Introduction
The receptor tyrosine kinase (TK), epidermal growth
factor receptor (EGFR) is involved in human physiological or
metabolic processes (1). Upon the
binding of the endogenous ligands such as epidermal growth factor
(EGF), transforming growth factor-α (TGF-α), amphiregulin,
heparin-binding EGF, or betacellulin to the EGFR, homo- or
hetero-dimerization of the EGFRs occur and lead to internalization
and autophosphoryla-tion of the intracytoplasmic EGFR tyrosine
kinase domains. The phosphorylated tyrosine kinase then activates
several downstream signaling pathways such as the
RAS-RAF-MEK-ERK-MAPK (RAS-MAPK), PI3K-AKT and JAK-STAT pathways,
which are responsible for regulating both apoptosis and
proliferation of cells (2).
However, an increased gene copy number or mutation of EGFR could
lead to the dysregulated activation of EGFR tyrosine kinase (TK),
which finally contributes to the increased survival, proliferation,
invasion and metastasis rate of tumor cells (3). In fact, more than 60% of metastatic
non-small cell lung cancer (NSCLC) patients have overexpression of
EGFR with poor prognosis (4).
Small molecule EGFR tyrosine kinases inhibitor (TKI)
can inhibit the autophosphorylation, activation and signal
transduction of EGFR. Erlotinib and gefitinib are the
first-generation oral synthetic anilinoquinazoline TKIs that bind
reversibly to the EGFR TK domain (2). New generation of EGFR TKIs such as
neratinib (5), afatinib (6) and dacomitinib (7) are irreversible inhibitors of the ErbB
family. EGFR mutations are commonly found in NSCLC and contribute
to the early development of lung cancer (1). However, it should be noted that the
sensitivity of the NSCLC to gefitinib and erlotinib is highly
dependent on the type of EGFR mutations. For example, NSCLC cells
with L858R mutant in EGFR are more sensitive to gefitinib than
those with G719S mutant (8).
Importantly, acquired drug resistance to gefitinib
and erlotinib is observed in NSCLC tumors with a point mutation in
the TK domain. For example, the threonine-790 to methionine (T790M)
substitution point mutation was found in approximately 50% of
cancer patients with acquired resistance to EGFR TKI therapy,
suggesting that T790M may work as a biomarker for identifying
patients who might be resistant to erlotinib or gefitinib
treatments (1). Further studies
supported the presence of T790M in gefitinib-sensitive cells
confers resistance to gefitinib treatment (9). Therefore, cancers with T790M or other
point mutations which may contribute to drug resistant of TKIs
remain as an important clinical challenge.
Autophagy is a well-known cellular maintenance
mechanism responsible for the degradation of unwanted organelles
and proteins to maintain normal cellular biosynthesis during
nutrient deprivation or metabolic stressful conditions. Disruption
on the autophagic function of cells can promote tumorigenesis. In
fact, various studies have demonstrated autophagy as a tumor
suppressor mechanism via accelerating the removal of damaged
organelles and proteins to maintain normal cell growth and
stability of genome (10). It was
showed that mice with deficient autophagic related gene beclin 1
were more susceptible to tumor development, suggesting the tumor
suppression role of autophagy (11). In fact, prolonged constitutive
activation of autophagy may eventually lead to the induction of
autophagic cell death, when turnover of cellular content overwhelm
the capacity of the cell (9).
Therefore, the induction of autophagic cell death has been an
attractive anti-cancers therapeutic approach recently. For example,
a novel small molecule (STF-62247) was demonstrated to promote
autophagic cell death in renal carcinoma cells, although the role
of autophagy in cancer therapy remained controversial (12).
Celastrol, a triterpene extracted from the herbal
plant Tripterygium wilfordii, has been shown to induce
apoptotic cell death in gefitinib-resistant non-small cell lung
cancer (NSCLC) cells (H1650 and H1975) with the loss of
mitochondria membrane potential, and promote degradation of two
well-known client proteins of Hsp90, EGFR and AKT (13). Although the pharmacological role of
celastrol has been identified in various diseases including
cancers, rheumatoid arthritis, lateral sclerosis, lupus
erythematosus, asthma and Alzheimer’s disease, the mechanisms
underlying celastrol-induced client proteins degradation remain
uninvestigated (13). In the
present study, we investigated the autophagic property of
celastrol, and further studied the mechanisms governing the
degradation of EGFR in gefitinib-resistant NSCLCs.
Our results demonstrated that celastrol induced
substantial cytotoxic effect and mobilized cytosolic calcium in
EGFR mutant NSCLCs. Celastrol also induced degradation of Hsp90
client protein in both wild-type and mutant EGFR NSCLCs via
mobilization of calcium and autophagy. Further analysis by
immunofluorescence staining confirmed that celastrol induced
autophagy and EGFR degradation simultaneously in H1975
gefitinib-resistant NSCLCs, suggesting the anticancer properties of
celastrol via promoting the degradation of EGFR through
calcium-mediated autophagy induction in resistant cancer cells.
Materials and methods
Reagents, chemicals and antibodies
All chemicals and reagents were purchased from
Sigma-Aldrich unless otherwise stated. The following regents were
used: 3-methyladenine (189490; Calbiochem, San Diego CA, USA),
BAPTA/AM (196419; Calbiochem), celastrol (China Chengdu MUST,
A0106), RIPA lysis buffer (9806; Cell Signaling Technology,
Danvers, MA, USA), Fluo-3, AM (F14218; Life Technologies, Carlsbad,
CA, USA), antibody against LC3B (2775; Cell Signaling Technology),
EGFR (5735S; Cell Signaling Technology), Akt (9272; Cell Signaling
Technology), anti-β-actin (sc-47778; Santa Cruz Biotechnology,
Santa Cruz, CA, USA), ZyMax™ TRITC conjugated anti-mouse secondary
antibody (PA1-28565; Invitrogen, Carlsbad, CA, USA). FITC
conjugated anti-rabbit secondary antibody (F-2765; Invitrogen).
Primer pairs for real-time PCR of EGFR: EGFR-forward,
TTGCCGCAAAGTGTGTAACG and EGFR-reverse, GAG ATCGCCACTGATGGAGG.
Cell culture
All cells were obtained from the American Type
Culture Collection (ATCC; Rockville, MD, USA) unless otherwise
specified. All media were cultured in RPMI-1640 medium supplemented
with 10% fetal bovine serum (FBS) and the antibiotics penicillin
(50 U/ml) and streptomycin (50 μg/ml; Invitrogen, Paisley, UK). All
cell cultures were incubated at 37°C in a 5% humidified
CO2 incubator.
Cytotoxicity assays
All test compounds were dissolved in dimethyl
sulfoxide (DMSO) at final concentrations of 50 mmol/l and stored at
−20°C before use. Cytotoxicity was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
(5.0 mg/ml) assay as previously described (14). Briefly, 4×103 cells were
seeded/well in 96-well plates before drug treatment. After
overnight culture, the cells were then exposed to different
concentrations of celastrol (0.039–100 μmol/l) for 72 h. Cells
without drug treatment were used as control. Subsequently, MTT (10
μl) was added to each well and incubated at 37°C for 4 h followed
by the addition of 100 μl solubilization buffer (10% SDS in 0.01
mol/l HCl) and overnight incubation. A570 nm was
determined from each well the next day. The percentage of cell
viability was calculated using the following formula: Cell
viability (%) = Atreated/Acontrol × 100. Data
were obtained from triplicate independent experiments.
Measurement of intracellular free
calcium
Changes in intracellular free calcium were measured
by a fluorescent dye, Fluo-3, AM as previously described (15). Briefly, NSCLC cells were washed
twice with RPMI-1640 media after 2 μM celastrol treatment for 0–4
h. The cell suspensions were then incubated with 5 μM Fluo-3, AM at
37°C for 30 min. After the cells were washed twice with HBSS, the
re-suspended cell samples were then subjected to FACS analysis. At
least 10,000 events were analyzed.
Endogenous LC3 and EGFR detection
The detection of endogenous LC3 was conducted using
immunofluorescence staining method as described below. In brief,
celastrol-treated cancer cells on coverslips were fixed with 4%
paraformaldehyde (Sigma-Aldrich) for 20 min at room temperature and
then rinsed with phosphate-buffered saline (PBS). Immerse
coverslips in methanol at room temperature for 2 min. After washing
with PBS, the cells were then incubated with anti-LC3 (1:200) in
TBST (100 mM Tris HCl, pH 7.5, 150 mM NaCl, 0.05% Tween-20 and 5%
BSA) overnight at 4°C. After washing with PBS, the cells were
incubated with anti-mouse secondary antibody (TRITC) 1:200 in TBST
containing 5% BSA at 37°C for 1 h in the dark. For detection of
EGFR, the cells were incubated with anti-EGFR (1:200) in TBST (100
mM Tris HCl, pH 7.5, 150 mM NaCl, 0.05% Tween-20 and 5% BSA)
overnight at 4°C. After washing with PBS, the cells were incubated
with anti-rabbit secondary antibody (FITC) 1:200 in TBST containing
5% BSA at 37°C for 1 h in the dark. The coverslips were then
mounted with FluorSave™ mounting media (Calbiochem) for
fluorescence imaging and localization of LC3 autophagosomes and
EGFR expression were captured under the API DeltaVision Live-cell
Imaging System (Applied Precision Inc., GE Healthcare Co.,
Issaquah, WA, USA). To quantify autophagy, guidelines were followed
to monitor autophagy (16), the
percentage of cells with punctuate LC3 immunofluorescence staining
was calculated by counting the number of the cells showing the
increased punctuate pattern of LC3 fluorescence (≥10 dots/cell) in
immunofluorescence-positive cells over the total number of cells in
the same field. A minimum of 1,000 cells from randomly selected
fields were scored. To quantify EGFR expression, the intensity of
green fluorescence FITC signal in cells was determined by API
DeltaVision Live-cell Imaging System.
Annexin V detection by flow cytometric
analysis
Apoptosis was detected by Annexin V staining kit (BD
Biosciences, San Jose, CA, USA). In brief, cells were exposed to
the indicated concentrations of celastrol for 24 h. Cells were then
harvested and analyzed by flow cytometry using FITC-Annexin V and
propidium iodide staining according to the manufacturer’s
instructions. Apoptotic cells were quantitatively counted by a flow
cytometer (FACSAria III; BD Biosciences). Data acquisition and
analysis were performed with the CellQuest (BD Biosciences) from
triple independent experiments.
Protein extraction and western
blotting
The celastrol treated cells were lysed with RIPA
lysis buffer. Protein concentrations were determined using the
Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The cell lysates of samples were subjected to electrophoresis
on SDS polyacrylamide gels and transferred to Hybond enhanced
chemiluminescence nitrocellulose membranes (Amersham Biosciences,
Piscataway, NJ, USA), which were then blocked with 5% non-fat dry
milk protein for 1 h. Membranes were then incubated with the
indicated primary antibodies overnight at 4°C. The binding of the
antibody was visualized by peroxidase-coupled secondary antibody
using the ECL Western Blotting detection reagents (Invitrogen).
Band intensities were quantified by using the software ImageJ (NIH,
Bethesda, MD, USA).
Reverse transcriptase-polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted from the cultured cancer
cells using FavorPrep™ Total RNA Purification Mini kit (Favorgen
Biotech Corp., Pingtung, Taiwan). The synthesis of the first strand
of cDNA was followed based on the instruction of
SuperScript® VILO™ Master Mix kit (Invitrogen, Grand
Island, NY, USA). DNase-treated total RNA (1 μg) from cells was
used for cDNA synthesis. Each RNA sample was incubated with the 2
μl random primers and 1 μl Oligo(dT) primer provided by the kit at
65°C for 10 min, and then cooled on ice for 2 min. Reaction mixture
which contains reaction buffer, RNase inhibitor and reverse
transcriptase was added to the tube and then incubated at 25°C for
10 min and 55°C for 30 min. The reaction was terminated by heating
at 70°C for 15 min. Quantitative real-time PCR were carried out on
ViiA™ 7 real-time PCR system (Applied Biosytems, Grand Island, NY,
USA) using the FastStart Universal SYBR-Green Master Rox (Roche
Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s
instructions. The PCR mixture is comprised of 10 μl SYBR Master
Mix, 0.3 μl forward and reverse primers, 2 μl template and 7.4 μl
ddH2O makes up to 20 μl. The procedure of RCR is 50°C
for 2 min, 95°C for 10 min, followed by 45 cycles of 95°C for 15
sec, 60°C for 60 sec. Expression of each EGFR transcription level
was normalized with β-actin. CT values were indicated in the bar
charts. Ranges of values obtained in three parallel analyses were
<5% of the means.
Statistical analysis
The results were expressed as means ± SD as
indicated. The difference was considered statistically significant
when the P-value was <0.05. Student’s t-test or one-way ANOVA
analysis was used for comparison among the different groups.
Results
Celastrol exhibits selective cytotoxic
effect and mobilizes cytosolic calcium towards EGFR mutant
NSCLCs
The natural triterpenoid compound celastrol is a
promising cytotoxic agent in a great variety of cancer models
(17–20). It was previously reported that
celastrol has the potency to combat gefitinib-resistant NSCLCs by
inducing apoptosis through caspase-dependent pathways and Hsp90
client protein degradation. However, its underlying mechanisms are
still rudimentary.
To investigate the detail mechanisms of the
anticancer effect of celastrol on gefitinib-resistant non-small
cell lung carcinoma (NSCLC) (17,21),
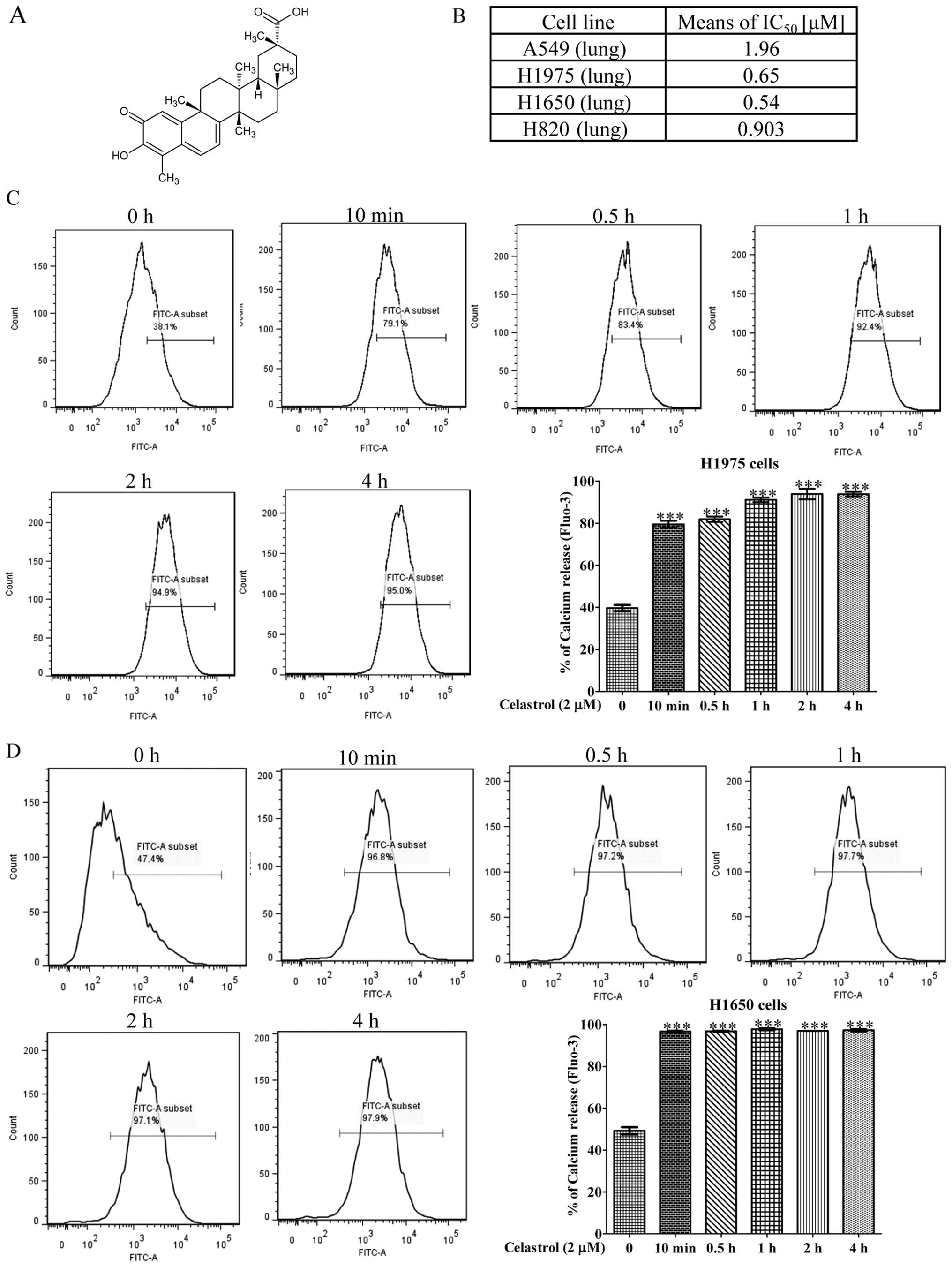
we examined the specific cytotoxicity of celastrol (Fig. 1A) towards four NSCLC cell lines,
including H1975, H1650, H820 and A549. Among these four NSCLCs,
H1975 harbors L858R and T790M double mutation on EGFR, while H1650
possesses the in-frame mutation in exon 19, delE746-A750, H820
harbors exon 19 in-frame deletion and T790M double mutation on
EGFR, while only A549 contains wild-type EGFR. As shown in Fig. 1B, celastrol displayed considerable
more cytotoxicity against H1975 and H1650 NSCLC cells than the
other NSCLC cell A549. The mean IC50 values were 0.65,
0.54 and 1.96 μM, respectively. The inhibitory effect of celastrol
on A549 gefitinib-sensitive human lung cancer was relative lower
implying that celastrol exhibited substantial cytotoxic effects
towards gefitinib-resistant NSCLCs.
A previous study demonstrated that celastrol could
induce cell death via calcium release from the endoplasmic
reticulum (22). To investigate
whether celastrol can mobilize cytosolic [Ca2+] in
NSCLCs, celastrol-treated H1975 and H1650 cells were stained with
Fluo 3-AM for determination of cellular [Ca2+] level
change. Flow cytometric analysis showed that H1975 and H1650 cells
loaded with Fluo 3-AM displayed a marked increase in fluorescence
intensity upon the treatment of 2 μM celastrol for 10 min (Fig. 1C and D) suggesting that celastrol
significantly increased the cellular calcium level within a short
time.
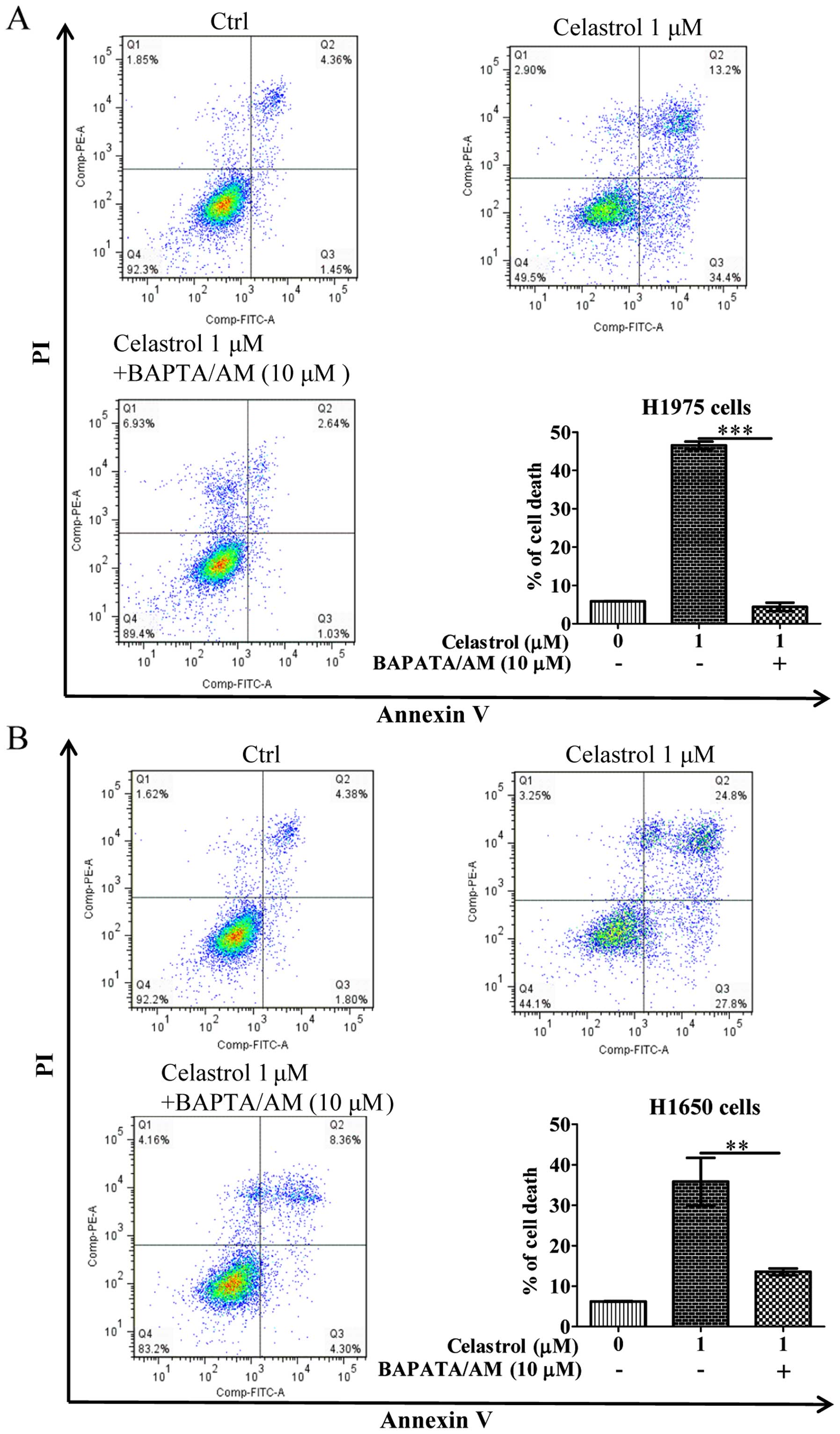
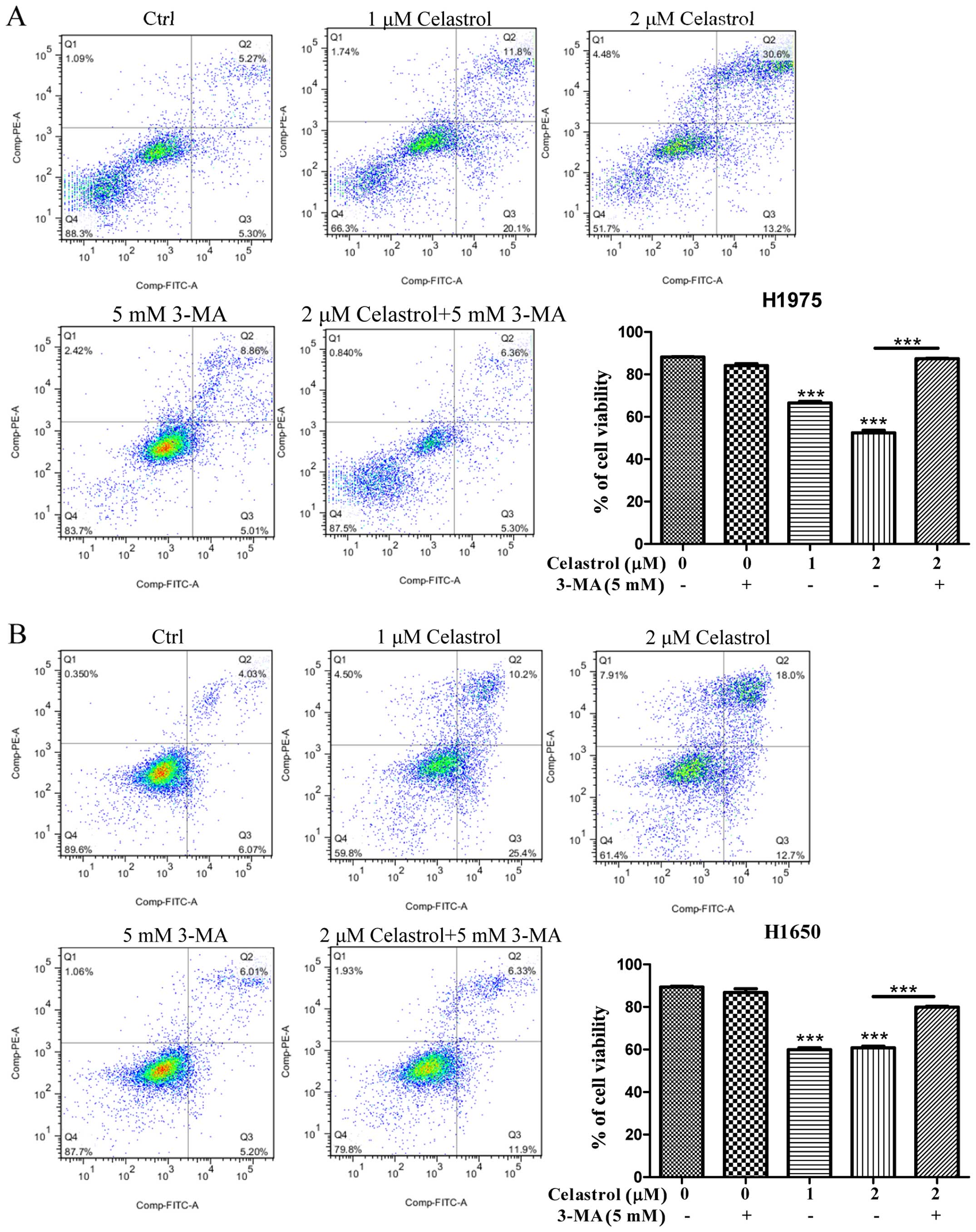
Celastrol induces cell death in
gefitinib-resistant NSCLCs via calcium mobilization
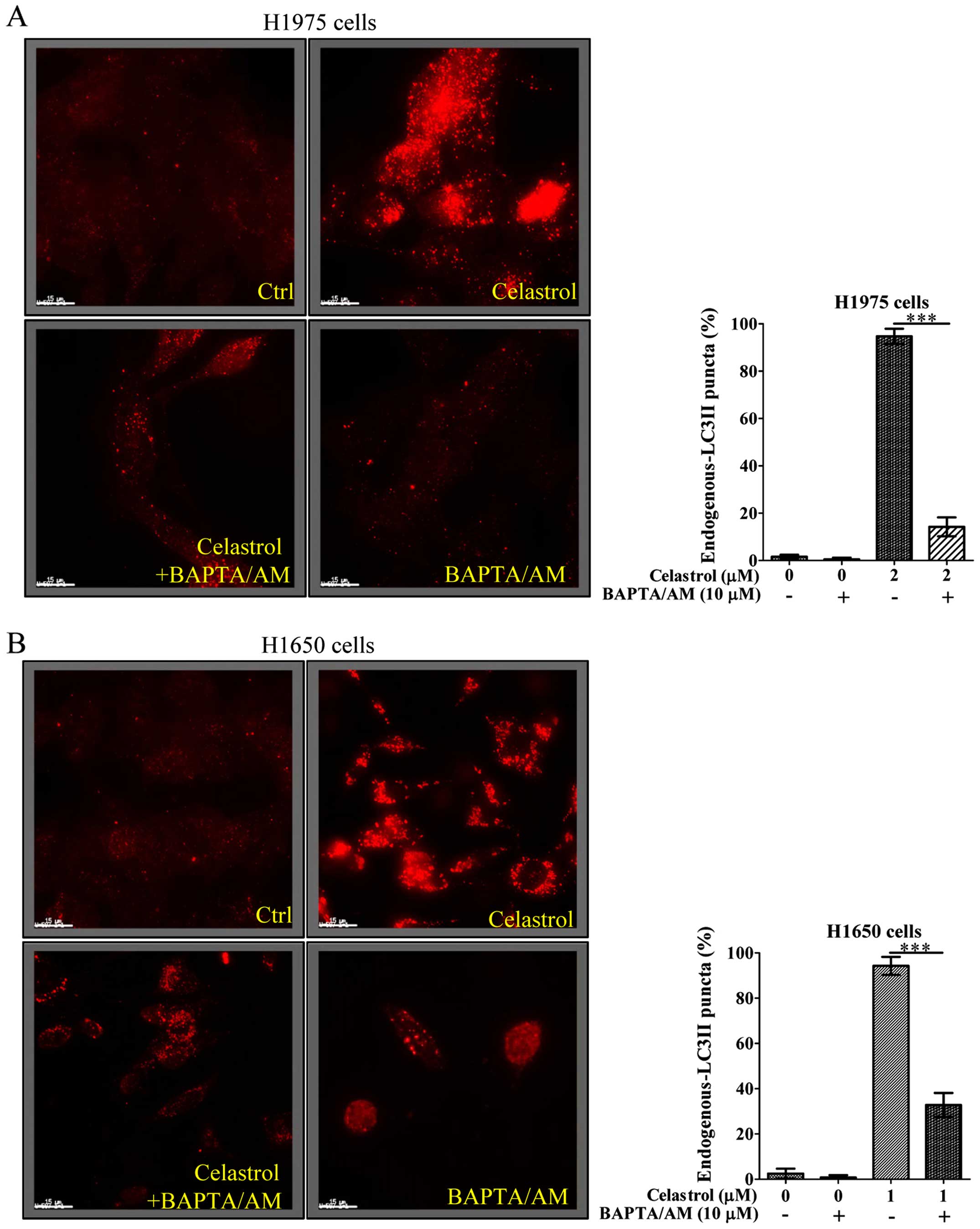
As the celastrol-induced autophagy in
gefitinib-resistant NSCLCs was due to calcium mobilization
(Fig. 2), we further addressed
whether celastrol-induced cell death is due to the increase of
[Ca2+] levels, we therefore examined its cytotoxicity
with the intracellular Ca2+ chelator (BAPTA/AM) using
Annexin V flow cytometry. As shown in Fig. 3, while celastrol significantly
induced cell death to >40% in H1975 and H1650 cancer cells,
addition of BAPTA/AM mostly recovered the celastrol-induced cell
death. These results suggested that the increase of
[Ca2+] level and its mediated autophagy were required
for celastrol-induced apoptosis.
Celastrol induces Hsp90 client protein
degradation in both EGFR wild-type and mutant NSCLCs via calcium
mobilization and autophagy induction
Gefitinib is a TKI of EGFR, it can specifically
block the activation of EGFR by binding to its ATP binding pocket,
resulting in EGFR kinase inhibition (24) and the downstream kinases like Akt
which hinder cancer cell growth and survival.
Various EGFR mutants have been found existing
amongst NSCLCs. Celastrol demonstrated marked inhibitory effect on
both EGFR wild-type and mutant NSCLC cells. Therefore, we
investigated whether this inhibitory effect on NSCLCs is due to the
degradation of EGFR. As shown in Fig.
4A, celastrol potently promoted the degradation of mutant EGFR
in H1975 cells. Moreover, the downstream targets of EGFR, Akt, were
also inhibited with concomitant increase of autophagic LC3-II
conversion. To determine whether the degradation of EGFR is
mutant-specific, we examined the degradation of wild-type EGFR from
A549 cells. As shown in Fig. 4B,
the wild-type EGFR was significantly degraded in A549 lung cancer
cells, suggesting there was no selectivity on celastrol-induced
EGFR degradation in NSCLCs.
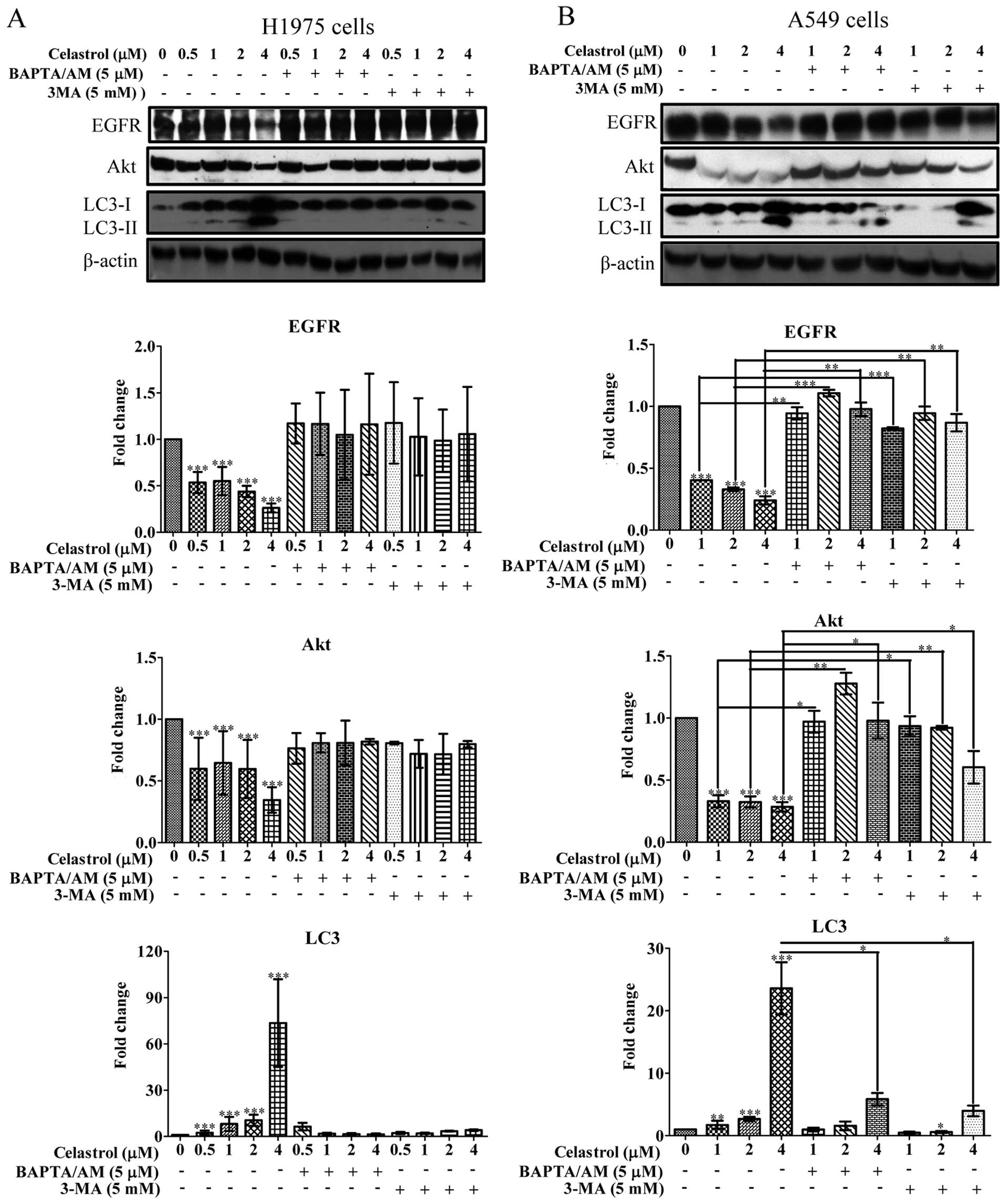 | Figure 4Celastrol induces Hsp90 client
protein degradation in both EGFR wild-type and mutant NSCLCs via
calcium mobilization and autophagy induction. (A and B) Both
calcium chelator BAPTA/AM and autophagic inhibitor 3-MA suppressed
celastrol-mediated degradation of Hsp90 client proteins: EGFR and
Akt, as well as LC3-II conversion on EGFR wild-type and mutant
NSCLCs. EGFR mutant H1975 cells were treated with DMSO or 0.5, 1, 2
and 4 μM of celastrol, whereas EGFR wild-type A549 cells were
treated with DMSO or 1, 2 and 4 μM of celastrol for 24 h in the
presence or absence of 5 μM BAPTA/AM or 5 mM 3-MA, respectively.
Cell lysates were then harvested and analyzed by western blot
detection of EGFR, AKT and LC3 conversion (LC3-I, 18 kDa; LC3-II,
16 kDa) and β-actin. Protein band intensities were quantified using
densitometry analysis and normalized to β-actin. Data were
expressed as a fold change relative to the DMSO-treated control.
Bar chart represents data of three independent experiments. Error
bars, SD. ***P<0.001. |
To verify whether the celastrol stimulated
degradation of EGFR through cytosolic calcium mobilization and
autophagy induction, we employed BAPTA/AM and 3-MA to co-incubate
with celastrol on both H1975 and A549 cells. BAPTA/AM and 3-MA
markedly prevented the celastrol-mediated degradation of either
wild-type or mutant EGFR, and abolished the celastrol-suppressed
Akt expression in both H1975 and A549 NSCLCs, revealing that the
inhibition on EGFR activation by celastrol benefit from calcium
mobilization and autophagy induction. As expected, BAPTA/AM or 3-MA
was able to suppress celastrol-induced LC3-II conversion in H1975
and A549 (Fig. 4). Taken together,
celastrol was able to significantly suppress the activity of
wild-type and mutant EGFR and Akt which is necessary for the
survival and growth of NSCLCs via calcium-mediated autophagy
induction.
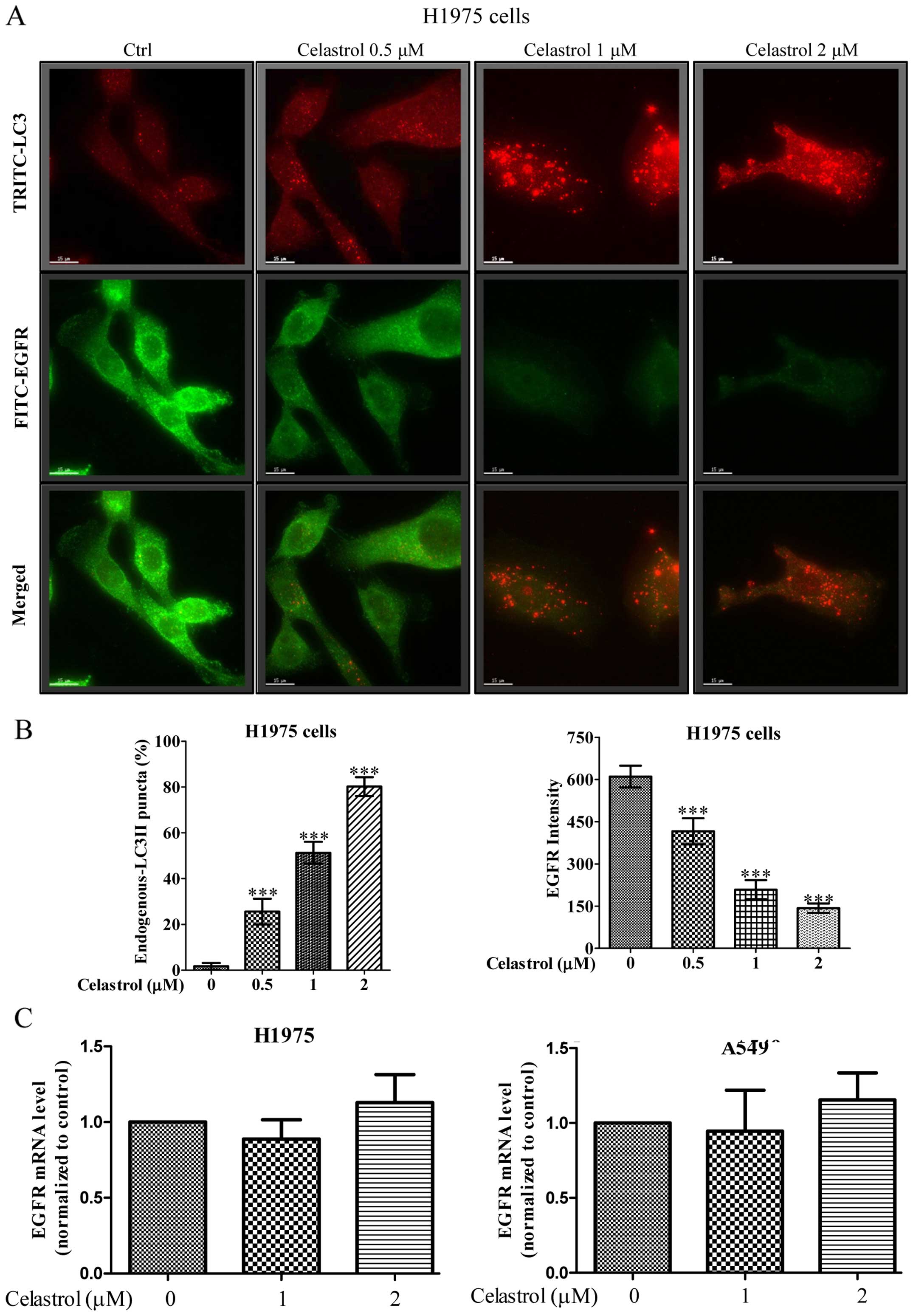
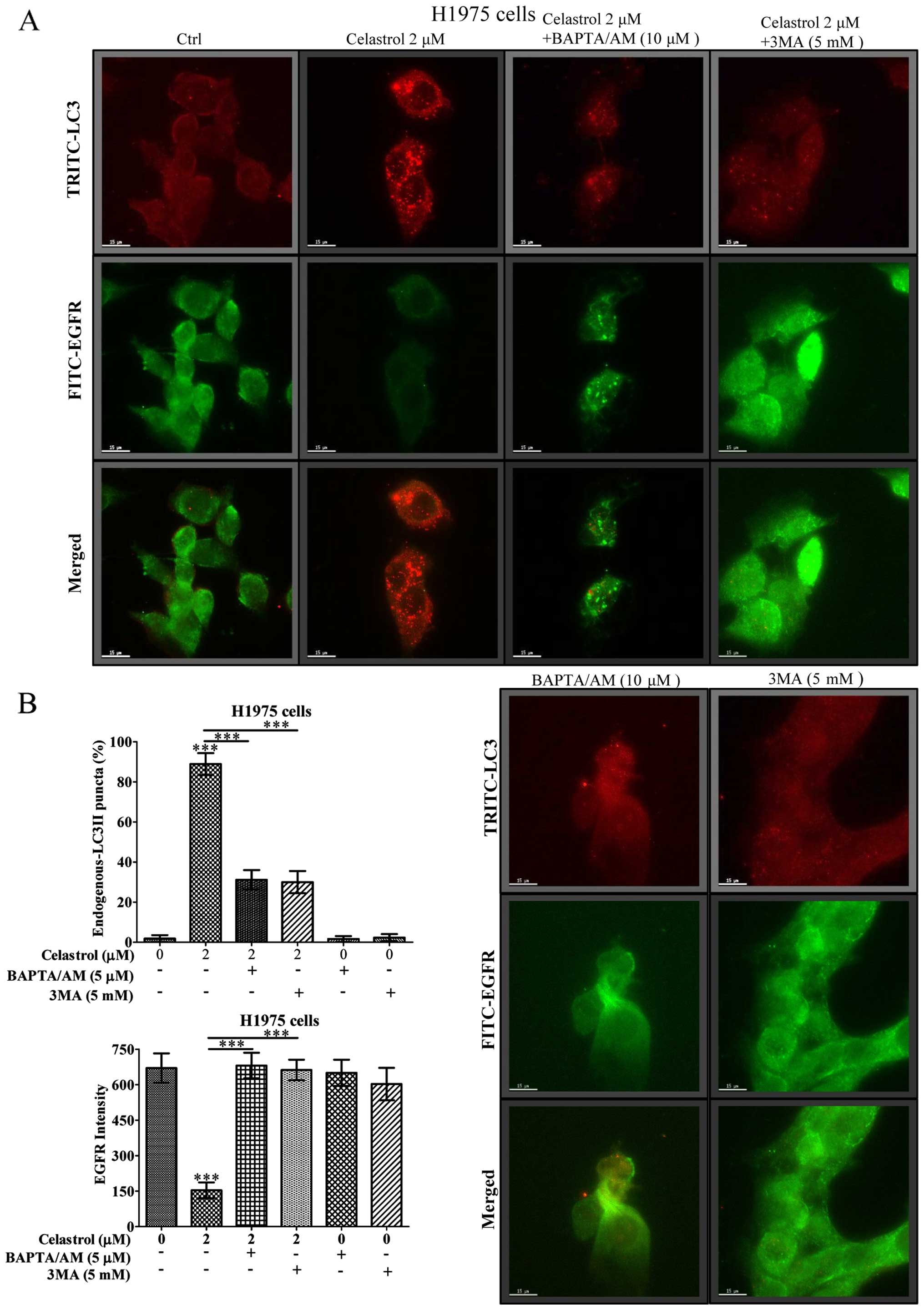
Immunofluorescence staining demonstrates
that celastrol simultaneously induces autophagy and EGFR
degradation in H1975 in dose-dependent manner
We further monitored the consequences of
celastrol-induced autophagic activity in EGFR degradation of H1975
NSCLCs using fluorescence microscopy. As shown in Fig. 5A, while the red puncta formation
(red TRITC signal) represented the endogenous LC3-II conversion
conferred by celastrol-induced autophagic activity, merged images
with green fluorescent image (green FITC signal) denoted as EGFR
expression indicated autophagic activity of celastrol was highly
associated with the EGFR degradation. Our results demonstrated a
dose-dependent increase in the percentage of cells with endogenous
LC3-II puncta formation (red TRITC signal) after celastrol
treatment, simultaneously accompanied a dose-dependent decrease of
fluorescence intensity of EGFR signal (Fig. 5B). In addition, real-time PCR
revealed the unchanged EGFR transcription in both wild-type and
mutant EGFR NSCLCs upon celastrol treatment (Fig. 5C), confirming that the
celastrol-induced autophagy in H1975 cells may contribute to EGFR
degradation.
Celastrol induces the degradation of EGFR
via calcium-mediated autophagy induction
We further addressed how calcium mobilization and
its mediated autophagic activity would eventually contribute to
EGFR degradation in H1975 NSCLCs using fluorescence microscopy. As
shown in Fig. 6A, while 2 μM of
celastrol induced vigorous autophagic effect in H1975 cancer cells
reflected by red TRITC endogenous LC3-II puncta formation signal,
the corresponding green fluorescent image (green FITC signal)
embodied in the same cell region had immensely faded. In contrast,
addition of the Ca2+ chelator (BAPTA/AM) and the
autophagic inhibitor 3-methyladenine (3-MA) fully recovered the
celastrol-induced EGFR depletion, as well as abolished the
compound-induced autophagy activity (Fig. 6B). Importantly, blockage of
celastrol-induced autophagy and its mediated EGFR degradation by
3-MA would eventually increase the survival rate of H1975 and H1650
cancer cells (Fig. 7). The above
evidence suggested that celastrol activates cytosolic
[Ca2+] mobilization to induce autophagy, thereby
promotes the EGFR degradation and circumvents the EGFR-resistant
phenotype in mutant EGFR NSCLCs.
Discussion
The present study demonstrated the potential of
celastrol in NSCLC intervention through direct and non-selective
EGFR clearance via induction of autophagy in an intracellular
calcium-dependent manner. The cytotoxicity induced by celastrol
upon our NSCLC cellular models is probably due to suppression of
EGFR and downstream survival signaling after induction of EGFR
loss. Since EGFR is responsible for eliciting downstream survival
signaling for DNA synthesis and cell proliferation (25), therapeutic EGFR protein degradation
effect induced by celastrol-mediated autophagy is likely playing
the causative role in mediating apoptosis. Moreover, EGFR loss is
unlikely associated with other transcriptional manipulations,
because pre-autophagy blockage significantly rescued EGFR
degradation and resumed the viability of the celastrol-treated
cancer cells. So et al (26) also illustrated that pharmaceutical
induction of autophagy, by protein kinase CK2 (CK2) inhibitor,
downregulated EGFR and led eventually to NSCLC cell death. Although
CK2 inhibitor may trigger autophagy through different signaling
pathways from celastrol, at least it provides supporting evidence
of the feasibility of using agent to induce direct
autophagic-mediated EGFR elimination.
Almost 90% of all histological types of lung cancers
belongs to NSCLC (27) [http://www.cancer.org/cancer/lungcancer-non-smallcell/detailedguide/non-small-cell-lung-cancer-what-is-non-small-cell-lung-cancer]
(3/11/2016), and around 60% of patients are overexpressing EGFR
(27). Therefore, developing an
effective intervention strategy targeting EGFR of NSCLC will
greatly enhance the survival of patients. In this connection, a
well-known EGFR molecular targeting strategy mainly based on the
use of EGFR inhibitors have been evolved, which is the use of a
small molecule TKI inhibitor. However, the clinical application of
these inhibitors encountered many constraints. For example, the
monocloncal antibodies cetuximab (erbitux) and panitumumab
(vectibix) have been suggested as pharmaceutical intervention for
NSCLC (28). These antibody
inhibitors target specifically to the mutated EGFR which in turn
shut down the downstream survival signaling for the cancer cells
(29–32). However, the antibody-dependent
cell-mediated cytotoxicity induced by the monoclonal immunoglobulin
G proteins could be complicated (33). The use of gefitinib (Iressa) and
erlotinib (Tarceva) to suppress the tyrosine kinase activities of
EGFR also faced several limitations. For example, drug-resistant
mutants of the cancer cells developed from compensation of parallel
signaling pathways has become a major concern for maintaining the
efficacy of these small molecules (24,34).
In addition, inhibitors which target the downstream signaling
molecules, rather than EGFR per se, by manipulating their
post-translational modifications, such as isoprenylation of RAS
(35,36), have been described. However, these
signaling proteins usually compose of multiple modifications which
greatly reduce the therapeutic potential of the post-translational
modifiers (37). Celastrol, when
compared with the above single molecules, could be a more efficient
therapeutic method targeting the EGF/EGFR pathway without the
mentioned practical drawbacks. In particular, the therapeutic
effects of celastrol is unrelated to the genetic variations of EGFR
making itself suitable for long-term treatment for cancer cells
easily developed with drug resistance. Of note, celastrol is a
bioactive component constituting the Chinese herbal medicine (CHM)
Radix tripterygii wilfordii (38). The herb is prescribed for a variety
of disorders associated with immunological dysfunctions (39), and tumorigenesis (40), implying the safety of using
celastrol for further clinical development.
Expression of both EGFR and Akt were hampered after
celastrol treatment. Since, nuclear EGFR is the transcription
factor for Akt, it could be a result of the loss of Akt expression
signaling after the treatment. However, it should not be neglected
that Akt is also an oncogenic protein and can be eliminated by the
celastrol-induced upregulation of autophagy. In fact, emerging
studies suggest the use of autophagy inducers for promoting
oncogenic protein degradation to ameliorate tumorigenesis. An
autophagy-deficient animal model demonstrated that the
autophagic-mediated removal of Nucleoporin p62 (p62) repressed
tumor progression of hepatocellular carcinoma (HCC) (10). Although, p62 functioned as an
adaptor protein assisting autophagy (41), it is a substrate of the catabolic
process (41–43) and an oncogenic protein mediating
the nuclear factor-κB (NF-κB) signaling to facilitate tumorigenesis
(44–46). In addition, HCC patients can be
diagnosed by the expression level of alpha-1 antitrypsin protein
(A1AT) in a blood sample (47).
The hepatic load of its mutant, alpha1-antitrypsin Z (ATZ), is
critical to the development of hepatic fibrosis which is the
clinical prerequisite of HCC. The autophagy-inducing drug
carbamazepine (CBZ) can reduce hepatic fibrosis by diminishing the
accumulation of ATZ in liver (48). In the case of colon cancer, the
natural flavonoid quercetin, a CHM-derived compound, that is
commonly found in fruits and vegetables (49–52),
exhibited similar clinical benefit. By targeting specifically to
the mutated Ras protein, the use of quercetin selectively degraded
the oncogenic form of Ras in colon adenocarcinoma rendering the
transformation and proliferation of the cancer cells in
vitro (53). The
quercetin-induced Ras degradation is mechanistically mediated by
the proteasomal system which may imply the co-involvement of
autophagy, since the two protein quality control machineries are
closely inter-twined. Another in vitro treatment study for
acute promyelocytic leukemia (APL) using all-trans retinoic acid
and arsenic trioxide also suggested the therapeutic efficacy of
autophagy-mediated oncogenic protein degradation. The oncogenic
protein promyelocytic leukemia/retinoic acid receptor alpha
(PML/RARA) critically underpinning the remission of APL were
markedly eliminated by autophagy triggered by the two compounds
(54). Gastrointestinal stromal
tumors (GIST) driven by mutated KIT proto-oncogene receptor
tyrosine kinase (KIT) is sensitive to autophagy enhancer treatment
as well. NVP-AUY922, a heat shock protein 90 (Hsp90) inhibitor,
induced specific KIT degradation through autophagy upregulation
which further suppress GIST cell growth (55). All these encouraging preclinical
findings, alongside our discoveries reported here, strongly
suggested the therapeutic potential of autophagy-mediated oncogenic
EGFR protein degradation in cancer therapy.
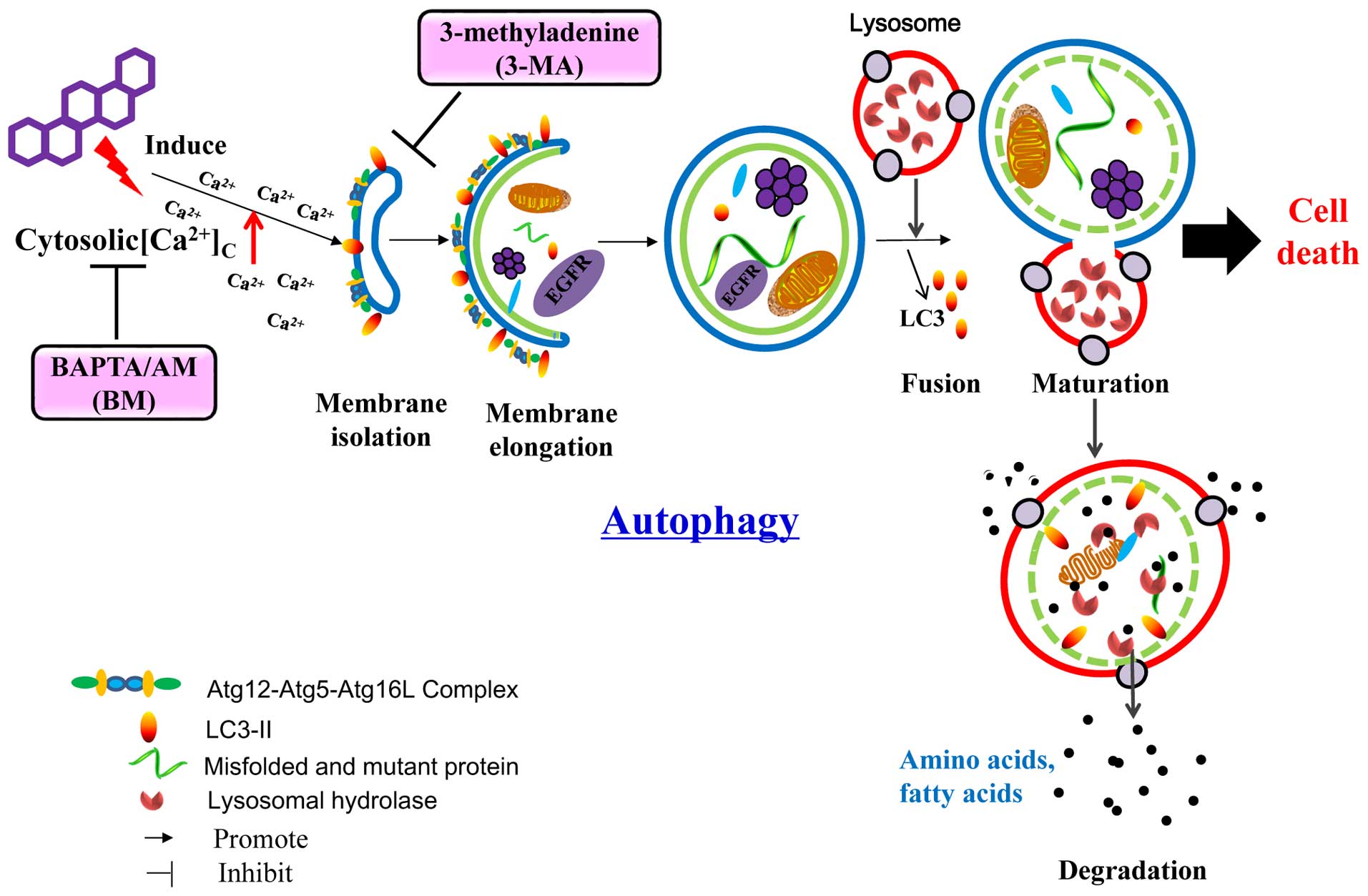
In conclusion, the CHM-derived celastrol represent a
new pharmaceutical candidate for NSCLC intervention which acted
through the direct autophagic degradation of EGFR (Fig. 8). The non-selective degradation
nature of celastrol targeting both mutant and wild-type EGFR
suggested that an in-depth examination, aimed to clarify the
effects of celastrol on normal cell, is needed. Our findings also
provided insight to the therapeutic development for other
EGFR-driven cancers, such as gastric and colorectal cancers, which
overcome the frequently and increasingly occurring drug-resistance
problems. Most importantly, we generally depicted the possibility
of managing tumorigenesis by degrading oncogenic proteins with the
use of autophagy enhancers. The CHM origin of celastrol also
encouraged the search of other novel autophagy enhancers from
medicinal herbs, a rich source of comparatively safe natural
compounds, for cancer therapy.
Acknowledgements
The present study was supported by a FDCT grant from
the Macao Science and Technology Development Fund (Project code:
084/2013/A3 & 005/2014/AMJ).
References
|
1
|
Gazdar AF: Activating and resistance
mutations of EGFR in non-small-cell lung cancer: Role in clinical
response to EGFR tyrosine kinase inhibitors. Oncogene. 28(Suppl 1):
S24–S31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roengvoraphoj M, Tsongalis GJ, Dragnev KH
and Rigas JR: Epidermal growth factor receptor tyrosine kinase
inhibitors as initial therapy for non-small cell lung cancer: Focus
on epidermal growth factor receptor mutation testing and
mutation-positive patients. Cancer Treat Rev. 39:839–850. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bose P and Ozer H: Neratinib: An oral,
irreversible dual EGFR/HER2 inhibitor for breast and non-small cell
lung cancer. Expert Opin Investig Drugs. 18:1735–1751. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li D, Ambrogio L, Shimamura T, Kubo S,
Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A,
Himmelsbach F, et al: BIBW2992, an irreversible EGFR/HER2 inhibitor
highly effective in preclinical lung cancer models. Oncogene.
27:4702–4711. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Engelman JA, Zejnullahu K, Gale CM,
Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov
GN, Bradner JE, et al: PF00299804, an irreversible pan-ERBB
inhibitor, is effective in lung cancer models with EGFR and ERBB2
mutations that are resistant to gefitinib. Cancer Res.
67:11924–11932. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang J, Greulich H, Jänne PA, Sellers WR,
Meyerson M and Griffin JD: Epidermal growth factor-independent
transformation of Ba/F3 cells with cancer-derived epidermal growth
factor receptor mutants induces gefitinib-sensitive cell cycle
progression. Cancer Res. 65:8968–8974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Greulich H, Chen TH, Feng W, Jänne PA,
Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR,
et al: Oncogenic transformation by inhibitor-sensitive and
-resistant EGFR mutants. PLoS Med. 2:e3132005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Turcotte S, Chan DA, Sutphin PD, Hay MP,
Denny WA and Giaccia AJ: A molecule targeting VHL-deficient renal
cell carcinoma that induces autophagy. Cancer Cell. 14:90–102.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fan XX, Li N, Wu JL, Zhou YL, He JX, Liu L
and Leung EL: Celastrol induces apoptosis in gefitinib-resistant
non-small cell lung cancer cells via caspases-dependent pathways
and Hsp90 client protein degradation. Molecules. 19:3508–3522.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wong VKW, Zhou H, Cheung SSF, Li T and Liu
L: Mechanistic study of saikosaponin-d (Ssd) on suppression of
murine T lymphocyte activation. J Cell Biochem. 107:303–315. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu MJ, Wang Z, Ju Y, Wong RNS and Wu QY:
Diosgenin induces cell cycle arrest and apoptosis in human leukemia
K562 cells with the disruption of Ca2+ homeostasis.
Cancer Chemother Pharmacol. 55:79–90. 2005. View Article : Google Scholar
|
|
16
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang WB, Feng LX, Yue QX, Wu WY, Guan SH,
Jiang BH, Yang M, Liu X and Guo DA: Paraptosis accompanied by
autophagy and apoptosis was induced by celastrol, a natural
compound with influence on proteasome, ER stress and Hsp90. J Cell
Physiol. 227:2196–2206. 2012. View Article : Google Scholar
|
|
18
|
Yang H, Chen D, Cui QC, Yuan X and Dou QP:
Celastrol, a triterpene extracted from the Chinese ‘Thunder of God
Vine’, is a potent proteasome inhibitor and suppresses human
prostate cancer growth in nude mice. Cancer Res. 66:4758–4765.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kannaiyan R, Shanmugam MK and Sethi G:
Molecular targets of celastrol derived from Thunder of God Vine:
Potential role in the treatment of inflammatory disorders and
cancer. Cancer Lett. 303:9–20. 2011. View Article : Google Scholar
|
|
20
|
Boridy S, Le PU, Petrecca K and Maysinger
D: Celastrol targets proteostasis and acts synergistically with a
heat-shock protein 90 inhibitor to kill human glioblastoma cells.
Cell Death Dis. 5:e12162014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng YN, Shi J, Liu J and Qu QM: Celastrol
protects human neuroblastoma SH-SY5Y cells from rotenone-induced
injury through induction of autophagy. Neurochem Int. 63:1–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY,
Kwon YJ and Choi KS: Release of Ca2+ from the
endoplasmic reticulum and its subsequent influx into mitochondria
trigger celastrol-induced paraptosis in cancer cells. Oncotarget.
5:6816–6831. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie
T, Zhang N and Ye ZM: Celastrol induces apoptosis and autophagy via
the ROS/JNK signaling pathway in human osteosarcoma cells: an in
vitro and in vivo study. Cell Death Dis. 6:e16042015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chan LY, Kosuri S and Endy D: Refactoring
bacteriophage T7. Mol Syst Biol. 1:00182005. View Article : Google Scholar
|
|
26
|
So KS, Kim CH, Rho JK, Kim SY, Choi YJ,
Song JS, Kim WS, Choi CM, Chun YJ and Lee JC:
Autophagosome-mediated EGFR down-regulation induced by the CK2
inhibitor enhances the efficacy of EGFR-TKI on EGFR-mutant lung
cancer cells with resistance by T790M. PLoS One. 9:e1140002014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
da Cunha Santos G, Shepherd FA and Tsao
MS: EGFR mutations and lung cancer. Annu Rev Pathol. 6:49–69. 2011.
View Article : Google Scholar
|
|
28
|
Pirker R and Filipits M: Cetuximab in
non-small-cell lung cancer. Transl Lung Cancer Res. 1:54–60.
2012.PubMed/NCBI
|
|
29
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al; West Japan Oncology Group. Gefitinib versus cisplatin plus
docetaxel in patients with non-small-cell lung cancer harbouring
mutations of the epidermal growth factor receptor (WJTOG3405): An
open label, randomised phase 3 trial. Lancet Oncol. 11:121–128.
2010. View Article : Google Scholar
|
|
30
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rosell R, Carcereny E, Gervais R,
Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R,
Pallares C, Sanchez JM, et al; Spanish Lung Cancer Group in
collaboration with Groupe Français de Pneumo-Cancérologie and
Associazione Italiana Oncologia Toracica. Erlotinib versus standard
chemotherapy as first-line treatment for European patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(EURTAC): A multicentre, open-label, randomised phase 3 trial.
Lancet Oncol. 13:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al; North-East Japan Study Group. Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yan L and Beckman RA: Pharmacogenetics and
pharma-cogenomics in oncology therapeutic antibody development.
Biotechniques. 39(S10): 565–568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kohl NE, Conner MW, Gibbs JB, Graham SL,
Hartman GD and Oliff A: Development of inhibitors of protein
farnesylation as potential chemotherapeutic agents. J Cell Biochem
Suppl. 22(S22): 145–150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ray D, Cuneo KC, Rehemtulla A, Lawrence TS
and Nyati MK: Inducing oncoprotein degradation to improve targeted
cancer therapy. Neoplasia. 17:697–703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vasan N, Boyer JL and Herbst RS: A RAS
renaissance: Emerging targeted therapies for KRAS-mutated non-small
cell lung cancer. Clin Cancer Res. 20:3921–3930. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu H, Straub A, Tian Z, Bassler N, Cheng J
and Peter K: Celastrol, a triterpene extracted from Tripterygium
wilfordii Hook F, inhibits platelet activation. J Cardiovasc
Pharmacol. 54:240–245. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tang W and Zuo JP: Immunosuppressant
discovery from Tripterygium wilfordii Hook f: The novel triptolide
analog (5R)-5-hydroxytriptolide (LLDT-8). Acta Pharmacol Sin.
33:1112–1118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kannaiyan R, Manu KA, Chen L, Li F,
Rajendran P, Subramaniam A, Lam P, Kumar AP and Sethi G: Celastrol
inhibits tumor cell proliferation and promotes apoptosis through
the activation of c-Jun N-terminal kinase and suppression of PI3
K/Akt signaling pathways. Apoptosis. 16:1028–1041. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ichimura Y, Kumanomidou T, Sou YS,
Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K and
Komatsu M: Structural basis for sorting mechanism of p62 in
selective autophagy. J Biol Chem. 283:22847–22857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Duran A, Linares JF, Galvez AS,
Wikenheiser K, Flores JM, Diaz-Meco MT and Moscat J: The signaling
adaptor p62 is an important NF-kappaB mediator in tumorigenesis.
Cancer Cell. 13:343–354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Komatsu M, Waguri S, Koike M, Sou YS, Ueno
T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al:
Homeostatic levels of p62 control cytoplasmic inclusion body
formation in autophagy-deficient mice. Cell. 131:1149–1163. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Takamura A, Komatsu M, Hara T, Sakamoto A,
Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K and Mizushima N:
Autophagy-deficient mice develop multiple liver tumors. Genes Dev.
25:795–800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Varela AS and López Sáez JJ: Utility of
plasmatic levels of alpha-1-antiprotease (A1AP) as a cancer marker.
Cancer Lett. 89:15–21. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hidvegi T, Ewing M, Hale P, Dippold C,
Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S,
et al: An autophagy-enhancing drug promotes degradation of mutant
α1-antitrypsin Z and reduces hepatic fibrosis. Science.
329:229–232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xianfei X, Xiaoqiang C, Shunying Z and
Guolin Z: Chemical composition and antimicrobial activity of
essential oils of Chaenomeles speciosa from China. Food Chem.
100:1312–1315. 2007. View Article : Google Scholar
|
|
50
|
Nahrstedt A and Butterweck V: Biologically
active and other chemical constituents of the herb of Hypericum
perforatum L. Pharmacopsychiatry. 30(Suppl 2): 129–134. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Luan L, Wang G and Lin R: Studies on the
chemical constituents of extract with water from Forsythia
suspensa. Zhong Yao Cai. 33:220–221. 2010.(In Chinese). PubMed/NCBI
|
|
52
|
Yang YB, Yang Y, Li X, Yang Z, Wu ZJ,
Zheng YL and Sun LN: Studies on the chemical constituents of
Chaenomeles speciosa. Zhong Yao Cai. 32:1388–1390. 2009.(In
Chinese). PubMed/NCBI
|
|
53
|
Psahoulia FH, Moumtzi S, Roberts ML,
Sasazuki T, Shirasawa S and Pintzas A: Quercetin mediates
preferential degradation of oncogenic Ras and causes autophagy in
Ha-RAS-transformed human colon cells. Carcinogenesis. 28:1021–1031.
2007. View Article : Google Scholar
|
|
54
|
Isakson P, Bjørås M, Bøe SO and Simonsen
A: Autophagy contributes to therapy-induced degradation of the
PML/RARA oncoprotein. Blood. 116:2324–2331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hsueh YS, Yen CC, Shih NY, Chiang NJ, Li
CF and Chen LT: Autophagy is involved in endogenous and
NVP-AUY922-induced KIT degradation in gastrointestinal stromal
tumors. Autophagy. 9:220–233. 2013. View Article : Google Scholar :
|