Introduction
Neuroblastoma is an embryonal tumor of the
sympathetic nervous system, arising during fetal or early postnatal
life from sympathetic cells derived from the neural crest (1). In children, it is the most common
extracranial solid tumor, representing approximately 7% of
childhood malignancies and up to 15% of childhood cancer mortality
(2). The median age of diagnosis
is 22 months (3) and it rarely
presents in adolescence and adulthood (4). Neuroblastoma is an extremely
heterogeneous disease (5); tumors
can spontaneously regress or mature, regardless of therapy, or
display a very aggressive, malignant phenotype that is poorly
responsive to current intensive, multimodal therapy (1). Management of this malignancy remains
a challenge. Recently, we have witnessed a rise in the incidence of
neuroblastoma with a very poor prognosis in children diagnosed
after the age of 2 years, where the 5-year survival rate is only
38% (2). The heterogeneous nature
of this disease makes treatment options tricky because different
treatment strategies are indicated on a case by case basis,
depending on the nature of the disease. Neuroblastoma prognosis
depends on various factors, including the child’s age, histological
characteristics of the tumor, magnitude of genetic abnormalities
and disease stage at diagnosis (6).
Various transcription factors have been implicated
in neuroblastoma pathogenesis contributing to its uncontrolled cell
proliferation, including MycN (2).
The MycN phosphoprotein is a member of the MYC family of
transcription factors, encoded by the MYCN oncogene (2), which drives cell proliferation. While
its expression is very abundant in early embryonic development and
in early neonatal life (7), its
expression in adult cells becomes generally confided to B
lymphocytes.
MycN expression arises again in many cancers and is
found to be amplified in up to 20% of neuroblastoma tumors
(8), and is associated with
advanced stages of disease, rapid tumor progression and poor
treatment outcome. As such and since its first identification in
1983 (5), MycN has gained a
reputation as being a powerful predictor of disease prognosis and
mortality in highly aggressive, malignant tumors, and therefore,
been the attractive target for therapeutic intervention in numerous
cancers including childhood neuroblastomas.
Another interesting molecule found to be expressed
in numerous tumors is the L1-cell adhesion molecule (L1-CAM). First
identified in 1984, the glycoprotein’s role in the development of
the nervous system has been well-established (9). L1-CAM participates in two different
but important physiological processes: on the one hand, it can act
as a cell adhesion molecule that forms the ‘glue’ between cells;
and on the other hand, it can promote cellular motility that drives
cell migration during neural development, but unfortunately also
induces metastasis of human cancers (10). L1-CAM feeds into the MAPK signal
transduction pathway to achieve its functions (11), and it also associates with casein
kinase 2 (12) which inhibits the
functions of the tumor suppressors PTEN and p53, implicated in
neurodegeneration and functional recovery after injury (13,14).
Upregulation of L1-CAM protein expression subsequently leads to the
downregulation of PTEN and p53 protein expression, thereby
promoting neurite outgrowth and neuronal survival (15).
Various cancers expressing high levels of L1-CAM
also exhibit enhanced survival and proliferation potential of the
cancer cells (16). L1-CAM works
together with various receptor tyrosine kinases to promote tumor
growth, metastasis and angiogenesis. In fact, the vascular
endothelial growth factor receptor (VEGFR) has been reported to
associate with L1-CAM and induce proliferation, migration and
angiogenesis (tube formation) in bovine aortic endothelial cells
(17). Furthermore, Schröder et
al (18), reported that
overexpression of L1-CAM in the high grade breast cancers
correlated with overexpression of VEGFR, human epidermal growth
receptor 2 (Her-2) and the plasminogen activator inhibitor 1
(PAI-1) and was a negative prognostic factor. In addition to
stimulating cancer proliferation and migration, L1-CAM has also
been reported to induce the maintenance of self-renewal and
pluripotency, two properties typical of stem cells (19).
Bao et al (20) demonstrated that the
CD133+ glioma stem cells also expressed higher levels of
L1-CAM compared to the CD133− non-stem glioma cells and
that targeting of L1-CAM for transcriptional knock-down led to the
decreased expression of the transcription factor Olig2 and
upregulated the expression of the p21WAF1/CIP1 tumor
suppressor. To determine its role on in vivo malignancy, the
authors injected the L1-CAM knocked-down cells into mice or
targeted L1-CAM for knock-down in mice with established in
vivo tumors. In both groups, tumor growth was inhibited and
survival in tumor-bearing mice was increased with L1-CAM knock-down
(20). Furthermore, L1-CAM was
reported in many cancers, to be concentrated in the peripheral
cells, sustaining invasion and metastasis. An abundance of L1-CAM,
therefore, is associated with poor patient prognosis (17).
In neuroblastomas of children, in contrast to most
adult cancers, the expression of L1-CAM induces improvement and not
worsening of prognosis (21).
However, other pediatric cancers such as osteosarcoma, showed a
negative correlation between the expression of L1-CAM and
prognosis, where higher expression of L1-CAM correlated with
disease progression and poorer prognosis in these children
(22).
In the present study, we investigated the role of
L1-CAM and the receptor tyrosine kinase inhibitor, sunitinib malate
(Sutent), on neuroblastoma cell migration, tumorsphere formation
and proliferation. We specifically explored its expression and
bio-function in the malignant, MycN-amplified human neuroblastoma
IMR-32 cells. We examined the effect of L1-CAM knock-down on the
expression of MycN and PTEN and the subsequent effect on
radio-resistance, cell proliferation, migration and tumorsphere
formation and self-renewal. Tumorsphere self-renewal in a limited
dilution assay is a characteristic of a cancer ‘initiating cell’
(23,24). In addition, these
anchorage-independent cells are usually resistant to conventional
antitumor therapy and can regrow large tumors both in vitro
and in vivo regardless of aggressive radio- or
chemotherapies (25). The
MycN-amplified human IMR-32 neuroblastoma cell line normally grows
as lightly adhered clusters of cell colonies and if cultured in
NeuroCult neural stem-cell enriching media, they would detach from
the culture flask and become fully anchorage-independent, forming
large tumorspheres that resist chemo- and radiotherapy (26). Chakrabarti et al (27) previously reported that
neuroblastoma cells grown in serum-free, NeuroCult stem cell media
supplemented with growth factors (EGF and FGF) underwent phenotypic
transformation from anchorage-dependent, adhered cells to
anchorage-independent floating tumorspheres that overexpressed
tumorigenic proteins and were treatment-resistant. The authors
reported that these cells use their plastic adaptive phenotypic
transformation as a tool to survive unfavorable selection pressure.
We attempted herein, to determine whether L1-CAM played a
significant role on the IMR-32 cells’ capability to undergo
anchorage-independence and form large tumorspheres that can
self-renew in a limited dilution assay. We further attempted to
determine if L1-CAM played a significant role on the IMR-32 cell
proliferation and migration.
Materials and methods
Human cell lines
The human neuroblastoma/neuro-epithelioma cell lines
IMR-32 (MycN-amplified) and SK-N-SH (non-MycN-amplified) were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). Cells were cultured in EMEM containing 10%
fetal bovine serum (FBS), 0.5% penicillin/streptomycin, 10%
L-glutamine.
Derivation of anchorage-independent
tumorspheres
Media
NeuroCult complete media was used to grow the IMR-32
cells as anchorage-independent tumorspheres. NeuroCult complete is
composed of NeuroCult neural stem cell (NSC) basal medium, 1/10
with NeuroCult NSC proliferation supplements, plus 20 ng/ml rh EGF,
10 ng/ml rh FGF-b and 2 μg/ml Heparin (StemCell Technologies, Inc.,
Vancouver, BC, Canada). EMEM (Sigma-Aldrich, St. Louis, MO, USA)
with 10% FBS was used to grow cells in a monolayer of
anchorage-dependent, adhered phenotype.
Tumorsphere formation and self-renewal
assay
Cells were grown in NeuroCult complete media until
large tumorspheres formed. Pelleted tumorspheres were dissociated
into a single-cell suspension, and cell viability determined on a
hemocytometer using trypan blue. Single cells were plated into
96-well plate and cultured in NeuroCult Complete or EMEM + 10% FBS
for 10–14 days. Self-renewal capacity was determined if a single
cell grown in NeuroCult media formed a large tumorsphere starting
as early as day 4–5 after reseeding and reaching full capacity by
days 10–14. Tumorsphere sizes were measured in several random field
images taken using a Zeiss camera mounted onto an inverted
microscope and the AxioVision Systems software (Carl Zeiss,
Oberkochen, Germany).
siRNA transfection
Four L1-CAM siRNA oligonucleotides were purchased
from Qiagen and used to knock-down L1-CAM protein expression in the
IMR-32 cells using Opti-MEM (Lonza, Bassel, Switzerland) and
HiPerFect (Qiagen, Hilden, Germany) transfection reagent. Cells
were grown in a 6-well plate until they reached 70–80% confluency
after which they were transfected with either L1-CAM siRNA at a
concentration of 2 μM for 6 h or with mock-transfection reagent.
The transfection media was then removed and cells were cultured in
their respective media for the length of the experiment.
Radiotherapy
Cells were grown to ~70–80% confluency in their
respective media, then collected and pelleted by centrifugation.
Cells were then irradiated using a single cycle of 2 Gy and then
reseeded in their respective media and grown in the standard
culture conditions. For western blot analysis of protein
expression, cells were lysed 48 h after radiotherapy. For viability
and proliferation studies, cells were plated in 96-well plates at a
density of 3×103 in triplicates and analyzed for the
respective measures at 24, 48, 72 and 96 h post-radiotherapy.
Sunitinib malate (Sutent)
treatment
Cells were grown to ~70–80% confluency in their
respective media, and then treated with varying doses of Sutent
(0.1, 0.2, 0.4, 0.8 and 1 μM) for 24, 48, 72 and 96 h to test cell
proliferation rate and tumorsphere formation over time. For cell
proliferation, the cells were seeded as described below in a
96-well plate and the absorbance of WST-1 was measured at the
indicated time-points. For tumorsphere renewal, 6 h after Sutent
treatment, cells were dissociated and reseeded in a
limited-dilution assay (LDA) in NeuroCult neural stem-cell media
with Sutent to determine tumorsphere self-renewal from single cells
over 7–14 days.
Cell proliferation and viability
assays
Proliferation rate was determined by the
colorimetric absorbance of the WST-1 assay (cat # ab155902; Abcam,
Cambridge MA, USA), at 24, 48, 72 and 96 h post-treatment. This
assay measures the cleavage of the tetrazolium salt WST-1 into
formazan by cellular mitochondrial dehydrogenases. This leads to
more formazan dye formation which can be quantified by measuring
the absorbance at 450 nm. The average absorbance of each group was
graphed using Microsoft Excel at 24, 48, 72 and 96 h
post-radiation. Cell viability was determined by hemocytometer cell
counting using trypan blue. The mean ± the standard deviation of
the mean of multiple experiments was graphed.
Cell migration assays
IMR-32 cells with or without L1-CAM KD were plated
in a tissue culture treated 6-well plate at a density of ~70–80%
confluence of semi-adhered monolayer, serum-deprived overnight and
the next day a cell ‘wound’ was created in the middle of the plate
using a 200 μl pipette. The cells scraped off were washed out (1x)
using serum-free media and fresh media was replenished containing
either 0 or 5% FBS. Images of 6–8 random fields in the scraped
‘wound’ were taken at the time of ‘wound’ induction (0 h) and again
8 h after. The area of the ‘wound’ in these fields was traced and
measured in μM2 using AxioVision systems. The average
area in μM2 of the ‘wound’ 8 h after induction was
subtracted from that at time of induction (0 h) and graphed using
Microsoft excel.
Immunoblot analysis
L1-CAM, PTEN and MycN protein expression was
confirmed by immunoblot as previously described (28). Briefly, whole cell lysates were
prepared from cells lysed in 200 μl of 1X cell lysis buffer (Cell
Signaling Technology). The protein concentration was determined
using the Bradford dye-binding assay (Bio-Rad Laboratories,
Hercules, CA, USA). An aliquot of the lysate was mixed with an
equal volume of 2X Laemmli sample buffer and heated at 97°C for 10
min. Between 20–40 μg of total protein concentration was
electrophoresed on a stain-free easy-cast 10% SDS-PAGE and
transferred onto a PVDF membrane (Bio-Rad Laboratories). Target
proteins were detected using primary antibodies for L1-CAM (rabbit
polyclonal ab123990), MycN (rabbit polyclonal ab24193) and PTEN
(rabbit polyclonal ab31392) with reactivity to mouse, rat and human
(dilution of 1:500; 1:250 and 1:500, respectively) (Abcam).
Beta-actin antibody (rabbit polyclonal anti-actin, sc-130656; Santa
Cruz Biotechnology, Dallas, TX, USA) was used as a loading control.
The blots were incubated with the primary antibody overnight at
4°C, washed (3x) 5 min each in TBS-Tween (0.1%) and incubated with
secondary antibody 1 h at room temperature using the anti-rabbit or
anti-mouse IgG HRP-conjugated (Bio-Rad Laboratories) at a dilution
of 1:2,000. The blots were then washed (4x) 15 min each with 1x
TBS-Tween (0.1%), incubated in Clarity Western ECL substrate (cat#
1705060; Bio-Rad Laboratories) and quantified by densitometric
analysis using Image Lab software from Bio-Rad Laboratories.
Protein expression of L1-CAM, PTEN and MycN was determined using
western blot analysis 24 h after L1-CAM siRNA transfection. Whereas
protein expression after radiotherapy was conducted 48 h after the
radiotherapy (2 Gy) treatment.
Statistical analysis
Experiments were conducted in triplicates and the
mean ± SD of all three experiments was calculated and plotted. A
two-sided Student’s t-test was used to determine statistical
significance between groups. The mean ± SD of three or more
experiments was derived and graphed using Microsoft Excel.
Statistical significance was set at P<0.05.
Results
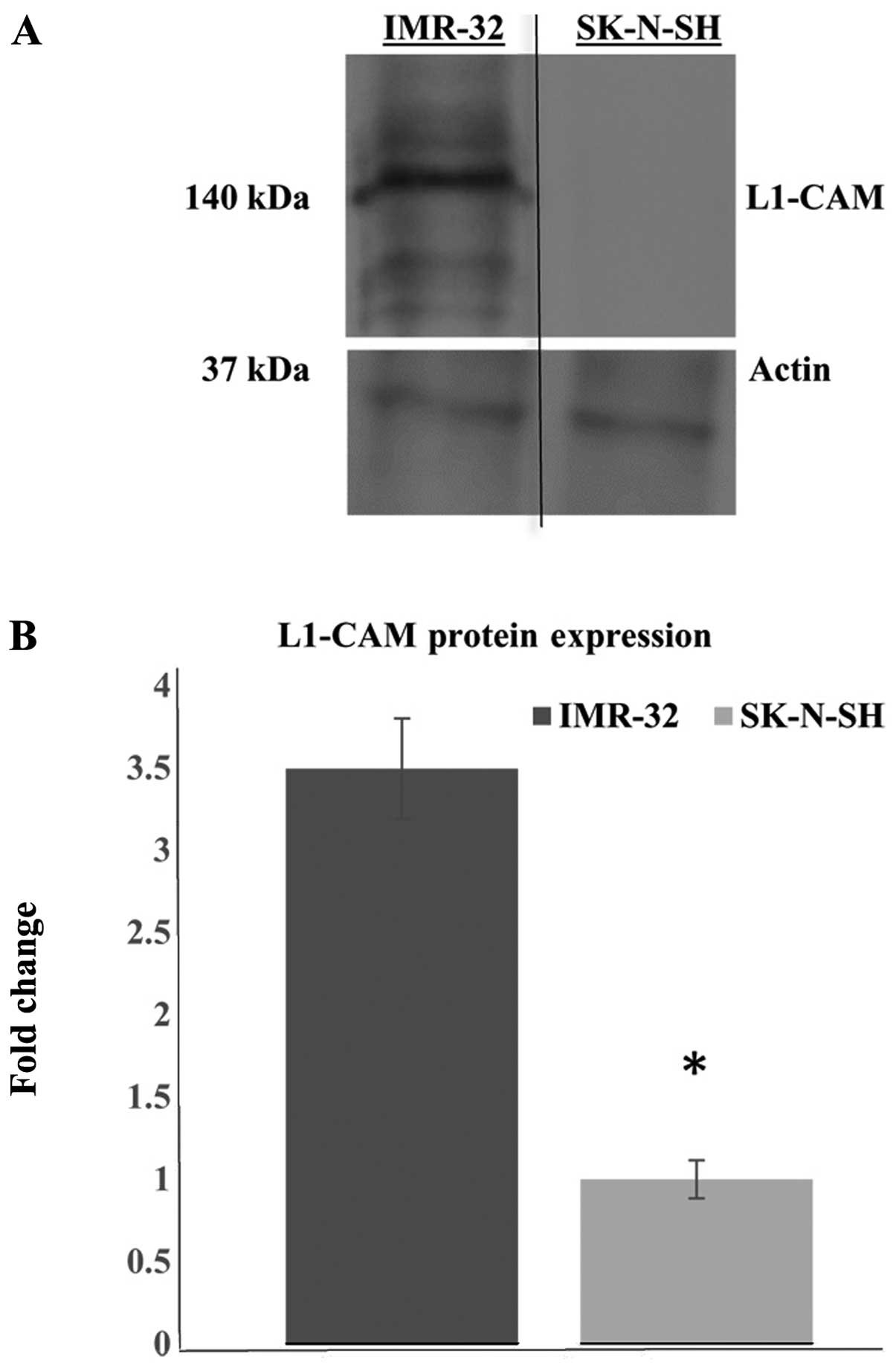
L1-CAM is upregulated in the
MycN-amplified IMR-32 cells compared to the non-MycN amplified
SK-N-SH cells
Western blot analysis revealed the protein
expression of L1-CAM to be upregulated in the MycN-amplified and
highly malignant IMR-32 human neuroblastoma cells, compared to the
less malignant, non-MycN amplified SK-N-SH human neuroblastoma
cells (Fig. 1A). Actin antibody
was used as a loading control. Multiple western blot analyses and
densitometric quantification of the protein bands showed a
statistically significant (P<0.05) upregulation of L1-CAM
protein expression (2.5-fold increase) in the IMR-32 compared to
the SK-N-SH cells (Fig. 1B).
L1-CAM protein expression correlates with
MycN, but inversely correlates with PTEN protein expression in
IMR-32 cells
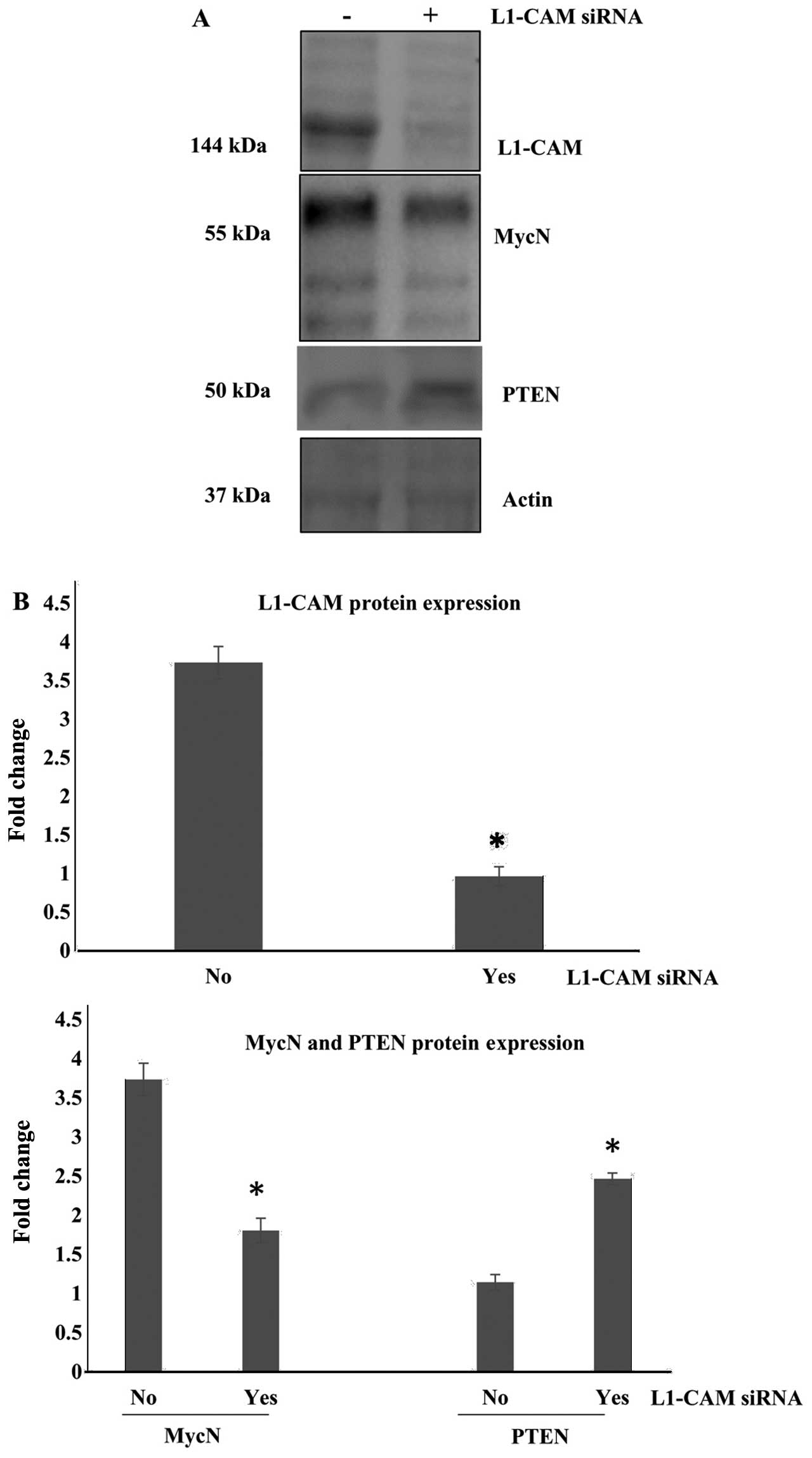
In an attempt to determine the role of L1-CAM
expression in our MycN-amplified IMR-32 cells, we sought to
knock-down (KD) the protein expression of L1-CAM in these cells and
examine the subsequent effect on cell behavior. Small interfering
RNA (siRNA) was used to KD the protein expression of L1-CAM in our
IMR-32 cells which was successfully inhibited compared to the
mock-transfected control cells (Fig.
2A). Notably, protein expression of MycN in these cells was
also abrogated after the siRNA KD of L1-CAM (Fig. 2). The phosphatase and tensin
homolog gene PTEN is one of the most frequently mutated tumor
suppressor genes in many human cancers. We previously reported that
PTEN expression is negatively regulated by the protein expression
and activation of the platelet-derived growth factor receptor beta
(PDGFRβ) in childhood medulloblastomas (28). We therefore sought to determine
whether PTEN expression would also be negatively regulated by the
protein expression of L1-CAM in our cells. We found that siRNA KD
of the L1-CAM protein expression in the IMR-32 cells, led to a
statistically significant (P<0.05) upregulation of PTEN protein
expression (Fig. 2).
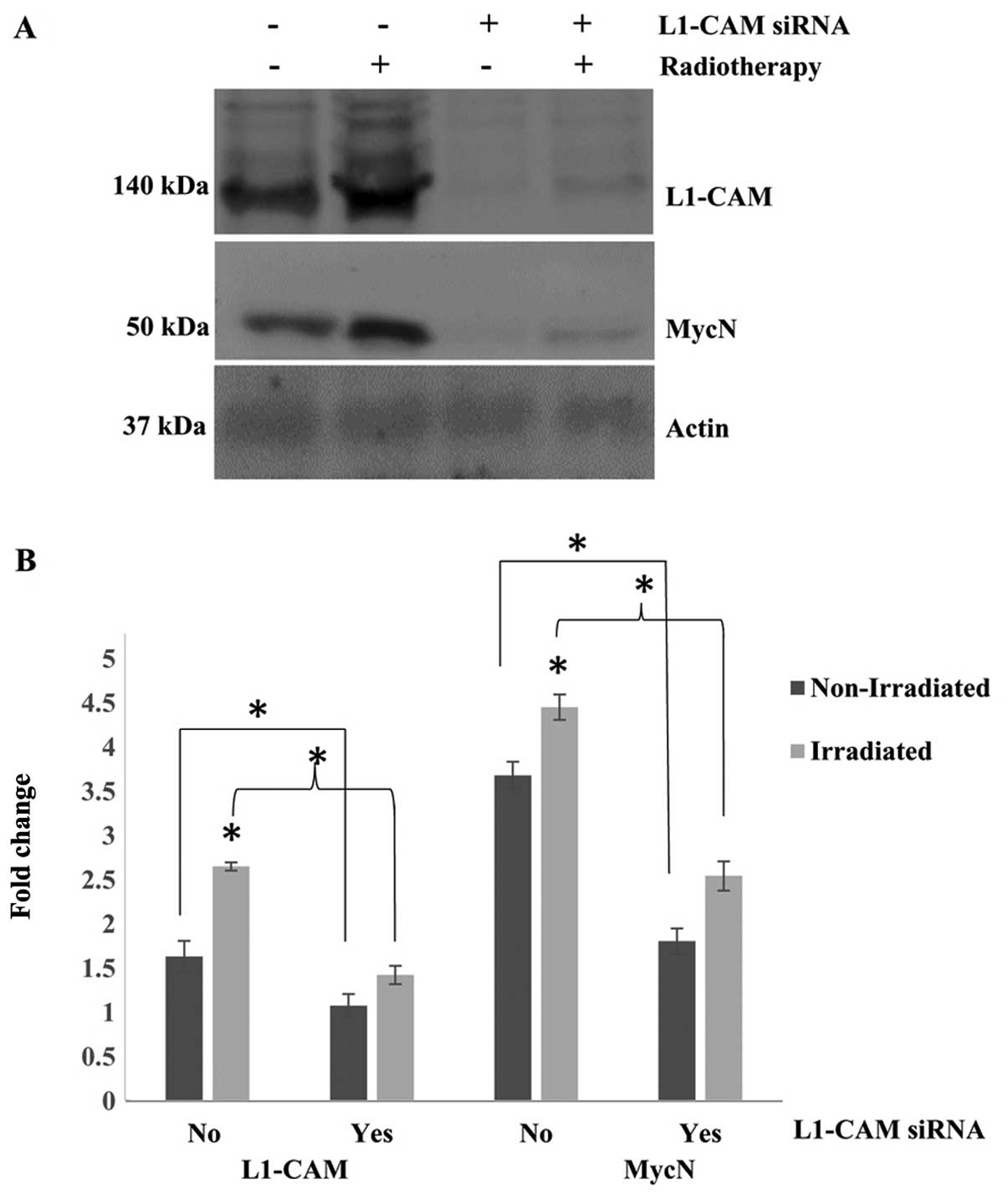
L1-CAM KD significantly inhibits the
radiotherapy-induced upregulation of L1-CAM and MycN protein
expression
Notably, exposing our cells to a single cycle of
2-Gy radiotherapy led to statistically significant upregulation
(P<0.05) of L1-CAM and MycN expression in the IMR-32 cells
(Fig. 3). This effect was observed
48 h after radiotherapy treatment. The radiotherapy-induced
overexpression of L1-CAM and MycN in our cells was abrogated after
L1-CAM siRNA KD of protein expression. Cells with L1-CAM KD were
treated with a single cycle of 2-Gy radiotherapy and then allowed
to grow in culture for an additional 48 h after radiotherapy
treatment. Western blot analysis 48 h after radiotherapy revealed a
statistically significant inhibition (P<0.05) in the
radiotherapy-induced upregulation of L1-CAM and MycN protein
expression in these cells after L1-CAM KD (Fig. 3).
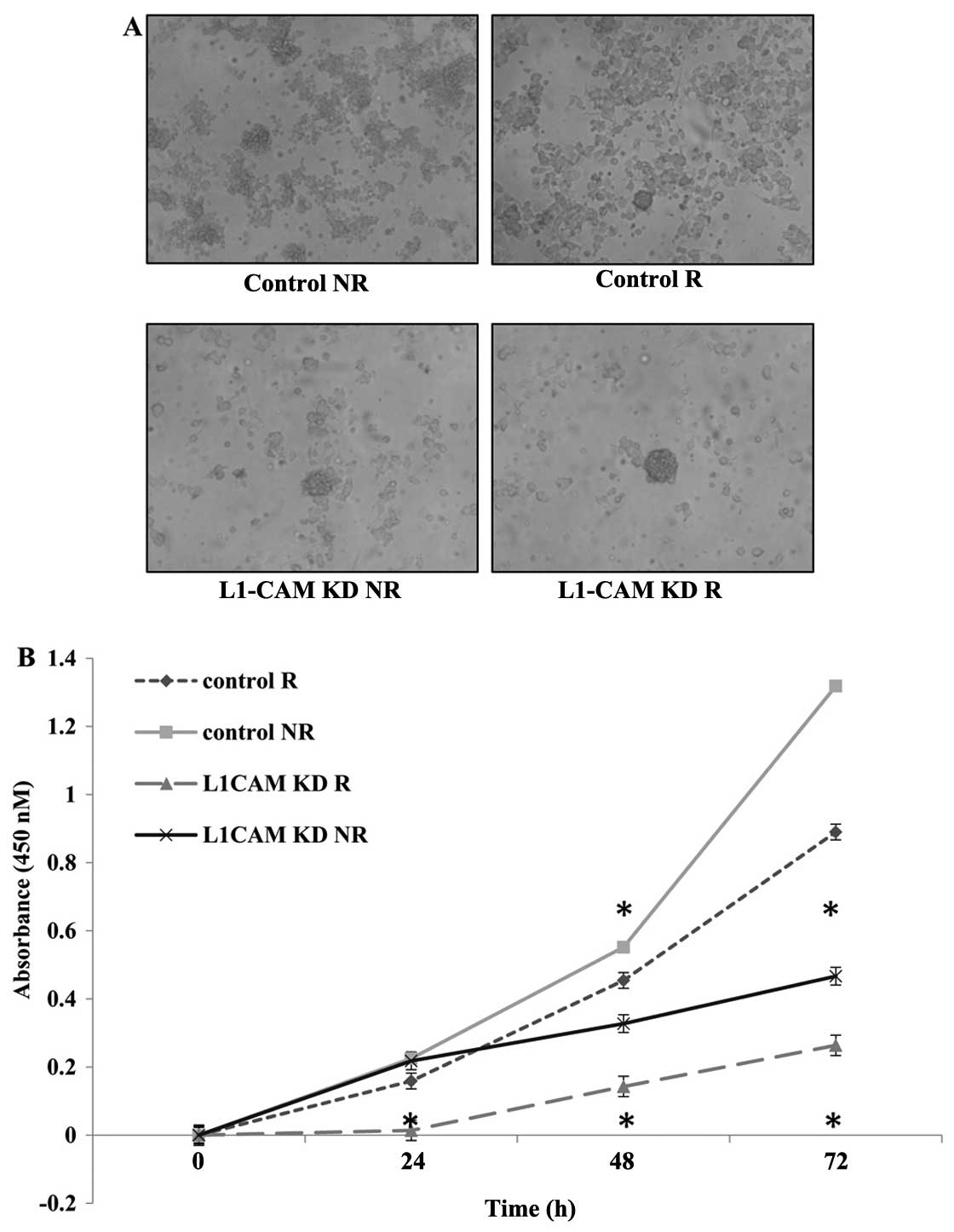
L1-CAM KD radiosensitizes IMR-32 cells by
inducing a synergistic inhibitory effect on cell proliferation
Next we aimed to measure the rate of cell
proliferation in IMR-32 cells after L1-CAM siRNA transfection
(L1-CAM KD) with (R) and without (NR) radiotherapy to determine if
L1-CAM KD would radiosensitize the cells. One cycle of radiotherapy
(2 Gy) in IMR-32 cells led to a statistically significant
inhibition (P<0.05) in the rate of cell proliferation 48 and 72
h post-treatment as measured using a WST-1 cell proliferation
assay. L1-CAM KD alone led to greater anti-proliferative effect in
our cells compared to radiotherapy alone. More importantly, L1-CAM
KD in combination with a single cycle of 2-Gy radiotherapy, led to
a statistically significant synergistic (P<0.001) inhibition on
the rate of cell proliferation compared to radiotherapy alone that
was evident as early as 24 h (P<0.05) post-radiotherapy. There
was no statistically significant difference in the rate of cell
proliferation between the treatment groups at 24 h post-therapy
except when L1-CAM KD was combined with radiotherapy (Fig. 4).
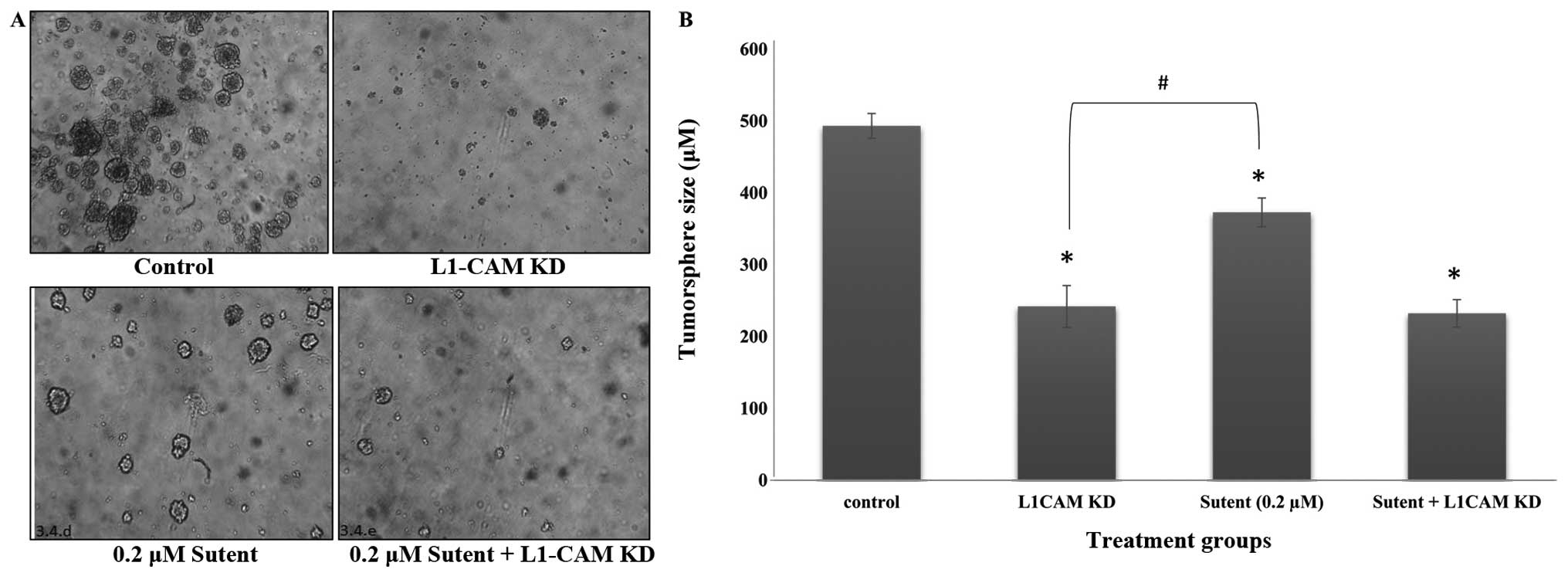
L1-CAM plays a more prominent role than
PDGFRβ or VEGFR on tumorsphere self-renewal in IMR-32 cells
To determine whether L1-CAM plays a significant role
on the formation of tumorspheres in the anchorage-independent
IMR-32 cells, we conducted a tumorsphere self-renewal assay after
L1-CAM KD in these cells. Cells were transfected with L1-CAM siRNA
and 6 h later, reseeded in a limited dilution assay in NeuroCult,
stem-cell enriching media supplemented with EGF and FGF.
Tumorsphere self-renewal was determined if a single cell
recapitulated a large tumorsphere within 5–10 days. IMR-32 cells
transfected with L1-CAM siRNA formed smaller and fewer tumorspheres
compared to the mock-transfected control cells, which formed more
and larger tumorspheres (Fig. 5A)
with a statistically significant (P<0.001) difference compared
to the L1-CAM siRNA transfected cells (Fig. 5B). In an attempt to determine
whether tumorsphere self-renewal is equally inhibited by
inactivation of the RTKs PDGFRβ and VEGFR, we treated the cells
with Sutent, the selective inhibitor of both PDGFR and VEGFR, and
tested tumorsphere self-renewal. Tumorsphere self-renewal capacity
was significantly (P<0.05) inhibited after Sutent treatment
(Fig. 5), but the inhibition
induced by L1-CAM KD was more significant (P<0.05) compared to
Sutent treatment. The combination of the two treatments yielded the
same outcome as that seen with L1-CAM KD alone (Fig. 5B).
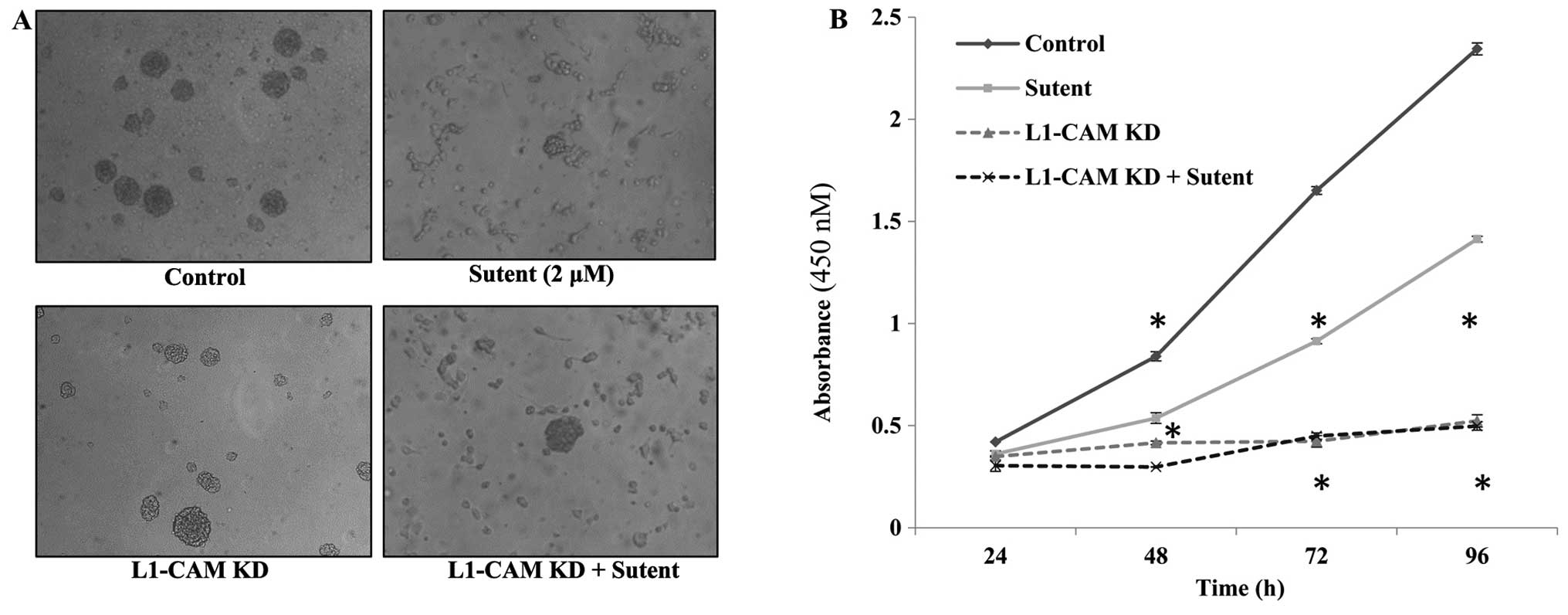
L1-CAM plays a more prominent role than
PDGFRβ or VEGFR on the rate of cellular proliferation in IMR-32
cells
The rate of cell proliferation was assessed after
L1-CAM KD, Sutent treatment or both in the IMR-32 cells compared to
mock-transfected or vehicle-treated control cells. L1-CAM KD led to
a statistically significant (P<0.01) inhibition in the rate of
cell proliferation at 48, 72 and 96 h post-transfection compared to
mock-transfected cells as assessed using a WST-1 cell proliferation
assay (Fig. 6). Sutent treatment
was used to determine whether the rate of cell proliferation was
dependent on the activity of the RTKs PDGFRβ and VEGFR. A single
dose of 0.2 μM Sutent led to a statistically significant inhibition
in the rate of cell proliferation (Fig. 6B); however, L1-CAM KD alone induced
a statistically more significant (P<0.005) reduction in the rate
of cell proliferation compared to Sutent treatment. There was no
added effect of dual L1-CAM KD and Sutent treatment on the rate of
cell proliferation compared to L1-CAM KD alone.
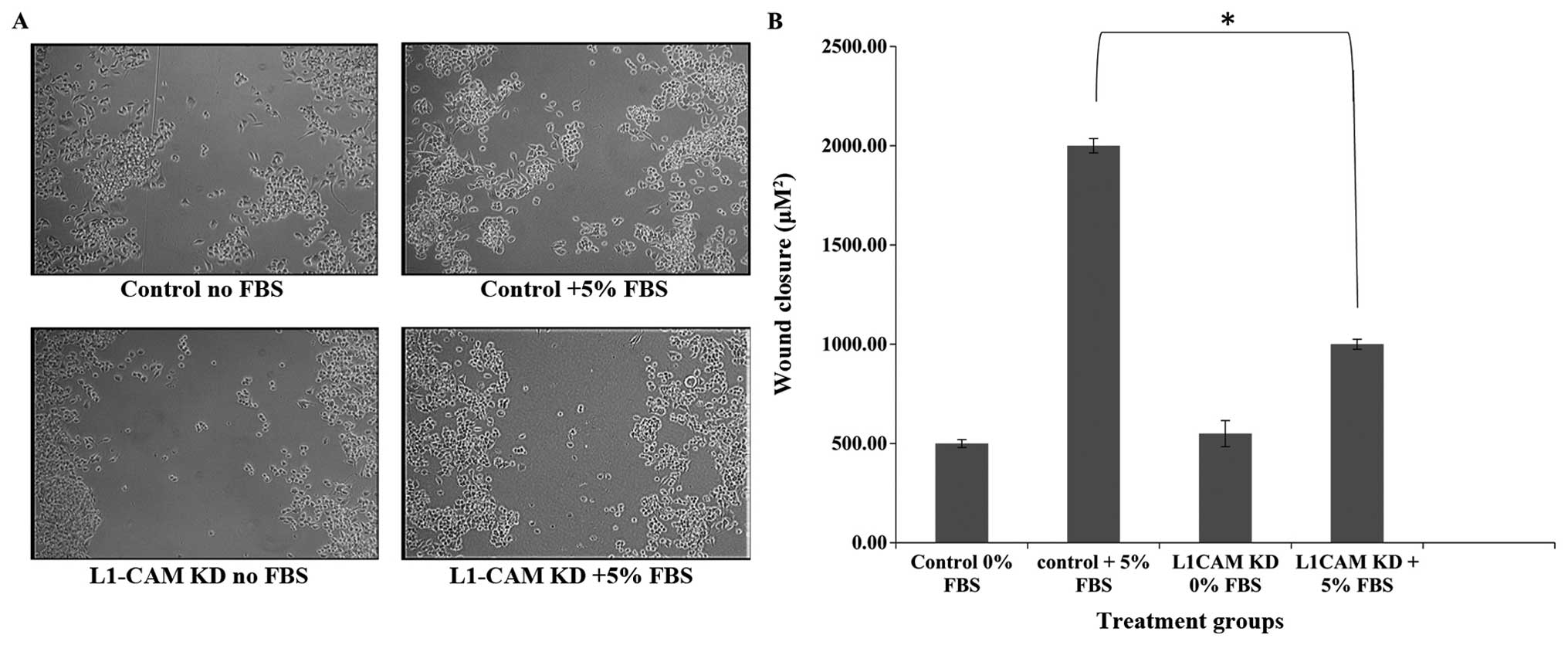
L1-CAM KD significantly inhibits
migration in IMR-32 cells
To determine whether L1-CAM expression is important
in IMR-32 cell migration, we used the ‘wound-healing’ assay to
measure IMR-32 cell migration before and after L1-CAM KD in the
presence of 5% FBS. L1-CAM KD led to a statistically significant
(P<0.005) inhibition in the migratory capability of IMR-32 cells
compared to mock-transfected controls as assessed by their
migration into and closure of the wound (Fig. 7). This effect was not due to a
decrease in the rate of cell proliferation because ‘wound-healing’
was assessed within 8 h of ‘wound-induction’ and only 24 h after
L1-CAM KD, while the rate of cell proliferation was not
significantly different between L1-CAM KD and mock-transfected
control cells until 48 h post-L1-CAM KD (Fig. 5).
Discussion
Of all the childhood malignancies, neuroblastoma
takes center stage, being the most common extracranial solid tumor,
with an incidence rate of approximately 10 cases per million each
year and a spectrum of stages ranging from very mild to very
severe. Children diagnosed with the mild, low-risk disease have an
excellent prognosis with a 5-year survival rate >95%, whereas
those diagnosed with the intermediate and high-risk disease have a
less favorable prognosis with a 5-year survival rate of 90–95 and
40–50%, respectively (29).
MycN-amplification in neuroblastoma is affiliated with poor
prognosis and treatment failure. In the present study, we sought to
determine the role played by L1-CAM in the MycN-amplified human
neuroblastoma cell line IMR-32. We found, using western blot
analysis, L1-CAM expression in the IMR-32 cells to be significantly
overexpressed compared to the non-MycN-amplified human SK-N-SH
cells. Originally identified in the nervous system, L1-CAM was
shown to be expressed in various human tumors and is involved in
cancer cell proliferation, progression and poor prognosis (30–32).
One of the first reports that demonstrated the involvement of
L1-CAM in cancer was the detection of L1-CAM expression in high and
low metastatic B16 melanoma cells (33). Other studies showed the expression
of L1-CAM only in metastatic melanoma cells compared to
non-metastatic cells, which correlated with high levels of
αv-integrin involved in tumor migration (34,35).
Using an antibody against the L1-CAM extracellular domain, L1-CAM
inhibition decreased the migration and invasion of melanoma cells
(36).
We report herein that transcriptional downregulation
of L1-CAM in IMR-32 cells using siRNA transfection led to a
statistically significant reduction in the rate of proliferation,
migration and tumorsphere formation. This effect may be partly due
to the simultaneous abrogation of MycN, and upregulation of PTEN in
these cells after L1-CAM KD. MycN is an onco-protein very well
known for its tumorigenic role in various cancers and particularly
neuroblastomas (8) where it has
been shown to drive uncontrolled cellular proliferation. PTEN, on
the other hand, is a powerful tumor suppressor whose expression and
activity are commonly found to be disrupted in various cancers
(37). PTEN acts a tumor
suppressor by negatively regulating the activity of PI3K/AKT/mTOR
pathway as well as promoting chromosomal stability and DNA repair.
Thus, the inhibition of proliferation in our cells after L1-CAM KD
may be due, in part, to the dysregulation of this pathway.
Previous studies in colon cancer cells showed that
L1-CAM overexpression increased proliferation and migration in
vivo, and metastasis formation upon injection of these cells
into nude mice (38). L1-CAM was
found overexpressed in the invasive front of the cancer (39) and correlated with poor prognosis
and formation of distant metastasis (39,40).
Other studies have shown that inhibiting L1-CAM in Capan-2
pancreatic cancer cells inhibited cell proliferation and invasion
and blocked the cell cycle (41),
while overexpressing L1-CAM in PT45-P1 pancreatic cells activated
proliferation and induced tumor growth in xenograft models
(11). L1-CAM was also detected in
breast and ovarian cancers, gastrointestinal stromal tumors, renal
carcinomas and Schwannomas (42).
Most interestingly, we report that a single cycle of
radiotherapy (2 Gy) led to a statistically significant upregulation
of both L1-CAM and MycN in our cells. Transcriptional KD of L1-CAM
in the IMR-32 cells abrogated this radiotherapy induced
upregulation of L1-CAM and MycN. Thus, there seems to be cross-talk
communication between L1-CAM and MycN in our cells. In support of
our findings, Keerthikumar et al (43) recently reported the co-expression
of L1-CAM among other highly tumorigenic proteins in the
MycN-amplified SK-N-BE2 neuroblastoma cells.
In glioma cells, a subpopulation of
CD133+ stem cells promotes high resistance to radio-and
chemotherapy. Bao et al (20) showed that L1-CAM expression
co-segregated with CD133. Knocking down L1-CAM in this tumor
subpopulation significantly decreased growth and neuro-sphere
formation and induced apoptosis of cells, showing that L1-CAM could
be potentially used to therapeutically target glioma cancer stem
cells. Cheng et al (44)
have also demonstrated that these stem cells activate the DNA
damage checkpoint through the translocation of L1-CAM intracellular
domain into the nucleus when exposed to radiation therapy. The
translocated domain activates the transcription of c-Myc, a member
of the Myc family, which upregulates the expression of NBS1, an
important factor involved in the checkpoint response. Inhibiting
L1-CAM using siRNA decreased c-Myc expression and the checkpoint
activation sensitizing the cells to radiation. Conversely, NBS1
overexpression rescued this decreased activation and
radioresistance. L1-CAM KD in our IMR-32 cells led to a
statistically significant synergistic inhibition on the rate of
cell proliferation when combined with radiotherapy compared to the
effect of radiotherapy alone in these cells.
We next sought to determine whether L1-CAM KD would
exert a synergistic effect on cell behavior when combined with the
tyrosine kinase inhibitor sunitinib malate (Sutent®), a
potent inhibitor of PDGFRβ and VEGFR. To that end, cells with and
without L1-CAM KD were treated with vehicle or 0.2 μM Sutent and
then seeded in a 96-well plate for the analysis of cell
proliferation and tumorsphere self-renewal over time. L1-CAM KD
alone was more prominent than Sutent treatment alone or combined
treatment on inhibiting tumorsphere self-renewal in our cells.
There was no synergistic effect of dual L1-CAM KD and Sutent
treatment on tumorsphere self-renewal capacity. Furthermore, there
was a greater reduction in the rate of cell proliferation after
L1-CAM KD compared to Sutent treatment alone, again demonstrating
the more prominent role played by L1-CAM on the rate of cell
proliferation compared to PDGFRβ and VEGFR. The combination of
L1-CAM KD and Sutent treatment yielded the same effect as L1-CAM KD
alone. Again we report that the lack of synergy when the two
treatments were combined highlights the prominent role played by
L1-CAM on these tumorigenic cellular behaviors. Cell migration, as
assessed using a ‘wound-healing’ assay was also significantly
inhibited after L1-CAM KD in our malignant, MycN-amplified
neuroblastoma cells.
Although the mechanism by which L1-CAM promotes
neuroblastoma growth is still not clear, the present study shows an
important interplay between L1-CAM, PTEN and N-MYC. L1-CAM has been
shown to mediate its signaling either through association with
receptor tyrosine kinases, activation of NF-κB inducing cell
proliferation and metastasis, or through intramembrane proteolysis.
The latter involves the release of L1-CAM extracellular domain to
interact with integrins promoting cell migration, and nuclear
translocation of its intracellular domain to activate gene
transcription (11). Other studies
have determined the involvement of L1-CAM in the MAPK-ERK pathway
where it activates ERK and interacts with Src protein kinase and
Ran-binding protein M (45–47).
It has been particularly found expressed on the invasive front edge
of many malignant tumors, thus, indicating its role in malignant
neoplasm invasion and metastasis, which coincide with poor patient
outcomes and increased mortality rates (47).
We report herein an important interaction between
L1-CAM, PTEN and MycN in the aggressive, MycN-amplified
neuroblastoma IMR-32 cells. Transcriptional downregulation of
L1-CAM led to the concurrent downregulation of MycN and
upregulation of PTEN protein expression. Cellular tumorigenic
behavior was inhibited after L1-CAM KD to a greater extent than
that observed with radiotherapy or Sutent treatment alone.
Moreover, L1-CAM KD led to a synergistic effect on
radiotherapy-induced inhibition of cell proliferation. We conclude
that L1-CAM KD radiosensitizes our IMR-32 cells partly by
downregulating MycN and upregulating PTEN protein expression. In
our future direction, we plan to interrogate the interplay between
PTEN, MycN and L1-CAM in our cells and investigate the molecular
mechanism of this interplay to determine the tumorigenic pathways
activated. We plan to concurrently knock-down the expression of
MycN and L1-CAM and examine the effect of this dual KD on cellular
tumorigenic behavior, radioresistance and PTEN expression and
activation. Moreover, we will examine if overexpression of L1-CAM
and MycN in the non-MycN-amplified SK-N-SH cells would render them
more malignant as assessed by their cellular behavior and
radioresistance.
Acknowledgements
The present study was supported in part by a
research grant funded by the Lebanese National Center for
Scientific Research (Grant # CNRS-632) and in part by the Lebanese
American University School of Pharmacy Research and Development
Fund (Grant # SRDC 2015-02). We would also like to acknowledge the
Radiotherapy Department of the Hopital Notre Dame De Secours in
Byblos, Lebanon where, through a mutual agreement between the
institutions, we have been granted unlimited access to their
radiation facility where we irradiated our cells for this
project.
References
|
1
|
Davidoff AM: Neuroblastoma. Semin Pediatr
Surg. 21:2–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stafman LL and Beierle EA: Cell
proliferation in neuroblastoma. Cancers (Basel). 8:E132016.
View Article : Google Scholar
|
|
3
|
Esiashvili N, Anderson C and Katzenstein
HM: Neuroblastoma. Curr Probl Cancer. 33:333–360. 2009. View Article : Google Scholar
|
|
4
|
Colon NC and Chung DH: Neuroblastoma. Adv
Pediatr. 58:297–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brodeur GM and Nakagawara A: Molecular
basis of clinical heterogeneity in neuroblastoma. Am J Pediatr
Hematol Oncol. 14:111–116. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bottino C, Dondero A, Bellora F, Moretta
L, Locatelli F, Pistoia V, Moretta A and Castriconi R: Natural
killer cells and neuroblastoma: tumor recognition, escape
mechanisms, and possible novel immunotherapeutic approaches. Front
Immunol. 5:562014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zimmerman KA, Yancopoulos GD, Collum RG,
Smith RK, Kohl NE, Denis KA, Nau MM, Witte ON, Toran-Allerand D,
Gee CE, et al: Differential expression of myc family genes during
murine development. Nature. 319:780–783. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang LL, Teshiba R, Ikegaki N, Tang XX,
Naranjo A, London WB, Hogarty MD, Gastier-Foster JM, Look AT, Park
JR, et al: Augmented expression of MYC and/or MYCN protein defines
highly aggressive MYC-driven neuroblastoma: A Children’s Oncology
Group study. Br J Cancer. 113:57–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schäfer MK and Altevogt P: L1CAM
malfunction in the nervous system and human carcinomas. Cell Mol
Life Sci. 67:2425–2437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kiefel H, Bondong S, Hazin J, Ridinger J,
Schirmer U, Riedle S and Altevogt P: L1CAM: A major driver for
tumor cell invasion and motility. Cell Adhes Migr. 6:374–384. 2012.
View Article : Google Scholar
|
|
11
|
Poplawski GH, Tranziska AK, Leshchyns’ka
I, Meier ID, Streichert T, Sytnyk V and Schachner M: L1CAM
increases MAP2 expression via the MAPK pathway to promote neurite
outgrowth. Mol Cell Neurosci. 50:169–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakata A and Kamiguchi H: Serine
phosphorylation by casein kinase II controls endocytic L1
trafficking and axon growth. J Neurosci Res. 85:723–734. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lewandowski G and Steward O:
AAVshRNA-mediated suppression of PTEN in adult rats in combination
with salmon fibrin administration enables regenerative growth of
corticospinal axons and enhances recovery of voluntary motor
function after cervical spinal cord injury. J Neurosci.
34:9951–9962. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schapira AH, Olanow CW, Greenamyre JT and
Bezard E: Slowing of neurodegeneration in Parkinson’s disease and
Huntington’s disease: Future therapeutic perspectives. Lancet.
384:545–555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y and Schachner M: The intracellular
domain of L1CAM binds to casein kinase 2α and is neuroprotective
via inhibition of the tumor suppressors PTEN and p53. J Neurochem.
133:828–843. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colombo F and Meldolesi J: L1-CAM and
N-CAM: From adhesion proteins to pharmacological targets. Trends
Pharmacol Sci. 36:769–781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Friedli A, Fischer E, Novak-Hofer I, Cohrs
S, Ballmer-Hofer K, Schubiger PA, Schibli R and Grünberg J: The
soluble form of the cancer-associated L1 cell adhesion molecule is
a pro-angiogenic factor. Int J Biochem Cell Biol. 41:1572–1580.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schröder C, Schumacher U, Fogel M,
Feuerhake F, Müller V, Wirtz RM, Altevogt P, Krenkel S, Jänicke F
and Milde-Langosch K: Expression and prognostic value of L1-CAM in
breast cancer. Oncol Rep. 22:1109–1117. 2009.PubMed/NCBI
|
|
19
|
Son YS, Seong RH, Ryu CJ, Cho YS, Bae KH,
Chung SJ, Lee B, Min JK and Hong HJ: Brief report: L1 cell adhesion
molecule, a novel surface molecule of human embryonic stem cells,
is essential for self-renewal and pluripotency. Stem Cells.
29:2094–2099. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bao S, Wu Q, Li Z, Sathornsumetee S, Wang
H, McLendon RE, Hjelmeland AB and Rich JN: Targeting cancer stem
cells through L1CAM suppresses glioma growth. Cancer Res.
68:6043–6048. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wachowiak R, Fiegel HC, Kaifi JT, Quaas A,
Krickhahn A, Schurr PG, Erttmann R, Schachner M, Kluth D, Sauter G,
et al: L1 is associated with favorable outcome in neuroblastomas in
contrast to adult tumors. Ann Surg Oncol. 14:3575–3580. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chong Y, Zhang J, Guo X, Li G, Zhang S, Li
C, Jiao Z and Shao M: MicroRNA-503 acts as a tumor suppressor in
osteosarcoma by targeting L1CAM. PLoS One. 9:e1145852014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weiswald LB, Bellet D and Dangles-Marie V:
Spherical cancer models in tumor biology. Neoplasia. 17:1–15. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mack SC, Hubert CG, Miller TE, Taylor MD
and Rich JN: An epigenetic gateway to brain tumor cell identity.
Nat Neurosci. 19:10–19. 2016. View
Article : Google Scholar
|
|
25
|
Xie Q, Flavahan WA, Bao S and Rich J: The
tailless root of glioma: cancer stem cells. Cell Stem Cell.
15:114–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chakrabarti L, Wang BD, Lee NH and Sandler
AD: A mechanism linking Id2-TGFβ crosstalk to reversible adaptive
plasticity in neuroblastoma. PLoS One. 8:e835212013. View Article : Google Scholar
|
|
27
|
Chakrabarti L, Abou-Antoun T, Vukmanovic S
and Sandler AD: Reversible adaptive plasticity: A mechanism for
neuroblastoma cell heterogeneity and chemo-resistance. Front Oncol.
2:822012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abouantoun TJ, Castellino RC and MacDonald
TJ: Sunitinib induces PTEN expression and inhibits PDGFR signaling
and migration of medulloblastoma cells. J Neurooncol. 101:215–226.
2011. View Article : Google Scholar
|
|
29
|
London WB, Castleberry RP, Matthay KK,
Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM,
Reynolds CP, et al: Evidence for an age cutoff greater than 365
days for neuroblastoma risk group stratification in the Children’s
Oncology Group. J Clin Oncol. 23:6459–6465. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bondong S, Kiefel H and Erbe-Hoffmann N:
Prognostic significance of L1CAM in ovarian cancer and its role in
constitutive NF-κB activation. Ann Oncol. 23:1795–1802. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li S, Jo YS, Lee JH, Min JK, Lee ES, Park
T, Kim JM and Hong HJ: L1 cell adhesion molecule is a novel
independent poor prognostic factor of extrahepatic
cholangiocarcinoma. Clin Cancer Res. 15:7345–7351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hai J, Zhu CQ, Bandarchi B, Wang YH, Navab
R, Shepherd FA, Jurisica I and Tsao MS: L1 cell adhesion molecule
promotes tumorigenicity and metastatic potential in non-small cell
lung cancer. Clin Cancer Res. 18:1914–1924. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Linnemann D and Bock E: Expression of the
cell adhesion molecules N-CAM and L1 in B16 melanoma cells. Med
Biol. 64:345–349. 1986.PubMed/NCBI
|
|
34
|
Fogel M, Mechtersheimer S, Huszar M,
Smirnov A, Abu-Dahi A, Tilgen W, Reichrath J, Georg T, Altevogt P
and Gutwein P: L1 adhesion molecule (CD 171) in development and
progression of human malignant melanoma. Cancer Lett. 189:237–247.
2003. View Article : Google Scholar
|
|
35
|
Linnemann D, Raz A and Bock E:
Differential expression of cell adhesion molecules in variants of
K1735 melanoma cells differing in metastatic capacity. Int J
Cancer. 43:709–712. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Thies A, Schachner M, Moll I, Berger J,
Schulze HJ, Brunner G and Schumacher U: Overexpression of the cell
adhesion molecule L1 is associated with metastasis in cutaneous
malignant melanoma. Eur J Cancer. 38:1708–1716. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dillon LM and Miller TW: Therapeutic
targeting of cancers with loss of PTEN function. Curr Drug Targets.
15:65–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gavert N, Sheffer M, Raveh S, Spaderna S,
Shtutman M, Brabletz T, Barany F, Paty P, Notterman D, Domany E, et
al: Expression of L1-CAM and ADAM10 in human colon cancer cells
induces metastasis. Cancer Res. 67:7703–7712. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boo YJ, Park JM, Kim J, Chae YS, Min BW,
Um JW and Moon HY: L1 expression as a marker for poor prognosis,
tumor progression, and short survival in patients with colorectal
cancer. Ann Surg Oncol. 14:1703–1711. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaifi JT, Reichelt U, Quaas A, Schurr PG,
Wachowiak R, Yekebas EF, Strate T, Schneider C, Pantel K, Schachner
M, et al: L1 is associated with micrometastatic spread and poor
outcome in colorectal cancer. Mod Pathol. 20:1183–1190. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ben Q, An W, Fei J, Xu M, Li G, Li Z and
Yuan Y: Downregulation of L1CAM inhibits proliferation, invasion
and arrests cell cycle progression in pancreatic cancer cells in
vitro. Exp Ther Med. 7:785–790. 2014.PubMed/NCBI
|
|
42
|
Raveh S, Gavert N and Ben-Ze’ev A: L1 cell
adhesion molecule (L1CAM) in invasive tumors. Cancer Lett.
282:137–145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Keerthikumar S, Gangoda L, Liem M, Fonseka
P, Atukorala I, Ozcitti C, Mechler A, Adda CG, Ang CS and
Mathivanan S: Proteogenomic analysis reveals exosomes are more
oncogenic than ectosomes. Oncotarget. 6:15375–15396. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cheng L, Wu Q, Huang Z, Guryanova OA,
Huang Q, Shou W, Rich JN and Bao S: L1CAM regulates DNA damage
checkpoint response of glioblastoma stem cells through NBS1. EMBO
J. 30:800–813. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schaefer AW, Kamiguchi H, Wong EV, Beach
CM, Landreth G and Lemmon V: Activation of the MAPK signal cascade
by the neural cell adhesion molecule L1 requires L1
internalization. J Biol Chem. 274:37965–37973. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng L, Lemmon S and Lemmon V: RanBPM is
an L1-interacting protein that regulates L1-mediated
mitogen-activated protein kinase activation. J Neurochem.
94:1102–1110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zecchini S, Bianchi M, Colombo N, Fasani
R, Goisis G, Casadio C, Viale G, Liu J, Herlyn M, Godwin AK, et al:
The differential role of L1 in ovarian carcinoma and normal ovarian
surface epithelium. Cancer Res. 68:1110–1118. 2008. View Article : Google Scholar : PubMed/NCBI
|