Introduction
Esophageal carcinoma, with a 5-year survival rate of
15–20% (1,2), is one of the most common causes of
cancer death worldwide and is rapidly increasing in incidence
(3). It has recently been reported
that the best outcomes are associated with early stage diagnosis
(4). The prognosis is often poor
for esophageal carcinomas due to early lymph node metastasis even
in the superficial stage and its invasiveness to neighboring organs
such as the aorta, trachea, and lung. Therefore, regulation of the
aggressive metastatic features of esophageal cancer can be
essential for improving patient survival. Various experimental
approaches have been implemented to identify the molecules involved
in metastasis processes including migration and invasion (5,6).
However, the underlying mechanisms remain unclear.
There is widespread belief that the phenomenon of
epithelial-mesenchymal transition (EMT) is strongly associated with
the acquisition of cancer metastasis and invasion. Epithelial cells
have sheet-like morphology and have close contact with neighboring
cells at cell junctions (7). Once
EMT occurs, cancer cells lose their tight contacts and become
isolated and motile, and modulate the organization of their
cytoskeletal systems (8,9). Afterwards, the cancer cells acquire
the ability to invade the basement membrane around the cells into
the blood vessels, when invasion and metastasis become possible
(10). However, invasion and
metastasis are exceedingly complex processes, and their mechanisms
remain incompletely understood. One of our focuses has been the
role of cadherins in cancer progression. Cadherin function is
critical in normal development, and its alteration has been
implicated in tumorigenesis (11).
It is well established that E-cadherin functions as a tumor
suppressor, while studies have shown that expression of an
inappropriate cadherin in epithelial cells is another way that
tumor cells can alter their adhesive function (12,13).
EMT is often accompanied by loss of E-cadherin function and
increased expression of other cadherins such as N-cadherin, which
is thought to play a fundamental role in the early steps of
invasion and metastasis of pancreatic cancer (14–16).
These data led us to ask whether blocking the function of
N-cadherin would prevent the malignant behavior of
N-cadherin-expressing esophageal cancer cells.
While iron is an essential trace element, its
overload induces some types of cancer, which suggests that its
manipulation can be a therapeutic target in cancer (17,18).
Iron deficiency has also been reported to suppress tumor growth
in vivo (19). However, its
efficacy as a single agent is not superior to standard
chemotherapy, and it seems unsuitable as a single-agent standard
therapeutic strategy. We previously reported that iron depletion
strongly suppressed tumor growth via cell cycle arrest when
combined with an ordinary molecular targeting drug (20). Recently, it has been reported that
transforming growth factor (TGF)-induced EMT via upregulation of
NDRG1 can be controlled by iron chelation, suggesting that iron
might be an essential element of EMT (21). However, the mechanisms have not
been elucidated and further investigation is required. We
hypothesize that iron depletion might prevent invasion and
migration of cancer cells by regulation of EMT-related molecules,
as well as inducing tumor regression by inhibiting cell
proliferation.
Here, we show a direct interaction between iron
metabolism and malignant phenotypes of cancer cells, with iron
chelation. This presents the possibility of a new therapeutic
strategy in cancer.
Materials and methods
Cell lines and cultures
The human esophageal cancer cell lines TE4, TE8,
TE10, and OE19 were used in this study. They were cultured in
RPMI-1640 medium (Sigma-Aldrich, MO, USA) at 37°C in humidified air
with 5% CO2. Medium was supplemented with 10% fetal calf
serum (FCS; Hyclone, Logan, UT, USA), 100 U/ml penicillin, and 100
mg/ml streptomycin (Sigma-Aldrich).
Reagents
Deferasirox, commercialized as EXJADE™,
was purchased from Novartis Pharma Co., Ltd. (Tokyo, Japan).
Cell viability/cytotoxicity assay
The proliferation of TE4, TE10 and OE19 cells were
evaluated using the XTT assay. Cell viability was determined using
a Cell Proliferation Kit II (Roche Molecular Biochemicals,
Indianapolis, IN, USA) according to the manufacturer’s protocol.
Cell cytotoxicity (dead cells) were evaluated using Live/Dead
Viability/Cytotoxicity assay kit (Molecular Probes, Eugene, OR,
USA) acording to the manufacturer’s protocol. TE4 and TE10 cells
(5×103) were seeded with 10% FCS. After 24-h incubation,
the medium was changed to serum-free medium and deferasirox added
at each concentrations. After 72-h incubation, each assay was
performed.
Migration assay
Cell migration was determined using 24-well BioCoat
cell culture inserts (BD Biosciences, Franklin Lakes, NJ, USA). The
TE4 and TE10 cells (5×104) were placed in the upper
chamber. After 24-h incubation with serum-free medium and
deferasirox, the cells on the outer surface of the bottom of the
filter were fixed in formaldehyde, stained with crystal violet
(Sigma-Aldrich), and counted with an Olympus IX71 Microscope
(Tokyo, Japan) at a magnification of ×100. Three randomly selected
fields were counted in each group, and the experiment was repeated
three times.
Invasion assay
Cell invasion was determined using 24-well BioCoat
cell culture inserts (BD Biosciences) with an 8-μm-porosity
polyethylene terephthalate membrane coated with Matrigel Basement
Membrane Matrix. TE4 and TE10 cells (5×104) were placed
in the upper chamber. RPMI-1640 medium with 10% FCS was added to
the lower chamber. After 24-h incubation with serum-free medium and
deferasirox, the cells on the outer surface of the membrane were
fixed in formaldehyde, stained with crystal violet (Sigma-Aldrich),
and counted with an Olympus IX71 Microscope at a magnification of
100. Three fields selected at random were counted in each group,
and the experiment was repeated three times.
Western blotting
The TE4 and TE10 cells (5×103) were
seeded with 10% FCS. After 24-h incubation, the medium was changed
for serum-free medium and deferasirox added. After 24-h incubation,
whole-cell lysates and nuclear protein were extracted using M-PER
buffer (Thermo Fisher Scientific, Rockford, IL, USA). The protein
concentrations in the supernatants were measured and equal amounts
of protein were electrophoresed under reducing conditions on
gradient polyacrylamide gels (ATTO, Tokyo, Japan) and then
transferred onto polyvinylidene difluoride filter membranes
(Millipore, Billerica, MA, USA). The membranes were incubated with
primary antibodies at 4°C overnight, followed by incubation with
secondary antibodies at room temperature for 1 h. The Amersham
chemiluminescent ECL Plus Western Blotting Detection system (GE
Healthcare, Piscataway, NJ, USA) was used for signal detection. The
following western blotting materials were used: E-cadherin (Cell
Signaling Technology, Inc., Danvers, MA, USA), N-cadherin (Takara
Bio Inc., Otsu, Japan), β-actin (Sigma-Aldrich), horseradish
peroxidase-conjugated rabbit anti-mouse IgG (Dako Cytomation,
Glostrup, Denmark), goat anti-rabbit IgG (American Qualex
Antibodies, La Mirada, CA, USA).
Quantitative real-time reverse
transcription PCR analysis
TE4 and TE10 cells (5×104) were seeded.
The cells were treated with serum-free medium and deferasirox (100
μM). After 1-, 2-, 3-, 6- and 12-h incubation, total RNA was
extracted from cells using a miRNeasy Mini kit (Qiagen, Venlo, The
Netherlands). The levels of E-cadherin and N-cadherin mRNA
expression were determined using quantitative real-time PCR and a
Step One Plus Real Time PCR System (Applied Biosystems, Foster
City, CA, USA). The relative levels of E-cadherin and N-cadherin
mRNA expression were calculated using the 2−ΔΔCt method
after normalization with reference to the expression of GAPDH mRNA
(22,32).
Sphere-forming assay
To determine migration and invasion ability on 3D
culture, the sphere-forming assay was used, as reported previously
(23,33). TE4 and TE8 cells (5×103)
were added into 96-well plates with 1.5% agarose and RPMI-1640
(1:1). After forming spheres, the cells were fed with serum-free
growth medium and deferasirox; spheres were photographed after 3
days.
N-cadherin small interference RNA
transfection
To confirm the effect of N-cadherin on migration and
invasion activity in esophageal cancer cells, we transfected small
interfering RNA (siRNA; 5 and 7.5 nM). We prepared pre-designed
N-cadherin targeting siRNA and unlabeled siRNA (Applied
Biosystems). TE4 and TE10 cells were transfected with siRNAs
according to the manufacturer’s instructions.
Animal experiments
The animal experimental protocol was approved by the
Ethics Review Committee for Animal Experimentation of Okayama
University, Okayama, Japan. All of the mice, the iron-deficient
diet, and the normal diet were purchased from Clea (Clea, Tokyo,
Japan). The 6-week-old male BALB/c nu/nu mice were randomized into
two groups of eight mice each: i) normal diet as a control, ii)
iron-deficient diet (Table I).
After 3 weeks, subcutaneous xenografts were produced on the backs
of mice by injecting 3×106 cells mixed with Matrigel (BD
Biosciences) at a 1:1 ratio. Water was provided to drink freely.
Tumor volume was measured weekly (1/2 × length ×
width2).
 | Table IContent of control and iron-deficient
diets. |
Table I
Content of control and iron-deficient
diets.
| g/kg diet |
|---|
|
|
|---|
| Control diet | Iron-deficient
diet |
|---|
| Corn starch | 610 | 610 |
| Casein | 220 | 220 |
| Celluose | 50 | 50 |
| Soybean oil | 40 | 40 |
| Vitamin
mixture | 10 | 10 |
| Mineral
mixture |
| Potassium | 17.3 | 17.3 |
| Phosphorus | 15 | 15 |
| Calcium | 13.55 | 13.55 |
| Magnesium | 8 | 8 |
| Corn starch | 8 | 9.9 |
| Sodium | 6 | 6 |
| Iron | 1.9 | |
| Manganese | 0.154 | 0.154 |
| Zinc | 0.06 | 0.06 |
| Iodine | 0.0154 | 0.0154 |
| Copper | 0.0126 | 0.0126 |
| Chloride | 0.004 | 0.004 |
Statistical analysis
A Student’s t-test was used to compare data between
the two groups. Data represent the mean ± SEM; p≤0.05 was
considered statistically significant.
Results
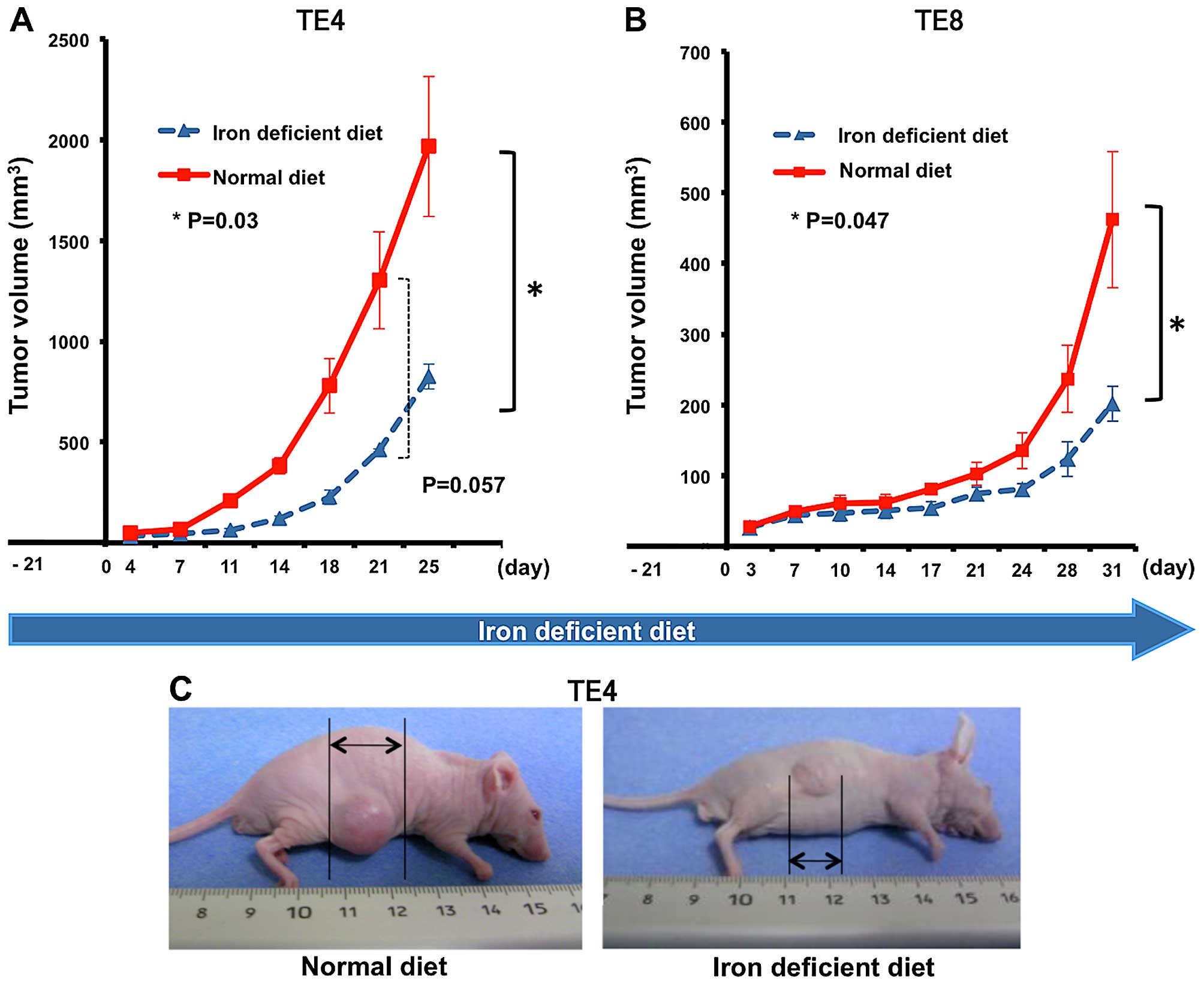
Esophageal tumor growth is suppressed
under decreased iron conditions
First, we made a decreased iron mouse model using
the iron-deficient diet as described previously (20). The iron-deficient and normal diets
were prepared as described in Table
I. We next investigated the growth of esophageal tumor under
the decreased iron conditions. Nude mice were divided into two
groups to receive normal or iron-deficient diet. TE4 subcutaneous
xenografts were produced on the backs of mice after 3 weeks of
iron-deficient diet feed. Tumor size was measured twice a week.
Tumor growth was significantly suppressed in the iron-deficient
group (tumor volume: normal diet vs. iron-deficient diet =
3,071.0±1,110.7 vs. 1,056.0±202.4 mm3; p=0.003). Tumors
in the iron-deficient group showed slower growth and their curve
rose more gradually compared with the normal-diet group (Fig. 1A and C). Similar results were
observed in TE8 (Fig. 1B). The
standard errors each day were also smaller due to overall poor
growth. No mice died and no significant side effects were observed
during the period of experimentation. Thus, esophageal tumor growth
is suppressed in decreased iron conditions in mouse models, similar
to our previous results with lung cancer tumors (20).
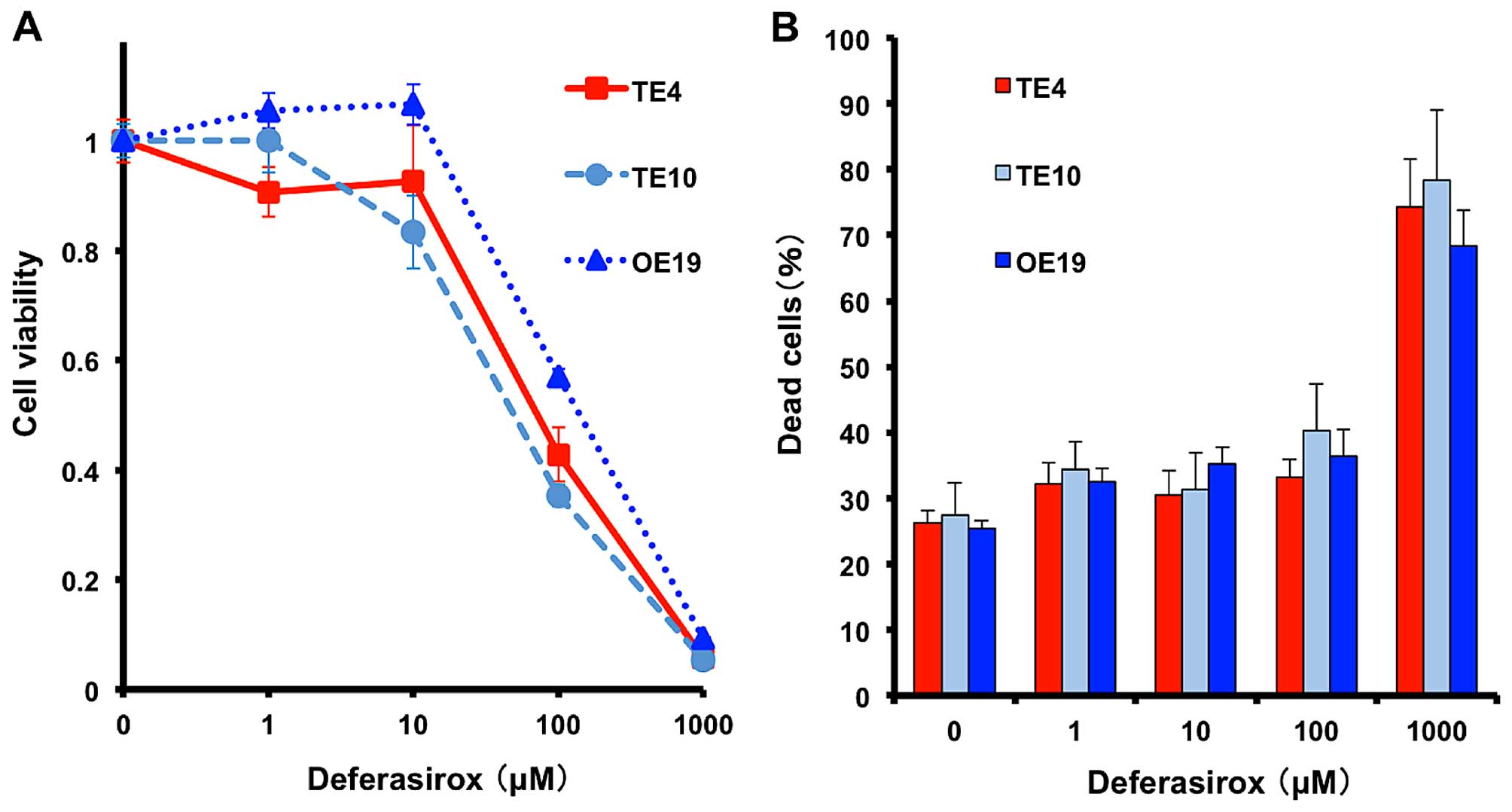
Decreased iron conditions inhibit
esophageal cancer cell proliferation in vitro
To reproduce the iron-deficient conditions in
vitro, the iron chelator deferasirox was used. We prepared
several esophageal cancer cell lines (TE4 and TE10, squamous-cell
carcinoma; OE19, adenocarcinoma). Cell viability was measured by
XTT assay and cytotoxicity was measured by Live/Dead assay after
72-h deferasirox treatment. Deferasirox suppressed proliferation of
all cell lines in a dose-dependent manner (Fig. 2A), whereas dead cells were
increased in high dose of deferasirox (1,000 μM) (Fig. 2B). These results suggested that
decreased iron condition effect rather on cell proliferation than
cytotoxicity.
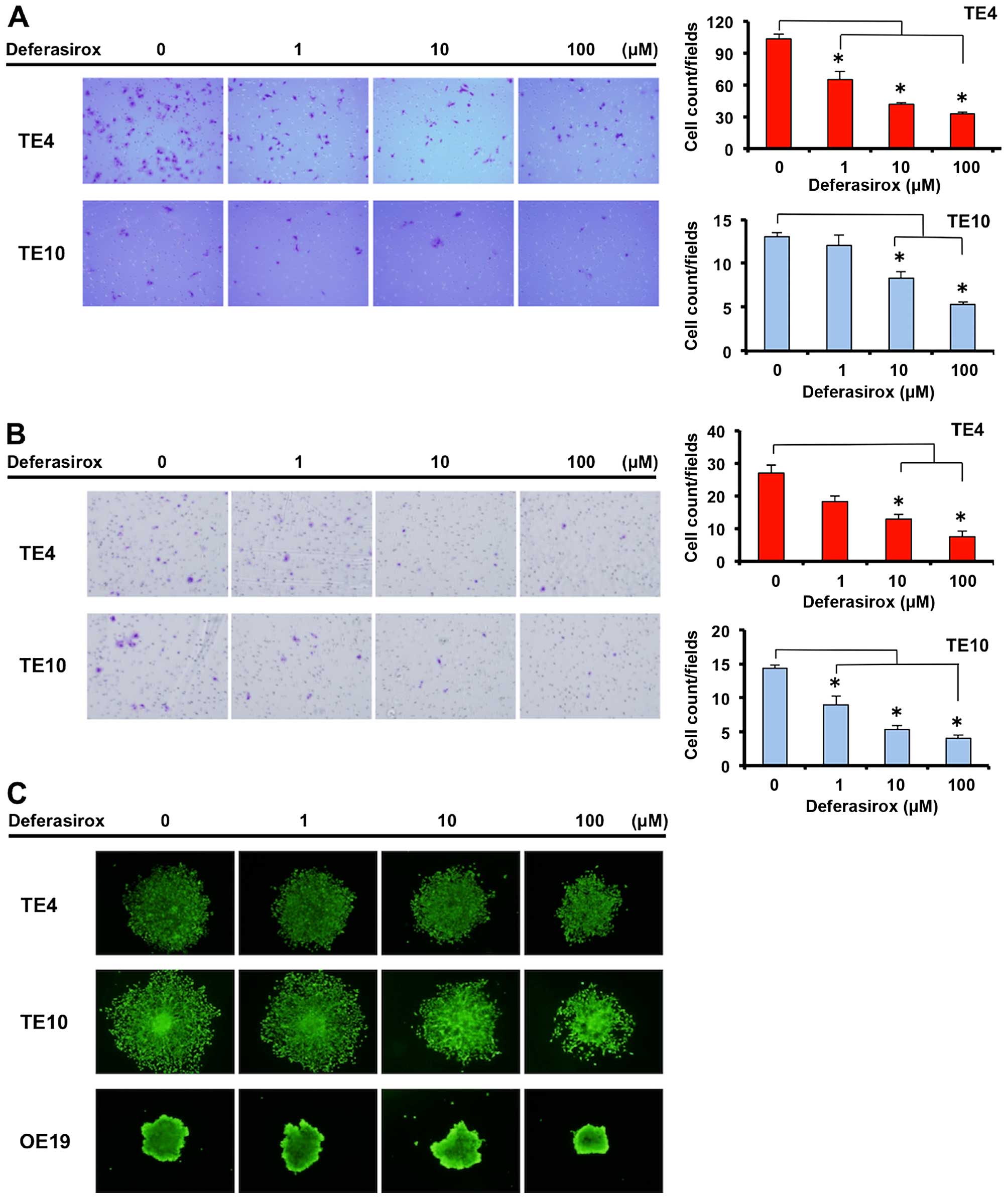
Decreased iron conditions suppress
migration and invasion abilities of esophageal cancer cells
To determine other anti-cancer effects concerning
the malignant abilities of cancer under decreased iron conditions,
we investigated the migration and invasion ability of esophageal
cancer cells. Migration and invasion abilities were measured using
double-layer chambers, migrating and invading esophageal cancer
cells were counted in the bottom chamber. The migration and
invasion abilities of TE4 and TE10 cells were suppressed by
deferasirox in a dose-dependent manner (Fig. 3A and B). Furthermore, these
abilities were confirmed in a three-dimensional sphere-forming
assay, which is more similar to biological conditions in a human.
Sphere formation of TE4 and TE10 cells was suppressed by
deferasirox in a dose-dependent manner (Fig. 3C) but OE19 cells were not affected
significantly by the deferasirox in this assay. These results
demonstrate that decreased iron conditions suppress migration and
invasion abilities of TE4 and TE10 cells.
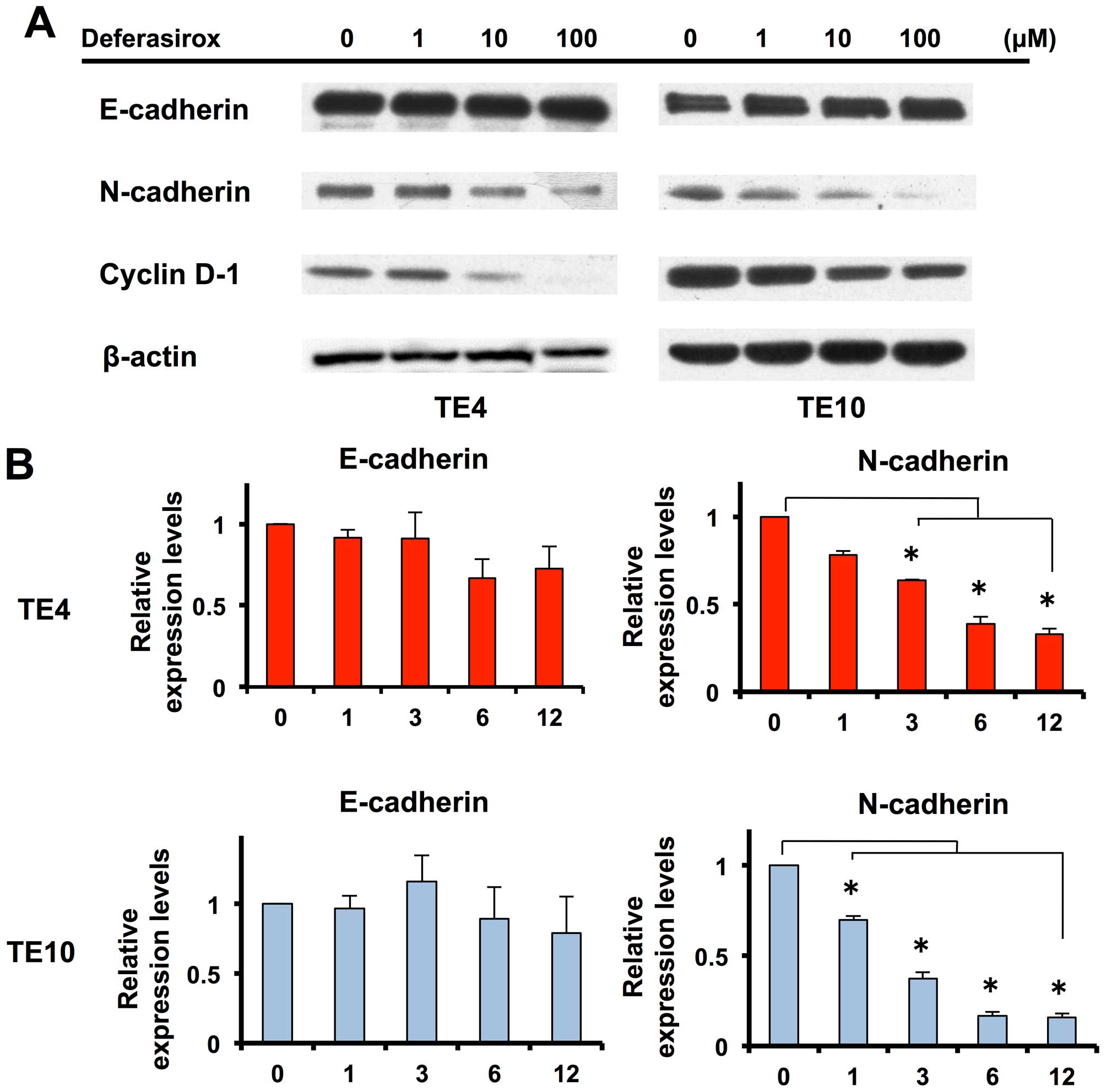
Migration and invasion abilities are
suppressed by inhibiting expression of N-cadherin under decreased
iron conditions
To identify the mechanism of suppression of
migration and invasion abilities under decreased iron conditions,
we focused on the intercellular adhesion molecule cadherin. Western
blot analysis showed that N-cadherin expression was suppressed in a
dose-dependent manner under decreased iron conditions (Fig. 4A), while E-cadherin expression was
unchanged. Next, we investigated the mRNA status of N-cadherin and
E-cadherin. Expression of N-cadherin mRNA was suppressed under
decreased iron conditions in a time-dependent manner (Fig. 4B). The mRNA expression response was
rapid and clearly observed after 1-h stimulation. Expression of
E-cadherin mRNA was unchanged, as in the protein assay. N-cadherin
has been reported to be a key molecule for migration and invasion
ability. Thus, these results suggest that migration and invasion
abilities were suppressed by inhibition of N-cadherin expression
under the decreased iron conditions.
N-cadherin knockdown specifically
inhibits migration and invasion abilities of esophageal cancer
cells
To confirm that N-cadherin ruled over the migration
and invasion abilities of esophageal cancer cells, we made
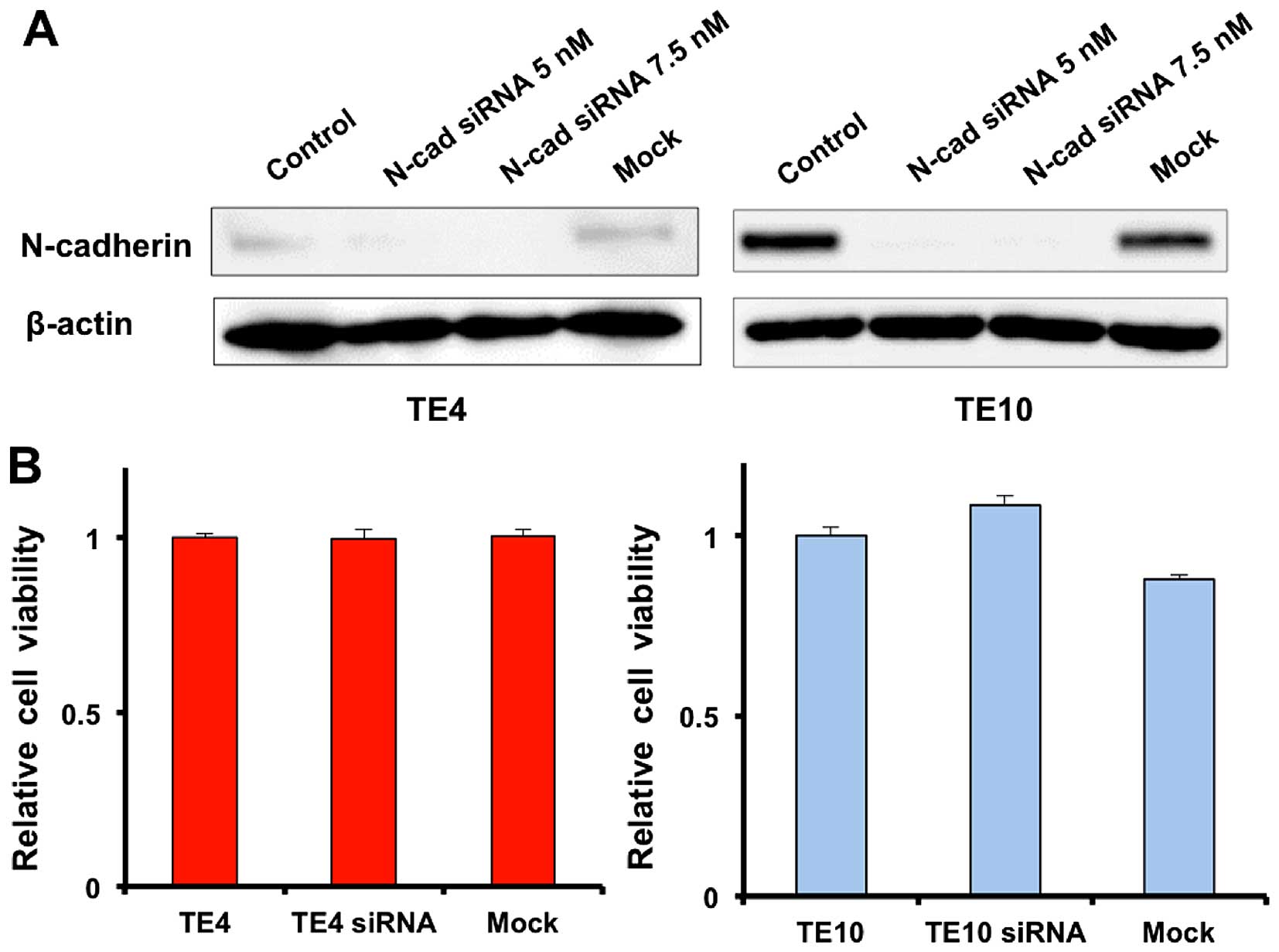
N-cadherin knockdown TE4 and TE10 cells using siRNA. Western blot
analysis proved that expression of N-cadherin was inhibited by
siRNA in both TE4 and TE10 cells (Fig.
5A). N-cadherin knockdown TE4 and TE10 cells had equivalent
proliferation ability to normal TE4 and TE10 cells (Fig. 5B), which suggests that N-cadherin
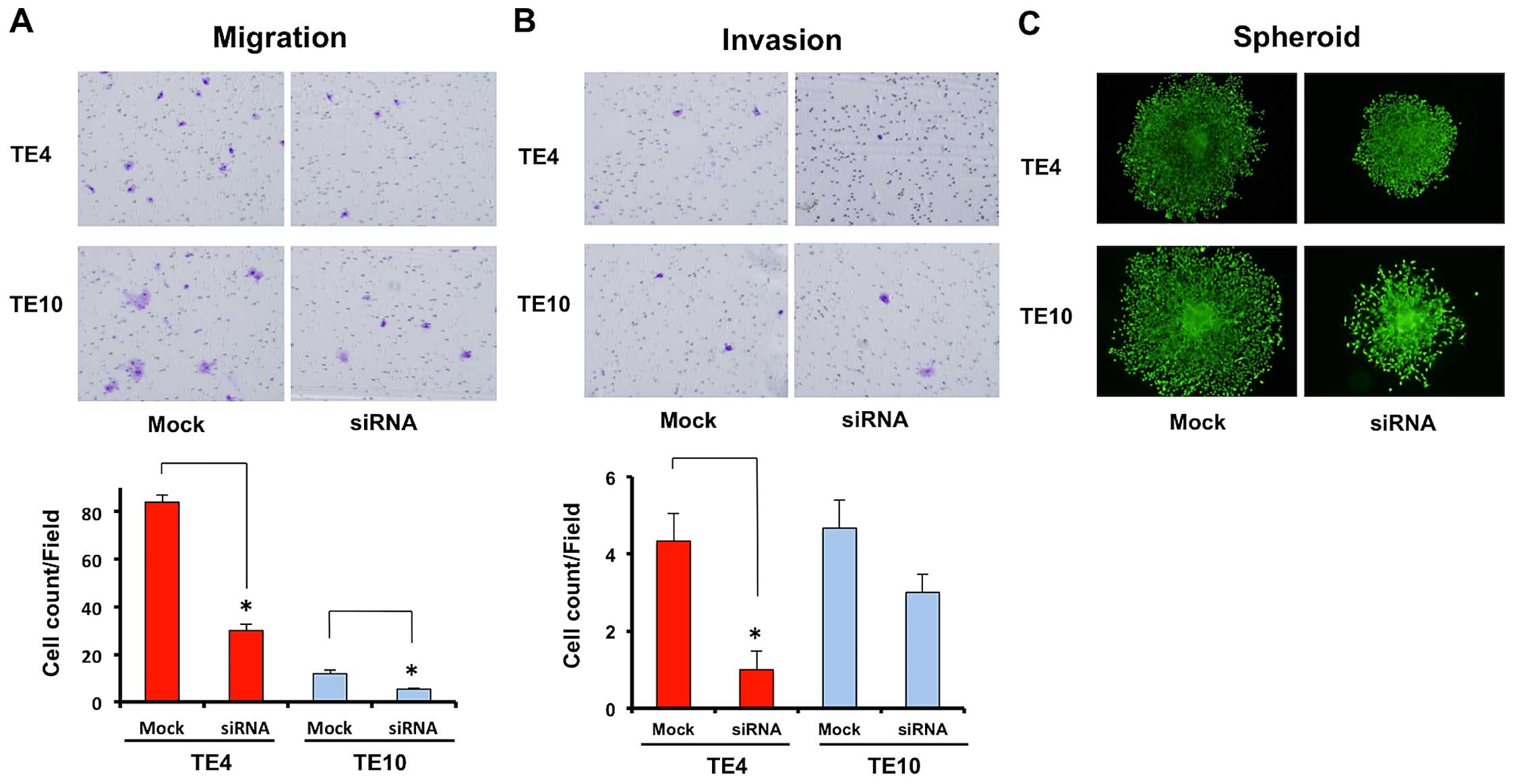
control is not related to cell proliferation. However, migration
and invasion assays showed the migration ability of N-cadherin
knockdown cancer cells to be inhibited in the same way as by
deferasirox, suggesting that such cell malignancy abilities
strongly depend on N-cadherin (N-cadherin knockdown TE4 vs.
negative control TE4 = 30.0±4.90 vs. 84.0±5.35/field; p=0.00046,
N-cadherin knockdown TE10 vs. negative control TE10 = 5.3±0.47 vs.
12.0±2.45/field; p=0.01944) (Fig. 6A
and B). Sphere formation by TE4 and TE10 cells was also clearly
suppressed by N-cadherin knockdown (Fig. 6C). These results suggest that
N-cadherin rules over the migration and invasion abilities of
esophageal cancer cells and that decreased iron conditions suppress
the migration and invasion abilities of esophageal cancer cell via
suppression of N-cadherin.
Discussion
Iron is an essential element for mammals, involved
in oxygen transport, intracellular DNA synthesis, and cell-cycle
progression (24,25). Mounting number of reports on in
vitro and in vivo experiments have suggested its
potential role in the carcinogenesis process (26–29).
Particularly in gastrointestinal tumors, breakdown of intracellular
iron regulation has been reported in esophageal (30) and colon cancers (31), where intracellular molecules
related to iron transport and metabolism were demonstrated to be
modified. All of these reports support iron’s key role in
carcinogenesis and its position as a target in cancer
regulation.
We have reported that iron depletion has an
inhibitory effect on cancer progression in lung cancer cells, and a
synergistic effect on anti-angiogenic therapy. The iron-depletion
conditions were induced by an iron-depletion diet or administration
of the iron chelator deferasirox (20). In this study, we adopted both
methods. This is the first report of an iron-depletion diet in an
esophageal cancer model. Iron has been reported to induce cell
cycle regulation (32); the same
report showed iron to be a sensitizer of chemotherapy. Similarly,
our data showed downregulation of cyclin D1, confirming that the
inhibitory effect is based on regulation of cell cycle
signaling.
Regarding the relationship between iron conditions
and cancer malignancy, many reports regarding EMT have been
published recently, showing that iron chelation inhibits
TGF-β-induced EMT (21). We used
deferasirox to analyze EMT-related proteins in esophageal cancer,
and discovered that iron chelation induced downregulation of
N-cadherin expression, upregulation of which is the well-known
cadherin-switch in EMT. However, other EMT-related proteins
represented by E-cadherin were not affected. It seems that iron
depletion does not regulate the whole EMT mechanism but at least
partially affects functional signaling via the key molecule
N-cadherin.
Although poor prognosis in esophageal cancer
patients led by tumor metastasis and local invasion, the mechanisms
have not yet been analyzed. As many studies have already
demonstrated that N-cadherin is directly related to and affects
cellular invasiveness or migration in several cancers (14–16),
we hypothesized that its expression is regulated by iron chelation
and directly related to those malignant features. Therefore, we
assessed whether cellular iron status affects the malignant
features of cancer cells, such as invasiveness and migration, that
are directly associated with mortality. As expected, in
vitro assays demonstrated that iron chelation decreased the
invasion and migration ability of cancer cells. Furthermore a
three-dimensional in vitro model, the sphere formation assay
(33), which is more
representative of human conditions, showed similar results to the
two-dimensional in vitro model. Invasion and migration seem
to depend on N-cadherin expression, confirmed by the fact that a
cell line that does not express N-cadherin is unaffected by iron
chelation and by the N-cadherin inhibition experiments with siRNA
techniques.
The importance of our discovery is highlighted by
the fact that deferasirox, originally designed to regulate iron
levels, has broad antitumor abilities to regulate cell
proliferation and mesenchymal properties, such as motile and
invasive abilities, simultaneously. Compared with current
molecularly targeted chemotherapy, chelating or controlling iron
might be less invasive, safer therapy. Considering these results,
our investigation provides a new chemotherapeutic strategy. As both
features might connect directly to patient damage and eventual
mortality, controlling iron has the potential to improve patient
survival and their quality of life post major treatment. Especially
in esophageal cancer, distal metastasis or local invasion is common
even during the early stages. We have presented the highly novel
finding that iron chelation has the potential to regulate cancer
progression by inhibition of proliferation and mesenchymal
features.
We note several limitations of this study. We still
need to look into other cancer cell lines as well as esophageal
cancer, and further analysis with metastatic models in vivo
is necessary to prove how to contribute to clinical prognosis. In
addition to N-cadherin regulation in iron chelation, other possible
means of regulation of the cell motility system must be examined.
In conclusion, we have shown that iron depletion leads to reduced
cancer cell growth, invasiveness, and migration through suppression
of N-cadherin expression. This novel observation is opening new
approaches for cancer therapy using nutrition.
Acknowledgements
We are grateful to Mr. Toru Tanida and Ms. Tae
Yamanishi for their technical assistance. We are also grateful to
Dr Fumiaki Kimura of Tamano City Hospital (Okayama, Japan) for
useful discussion. This study was supported by grants-in-aid from
the Ministry of Education Culture, Sports, Science and Technology,
Japan (Kazuhiro Noma) and grants from the Ministry of Health, Labor
and Welfare, Japan (Toshiyoshi Fujiwara).
Abbreviations:
|
EMT
|
epithelial mesenchymal transition
|
|
FCS
|
fetal calf serum
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
NDRG1
|
N-myc downstream regulated gene 1
|
|
RPMI
|
Roswell Park Memorial Institute
medium
|
|
3D
|
three dimensions
|
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lepage C, Rachet B, Jooste V, Faivre J and
Coleman MP: Continuing rapid increase in esophageal adenocarcinoma
in England and Wales. Am J Gastroenterol. 103:2694–2699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pennathur A, Farkas A, Krasinskas AM,
Ferson PF, Gooding WE, Gibson MK, Schuchert MJ, Landreneau RJ and
Luketich JD: Esophagectomy for T1 esophageal cancer: Outcomes in
100 patients and implications for endoscopic therapy. Ann Thorac
Surg. 87:1048–1054; discussion 1054–1055. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ito T, Shimada Y, Hashimoto Y, Kaganoi J,
Kan T, Watanabe G, Murakami Y and Imamura M: Involvement of TSLC1
in progression of esophageal squamous cell carcinoma. Cancer Res.
63:6320–6326. 2003.PubMed/NCBI
|
|
6
|
Qian H, Lu N, Xue L, Liang X, Zhang X, Fu
M, Xie Y, Zhan Q, Liu Z and Lin C: Reduced MTA1 expression by RNAi
inhibits in vitro invasion and migration of esophageal squamous
cell carcinoma cell line. Clin Exp Metastasis. 22:653–662. 2005.
View Article : Google Scholar
|
|
7
|
De Wever O, Pauwels P, De Craene B, Sabbah
M, Emami S, Redeuilh G, Gespach C, Bracke M and Berx G: Molecular
and pathological signatures of epithelial-mesenchymal transitions
at the cancer invasion front. Histochem Cell Biol. 130:481–494.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ruiz P and Günthert U: The cellular basis
of metastasis. World J Urol. 14:141–150. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boyer B, Vallés AM and Edme N: Induction
and regulation of epithelial-mesenchymal transitions. Biochem
Pharmacol. 60:1091–1099. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Birchmeier C, Birchmeier W and
Brand-Saberi B: Epithelial-mesenchymal transitions in cancer
progression. Acta Anat (Basel). 156:217–226. 1996. View Article : Google Scholar
|
|
11
|
Cavallaro U, Schaffhauser B and
Christofori G: Cadherins and the tumour progression: Is it all in a
switch? Cancer Lett. 176:123–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tomita K, van Bokhoven A, van Leenders GJ,
Ruijter ET, Jansen CF, Bussemakers MJ and Schalken JA: Cadherin
switching in human prostate cancer progression. Cancer Res.
60:3650–3654. 2000.PubMed/NCBI
|
|
14
|
Nakajima S, Doi R, Toyoda E, Tsuji S, Wada
M, Koizumi M, Tulachan SS, Ito D, Kami K, Mori T, et al: N-cadherin
expression and epithelial-mesenchymal transition in pancreatic
carcinoma. Clin Cancer Res. 10:4125–4133. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Derycke LD and Bracke ME: N-cadherin in
the spotlight of cell-cell adhesion, differentiation,
embryogenesis, invasion and signalling. Int J Dev Biol. 48:463–476.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shintani Y, Hollingsworth MA, Wheelock MJ
and Johnson KR: Collagen I promotes metastasis in pancreatic cancer
by activating c-Jun NH(2)-terminal kinase 1 and up-regulating
N-cadherin expression. Cancer Res. 66:11745–11753. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Richmond HG: Induction of sarcoma in the
rat by iron-dextran complex. BMJ. 1:947–949. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okada S, Hamazaki S, Toyokuni S and
Midorikawa O: Induction of mesothelioma by intraperitoneal
injections of ferric saccharate in male Wistar rats. Br J Cancer.
60:708–711. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hann HW, Stahlhut MW and Blumberg BS: Iron
nutrition and tumor growth: Decreased tumor growth in
iron-deficient mice. Cancer Res. 48:4168–4170. 1988.PubMed/NCBI
|
|
20
|
Ohara T, Noma K, Urano S, Watanabe S,
Nishitani S, Tomono Y, Kimura F, Kagawa S, Shirakawa Y and Fujiwara
T: A novel synergistic effect of iron depletion on antiangiogenic
cancer therapy. Int J Cancer. 132:2705–2713. 2013. View Article : Google Scholar
|
|
21
|
Chen Z, Zhang D, Yue F, Zheng M, Kovacevic
Z and Richardson DR: The iron chelators Dp44mT and DFO inhibit
TGF-β-induced epithelial-mesenchymal transition via up-regulation
of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem.
287:17016–17028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takaoka M, Harada H, Andl CD, Oyama K,
Naomoto Y, Dempsey KL, Klein-Szanto AJ, El-Deiry WS, Grimberg A and
Nakagawa H: Epidermal growth factor receptor regulates aberrant
expression of insulin-like growth factor-binding protein 3. Cancer
Res. 64:7711–7723. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yano S, Tazawa H, Hashimoto Y, Shirakawa
Y, Kuroda S, Nishizaki M, Kishimoto H, Uno F, Nagasaka T, Urata Y,
et al: A genetically engineered oncolytic adenovirus decoys and
lethally traps quiescent cancer stem-like cells in S/G2/M phases.
Clin Cancer Res. 19:6495–6505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kalinowski DS and Richardson DR: The
evolution of iron chelators for the treatment of iron overload
disease and cancer. Pharmacol Rev. 57:547–583. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miller LD, Coffman LG, Chou JW, Black MA,
Bergh J, D’Agostino R Jr, Torti SV and Torti FM: An iron regulatory
gene signature predicts outcome in breast cancer. Cancer Res.
71:6728–6737. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kikyo N, Suda M, Kikyo N, Hagiwara K,
Yasukawa K, Fujisawa M, Yazaki Y and Okabe T: Purification and
characterization of a cell growth factor from a human leukemia cell
line: Immunological identity with ferritin. Cancer Res. 54:268–271.
1994.PubMed/NCBI
|
|
27
|
Yu Y, Kovacevic Z and Richardson DR:
Tuning cell cycle regulation with an iron key. Cell Cycle.
6:1982–1994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nurtjahja-Tjendraputra E, Fu D, Phang JM
and Richardson DR: Iron chelation regulates cyclin D1 expression
via the proteasome: A link to iron deficiency-mediated growth
suppression. Blood. 109:4045–4054. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chaston TB, Lovejoy DB, Watts RN and
Richardson DR: Examination of the antiproliferative activity of
iron chelators: Multiple cellular targets and the different
mechanism of action of triapine compared with desferrioxamine and
the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin
Cancer Res. 9:402–414. 2003.PubMed/NCBI
|
|
30
|
Boult J, Roberts K, Brookes MJ, Hughes S,
Bury JP, Cross SS, Anderson GJ, Spychal R, Iqbal T and Tselepis C:
Overexpression of cellular iron import proteins is associated with
malignant progression of esophageal adenocarcinoma. Clin Cancer
Res. 14:379–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brookes MJ, Hughes S, Turner FE, Reynolds
G, Sharma N, Ismail T, Berx G, McKie AT, Hotchin N, Anderson GJ, et
al: Modulation of iron transport proteins in human colorectal
carcinogenesis. Gut. 55:1449–1460. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ford SJ, Obeidy P, Lovejoy DB, Bedford M,
Nichols L, Chadwick C, Tucker O, Lui GY, Kalinowski DS, Jansson PJ,
et al: Deferasirox (ICL670A) effectively inhibits oesophageal
cancer growth in vitro and in vivo. Br J Pharmacol. 168:1316–1328.
2013. View Article : Google Scholar :
|
|
33
|
Haass NK, Sproesser K, Nguyen TK,
Contractor R, Medina CA, Nathanson KL, Herlyn M and Smalley KS: The
mitogen-activated protein/extracellular signal-regulated kinase
kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in
melanoma cells and tumor regression when combined with docetaxel.
Clin Cancer Res. 14:230–239. 2008. View Article : Google Scholar : PubMed/NCBI
|