Introduction
Hepatic fibrosis is the wound healing response of
the liver to chronic hepatic injury caused by virus infection,
alcohol ingestion and biliary dysfunction. Several studies have
elucidated the various types of effector cells involved in
fibrogenesis and molecular mechanisms regulating these effector
cells (1–3). Hepatic stellate cells (HSCs) are
generally considered as the most prominent cell type involved in
liver fibrogenesis. These cells, upon myofibroblastic transition
(or activation) in response to fibrogenic stimuli, produce large
amounts of ECM proteins, which in turn lead to increased stiffness
of the liver (4).
Although HSC is recognized as a major player in
liver fibrosis, its myofibroblastic transition is regulated by
multiple factors such as inflammation and vascular remodeling. In
the initial stage of fibrogenesis, injured hepatocytes elicit an
inflammatory response that initiates pro-fibrotic cascades
(3,5). For example, infiltrated macrophages
release not only pro-inflammatory cytokines such as tumor necrosis
factor (TNF)-α, interleukin (IL)-6, and IL-1β, but also generate
pro-fibrotic growth factors such as TGF-β1 and platelet-derived
growth factor (PDGF). At the same time, liver sinusoidal
endothelial cells (LSECs) undergo vascular remodeling under
fibrogenic conditions. The vascular remodeling includes
angiogenesis as well as ‘capillarization’ in which LSECs start to
have a basement membrane typical to capillaries in other organs.
Such vascular remodeling is known to make LSECs lose their normal
function of inhibiting HSC activation, which in turn, promotes
fibrosis (6–8).
AMPK is a cellular sensor of energy metabolism that
maintains energy homeostasis of the cell (9). Since many human disorders including
type 2 diabetes, cancer, and inflammatory disease are related to
impaired energy balance, AMPK is becoming a promising drug target
for a wide range of diseases (10–12).
Several lines of studies also reported the importance of AMPK
signaling in hepatic fibrosis. For instance, some AMPK activators
such as 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR),
metformin, and berberine have shown anti-fibrotic effects in animal
models of liver fibrosis (13,14)
and inhibit TGF-β1-induced activation of cultured HSCs (15). Moreover, activation of AMPK
prevents the activation of macrophages in culture (16) and inhibits inflammation either in
liver or in adipose tissues (16,17).
Therefore, AMPK signaling is unlikely to target a single cell type;
rather, it affects multiple cell types.
HL156A is a novel derivative of phenyl biguanide
that is capable of inducing AMPK phosphorylation more potently than
metformin or AICAR (18).
Recently, HL156A has been reported to possess anti-fibrotic
activity in an animal model of peritoneal fibrosis (18). Based on the recent finding on the
HL156A effects on peritoneal fibrosis and the importance of AMPK
signaling in liver fibrogenesis, we hypothesized that HL156A may
have therapeutic potential for liver fibrosis. In the present
study, we therefore explored the anti-fibrotic effect of HL156A in
a mouse model of TAA-induced liver fibrosis as well as in cultured
HSCs and macrophages.
Materials and methods
Animal treatment
All animal procedures conducted in this study were
approved by the Committee for Care and Use of Laboratory Animals at
Seoul National University, according to the Guide for Animal
Experiments edited by the Korean Academy for Medical Sciences. Male
8-week-old C57BL/6 mice were purchased from Orient Bio (Seoul,
Korea). The mice were randomly assigned to four groups. The vehicle
group (n=4) received saline, and the TAA group (n=4) received
thioacetamide intraperitoneally three times a week for a total
duration of 6 weeks. The injection doses of TAA (Sigma-Aldrich, St.
Louis, MO, USA) were 50 mg/kg for the first injection, 100 mg/kg
for the second injection and 150 mg/kg for third to sixth
injections and 300 mg/kg for the rest of the injections. The TAA
and HL156A co-treatment groups (n=5 each) received the same TAA as
the TAA group and HL156A was intraperitoneally injected on the
alternative days of TAA injection at a dose of either 2 mg/kg or 10
mg/kg. After 6 weeks of treatment, mice were euthanized via deep
anesthesia followed by cardiac perfusion.
Histological analysis and
immunohistochemistry
The paraffin sections were de-paraffinized and
stained with hematoxyline and eosin (H&E) or Picro-Sirius Red
(Abcam, Cambridge, MA, USA) according to manufacturer’s
instructions. Immunohistochemistry was performed as previously
described (19). Briefly, paraffin
sections were subjected to antigen retrieval by incubating in
Tris-EDTA buffer (10 mM Tris Base, 1 mM EDTA, 0.05% Tween-20, pH
9.0) for 40 min at 95°C. After blocking with 5% normal donkey serum
(Sigma-Aldrich)/PBS solution, the sections were incubated overnight
at 4°C with primary antibodies for α-smooth muscle actin (α-SMA)
(1:200, Dako, San Diego, CA, USA), Desmin (1:200, Abcam), and
Laminin (1:200, Sigma). After extensive washing in PBS/0.1%
Tween-20 solution, the sections were treated with Alexa-488 or
546-conjugated secondary antibodies (1:750, Invitrogen, Carlsbad,
CA, USA) for 1 h at room temperature followed by counter staining
with Hoechst (Sigma). Fluorescent images were taken under confocal
microscope (Carl Zeiss AG, Oberkochen, Germany), and
immuno-positive areas were quantified by ImageJ software.
Cell culture
HSC-T6 cells were kindly provided by Dr Scott L.
Friedman of Icahn School of Medicine at Mount Sinai. HSC-T6 cells
were maintained in Dulbecco’s modified Eagle’s medium (DMEM;
GenDEPOT, Barker, TX, USA) containing 10% fetal bovine serum (FBS,
GenDEPOT), 100 U/ml penicillin and 100 μg/ml streptomycin
(GenDEPOT). To investigate the effect of HL156A on HSC activation,
cells were pre-treated with HL156A at indicated doses for 2 h and
then treated with TGF-β1 (Peprotech, Rocky Hill, NJ, USA) for 16 h
in serum-free condition. Raw264.7 cells were purchased from Korean
Cell Line Bank (KCLB, Seoul, Korea) and maintained in DMEM
containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml
streptomycin. The cells were pre-treated with HL156A for 2 h at
indicated doses and with LPS (Sigma) for another 4 h. Primary
macrophages were obtained from cultures of bone marrow cells from
femora and tibiae of 6 to 8-week-old male C57BL/6 mice. In brief,
mouse total bone marrow cells were isolated by flushing the
diaphysis of femora and tibiae with cold PBS and incubating
overnight in cell culture dishes in α-modified Eagle medium (α-MEM;
Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10%
FBS, to remove non-hematopoietic lineage cells. After discarding
adherent cells, floating cells were further incubated in α-MEM
supplemented with 10% FBS and macrophage colony-stimulating factor
(M-CSF) (30 ng/ml, BioLegend, San Diego, CA, USA) in Petri dishes.
After 6 days, non-adherent cells were removed and bone marrow
macrophages (BMMs) were re-plated and used in the experiments.
RNA extraction and RT-qPCR
Liver tissues were incubated in Trizol Reagent
(Invitrogen) for 5 min followed by homogenization using Tissue
Lyser II (Qiagen, Hilden, Germany). Total RNA was isolated
according to the manufacturer’s instructions. Total RNA (2 μg) from
each sample was reverse-transcribed with Moloney murine leukemia
virus (MMLV) reverse transcriptase (Promega Corp., Madison, WI,
USA). Quantitative real-time PCR was then performed using
StepOnePlus real-time PCR system (Applied Biosystems, Foster city,
CA, USA) with RealHelix qPCR kit (NanoHelix, Seoul, Korea). The
relative mRNA levels were normalized using glyceraldehyde
3-phosphate dehydrogenase (GAPDH) as an internal control. Primer
sequences used for PCR are summarized in Table I.
 | Table IPrimer sequences used in this
study. |
Table I
Primer sequences used in this
study.
| Primers | Sequences
(5′→3′) |
|---|
| col1a1
(sense) |
CATGTTCAGCTTTGTGGACCT |
| col1a1
(antisense) |
GCAGCTGACTTCAGGGATGT |
| col3a1
(sense) |
TCCCCTGGAATCTGTGAATC |
| col3a1
(antisense) |
TGAGTCGAATTGGGGAGAAT |
| tgf-β1
(sense) |
TTGCTTCAGCTCCACAGAGA |
| tgf-β1
(antisense) |
TGGTTGTAGAGGGCAAGGAC |
| IL-6
(sense) |
TTCCATCCAGTTGCCTTCTTG |
| IL-6
(antisense) |
GGGAGTGGTATCCTCTGTGAAGTC |
| IL-1β
(sense) |
CTACAGGCTCCGAGATGAACAAC |
| IL-1β
(antisense) |
TCCATTGAGGTGGAGAGCTTTC |
| gapdh
(sense) |
TGAACGGGAAGCTCACTGG |
| gapdh
(antisense) |
TCCACCACCCTGTTGCTGTA |
Western blotting
Cells were lysed in radioimmunoprecipitation assay
(RIPA) buffer containing 25 mM Tris pH 7.4, 150 mM NaCl, 5 mM
MgCl2, 0.5% NP-40, phosphatase inhibitor cocktail
(Sigma) and proteinase inhibitor cocktail (Calbiochem, Billerica,
MA, USA). Protein concentrations were determined using a
bicinchoninic acid (BCA) Assay kit (Thermo). Lysates (20 to 30 μg)
were resolved on poly acrylamide gel and then immunoblotted as
previously described (20). The
following antibodies were used; mouse anti-α-SMA (Dako, Glostrup,
Denmark), rabbit anti-vinculin (Santa Cruz Biotechnology, Dallas,
TX, USA), rabbit anti-phosphoAMPK Thr172 (Cell Signaling
Technology, Beverly, MA, USA), and rabbit anti-phosphoSmad2 (Cell
Signaling Technology).
Nitric Oxide (NO) assay
NO assay was carried out in Raw264.7 cells as
previously described (20). In
brief, sub-confluent cells were first treated with HL156A for 2 h
and then incubated in the presence or absence of lipopolysaccharide
(LPS) (100 ng/ml) for 24 h. The conditioned medium was then
collected and mixed with the same volume of Griess reagent (1:1
mixture of 1% sulfanilamide in 30% acetate and 0.1%
N-1-aphthylethylenediamine dihydrochloride in 60% acetate) at room
temperature for 10 min. The absorbance of the incubated samples was
measured by using microplate reader at 540 nm. The concentration of
nitrite in each sample was calculated based on a standard curve
built with known concentrations of sodium nitrite.
Cell viability assay
Cells were plated on 96-well plates and treated with
serial doses of HL156A for 24 h to investigate its effect on cell
viability. Relative cell viability was measured using CellTiter
96® AQueous One Solution Cell Proliferation Assay System
(Promega Corp.) according to manufacturer’s protocol.
HL156A synthesis
HL156A is a derivative of phenyl biguanide, which is
designed and synthesized by Hanall Biopharma Inc. (Seoul, Korea).
The detailed procedure of HL156A synthesis was described in a
previous study (18).
Statistics
Data were expressed as mean ± SEM. One-way ANOVA
followed by Tukey’s test or two-tailed Student’s t-test was used
for statistical analysis. Differences were considered significant
at a P-value <0.05.
Results
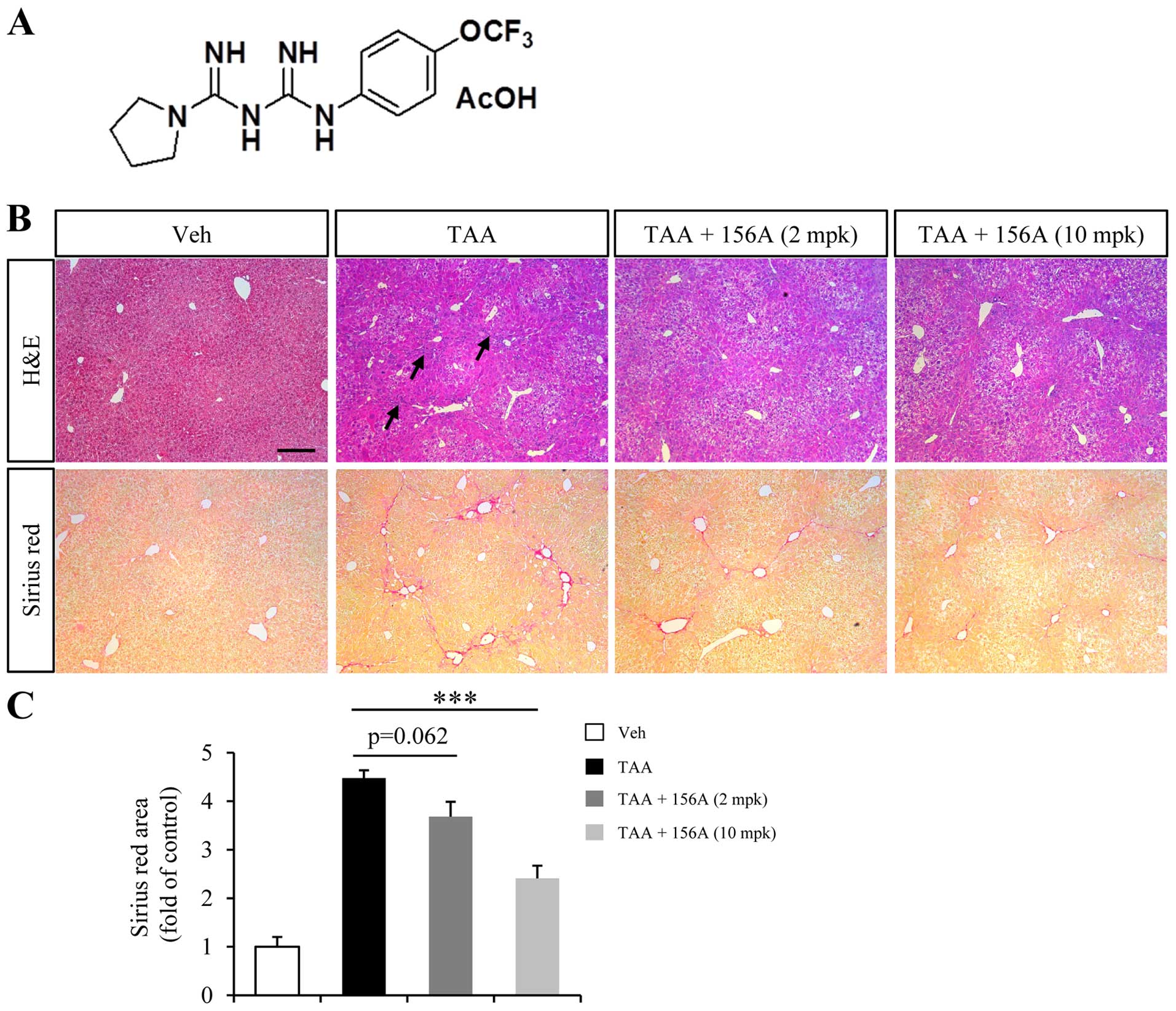
HL156A reduces TAA-induced liver fibrosis
in mice
TAA is one of the common hepatotoxins used in
experimental liver fibrosis that causes centrilobular fibrosis
(21). Since TAA-induced liver
fibrosis is reversible by the withdrawal of TAA exposure, mice were
treated with HL156A and TAA together to investigate the
anti-fibrotic effect of HL156A in liver. Low (2 mg/kg) or high (10
mg/kg) dose of treatment was decided based on a previous study done
in rodents (18). Repeated TAA
injection for a total duration of 6 weeks resulted in inflammation
and alteration of liver histology (Fig. 1B, arrows in upper row). Massive ECM
deposition was also obvious in TAA group as revealed by Sirius Red
staining (Fig. 1B, lower row).
Co-treatment with low dose HL156A slightly reduced ECM deposition
but the difference relative to TAA alone group was not
statistically significant. High dose of HL156A, however,
significantly reduced TAA-induced ECM deposition by approximately
40% when compared to TAA group (Fig.
1B and C).
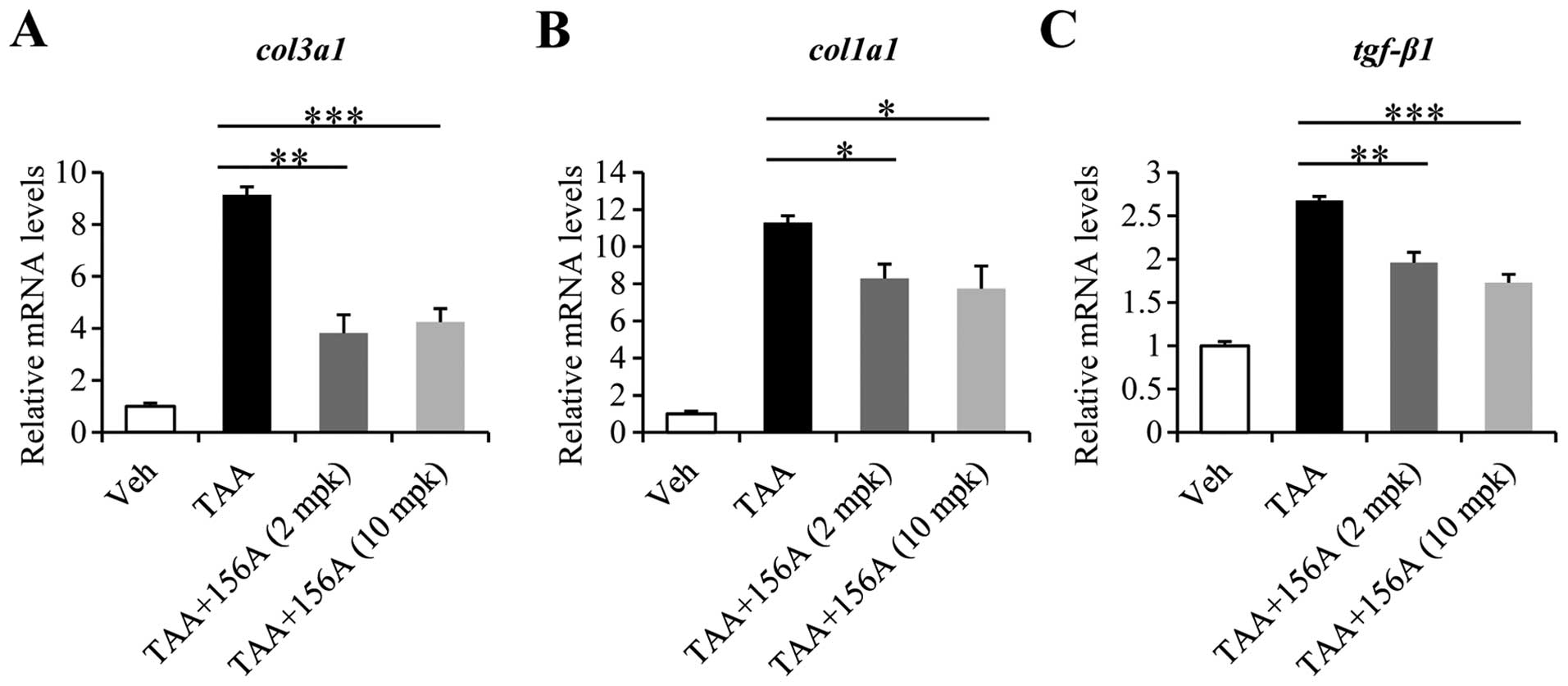
HL156A decreases mRNA levels of collagens
and tgf-β1 induced by liver fibrosis
To analyze the alterations in gene expression caused
by HL156A, RNA was extracted from liver tissues of each
experimental group and subjected to RT-qPCR. Among the several ECM
components, the mRNA expressions of col1a1 and col3a1
were drastically increased by up to 10-fold by TAA. Co-treatment
with HL156A reduced col3a1 levels by up to 60% and those of
col1a1 by up to 30% at either 2 mg/kg or 10 mg/kg dose
(Fig. 2A and B). Of note, the mRNA
level of tgf-β1, a potent profibrogenic factor was also
significantly decreased by HL156A co-treatment (Fig. 2C).
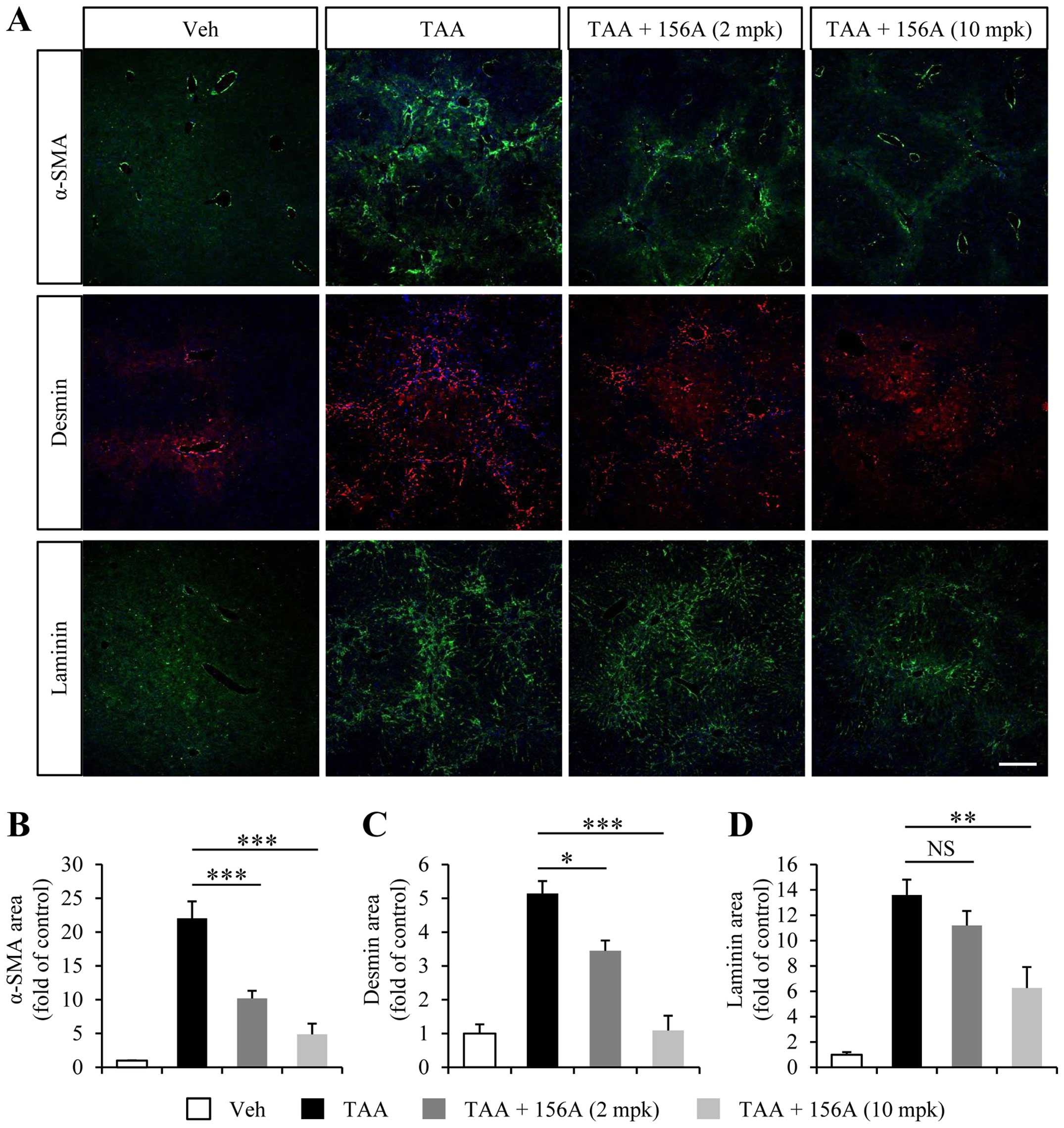
TAA-induced HSC activation and
endothelial capillarization are reversed by HL156A
One major feature of hepatic fibrosis is the
activation of hepatic stellate cells, which is characterized by the
expression of myofibroblast markers such as α-SMA and desmin. To
address cellular events regulated by HL156A, we analyzed liver
histology using myofibroblast markers. As shown earlier, TAA
administration led to upregulation of α-SMA- as well as
desmin-positive cell populations (Fig.
3A). HL156A significantly and dose-dependently diminished both
α-SMA and desmin-positive immunoreactivity (Fig. 3A–C). These results indicate that
HL156A inhibits HSC activation, which is a critical step in hepatic
fibrogenesis.
Several lines of evidence revealed that liver
sinusoidal endothelium underwent vascular remodeling upon
fibrogenesis (6,22). The loss of liver sinusoid
characteristics, referred to as capillarization, is known to
accelerate HSC activation (7). As
previously reported (23), we
observed an increase in laminin-positive area in close proximity to
fibrosis area in the liver sections of TAA treated animals
(Fig. 3A). High dose of HL156A (10
mg/kg) significantly reduced laminin area while there was no
significant reduction in the laminin immunoreactivity at low doses
of HL156A (2 mg/kg).
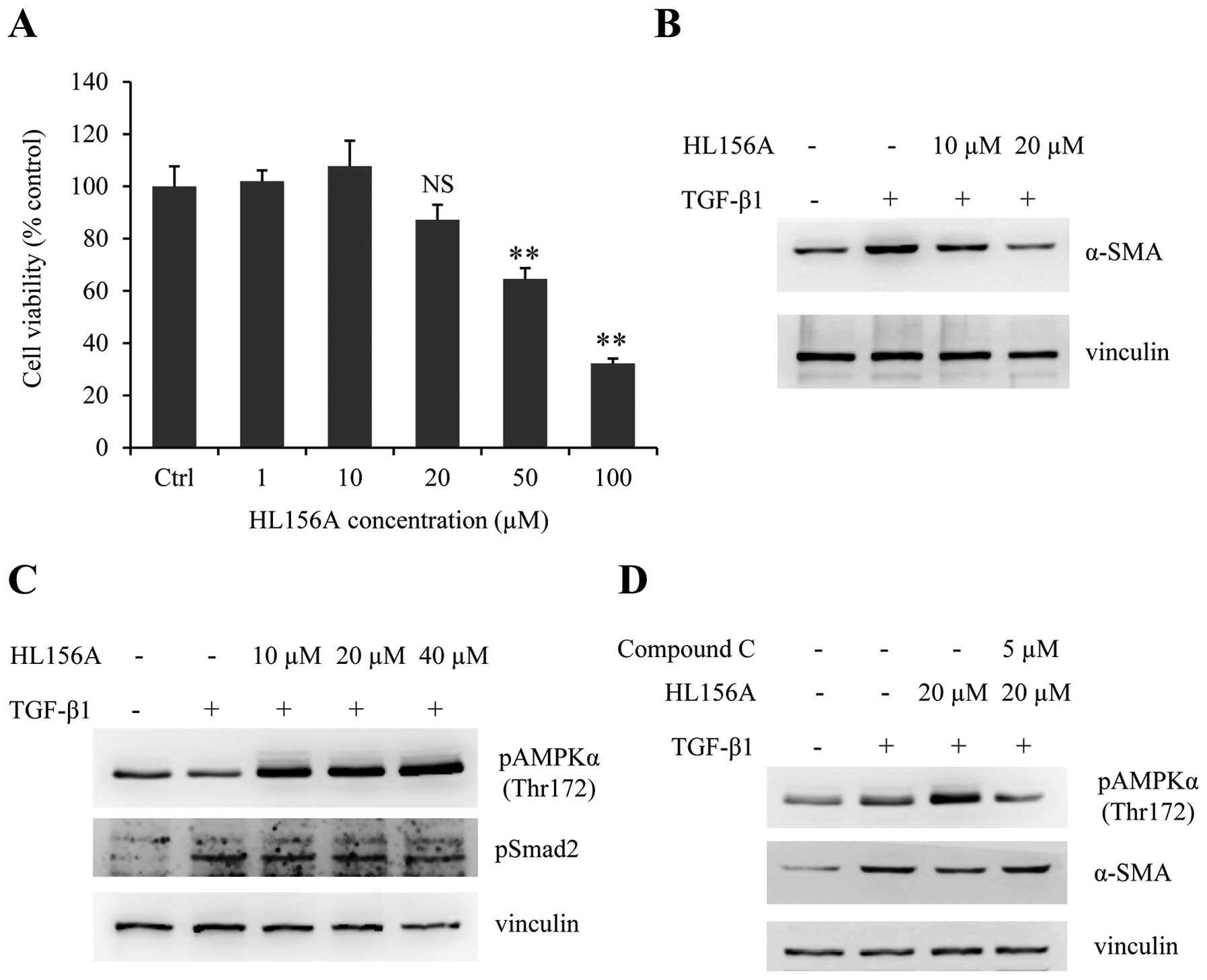
HL156A attenuates TGF-β1-mediated HSC
activation via the activation of AMPK
To further study the anti-fibrotic effect of HL156A
in vitro, we took advantage of the rat HSC cell line HSC-T6.
MTS assay was carried out to determine proper doses of HL156A
without cytotoxicity. HSC-T6 cells were treated with serial doses
of HL156A ranging from 1 to 100 μM for 24 h and relative cell
viabilities were measured. As shown in Fig. 4A, no significant cytotoxicity was
observed up to 20 μM while HL156A concentrations over 50 μM showed
some cytotoxicity. Preliminary experiments revealed that there is
no or limited effect of HL156A on TGF-β1-induced HSC activation at
concentrations <10 μM (data not shown). The treatment of HSC-T6
cells with TGF-β1 resulted in induction of α-SMA; simultaneous
treatment with HL156A attenuated TGF-β1-mediated α-SMA induction at
doses of 10 to 20 μM (Fig. 4B).
Since HL156A is a new AMPK activator, we next investigated whether
or not the inhibitory effect of HL156A on HSC activation involved
AMPK activation. Treatment with HL156A promoted AMPK
phosphorylation in a dose-dependent manner, whereas Smad
phosphorylation by TGF-β1 was not affected by HL156A (Fig. 4C). Moreover, the inhibitory effect
of HL156A on α-SMA expression was reversed by co-treatment with the
AMPK inhibitor, compound C (Fig.
D). These data revealed that HL156A has an inhibitory effect on
HSC activation, probably via the stimulation of AMPK signaling.
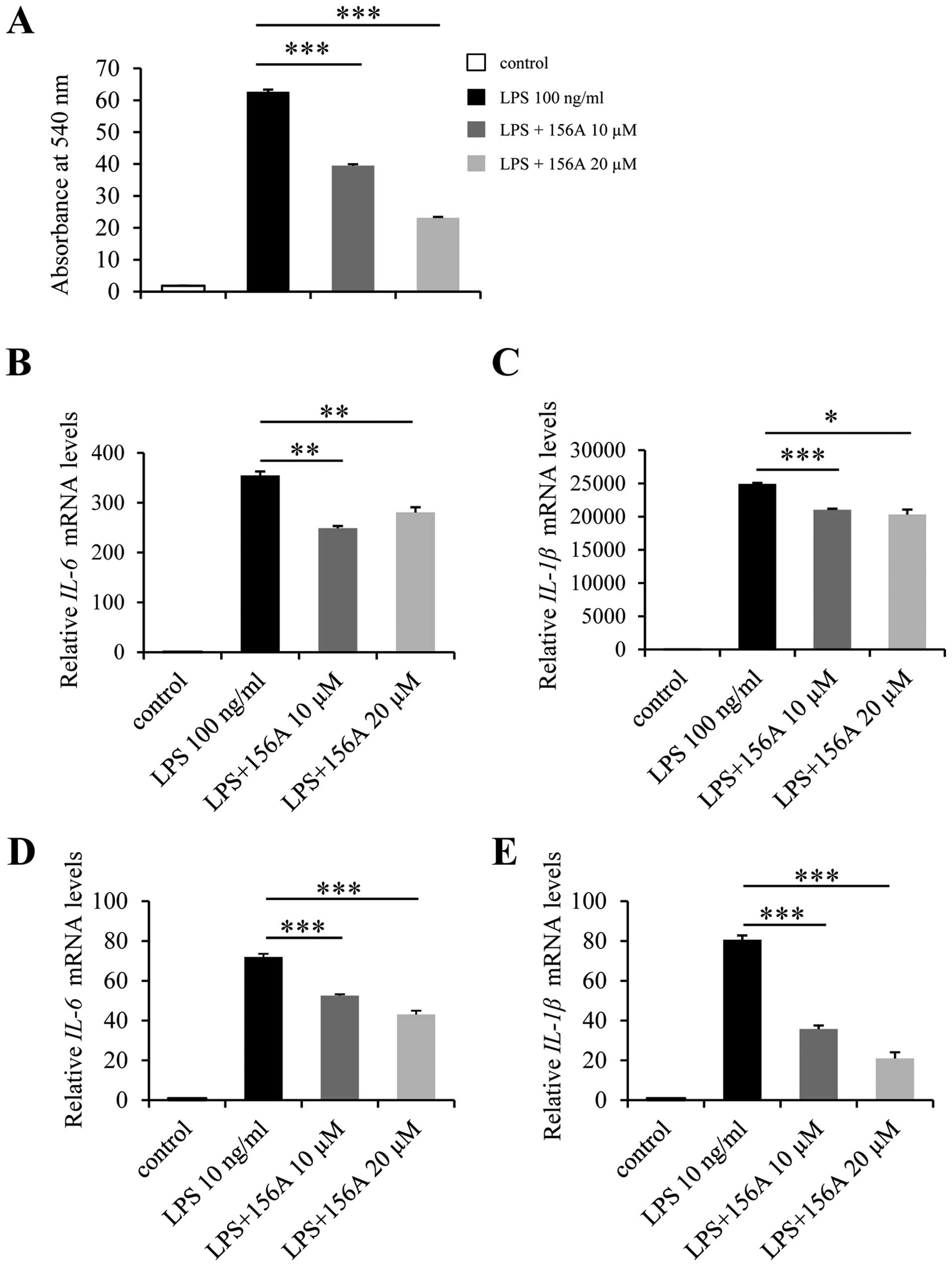
HL156A diminishes LPS-induced macrophage
activation
Inflammation is closely associated with liver
fibrosis (3) and there are several
studies reporting protective effects of AMPK signaling in
inflammation (11,16). To examine the anti-inflammatory
effect of HL156A, LPS, a potent proinflammatory agent was added to
Raw264.7 cell culture, which is a widely used mouse macrophage cell
line. As expected, LPS treatment of Raw264.7 cells increased the
production and release of NO by up to 60-fold. This LPS-induced NO
release was diminished by HL156A in a dose-dependent manner
(Fig. 5A). We next examined if
HL156A was capable of regulating proinflammatory cytokines.
Raw264.7 cells were treated with LPS for 4 h with or without
HL156A, and the relative mRNA levels of proinflammatory cytokines
were analyzed. A single treatment of inactivated Raw264.7 cells
with LPS resulted in up to 350-fold increase in IL-6 and
25,000-fold increase in IL-1β transcripts. However, HL156A
decreased the levels of IL-6 and IL-1β transcripts by approximately
30 and 20%, respectively (Fig. 5B and
C). The inhibitory effect of HL156A on LPS-induced inflammation
was further confirmed in primary macrophages, which were isolated
and differentiated from mouse bone marrow. HL156A reduced the
levels of both IL-6 and IL-1β mRNA in primary macrophages also and
more efficiently than in Raw264.7 cells (Fig. 5D and E). Together, these results
demonstrate that HL156A inhibits inflammation, which is probably
responsible for its anti-fibrotic activity, at least in part.
Discussion
In the present study, we demonstrate the potential
therapeutic effect of a novel AMPK activator, HL156A, in hepatic
fibrosis. We found that HL156A inhibited TAA-induced liver fibrosis
in mice. Specifically, it reduced ECM deposition and TGF-β1
expression, which are induced by TAA administration. Histological
analysis also revealed that the activation of HSCs and
capillarization of LSECs are diminished by HL156A. In vitro
experiments using rat HSC cell line and cultured macrophages
further confirmed the inhibitory effects of HL156A on both
TGF-β1-induced HSC activation and LPS-induced inflammation of
macrophages. As AMPK is expressed ubiquitously in almost all cells
and organs, HL156A seems to target multiple cell types in
inhibiting hepatic fibrosis. Our results also imply that HL156A may
affect multiple responses to fibrotic insult such as inflammation,
capillarization of LSECs, and myofibroblastic transition of HSCs.
However, since these multiple responses influence each other,
further studies would be needed to figure out the exact point of
action of HL156A in in vivo settings.
Fibrosis is a wound healing response that can be
considered as a part of innate immunity. Therefore, multiple organs
including the liver, lung, kidney, and peritoneum commonly develop
fibrosis upon chronic tissue injuries (24). There are common molecular
mechanisms involved in fibrosis of multiple organs such as αv
integrin and TGF-β signaling pathways (25,26).
In this context, it is noteworthy that HL156A has been recently
reported to show inhibitory effects in peritoneal fibrosis. In
light of the results of our present work on liver fibrosis and
previous ones on peritoneal fibrosis (18), it would
be worthwhile to explore the anti-fibrotic effects of HL156A in
other organ systems such as pulmonary fibrosis and renal fibrosis
to determine whether HL156A can be developed as an anti-fibrosis
drug targeting multiple organs.
Since metabolic derangement is considered as one of
the major causes of many human cancers, the pharmacological
activators of AMPK are being revisited for cancer therapy (27). The importance of metabolic
dysregulation in liver cancer has been studied extensively. The
components of the AMPK signaling pathway, likely, play a crucial
role in hepatocellular carcinogenesis (28–30).
Among organs that develop fibrosis, the liver is the one in which
fibrosis is the most closely associated with cancer (31). Approximately 90% of hepatocellular
carcinoma (HCC) cases arise in cirrhotic livers (32) and the incidence of HCC is
approximately 15% in HBV patients with cirrhosis (33). Thus, targeting liver fibrosis and
HCC simultaneously would be an efficient strategy rather than
treating either fibrosis or HCC in isolation. HL156A, as an AMPK
activator with an anti-fibrotic effect, may benefit the development
of new strategies targeting liver cancer with cirrhosis.
Acknowledgements
This work was supported by the Global Research
Laboratory Program (2011-0021874), the Global Core Research Center
(GCRC) Program (2011-0030001) through the National Research
Foundation (NRF) funded by the Korean Ministry of Science, ICT and
Future Planning (MSIP), and the NRF grant funded by the Korea
government (MSIP) (2015R1C1A2A01054446).
References
|
1
|
Lee SJ, Kim KH and Park KK: Mechanisms of
fibrogenesis in liver cirrhosis: The molecular aspects of
epithelial-mesenchymal transition. World J Hepatol. 6:207–216.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seki E and Schwabe RF: Hepatic
inflammation and fibrosis: Functional links and key pathways.
Hepatology. 61:1066–1079. 2015. View Article : Google Scholar :
|
|
3
|
Pellicoro A, Ramachandran P, Iredale JP
and Fallowfield JA: Liver fibrosis and repair: Immune regulation of
wound healing in a solid organ. Nat Rev Immunol. 14:181–194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iwakiri Y, Shah V and Rockey DC: Vascular
pathobiology in chronic liver disease and cirrhosis - current
status and future directions. J Hepatol. 61:912–924. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Langer DA, Das A, Semela D, Kang-Decker N,
Hendrickson H, Bronk SF, Katusic ZS, Gores GJ and Shah VH: Nitric
oxide promotes caspase-independent hepatic stellate cell apoptosis
through the generation of reactive oxygen species. Hepatology.
47:1983–1993. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deleve LD, Wang X and Guo Y: Sinusoidal
endothelial cells prevent rat stellate cell activation and promote
reversion to quiescence. Hepatology. 48:920–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grahame Hardie D: Regulation of
AMP-activated protein kinase by natural and synthetic activators.
Acta Pharm Sin B. 6:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vallianou NG, Evangelopoulos A and Kazazis
C: Metformin and cancer. Rev Diabet Stud. 10:228–235. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Salt IP and Palmer TM: Exploiting the
anti-inflammatory effects of AMP-activated protein kinase
activation. Expert Opin Investig Drugs. 21:1155–1167. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Salminen A, Kaarniranta K, Haapasalo A,
Soininen H and Hiltunen M: AMP-activated protein kinase: A
potential player in Alzheimer’s disease. J Neurochem. 118:460–474.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J, Pan Y, Kan M, Xiao X, Wang Y, Guan
F, Zhang X and Chen L: Hepatoprotective effects of berberine on
liver fibrosis via activation of AMP-activated protein kinase. Life
Sci. 98:24–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tripathi DM, Erice E, Lafoz E,
García-Calderó H, Sarin SK, Bosch J, Gracia-Sancho J and
García-Pagán JC: Metformin reduces hepatic resistance and portal
pressure in cirrhotic rats. Am J Physiol Gastrointest Liver
Physiol. 309:G301–G309. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lim JY, Oh MA, Kim WH, Sohn HY and Park
SI: AMP-activated protein kinase inhibits TGF-β-induced fibrogenic
responses of hepatic stellate cells by targeting transcriptional
coactivator p300. J Cell Physiol. 227:1081–1089. 2012. View Article : Google Scholar
|
|
16
|
Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY,
Shin HJ, Kim WS and Kim JB: Berberine suppresses proinflammatory
responses through AMPK activation in macrophages. Am J Physiol
Endocrinol Metab. 296:E955–E964. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo T, Woo SL, Guo X, Li H, Zheng J,
Botchlett R, Liu M, Pei Y, Xu H, Cai Y, et al: Berberine
ameliorates hepatic steatosis and suppresses liver and adipose
tissue inflammation in mice with diet-induced obesity. Sci Rep.
6:226122016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ju KD, Kim HJ, Tsogbadrakh B, Lee J, Ryu
H, Cho EJ, Hwang YH, Kim K, Yang J, Ahn C, et al: HL156A, a novel
AMP-activated protein kinase activator, is protective against
peritoneal fibrosis in an in vivo and in vitro model of peritoneal
fibrosis. Am J Physiol Renal Physiol. 310:F342–F350. 2016.
View Article : Google Scholar
|
|
19
|
Mobley AK, Tchaicha JH, Shin J, Hossain MG
and McCarty JH: Beta8 integrin regulates neurogenesis and
neurovascular homeostasis in the adult brain. J Cell Sci.
122:1842–1851. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin MW, Bae SJ, Wee HJ, Lee HJ, Ahn BJ,
Le H, Lee EJ, Kim RH, Lee HS, Seo JH, et al: Ninjurin1 regulates
lipopolysaccharide-induced inflammation through direct binding. Int
J Oncol. 48:821–828. 2016.
|
|
21
|
Liu Y, Meyer C, Xu C, Weng H, Hellerbrand
C, ten Dijke P and Dooley S: Animal models of chronic liver
diseases. Am J Physiol Gastrointest Liver Physiol. 304:G449–G468.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
DeLeve LD: Liver sinusoidal endothelial
cells in hepatic fibrosis. Hepatology. 61:1740–1746. 2015.
View Article : Google Scholar :
|
|
23
|
Straub AC, Stolz DB, Ross MA,
Hernández-Zavala A, Soucy NV, Klei LR and Barchowsky A: Arsenic
stimulates sinusoidal endothelial cell capillarization and vessel
remodeling in mouse liver. Hepatology. 45:205–212. 2007. View Article : Google Scholar :
|
|
24
|
Rockey DC: Current and future
anti-fibrotic therapies for chronic liver disease. Clin Liver Dis.
12:939–962. xi2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Henderson NC, Arnold TD, Katamura Y,
Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E,
Betsholtz C, Ruminski PG, et al: Targeting of αv integrin
identifies a core molecular pathway that regulates fibrosis in
several organs. Nat Med. 19:1617–1624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Munger JS, Huang X, Kawakatsu H, Griffiths
MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et
al: The integrin alpha v beta 6 binds and activates latent TGF beta
1: A mechanism for regulating pulmonary inflammation and fibrosis.
Cell. 96:319–328. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuhajda FP: AMP-activated protein kinase
and human cancer: Cancer metabolism revisited. Int J Obes. 32(Suppl
4): S36–S41. 2008. View Article : Google Scholar
|
|
28
|
Ferretti AC, Tonucci FM, Hidalgo F, Almada
E, Larocca MC and Favre C: AMPK and PKA interaction in the
regulation of survival of liver cancer cells subjected to glucose
starvation. Oncotarget. Feb 15–2016.(Epub ahead of print).
View Article : Google Scholar :
|
|
29
|
Park SY, Lee YK, Kim HJ, Park OJ and Kim
YM: AMPK interacts with β-catenin in the regulation of
hepatocellular carcinoma cell proliferation and survival with
selenium treatment. Oncol Rep. 35:1566–1572. 2016.
|
|
30
|
Yang CC, Chang SF, Chao JK, Lai YL, Chang
WE, Hsu WH and Kuo WH: Activation of AMP-activated protein kinase
attenuates hepatocellular carcinoma cell adhesion stimulated by
adipokine resistin. BMC Cancer. 14:1122014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang DY and Friedman SL:
Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology.
56:769–775. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seitz HK and Stickel F: Risk factors and
mechanisms of hepatocarcinogenesis with special emphasis on alcohol
and oxidative stress. Biol Chem. 387:349–360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fattovich G, Stroffolini T, Zagni I and
Donato F: Hepatocellular carcinoma in cir rhosis: Incidence and
risk factors. Gastroenterology. 127(Suppl 1): S35–S50. 2004.
View Article : Google Scholar : PubMed/NCBI
|