01668821) and PIK3CD
(QT00091840) were used with SensiMix SYBR Low-ROX kit (Bioline,
London, UK). The qPCR was run on the Applied Biosystems 7500
Real-Time PCR system (Applied Biosystems, Auckland, New Zealand)
with the following parameters: Stage 1: 94°C 15 min, Stage 2 (40
repeats): 94°C 15 sec, 55°C 30 sec, 72°C 35 sec. Each QuanTitect
Primer Assay is pre-validated to have equivalent amplification
efficiency, thus, direct normalisation to HPRT and 18s was carried
out via the ΔΔCt method. Ct (cycle threshold) values were exported
from the 7500 System software into an Excel file (Microsoft,
Redmond, WA, USA) and all following calculations were done using
Excel software. The triplicate Ct values for each test primer pair
were averaged and then normalized to the average Ct value of 18S
primer pair for each sample to give the ΔCt value. Then the
difference between the ΔCt value for the control and ΔCt value for
the treated cells was calculated to give a ΔΔCt value. The fold
change in gene expression was calculated using
2−ΔΔCt.
Growth factor signalling
Two million cells were plated in SCM (08/04) or
complete RPMI-1640 (LN18) and allowed to proliferate for 2 days.
Media were replaced with NeuroCult NS-A Medium with proliferation
supplement and heparin, but without EGF or FGF (08/04), or
RPMI-1640 without serum (LN18). After 16-h growth factor
withdrawal, cells were treated with PI3K inhibitor or vehicle
control for 1 h before addition of 20 ng/ml hEGF and 10 ng/ml
hFGF-β (08/04) or 10% FBS v/v (LN18) to re-stimulate PI3K activity.
Cells were incubated for 15 min then harvested for analysis by
western blot analyis.
Western blotting
Cells were lysed in 70 mM NaCl, 20 mM Tris, 0.1%
Tween with 1X protease inhibitor (Complete ULTRA EDTA free; Roche,
Auckland, New Zealand) and 1X phosphatase inhibitor (PhosStop;
Roche) and maintained on ice for all subsequent steps. Soluble
material was retained by centrifugation of lysates and total
protein quantified. Protein (50 μg) was electrophoresed with
Amersham GE Precast gels, transferred to PVDF membrane (Bio-Rad
Laboratories) and blocked in 5% BSA (ICP Bio, Auckland, New
Zealand) in PBS. The primary antibodies used were: polyclonal
rabbit anti-human pAKT Ser473, monoclonal rabbit anti-human pAKT
Thr308 (C31E5E), polyclonal rabbit anti-human AKT (Cell Signaling
Technology, Inc., Danvers, MA, USA) and monoclonal, mouse
anti-human α-tubulin (Sigma-Aldrich, Auckland, New Zealand).
Primary antibodies were sequentially blotted at a concentration of
1:1,000 overnight followed by the appropriate anti-mouse or
anti-rabbit horseradish peroxidase antibody at a concentration of
1:7,000 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and
detected by enhanced chemiluminescence (SuperSignal Pico; Pierce,
Auckland, New Zealand). Chemiluminescent images were captured by
the Carestream Gel Logic 4000 Pro using Carestream MI NE software
(Carestream, Rochester, NY, USA). Membranes were stripped (Restore
Western Blot stripping buffer; Thermo Fisher Scientific, Auckland,
New Zealand) and re-imaged between antibodies to confirm complete
signal ablation.
MTS assay
08/04 or LN18 cells were seeded at 25,000 cell/well
on a 96-well plate in the appropriate complete growth media. Cells
were allowed to establish for 24 h before addition of PI3K
inhibitors, which were refreshed every 48 h. Five days following
treatment, 20 μl of
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2H-tetrazolium
(MTS) reagent per well (CellTiter 96 AQueous One Solution; Promega,
Madison, WI, USA) was added and incubated at 37°C, 5%
CO2 for 4 h before absorbance at 490 nm was measured.
Data were normalised to the media only control.
Migration assay
08/04 cells were seeded at 100,000 cells/well on a
24-well plate in SCM and allowed to become confluent (48–72 h),
then pre-treated with PI3K inhibitor for 1 h. A scratch injury was
formed in the monolayer and photographed using the ×10 objective
lens every 2 h for a total of 16 h using the Olympus IX51 inverted
microscope with ColorView III 5 MP camera and Cell A software
(Olympus, Auckland, New Zealand). Cell migration was assessed at
each time-point by area measurements generated in ImageJ software
(33).
Differentiation assay
08/04 cells were seeded in SCM at 96,000 cells/well
on a 6-well plate and allowed to establish for 2 days before
addition of PI3K inhibitor, vehicle control, media only control or
10% FBS v/v to induce differentiation. Cells were incubated for 5
days with inhibitors renewed at day 3, then harvested for RT-qPCR
analysis of embryonic and neural stem cell genes.
Immunofluorescence of GFAP
08/04 cells were seeded at 96,000 cells/well on
coverslips in 6-well plates in SCM and treated for 5 days with PI3K
inhibitor, vehicle control, media only control or 10% FBS v/v to
induce differentiation. Cells were fixed with 95% ethanol and 5%
acetic acid v/v before permeabilisation with PBS/0.5% Tween. Cells
were blocked in 2% BSA in PBS with 0.1% Tween, probed with
monoclonal mouse anti-GFAP (GA5; Cell Signaling Technology,
Auckland, New Zealand) at 4°C overnight. Slides were washed then
incubated with Alexa Fluor 488 goat anti-mouse IgG (Life
Technologies, Auckland, New Zealand) at room temperature for 1 h.
Slides were washed, DAPI stained (UltraCruz mounting media; Santa
Cruz Biotechnology) then imaged by the Olympus BX51TF fluorescent
compound microscope (Olympus). Representative pictures of each
condition were taken in brightfield, DAPI and UV channels and
combined using Pixelmator software for Mac (Pixelmator, Vilnius,
Lithuania).
Neurosphere induction
LN18 cells were pre-treated with inhibitor for 5
days in complete RPMI-1640, with drug refreshed at day 3. Cells
were then seeded into 24-well plate format at 500 cells/well in SCM
containing PI3K inhibitor. After 7 days in culture, developed
neurospheres were harvested and prepared for analysis of embryonic
and neural stem cell genes by RT-qPCR.
Statistical analysis of in vitro
experiments
All statistical analyses were performed using
GraphPad Prism version 4.00 (Graphpad Software, Inc., La Jolla, CA,
USA). Student's unpaired, two-tailed t-tests were performed in
sequence to assess statistical significance between two groups,
with P-values of <0.05 considered as statistically
significant.
Results
PI3K p110α, p110β and p110δ are expressed
but differentially active in 08/04 cells
To determine which isoforms of p110 were expressed
in the GBM stem cell model 08/04, expression of PIK3CA,
PIK3CB and PIK3CD was measured by quantitative
RT-PCR. This confirmed that all isoforms were expressed, although
PIK3CD transcript was present at a lower level than the
other two (Table I).
 | Table IThe PI3K catalytic subunits expressed
in 08/04 cells. |
Table I
The PI3K catalytic subunits expressed
in 08/04 cells.
| α subunit | β subunit | δ subunit |
|---|
| Cycle threshold
(Ct) | 27.04 | 28.86 | 34.04 |
| ΔCt (18s) | 12.86 | 14.69 | 19.86 |
To determine which isoform/s contributed to growth
factor-driven signal transduction in the 0904-SC models, cells were
pre-treated with a specific subunit inhibitor, either A66 (p110α),
TGX221 (p110β) or IC87114 (p110δ) and the effect on growth
factor-driven signal transduction measured by phosphorylation of
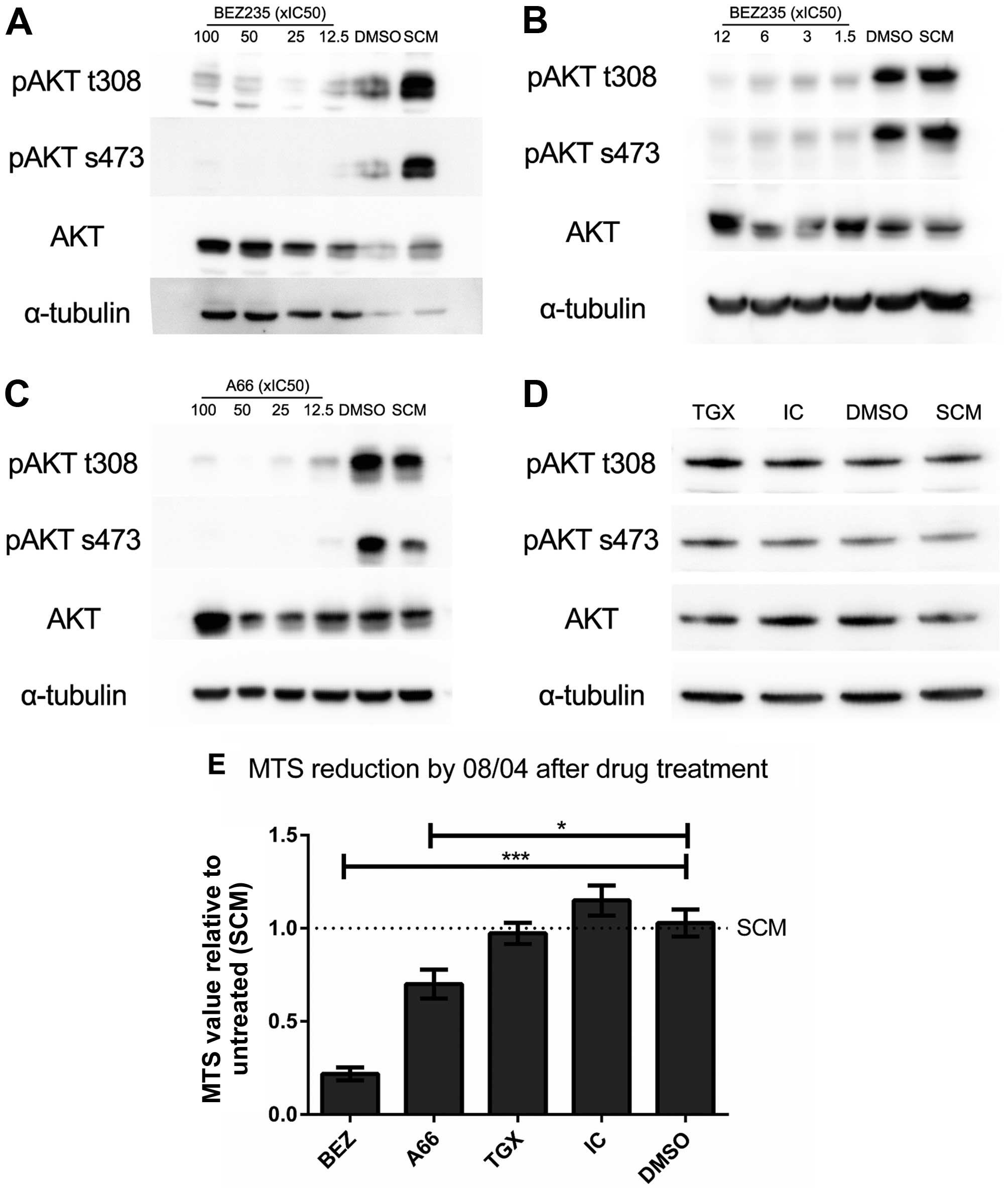
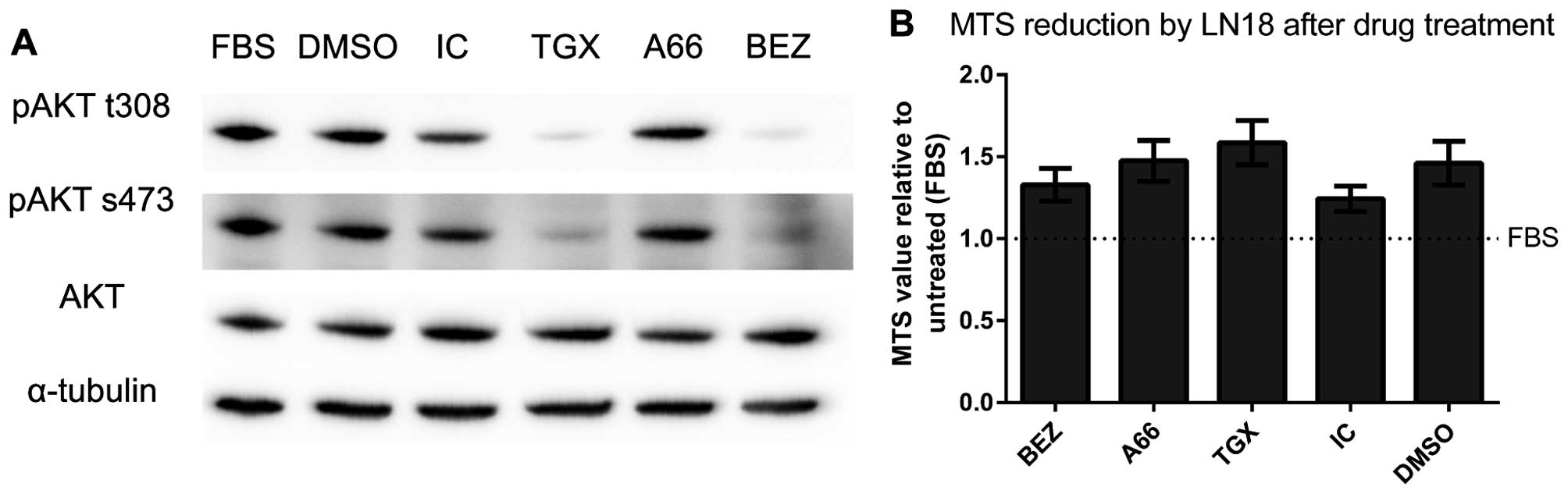
AKT (Fig. 1). The initial dose
chosen was 100× the IC50 of each isoform in a cell-free
kinase assay. For each inhibitor this was 10–20× the
IC50 in cell-based assays, and each was well within the
specific range (38–41). The pan-PI3K/mTOR inhibitor BEZ235
was used as a positive control for lost AKT phosphorylation, and to
compare specific isoform inhibition to broad-spectrum inhibition.
The BEZ235 dose was titrated from 100× to 1.5× IC50. At
this concentration, substantial inhibition of phospho-AKT was
observed (Fig. 1A and B), but
cellular viability and processes were sufficiently conserved to
purify intact RNA from treated cells. The dose of 112.5 nM was used
throughout. A similar titration of A66 was performed, and a dose of
12.5× IC50 (537.5 nM) was identified. Inhibition of
p110α by A66 produced a significant reduction in AKT
phosphorylation at all doses tested, with no detectable signal for
serine 473 and a marked reduction of threonine 308 phosphorylation
(Fig. 1C). In contrast, inhibition
by TGX221 and IC87114 did not change phosphorylation at either
residue at the highest starting dose (Fig. 1D) and had no observable impact on
viability, indicating that PI3K isoforms p110β and p110δ did not
significantly contribute to signalling through AKT in the 08/04
model.
Inhibition of p110α, but not p110β or
p110δ, inhibits proliferation in 08/04 cells
To examine the functional consequence of selective
PI3K p110 inhibition, cytoplasmic reduction of NAD(P)H was measured
using MTS reduction (42,43) following 5 days treatment with each
PI3K inhibitor. Based on the dose titration, treatment was not
accompanied by widespread cell death so the MTS value directly
correlated with cell number, and changes in value reflected
cellular proliferation. Consistent with the reduction of pAKT,
inhibition of p110α by A66, and PI3K/mTOR inhibition by BEZ235
significantly reduced final cell number, and hence proliferation,
for 08/04 in contrast to untreated and vehicle treated cells
(Fig. 1E). Neither TGX221 nor
IC87114 affected final cell number, again consistent with the
observation that p110β and p110δ did not contribute to signalling
through AKT in this model. These data were indicative of a
cytostatic, rather than toxic, effect of inhibition, as observed
previously in inhibition of the PI3K/AKT/mTOR axis (15,44).
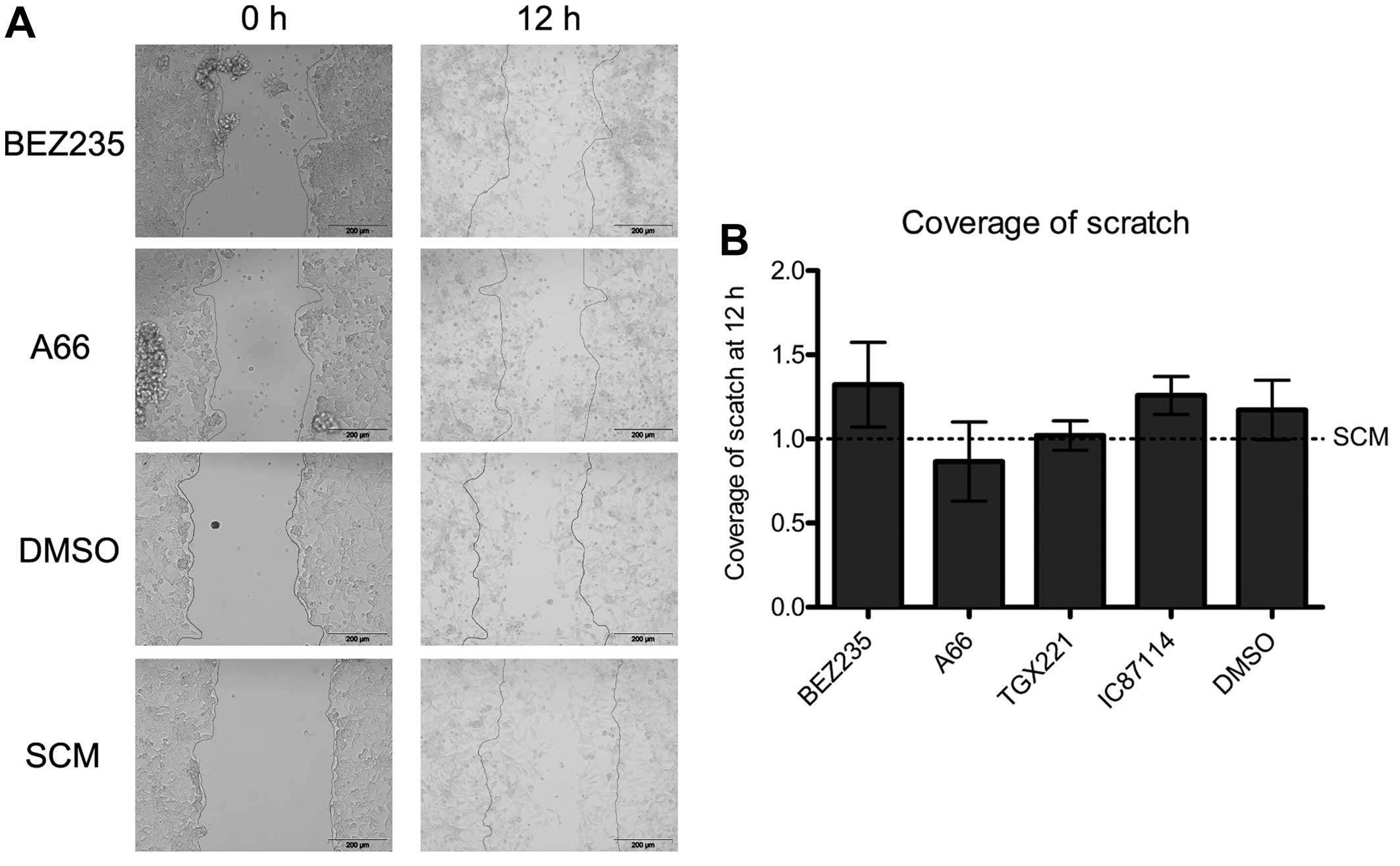
PI3K signalling does not drive migration
in 08/04 cells
Given the potential for CSC to contribute to the
invasive nature of glioblastoma in vivo, the effect of
isoform selective PI3K inhibition on migration was assessed with
the scratch assay. All cells treated with PI3K inhibitors remained
highly migratory, similar to vehicle treated cells (Fig. 2) and despite reduced AKT
phosphorylation in the presence of A66 or BEZ235. No difference in
migration was observed at any of multiple time-points, with all
scratches filled in completely by 16 h. Migration data from 12 h is
shown.
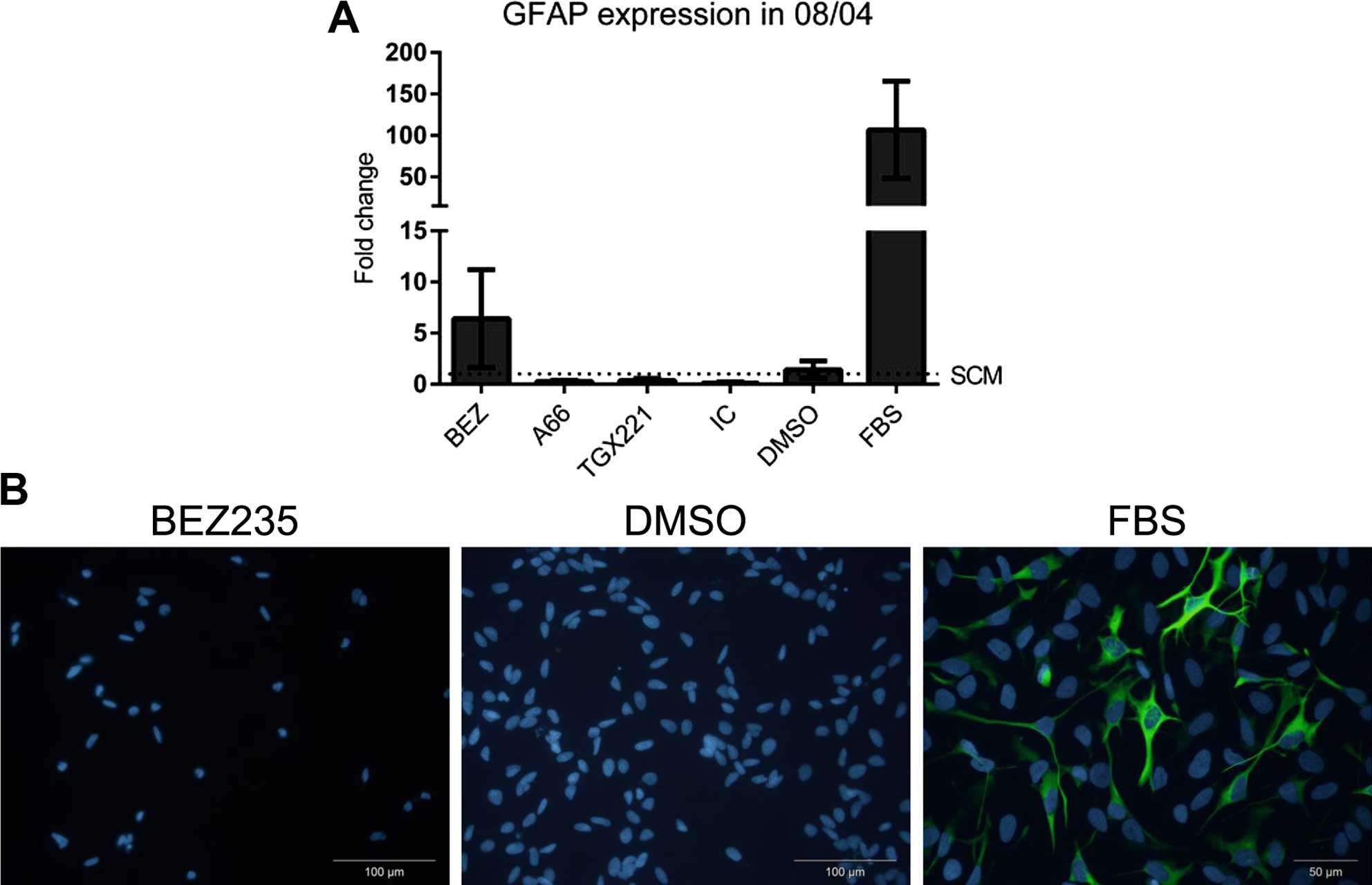
PI3K inhibition does not induce
differentiation of 08/04 cells
Inhibition of either p110α or the PI3K/mTOR pathway
suppressed proliferation of 08/04 cells through a cytostatic
mechanism, as no cell death was observed. We next determined the
effect of isoform selective PI3K inhibition on differentiation of
cells. GFAP is an intermediate filament protein of lineage
committed glial cells, and expression indicates differentiation
down the glial lineage. Using GFAP as a marker for differentiation,
08/04 cells exposed to 10% FBS had 100-fold upregulation of GFAP
mRNA (Fig. 3A) and increased
protein expression (Fig. 3B).
However, isoform selective inhibition did not upregulate the GFAP
transcript (Fig. 3A) and neither
BEZ235 nor A66 induced expression of GFAP protein (Fig. 3B). This suggested that PI3K
inhibition was not associated with increased differentiation of
glioblastoma cancer stem cells.
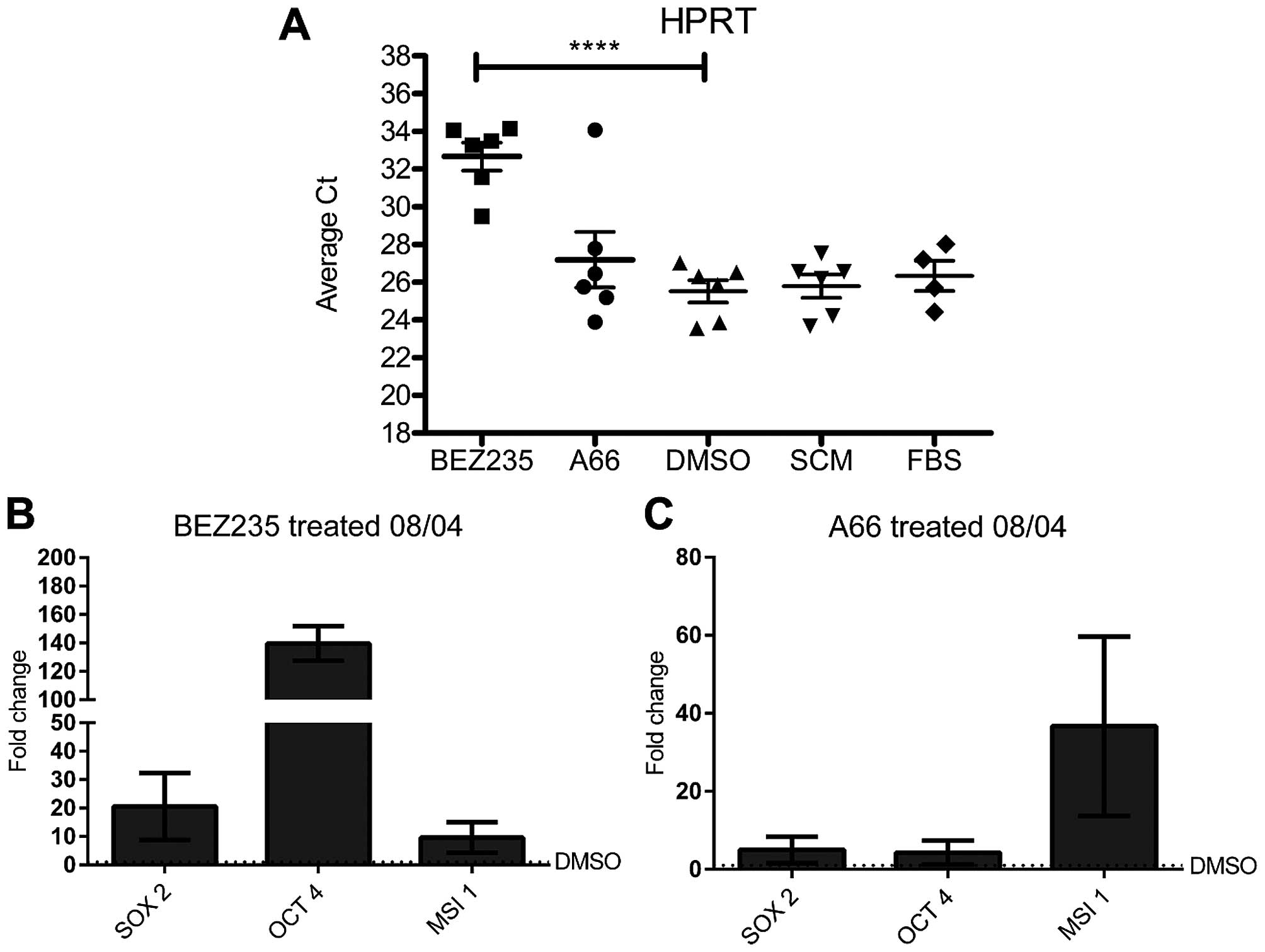
Surprisingly, inhibition of PI3K by BEZ235
reproducibly appeared to upregulate GFAP mRNA, in contrast to the
lack of GFAP protein detected by immunofluorescence. On further
investigation, treatment by BEZ235 was found to decrease the
relative transcript abundance of the gene used for normalisation of
gene expression, HPRT (Fig. 4A) as
well as a number of other genes commonly used for normalisation
(18s rRNA, TBP, β-actin; data not shown). This led to the apparent
increase in GFAP transcription. Importantly, the effect was
restricted to BEZ235 treatment (Fig.
4A). This prevented direct comparison of gene expression
between BEZ235 treated cells and other inhibitor treatments.
However, within a treatment group, gene expression data were
directly comparable, and this approach was used for all further
qRT-PCR analysis.
PI3K inhibition increased expression of
CSC genes in 08/04 cells
We next examined expression of stem cell genes in
our GBM CSC model. Cells were treated with inhibitor for 5 days and
expression of the embryonic and neural stem cell transcription
factors SOX2, OCT4 and MSI1 was measured. A surprising but
consistent pattern of upregulation of CSC gene expression was
observed. When compared to vehicle treated cells, A66
treatment led to an average 5-fold increase in SOX2 and
OCT4, and a 30-fold average increase in MSI1 mRNA (Fig. 4C). Treatment with BEZ235 also
increased expression of SOX2 and MSI1, with a notable
increase in OCT4 expression (Fig. 4B).
PI3K p110α, p110β and p110δ are expressed
but differentially active in LN18
The 08/04 cells effectively model maintenance of an
established cancer stem cell phenotype. In order to determine the
effect of PI3K p110 inhibition on the acquisition of CSC
characteristics, a neurosphere formation model was used. LN18 cells
proliferate as an adherent monolayer when cultured in 10% FBS, but
form neurospheres on transfer to neural stem cell media at low cell
density (37). This acquisition of
a stem cell phenotype, or de-differentiation, is a model of cancer
stem cell initiation.
Basal expression of PI3K p110α, p110β and p110δ in
serum-grown GBM cell line LN18 was established by qRT-PCR and the
isoforms found to be of equal abundance (Table II). Analysis of signalling through
the PI3K/AKT/mTOR axis revealed a difference in the dominant
isoform between LN18 and 08/04. In contrast to 08/04, inhibition of
catalytic subunit p110α by A66 had no effect on pAKT relative to
vehicle control (Fig. 5A).
Instead, inhibition of p110β by TGX221 suppressed phosphorylation
at both sites. Inhibition of p110δ by IC87114 had no significant
effect on the phosphorylation of AKT at either residue, similar to
08/04. As expected, inhibition of PI3K/mTOR activity by BEZ235
supressed phosphorylation of AKT at both threonine 308 and serine
473. Despite suppressed AKT phosphorylation by BEZ235 and TGX221,
there was no significant difference in final number of LN18 cells
(Fig. 5B) following PI3K
inhibition, implying no loss of proliferation or increase in cell
death.
| Table IIThe PI3K catalytic subunits expressed
in LN18. |
Table II
The PI3K catalytic subunits expressed
in LN18.
| α subunit | β subunit | δ subunit |
|---|
| Cycle threshold
(Ct) | 27.98 | 29.65 | 29.55 |
| ΔCt (HPRT) | 1.81 | 3.91 | 4.53 |
PI3K inhibition prior to neurosphere
formation enhances expression of neural and embryonic transcription
factors
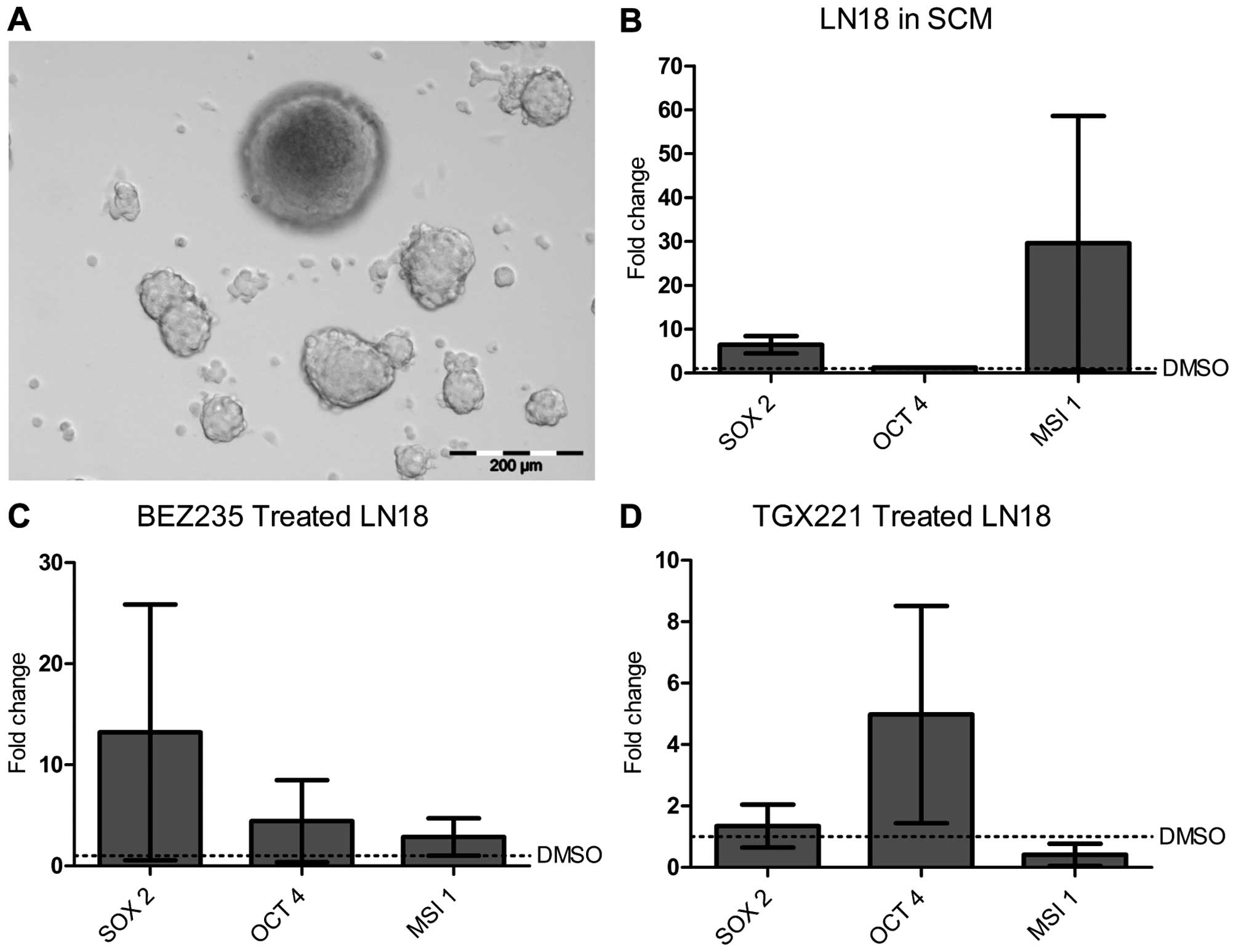
Formation of LN18 neurospheres was previously
characterised by upregulation of embryonic and neural stem cell
gene transcription (37). LN18
cells were pre-treated with TGX221, BEZ235 or DMSO, then
transferred to serum-free stem cell media to induce sphere
formation (Fig. 6A). The effect of
sphere formation on expression of the CSC gene panel was compared
between DMSO and inhibitor treated cells, either BEZ235 (Fig. 6C) or TGX221 (Fig. 6D). There was no significant change
in the rate of sphere formation (data not shown), but in each case,
expression of at least 1 stem cell gene increased with PI3K
inhibition, with a notable increase in OCT4 expression
compared to untreated cells, similar to the 08/04 data (Fig. 4B). Also similar to PI3K inhibition
of 08/04, increased gene expression was more pronounced with
pan-PI3K/mTOR inhibition (Fig.
6C), again suggesting that multiple kinases regulated CSC gene
expression.
Discussion
Activation of the PI3K/AKT/mTOR signalling pathway
reportedly enriches for highly tumorigenic stem-like cancer cells
(45,46) and PI3K inhibition promotes
differentiation, potentially reducing the self-renewal potential of
cancer stem cells within tumours. The different isoforms of the
PI3K catalytic subunit, p110α, β and δ, have been suggested to have
differential roles in pathway activation, and differential effects
on proliferation, migration and differentiation. Based on the
de-differentiated, self-renewing nature of glioblastoma cancer stem
cells, we hypothesised that isoform-specific PI3K inhibition might
specifically target components of cancer stem cell activity as has
been found for embryonic stem cells (47). We looked at activity of the PI3K
catalytic subunits in two different GBM cancer stem cell models,
using specific inhibitors to look first at any role in cancer stem
cell proliferation, and secondly in the acquisition of a stem-like
phenotype. The origin of the GBM CSC is generally thought to be the
acquisition of key stem cell characteristics by a lineage-committed
cell (48). The LN18 sphere
formation assay was used to look specifically at PI3K activity in
the acquisition of stem cell gene expression, as a marker of
de-differentiation of a lineage-committed cell.
The specificity and selectivity of the PI3K
inhibitors was critical to these experiments. The dose titrations
started at 100× the IC50 for a cell-free kinase assay.
At this dose, there was no effect of p110δ inhibition in either
cell line with IC87114, which we interpreted as no activity. This
was equivalent to 12× the cellular IC50. Similarly,
there was no effect of TGX221 on 08/04, at 17× the cellular
IC50. While we have not formally excluded that either
inhibitor would not have been effective at higher concentration
this seems unlikely. There is also the possibility of non-specific
activity if an inhibitor is used at a high dose. There was no
non-specific activity of TGX221 and IC87114, but this could
potentially have been observed with the p110α inhibitor. However,
selectivity of A66 is very high. At 537.5 nM, the dose used in this
study, there is >40-fold higher concentration needed to inhibit
the δ isoform, and >400-fold for inhibition of β (38). Hence, we were well within the
selective range.
While activation of the overall PI3K pathway led to
constitutive AKT phosphorylation in both GBM CSC models, the
activity of the p110 catalytic sub-units were different in each
model. A single dominant isoform was identified in each model,
which differed between the two models, p110α was active in 08/04,
and p110β in the LN18 neurosphere model. Inhibition of PI3K,
whether specific to the dominant p110 isoform, or pan-PI3K/mTOR
inhibition, had similar effects on proliferation in each model.
PI3K inhibition blocked AKT phosphorylation and reduced
proliferation in 08/04. However, neither inhibitor had any effect
on LN18 proliferation despite blocking AKT phosphorylation. The two
GBM cell lines used are very similar in appearance and behaviour
(37), but these data indicate
that there are fundamental differences underlying their PI3K
biology. This could include differences in the mutational status of
the PI3K pathway, such as PTEN loss of function or activating PI3K
mutations. In this regard, LN18 cells are known to have wild-type
PTEN (49), but the genetic
background of the 08/04 line is completely unknown. As differential
expression of targets obviously leads to differential efficacy of
targeted therapies, these data highlight the importance of further
study into the PI3K pathway in GBM before any successful
implementation of PI3K inhibition for cancer stem cell
targeting.
A recently proposed competition model for p110
association with activated receptor tyrosine kinases (RTKs)
(18) highlights the differential
role of the isoforms in the regulation of PI3K activity.
Differences in both lipid- and protein-kinase activities have been
described for PI3K p110 isoforms (50,51),
which allow precise control over downstream transduction in
response to activating signals. In addition, rebound signalling in
other pathways following inhibition of dominant signalling isoforms
has been identified in various cell lines (52,53),
demonstrating the adaptive nature of cancer cell signalling
pathways and consequent cell survival.
Inhibition of either PI3K/mTOR or the dominant
isoform altered expression of embryonic and neural stem cell genes
in both CSC models, with overall increased stem cell gene
expression. The precise effect of PI3K inhibition on stem cell gene
expression varied between inhibitors. Whether this difference
between pan-PI3K/mTOR and dominant isoform inhibition indicates a
link between a given isoform and a corresponding specific CSC gene
is not clear. Any contributory role of minor p110 isoforms, or of
other signalling pathways to CSC gene expression requires further
analysis.
Given that PI3K/AKT activation was previously
reported to increase cancer stem cell gene expression (45,46),
the increased stem cell gene expression with PI3K inhibition was
unexpected and inconsistent with those previous reports. Instead,
it reflected data on PI3K inhibition in embryonic stem cells
(47). It is possible that in our
models, PI3K inhibition directly increased cancer stem cell gene
expression, and simply reflected de-differentiation and acquisition
of a more stem-like phenotype. Alternatively, the increase we
observed could result from a more complex disruption to the stem
cell phenotype. We speculate that while PI3K inhibition attempted
to force cells to differentiate, another signalling pathway
intervened to push stem gene expression back and restore the CSC
phenotype, limiting the effect. In this circumstance, the increased
stem cell gene expression we observed could be an ‘over-shoot’
phenomenon. Signal transduction in the PI3K/AKT/mTOR axis is
partially mediated through a cross-inhibitory relationship with the
MEK/ERK pathway in GBM (54).
MEK/ERK signalling is highly integrated with the downstream
transcriptional activation of embryonic and neural stem cell genes
(55) and it is plausible that
PI3K/MEK/ERK cross-talk plays a role in cancer stem cell gene
expression and activity (56,57).
Regardless of the mechanism, increased expression of
genes that drive a cancer stem cell phenotype would make PI3K
inhibition counter-productive for cancer stem cell targeting, and
for treatment of GBM. These data illustrate the complexity of PI3K
biology in the cancer stem cell phenotype. A careful analysis of
the role of other signalling pathways in the response to specific
isoform inhibition will be necessary before these inhibitors can be
successfully deployed against GBM cancer stem cells, or other
tumours with a cancer stem cell component.
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al; European Organisation for Research and Treatment
of Cancer Brain Tumour and Radiation Oncology Groups; National
Cancer Institute of Canada Clinical Trials Group. Effects of
radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised
phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet
Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maugeri-Saccà M, Di Martino S and De Maria
R: Biological and clinical implications of cancer stem cells in
primary brain tumors. Front Oncol. 3:62013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cruceru ML, Neagu M, Demoulin JB and
Constantinescu SN: Therapy targets in glioblastoma and cancer stem
cells: Lessons from haematopoietic neoplasms. J Cell Mol Med.
17:1218–1235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lathia JD: Cancer stem cells: Moving past
the controversy. CNS Oncol. 2:465–467. 2013. View Article : Google Scholar
|
|
6
|
Hale JS, Sinyuk M, Rich JN and Lathia JD:
Decoding the cancer stem cell hypothesis in glioblastoma. CNS
Oncol. 2:319–330. 2013. View Article : Google Scholar
|
|
7
|
Beier D, Schulz JB and Beier CP:
Chemoresistance of glioblastoma cancer stem cells - much more
complex than expected. Mol Cancer. 10:1282011. View Article : Google Scholar :
|
|
8
|
Yan K, Yang K and Rich JN: The evolving
landscape of glioblastoma stem cells. Curr Opin Neurol. 26:701–707.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Molina JR, Hayashi Y, Stephens C and
Georgescu MM: Invasive glioblastoma cells acquire stemness and
increased Akt activation. Neoplasia. 12:453–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cantley LC and Neel BG: New insights into
tumor suppression: PTEN suppresses tumor formation by restraining
the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA.
96:4240–4245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kwiatkowska A, Kijewska M, Lipko M, Hibner
U and Kaminska B: Downregulation of Akt and FAK phosphorylation
reduces invasion of glioblastoma cells by impairment of MT1-MMP
shuttling to lamellipodia and downregulates MMPs expression.
Biochim Biophys Acta. 1813:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakada M, Nakada S, Demuth T, Tran NL,
Hoelzinger DB and Berens ME: Molecular targets of glioma invasion.
Cell Mol Life Sci. 64:458–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Westhoff MA, Karpel-Massler G, Brühl O,
Enzenmüller S, La Ferla-Brühl K, Siegelin MD, Nonnenmacher L and
Debatin KM: A critical evaluation of PI3K inhibition in
Glioblastoma and Neuroblastoma therapy. Mol Cell Ther. 2:322014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan QW, Knight ZA, Goldenberg DD, Yu W,
Mostov KE, Stokoe D, Shokat KM and Weiss WA: A dual PI3 kinase/mTOR
inhibitor reveals emergent efficacy in glioma. Cancer Cell.
9:341–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lefranc F, Brotchi J and Kiss R: Possible
future issues in the treatment of glioblastomas: Special emphasis
on cell migration and the resistance of migrating glioblastoma
cells to apoptosis. J Clin Oncol. 23:2411–2422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stegh AH, Chin L, Louis DN and DePinho RA:
What drives intense apoptosis resistance and propensity for
necrosis in glioblastoma? A role for Bcl2L12 as a multifunctional
cell death regulator. Cell Cycle. 7:2833–2839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Utermark T, Rao T, Cheng H, Wang Q, Lee
SH, Wang ZC, Iglehart JD, Roberts TM, Muller WJ and Zhao JJ: The
p110α and p110β isoforms of PI3K play divergent roles in mammary
gland development and tumorigenesis. Genes Dev. 26:1573–1586. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Knight ZA, Gonzalez B, Feldman ME, Zunder
ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth
B, et al: A pharmacological map of the PI3-K family defines a role
for p110alpha in insulin signaling. Cell. 125:733–747. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chaussade C, Rewcastle GW, Kendall JD,
Denny WA, Cho K, Grønning LM, Chong ML, Anagnostou SH, Jackson SP,
Daniele N, et al: Evidence for functional redundancy of class IA
PI3K isoforms in insulin signalling. Biochem J. 404:449–458. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup
OV, Mikami A and Roberts TM: The p110alpha isoform of PI3K is
essential for proper growth factor signaling and oncogenic
transformation. Proc Natl Acad Sci USA. 103:16296–16300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Foukas LC, Claret M, Pearce W, Okkenhaug
K, Meek S, Peskett E, Sancho S, Smith AJ, Withers DJ and
Vanhaesebroeck B: Critical role for the p110alpha
phosphoinositide-3-OH kinase in growth and metabolic regulation.
Nature. 441:366–370. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Graupera M, Guillermet-Guibert J, Foukas
LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J,
Cutillas PR, et al: Angiogenesis selectively requires the p110alpha
isoform of PI3K to control endothelial cell migration. Nature.
453:662–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Okkenhaug K, Turner M and Gold MR: PI3K
signaling in B cell and T cell biology. Front Immunol. 5:5572014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schmit F, Utermark T, Zhang S, Wang Q, Von
T, Roberts TM and Zhao JJ: PI3K isoform dependence of
PTEN-deficient tumors can be altered by the genetic context. Proc
Natl Acad Sci USA. 111:6395–6400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar :
|
|
27
|
Gallia GL, Rand V, Siu IM, Eberhart CG,
James CD, Marie SK, Oba-Shinjo SM, Carlotti CG, Caballero OL,
Simpson AJ, et al: PIK3CA gene mutations in pediatric and adult
glioblastoma multiforme. Mol Cancer Res. 4:709–714. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samuels Y, Wang Z, Bardelli A, Silliman N,
Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al:
High frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:5542004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizoguchi M, Nutt CL, Mohapatra G and
Louis DN: Genetic alterations of phosphoinositide 3-kinase subunit
genes in human glioblastomas. Brain Pathol. 14:372–377. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hui AB, Lo KW, Yin XL, Poon WS and Ng HK:
Detection of multiple gene amplifications in glioblastoma
multiforme using array-based comparative genomic hybridization. Lab
Invest. 81:717–723. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jia S, Roberts TM and Zhao JJ: Should
individual PI3 kinase isoforms be targeted in cancer? Curr Opin
Cell Biol. 21:199–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Filbin MG, Dabral SK, Pazyra-Murphy MF,
Ramkissoon S, Kung AL, Pak E, Chung J, Theisen MA, Sun Y,
Franchetti Y, et al: Coordinate activation of Shh and PI3K
signaling in PTEN-deficient glioblastoma: New therapeutic
opportunities. Nat Med. 19:1518–1523. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jhanwar-Uniyal M, Albert L, McKenna E,
Karsy M, Rajdev P, Braun A and Murali R: Deciphering the signaling
pathways of cancer stem cells of glioblastoma multiforme: Role of
Akt/mTOR and MAPK pathways. Adv Enzyme Regul. 51:164–170. 2011.
View Article : Google Scholar
|
|
35
|
Paul-Samojedny M, Pudełko A, Suchanek-Raif
R, Kowalczyk M, Fila-Daniłow A, Borkowska P and Kowalski J:
Knockdown of the AKT3 (PKBγ), PI3KCA, and VEGFR2 genes by RNA
interference suppresses glioblastoma multiforme T98G cells
invasiveness in vitro. Tumour Biol. 36:3263–3277. 2015. View Article : Google Scholar
|
|
36
|
Höland K, Boller D, Hagel C, Dolski S,
Treszl A, Pardo OE, Cwiek P, Salm F, Leni Z, Shepherd PR, et al:
Targeting class IA PI3K isoforms selectively impairs cell growth,
survival, and migration in glioblastoma. PLoS One. 9:e941322014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Broadley KW, Hunn MK, Farrand KJ, Price
KM, Grasso C, Miller RJ, Hermans IF and McConnell MJ: Side
population is not necessary or sufficient for a cancer stem cell
phenotype in glioblastoma multiforme. Stem Cells. 29:452–461. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jamieson S, Flanagan JU, Kolekar S,
Buchanan C, Kendall JD, Lee WJ, Rewcastle GW, Denny WA, Singh R,
Dickson J, et al: A drug targeting only p110α can block
phosphoinositide 3-kinase signalling and tumour growth in certain
cell types. Biochem J. 438:53–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jackson SP, Schoenwaelder SM, Goncalves I,
Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM,
Yuan Y, et al: PI 3-kinase p110beta: A new target for
antithrombotic therapy. Nat Med. 11:507–514. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sadhu C, Masinovsky B, Dick K, Sowell CG
and Staunton DE: Essential role of phosphoinositide 3-kinase delta
in neutrophil directional movement. J Immunol. 170:2647–2654. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Maira SM, Stauffer F, Brueggen J, Furet P,
Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker
K, et al: Identification and characterization of NVP-BEZ235, a new
orally available dual phosphatidylinositol 3-kinase/mammalian
target of rapamycin inhibitor with potent in vivo antitumor
activity. Mol Cancer Ther. 7:1851–1863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Berridge MV, Herst PM and Tan AS:
Tetrazolium dyes as tools in cell biology: New insights into their
cellular reduction. Biotechnol Annu Rev. 11:127–152. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Berridge MV and Tan AS: Characterization
of the cellular reduction of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT):
Subcellular localization, substrate dependence, and involvement of
mitochondrial electron transport in MTT reduction. Arch Biochem
Biophys. 303:474–482. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fan QW, Cheng CK, Nicolaides TP, Hackett
CS, Knight ZA, Shokat KM and Weiss WA: A dual
phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with
blockade of epidermal growth factor receptor in PTEN-mutant glioma.
Cancer Res. 67:7960–7965. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
He K, Xu T, Xu Y, Ring A, Kahn M and
Goldkorn A: Cancer cells acquire a drug resistant, highly
tumorigenic, cancer stem-like phenotype through modulation of the
PI3K/Akt/β-catenin/CBP pathway. Int J Cancer. 134:43–54. 2014.
View Article : Google Scholar
|
|
46
|
Matsubara S, Ding Q, Miyazaki Y, Kuwahata
T, Tsukasa K and Takao S: mTOR plays critical roles in pancreatic
cancer stem cells through specific and stemness-related functions.
Sci Rep. 3:32302013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kingham E and Welham M: Distinct roles for
isoforms of the catalytic subunit of class-IA PI3K in the
regulation of behaviour of murine embryonic stem cells. J Cell Sci.
122:2311–2321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang R, Banik NL and Ray SK: Differential
sensitivity of human glioblastoma LN18 (PTEN-positive) and A172
(PTEN-negative) cells to Taxol for apoptosis. Brain Res.
1239:216–225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Meier TI, Cook JA, Thomas JE, Radding JA,
Horn C, Lingaraj T and Smith MC: Cloning, expression, purification,
and characterization of the human class Ia phosphoinositide
3-kinase isoforms. Protein Expr Purif. 35:218–224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Beeton CA, Chance EM, Foukas LC and
Shepherd PR: Comparison of the kinetic properties of the lipid- and
protein-kinase activities of the p110alpha and p110beta catalytic
subunits of class-Ia phosphoinositide 3-kinases. Biochem J.
350:353–359. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schwartz S, Wongvipat J, Trigwell CB,
Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de
Stanchina E, Baselga J, et al: Feedback suppression of PI3Kα
signaling in PTEN-mutated tumors is relieved by selective
inhibition of PI3Kβ. Cancer Cell. 27:109–122. 2015. View Article : Google Scholar
|
|
53
|
Costa C, Ebi H, Martini M, Beausoleil SA,
Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, et al:
Measurement of PIP3 levels reveals an unexpected role for p110β in
early adaptive responses to p110α-specific inhibitors in luminal
breast cancer. Cancer Cell. 27:97–108. 2015. View Article : Google Scholar
|
|
54
|
Sunayama J, Matsuda K, Sato A, Tachibana
K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A,
et al: Crosstalk between the PI3K/mTOR and MEK/ERK pathways
involved in the maintenance of self-renewal and tumorigenicity of
glioblastoma stem-like cells. Stem Cells. 28:1930–1939. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gough DJ, Koetz L and Levy DE: The MEK-ERK
pathway is necessary for serine phosphorylation of mitochondrial
STAT3 and Ras-mediated transformation. PLoS One. 8:e833952013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Soares HP, Ming M, Mellon M, Young SH, Han
L, Sinnet-Smith J and Rozengurt E: Dual PI3K/mTOR inhibitors induce
rapid overactivation of the MEK/ERK pathway in human pancreatic
cancer cells through suppression of mTORC2. Mol Cancer Ther.
14:1014–1023. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Toulany M, Minjgee M, Saki M, Holler M,
Meier F, Eicheler W and Rodemann HP: ERK2-dependent reactivation of
Akt mediates the limited response of tumor cells with constitutive
K-RAS activity to PI3K inhibition. Cancer Biol Ther. 15:317–328.
2014. View Article : Google Scholar :
|