Introduction
Oncolytic viruses (OV) exhibit unique features such
as: i) outstanding safety profiles (especially when vaccine-derived
viral vectors are coming to application), ii) high levels of tumor
selectivity, iii) an incomparable self-amplification property, iv)
lack of cross-resistance with other anticancer drugs (e.g.,
chemotherapeutic compounds), v) superior capabilities of targeting
cancer stem cells as well as, vi) distant metastases and the
possibility, vii) to significantly impair the blood supply to tumor
beds (1–5).
Recently, OV have made their breakthrough with
respect to their implementation in daily clinical practice. Due to
the favourable results of the herpes simplex virus type 1
(HSV-1)-derived virotherapeutic vector Imlygic® in a
recent phase III clinical trial with patients exhibiting advanced
stage melanoma (6), its approval
has to be regarded as a hallmark in the clinical development of
virotherapy (7). Beyond that, the
unique properties of OV as self-amplifying agents that selectively
infect and kill cancer cells have been successfully exploited in
the treatment of patients suffering from multiple myeloma resulting
in an impressive case with long-term tumor remission following a
single shot, high-dose application of a marker gene-encoding
recombinant measles vaccine virus (MeV-NIS) (8).
Despite these promising results, OV still have to
face several limitations before taking full advantage of their
great potential to kill cancer cells. On the one hand, OV like any
other viruses are recognized as pathogens facing efficient
elimination by the host immune system (9). On the other hand, numerous cancer
cell types have been shown to be resistant toward virus-mediated
oncolysis due to features such as entry receptor down-regulation as
well as an insufficient extent of inactivation of anti-viral
signaling pathways in the tumor cells (10–12).
In this context, we have revealed that 50% of the cell lines being
represented in the well-characterized NCI-60 tumor cell panel
display unwanted mid and high grade resistance toward MeV-mediated
oncolysis (13).
In order to address the limitations and with further
respect to the advantage that there are no cross-resistances of OV
with other therapeutic regimens, researchers have been prompted by
the rationale of combining OV with other anticancer agents,
including histone deacetylase inhibitors (HDACi) (recently reviewed
in refs. 14,15). The impact of HDACi on cancer cells
was found to be highly diversified in terms of mechanisms of
action, eliciting induction of apoptosis, causing accumulation of
reactive oxygen species as well as inhibiting angiogenesis and
metastasis (16,17). Thereby, HDACi almost selectively
affect tumor tissues, while sparing healthy cells (17). As a consequence, a reciprocal
amplification of antitumoral effects was hypothesized for putative
HDACi plus OV combination regimens. In line with this,
epi-virotherapeutic strategies already have proven to effectively
boost tumor cell killing when compared with either monotherapeutic
efficiencies (14,18–23),
raising the novel term ‘epi-virotherapeutic approach’ (24).
Regarding the underlying molecular mechanisms of
such epi-virotherapy concepts, several steps of virus-mediated
oncolysis can be augmented by HDACi (Fig. 1). Among them, an HDACi-induced
impairment of a proper anti-viral immune response is discussed as a
potential synergistic mechanism as it is assumed to highly
facilitate both virus replication and virus spreading (14). Since HDAC activity is involved in
almost every step of the interferon (IFN) pathway, particularly in
the transcription of IFN-β, activation of signal transducers and
activators of transcription (STAT) proteins, IFN-stimulated gene
factor 3 (ISGF3) formation and ultimately expression of
IFN-stimulated genes (ISGs) (25–29),
HDACi like VPA and TSA were shown to blunt this cellular anti-viral
response (30,31). Moreover, in xenograft models it was
shown that T-cell and NK-cell mediated anti-viral immune responses
can be significantly impaired by concomitant treatment with
entinostat (MS-275) and VPA (21,23).
Since virus entry receptors are often epigenetically downregulated
in different tumor cells, HDACi were shown to restore coxsackie-
and adenovirus receptor (CAR) as well as the human reovirus
receptor junctional adhesion molecule-1 (JAM-1) on tumor cell
surfaces, thereby significantly increasing rates of primary
infections with OV (14,32–34).
Furthermore, several OV including vesicular stomatitis virus (VSV)
and MeV have been spotted to profit from an HDACi-related
enhancement of autophagy (35–37),
displaying a cellular catabolic process that serves: i) for the
degradation of cellular components being no longer in general use
and ii) for the maintenance of energy levels in times of starvation
and cellular stress (38). At
last, both the translocation of OV genomes to the cell nucleus via
microtubules and the expression of viral genes can be amplified by
concomitant HDACi treatment (15,30,39).
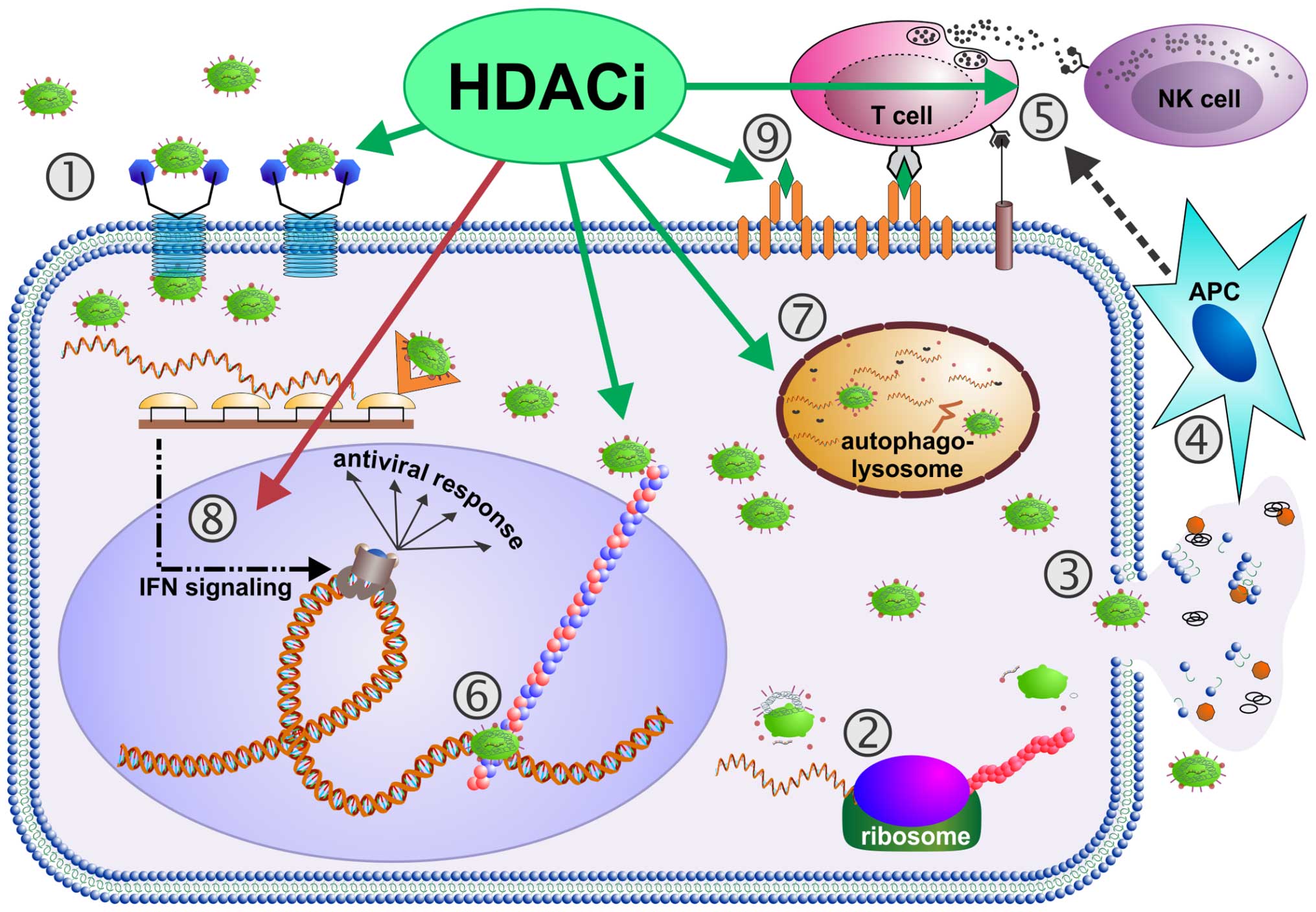 | Figure 1Virus-induced oncolysis augmented by
histone deacetylase inhibitors (HDACi). Following receptor binding
(1), oncolytic viruses (OV) infect tumor cells. Then, host cell
ribosomes are occupied with translation of viral RNA into viral
structural/functional proteins (2), resulting in generation of
numerous progeny viral particles per single host tumor cell. This
enormous replicative process ends up in complete exhaustion of host
tumor cells, inescapably leading to tumor cell disintegration,
i.e., viral oncolysis. Thereby, not only newly produced viral
particles are released (3), but also tumor-associated antigens
(TAAs) and damage-associated molecular pattern molecules (DAMPs),
which are detected by antigen-presenting cells (APCs) (4) (68). Concurrently, OV infection induces
production of pro-inflammatory, immune cell-attracting cytokines,
which also exert potent anti tumor activities (61). Further more, OV infections
upregulate MHC-I expression on tumor cells (69). Altogether, APC activation,
production of antitumor cytokines and upregulation of
antigen-presenting receptors are assumed to initiate a powerful T
cell-mediated antitumor immune response (5) (68,70).
Notably, all these steps of the viral oncolytic cycle (1–5) can
be influenced by histone deacetylase inhibitors (HDACi): first of
all, HDACi can upregulate expression of viral entry receptors on
tumor cells leading to increased rates of tumor cell infection by
OV (1) (14,32,33).
Further, trans location of OV genomes, i.e., post-entry shuttling
to the nucleus via microtubules (6) can be increased by HDACi (39). Next, expression of viral genes can
be augmented by HDACi in tumor cells (15,30,39).
HDACi-enhanced tumor cell autophagy (7) can result in increased OV-mediated
oncolysis and enhanced induction of tumor cell apoptosis (36). Remarkably, HDACi were also found to
be able to dampen antiviral IFN responses being characteristic for
OV-resistant tumor cells (8)
(11,12), thereby significantly facilitating
OV replication and spread (14,24,30,59).
In terms of boosting immune cell-mediated antitumor response, HDACi
can raise the levels of cytokines being supportive for the
functional development of tumor antigen-specific CD8+ T
cells (5) (21). Beyond that, HDACi can also inhibit
T- and NK cell-mediated antiviral responses, supporting an
unimpaired OV replication and propagation in tumor cells (21,23).
As HDACi can also cause upregulation of MHC-I molecules,
co-stimulatory receptors as well as TAAs (9) (17,71,72),
it is tempting to speculate that combined
epi(HDACi)-virotherapeutic approaches might further amplify the
magnitude of antitumor immune response. Taken together, OV-induced
oncolysis can be augmented by HDACi in many steps and on numerous
levels of the interaction between host tumor cells and OV. |
OV derived from the measles virus vaccine strain
Edmonston have been extensively investigated in numerous
preclinical and clinical studies and have been found to constitute
well suited anticancer agents (40–42).
During the decades-long use as a vaccine, its safety has been
comprehensively verified, whilst a reversion to wild-type MeV
followed by any potential outbreak of harmful MeV infections has
not been documented at any time in history (43).
We studied the antitumoral potential of combining
the oral HDACi resminostat with oncolytic MeV in terms of a future
epi-virotherapy of advanced pancreatic adenocarcinoma, a tumor
entity which is still tainted with a poor prognosis (44). Resminostat is a hydroxamic
acid-based HDACi, inhibiting selectively class I, IIb and IV HDAC
enzymes and has already been subject of different successful
clinical trials, underlining not only its efficiency, but also its
safety and tolerability (42,45,46).
We further report that our novel epi-virotherapeutic combination of
resminostat with oncolytic MeV resulted in an enhanced tumor cell
killing in human pancreatic cancer cells. Most interestingly and in
contrast to the hitherto prevailing opinion, this boostering effect
was found not to be related to a resminostat-induced impairment of
the anti-viral IFN response.
Materials and methods
Cell culture and non-viral compounds
Human pancreatic cancer cell lines AsPC-1, MIA
PaCa-2, and PANC-1 were purchased from the European Collection of
Authenticated Cell Cultures (ECACC); cell line BxPC-3 was obtained
from the American Type Culture Collection (ATCC); Vero cells were
obtained from the German Collection of Microorganisms and Cell
Cultures (DSMZ, Braunschweig, Germany). The cells were kept in a
humidified incubator at 37°C, containing 5% CO2 and
cultured in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich, Munich, Germany) supplemented with 10% fetal calf
serum. Resminostat was kindly provided by 4SC AG
(Planegg-Martinsried, Germany).
Propagation and titration of measles
vaccine virus
Construction of recombinant measles vectors MeV-GFP
(measles vector encoding for green-fluorescent protein as a marker
gene integrated into the viral genome) has been described (47). Virus stocks were prepared in Vero
cells. For this purpose, 1×107 Vero cells were seeded in
15-cm plates. The next day, cells were washed once with
phosphate-buffered saline (PBS; Sigma-Aldrich) and infected for 3 h
at a MOI of 0.03 in infection medium (Opti-MEM; Gibco; Grand
Island, NY, USA). Subsequently, medium was replaced with DMEM
containing 10% FBS. After an incubation period of 54 h, when most
of the cells were infected, medium was removed and attached Vero
cells were scraped into 1 ml Opti-MEM. Release of virus was
achieved by one freeze-thaw cycle. After centrifugation (1,900 × g,
for 15 min at 4°C), supernatants were stored at −80°C. Viral titers
were determined on Vero cells according to the method of Spearman
(48) and Kärber (49).
Infection of cells with measles vaccine
virus
Cells were seeded in 6- or 24-well plates the day
before virus infection. Then, culture medium was removed, cells
were washed with PBS and subsequently virus was diluted in Opti-MEM
at required multiplicities of infection (50) was added. After 3 h of incubation,
the inoculum was removed and DMEM supplemented with 10% FCS and, if
required additionally resminostat was added.
SRB assay
For SRB assay cells were seeded in 24-well plates
with cell numbers ranging from 2×104 for MIA PaCa-2 to
3×104 for PANC-1 and 4×104 per well for
AsPC-1 and BxPC-3. Experiments were stopped at required time-points
after treatment by removing medium, washing with PBS and
subsequently fixing with trichloroacetic acid (10%, 4°C for 30
min). Afterwards, fixed cells were washed four times with tap
water, dried and then stained with SRB solution (0.4% in 1% acetic
acid) for 10 min at room temperature. After washing with 1% acetic
acid and drying again bound SRB was dissolved in 10 mM Tris base
(pH 10.5) and the optical density was measured at a wavelength of
550 nm using a microplate reader (Tecan Genios Plus). The mean of
mock-treated controls was set to 100% and treated samples were
stated in percent of this control.
Real-time cell proliferation assay
Cells were seeded in 96-well plates (E-Plate 96,
Roche Applied Science, Mannheim, Germany) in different
concentrations according to their particular proliferation
characteristics (AsPC-1: 1×104 cells/well; MIA PaCa-2:
7.5×103 cells/well; PANC-1: 5×103
cells/well). Real-time dynamic cell proliferation was monitored in
30-min intervals during a 120-h observation period using the
xCELLigence RTCA SP system (Roche Applied Science). Cell index
values were calculated using the RTCA Software (1.0.0.0805). At 21
h after seeding, cells were infected with MeV-GFP diluted in
Opti-MEM and at 3 hpi resminostat was added in required
concentrations. All values were normalized to the beginning of the
treatment period (24 h after seeding) (51,52).
Viral growth curves
Cells were infected with MeV-GFP in 24-well plates.
At 3 hpi the inoculum was removed and cells were washed three times
with PBS. Then 0.5 ml medium or medium with resminostat were added.
At 3, 24, 48, 72 and 96 hpi supernatants were harvested and cells
were scraped off in 0.5 ml Opti-MEM. Cell lysis was performed by
one freeze-thaw cycle and subsequently virus titers were determined
by titrating samples on Vero cells following Spearman (48) and Kärber (49). Therefore, Vero cells were seeded in
a density of 1×104 cells per well in 96-well plates in
DMEM containing 5% FCS. Twenty-four hours later, cells were
infected with 1:10 dilution series generated from cell lysate and
supernatant samples. Tissue culture infective dose
(TDC50) was calculated by observing measles-induced
cytopathic effect with a fluorescence microscope and converted into
plaque forming units per ml (pfu/ml).
Immunoblotting
Protein samples were obtained by seeding, infecting
and treating pancreatic cancer cells in 6-well plates. At required
time-points, medium was removed, cells were washed with PBS and
afterwards harvested in lysis buffer (50 mM Tris-HCl pH 7.6, 150 mM
NaCl, 1% NP40). Cell lysis was performed by three freeze-thaw
cycles. Lysates were then cleared by centrifugation at 13,000 rpm
for 10 min. Protein concentrations in the supernatants were
determined by Bradford protein assay (Bio-Rad, Hercules, CA,
USA).
Each sample (70 μg) was mixed with 6-fold Roti Load
buffer and boiled at 95°C for 5 min. Proteins were separated on a
8% polyacrylamide gel and blotted on a polyvinylidene difluoride
(PVDF) membrane (Amersham Hybond P, GE Healthcare). Membranes were
blocked in 5% powdered milk in Tris-buffered saline containing
0.02% Tween-20 (TBS-T) and then incubated with primary antibodies
(anti-IFIT1: GTX103452; 1:1,000; GeneTex, Irvine, CA, USA;
anti-phospho-Stat1: 58D61; 1:1,000; Cell Signaling Technology,
Danvers, MA, USA; anti-Stat1: sc-591; 1:500; Santa Cruz
Biotechnology, Santa Cruz, CA, USA; anti-β-actin: A 4700; 1:6,000;
Sigma-Aldrich) overnight. After washing three times with TBS-T,
membranes were exposed to the secondary antibody (goat anti-rabbit
IgG; goat anti-mouse IgG; HRP-coupled; Abcam Ltd., Cambridgeshire,
UK). After washing three times with TBS-T again proteins were
detected by enhanced chemiluminescence western blotting detection
reagent (GE Healthcare).
qPCR
Cells were treated with resminostat, MeV-GFP or the
combination and subsequently RNA was isolated using the
NucleoSpin® RNA kit (Macherey-Nagel, Dueren, Germany)
according to the manufacturer's instructions.
Each RNA sample (500 ng) was mixed with 2 μl M-MLV
RT buffer (Promega, Madison, WI, USA), 1 μl RNase-inhibitor RNasin
Plus (Promega), 1 μl oligo-dT-Primer (0.5 μg/μl) (TIB MolBio,
Berlin, Germany), 0.5 μl dNTP mix (Roti-Mix PCR3, Carl Roth) and
added up to a total volume of 9.6 μl in RNAse-free water. Samples
were then incubated at 70°C for 2 min. After adding 0.4 μl
reverse-transcriptase M-MLV RT H(−) Point Mutant (Promega), samples
were incubated at 42°C for 60 min.
The cDNA samples diluted (1/40) with
tRNA-H2O; primers were used in a concentration of 500
nM. PCR was carried out in an iCycler (Bio-Rad) with iQ5 Multicolor
Real-time Detection system (Bio-Rad), using the following setup: 10
μl iQSYBR Green PCR Master Mix (Promega), 0.1 μl of each primer
(100 μM stock), 5.8 μl H2O and 4 μl cDNA (diluted 1/40).
The following primer pairs were used: zfp64 (splicing variants
1,3,4) forward, ACCTGCCCACGGAA AGTAAT; zfp64 (splicing variants
1,3,4) reverse, TATGGGG TTTGTCTCCCGTG; RPS18 (housekeeping gene)
forward, GAGGATGAGGTGGAACGTGT; RPS18 reverse, TCTTCAG
TCGCTCCAGGTCT. PCR was carried out with the following thermal
profile: 3 min at 95°C with subsequently 40 cycles for 15 sec at
95°C, 20 sec at 58°C, and 15 sec at 62°C. Heating up for 1 min at
95°C was followed by 1 min at 65°C and 81 cycles at 65°C cooling
down to 20°C. Target gene expression was evaluated via the
2−ΔCt method and normalized to the housekeeping gene
RPS18 and sub sequently graphed relative to the respective mock
sample for each time-point and expressed as ‘relative gene
expression’.
Statistical analysis
The influence of measles and resminostat on the
decadic logarithm of cell mass (in % of the mean of the cell line
control) was examined by performing a two-way analysis of variance
(ANOVA). Additionally, an interaction of measles and resminostat
was used in the ANOVA. Calculations were done by the JMP software
for windows. P-values <0.01 were considered to be statistically
significant. Graphs including error bars were imaged with GraphPad
Prism 4 for windows.
Results
Dose- and time-dependent effects of
resminostat and MeV on pancreatic cancer cells
Following our encouraging results recently obtained
in the epi-virotherapeutic treatment of human hepatoma cells
(24), we now examined the
antitumor potential of the epi-virotherapeutic approach consisting
of the oral HDACi resminostat and MeV for the therapy of pancreatic
ductal adenocarcinoma. For this purpose, we first determined the
antitumor effects elicited by each agent in monotreatment on a
panel of four human pancreatic cancer cell lines (AsPC-1, BxPC-3,
MIA PaCa-2, and PANC-1).
First, tumor cells were treated with resminostat at
different concentrations ranging from 0 to 10 μM. Experiments were
stopped at different time-points and tumor cell viabilities were
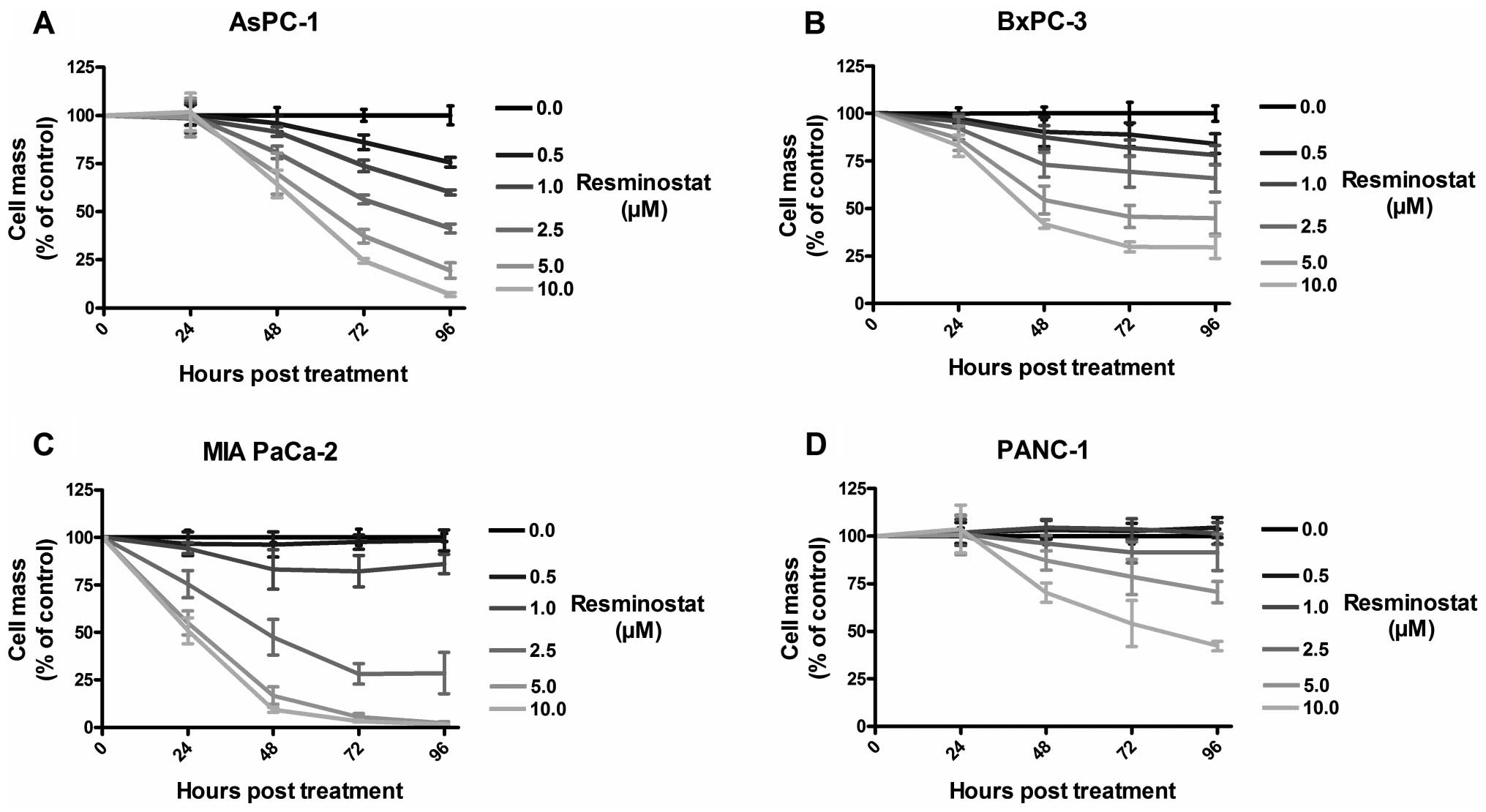
subsequently examined by sulforhodamine B (SRB) assays (Fig. 2).
As a result, treatment with resminostat displayed a
dose- and time-dependent cytoreductive effect in all investigated
human pancreatic cancer cell lines. In detail, MIA PaCa-2 cells
were shown to be most sensitive exhibiting a tumor cell mass
reduction of almost 100% at 5 μM resminostat at 96 h post-treatment
(hpt). In contrast, PANC-1 cells were shown to be most resistant
with a remaining tumor cell mass of ~70% with 5 μM resminostat at
96 hpt (Fig. 2D). For further
experiments, concentrations of resminostat were adjusted in a tumor
cell line-specific manner ensuring remaining tumor cell masses ~75%
at 96 hpt with resminostat only: 1 μM resminostat for tumor cell
lines MIA PaCa-2 and AsPC-1, 2.5 μM for BxPC-3 tumor cells, and 5
μM for PANC-1 tumor cells, respectively.
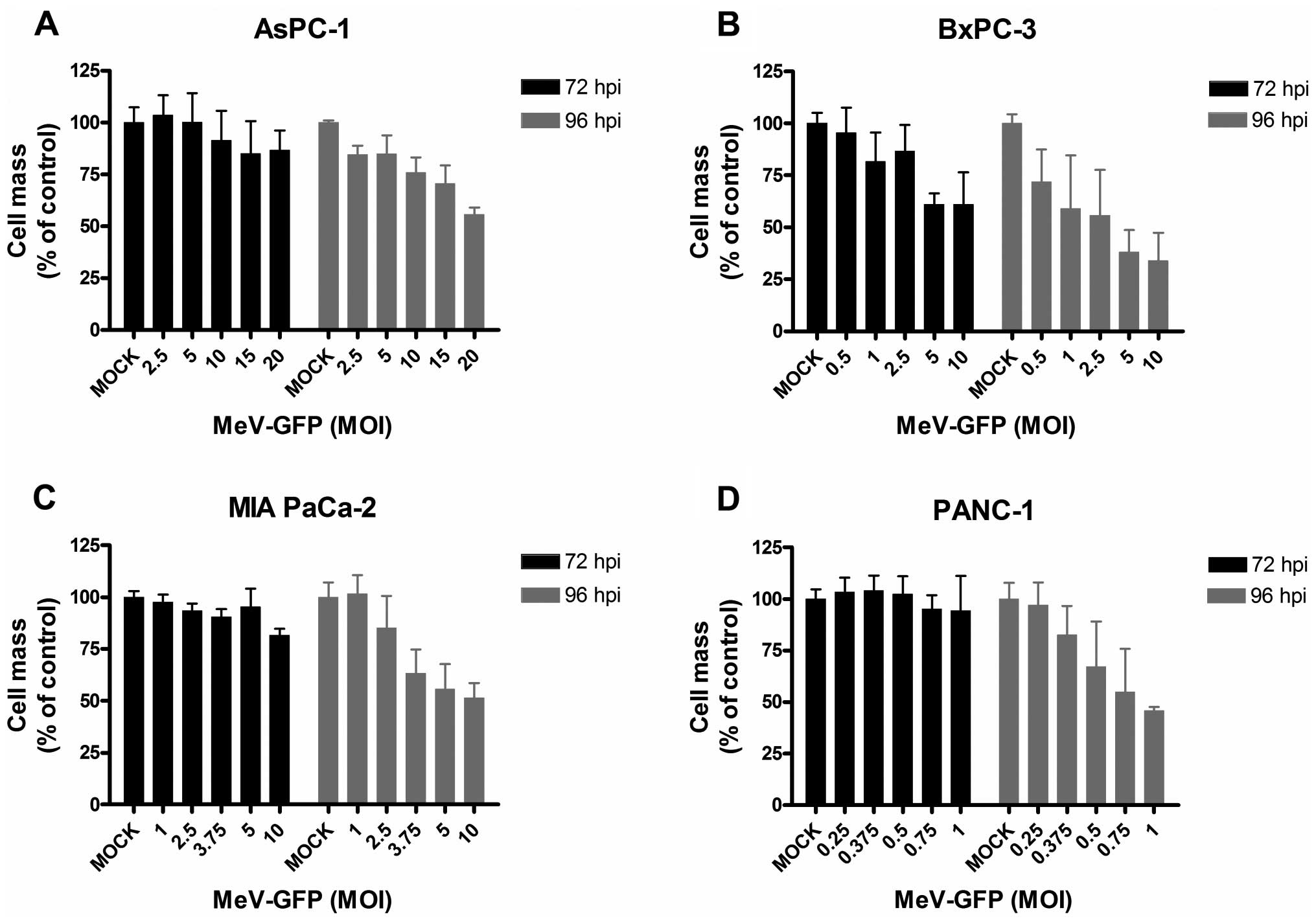
Next, all four human pancreatic cancer cell lines
were infected with a GFP marker gene-encoding oncolytic measles
vaccine virus vector (MeV-GFP) at different multiplicities of
infection (50) ranging from 0.25
to 20 (i.e., using a ratio of 0.25–20 virus particles per single
tumor cell to be infected) (Fig.
3). Again, tumor cell viabilities were determined by SRB
assays, now at both 72 and 96 h post infection (hpi). As a result,
pancreatic cancer cells displayed great differences in
susceptibility towards MeV-GFP-mediated oncolysis. The tumor cell
line being most sensitive to MeV-GFP-mediated oncolysis was PANC-1
(Fig. 3D), whereas AsPC-1 tumor
cells (Fig. 3A) were found to be
most resistant. Considering this, >50% of AsPC-1 cells survived
virus infections at a MOI of as high as 20. In contrast, a tumor
cell mass reduction of 50% was obtained by infecting PANC-1 cells
at a MOI of as low as 1. For further experiments, MOIs were
adjusted in a tumor cell line-specific manner resulting in
remaining tumor cell masses ~75% at 96 hpi: MOIs of 2.5 and 5 for
MIA PaCa-2 and AsPC-1; MOIs of 0.5 and 1 for BxPC-3, and MOIs of
0.25 and 0.375 for PANC-1.
Addressing the question whether there is
cross-resistance between resminostat and MeV-GFP, a remarkable
trend could be observed. Tumor cell lines, which had been
identified to be more resistant toward resminostat exhibited a
relatively strong sensitivity toward MeV-GFP-mediated oncolysis and
vice versa. The largest difference in tumor cell susceptibility was
obtained in experiments with the PANC-1 tumor cell line being most
resistant against resminostat treatment, but most sensitive towards
MeV-GFP-mediated oncolysis (Figs.
2D and 3D).
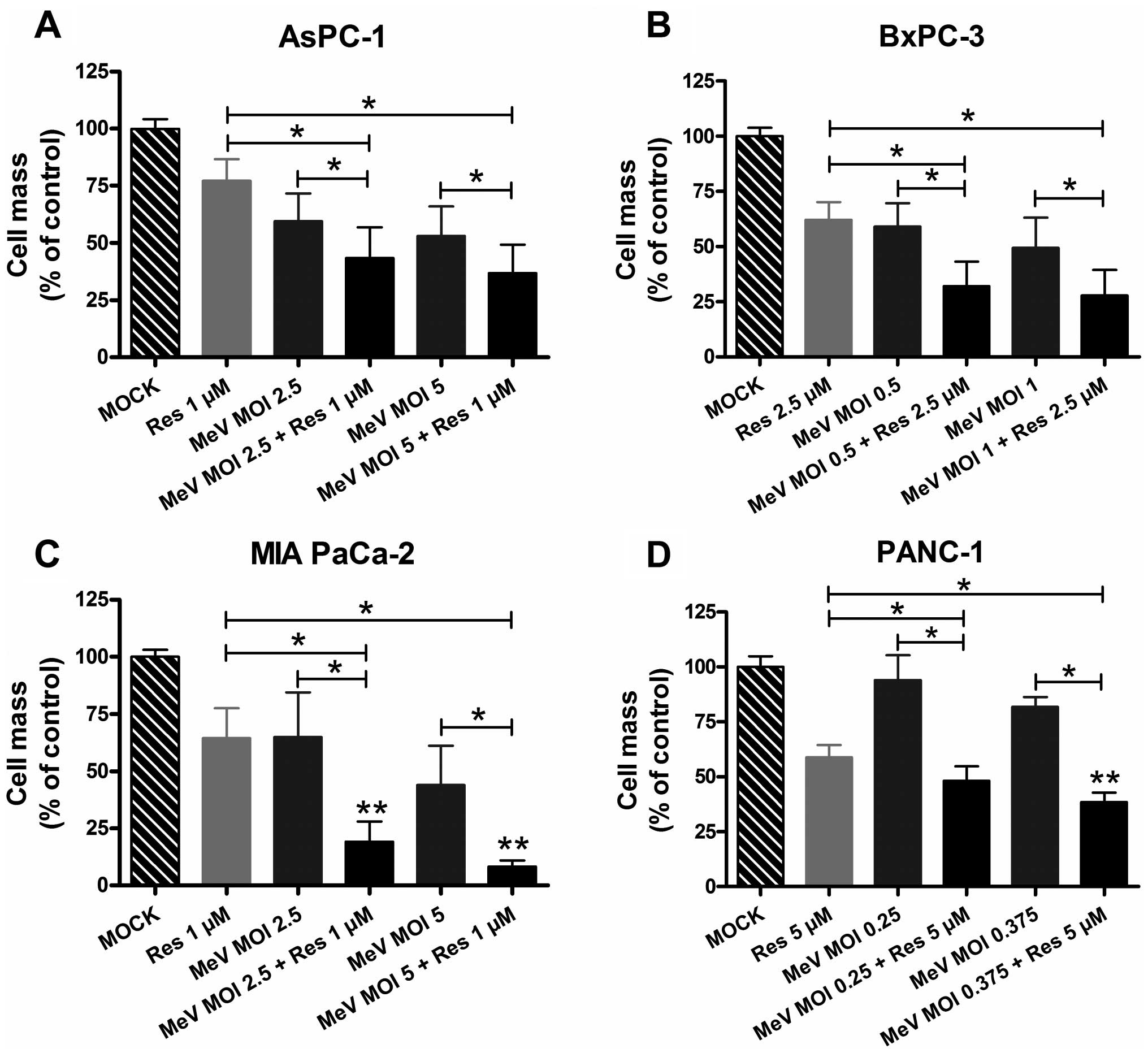
Enhanced tumor cell-killing by
epi-virotherapeutic co-treatment
To further determine whether resminostat and
oncolytic MeV operate beneficially when administered in
combination, pancreatic cancer cells were initially infected with
MeV-GFP; then, resminostat was added following the regular change
of infection culture medium at 3 hpi (Fig. 4). Tumor cell line adjusted MOIs of
MeV-GFP and concentrations of resminostat were used as determined
prior in the monotherapy settings.
As a result, supplementation of oncolytic MeV-GFP by
resminostat resulted in beneficial effects on rates of tumor cell
mass reduction in all tested pancreatic cancer cell lines. With
regard to MOIs of MeV-GFP and concentrations of resminostat
employed in later experiments, the reduction in tumor cell mass
could be amplified from 53 to 37% for AsPC-1 (MOI 5), from 60 to
32% for BxPC-3 (MOI 0.5), from 65 to 19% for MIA PaCa-2 (MOI 2.5),
and from 93 to 48% for PANC-1 (MOI 0.25) (Fig. 4). Considering that HDACi per
se induce a reduction in pancreatic cancer cell masses, the
most striking benefit could be obtained in the treatment of MIA
PaCa-2 cells, achieving a further 45% reduction in tumor cell mass
(Fig. 4C, comparison of bars 2 and
6). Whereas both agents in monotherapy reduced tumor cell viability
each by 35% in comparison to the mock control, the combination led
to a tumor cell mass reduction of >80% in comparison to the mock
control (Fig. 4C, comparison of
bars 1 and 6).
A statistical analysis was carried out to
investigate whether an interaction between MeV-GFP and resminostat
is verifiable that caused a more pronounced effect on tumor cell
mass reduction than expected from a simple additive effect. The
interaction term in the ANOVA on the logarithms of tumor cell mass
in % of control confirmed a clear significant synergistic antitumor
effect for the treatment of MIA PaCa-2 cells (Fig. 4C) as compared to the cytotoxic
effect that would be expected from an additive effect. With regard
to the other pancreatic cancer cell lines, synergistic tumor cell
killing could be significantly revealed in PANC-1 cells for only
one of the two combinations (MeV MOI 0.375 and 5 μM resminostat;
Fig. 4D); in contrast, no
synergistic effects were found for AsPC-1 and BxPC-3 tumor cells
(Fig. 4A and B), suggesting that
the epi-virotherapeutic approach does not elicit synergistic
effects in all pancreatic cancer cell entities, presumably as a
result of tumor cell specific features.
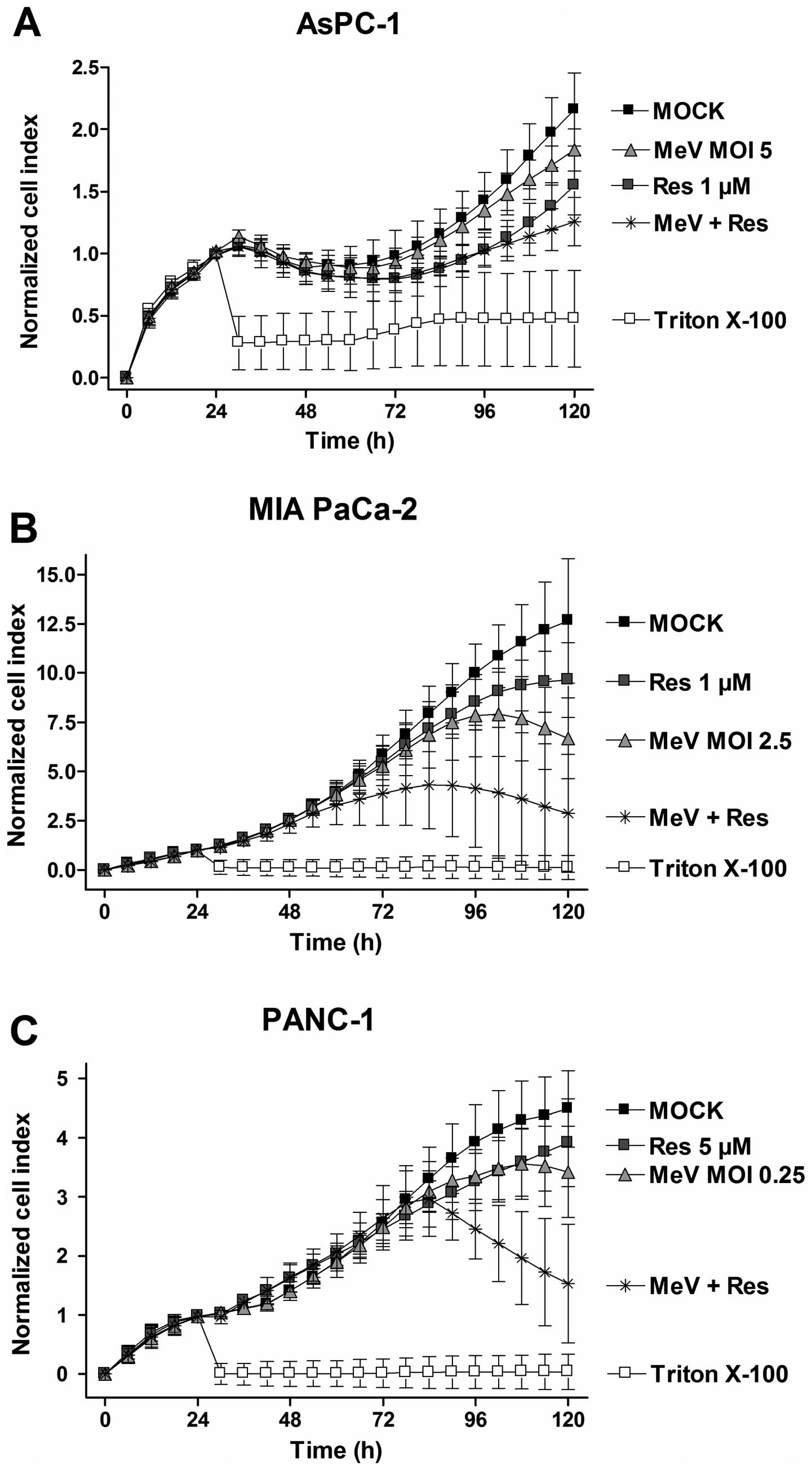
To confirm our results from the SRB viability assays
and to gain more precise information on the entire treatment time
course, real-time pancreatic cancer cell proliferation was
determined using the xCELLigence system (Fig. 5). The acquired data revealed that
our epi-virotherapeutic treatment elicited beneficial effects on
tumor cell viabilities in three out of the four tested pancreatic
cancer cell lines (Fig. 5). Taken
together, these findings underline that: i) our specific
epi-virotherapeutic treatment is much more valuable for MIA PaCa-2
and PANC-1 tumor cells than for AsPC-1 cells (BxPC-3 tumor cells
were not included in this specific testing) and ii) the mode of
synergistic tumor cell killing is first observed at 72 hpi in all
tested pancreatic cancer cell lines (going along with MeV-mediated
oncolysis phenomena taking place at this time-point).
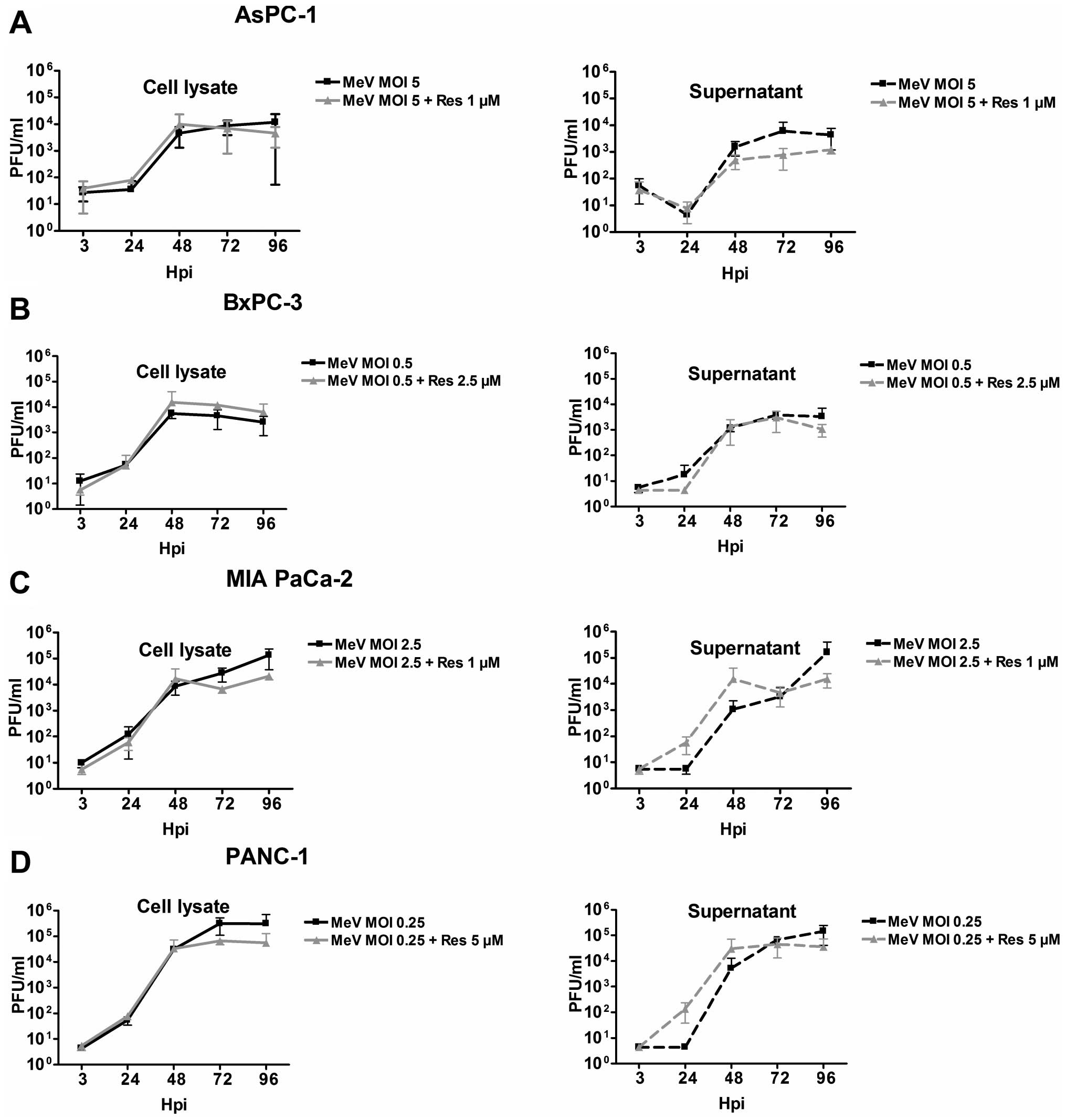
Absence of alterations in virus growth
kinetics under continuous treatment with resminostat
To examine whether the resminostat-related
enhancement of MeV-GFP-mediated oncolysis is based on an
accelerated virus replication and spread, virus growth kinetics
were analyzed for the four tested pancreatic cancer cell lines in
presence and absence of resminostat at five different time-points
(at 3, 24, 48, 72, and 96 hpi). For this purpose, tumor cells were
infected with indicated MOIs and treated continuously with
different concentrations of resminostat (Fig. 6). Comparing the virus growth curves
of MeV-GFP monotreatment with those of co-treatment with
resminostat, no relevant differences were obtained.
The highest virus titers were reached in PANC-1 and
MIA PaCa-2 tumor cells (Fig. 6C and
D), amounting to 105 pfu/ml whereas in AsPC-1 and
BxPC-3 titers of only 104 pfu/ml were detected (Fig. 6A and B). In all tumor cell lines
viral titers in supernatants were almost equal to those still bound
inside tumor cells. Thus, there was no clear correlation between
the susceptibility of the tumor cell lines toward measles vaccine
virus-mediated oncolysis and virus titers.
At later time-points (at 72 and 96 hpi) viral titers
were slightly lower in supernatants as well as in tumor cell
lysates in the presence of resminostat. This may be due to a
greater tumor cell mass reduction induced by the combination
treatment at later time-points, so that fewer tumor cells were
present in the cultures at these later time-points resulting in a
significantly lower cellular capacity for production of viral
progeny particles.
In conclusion, enhanced oncolytic effects by the
combined treatment of MeV-GFP and resminostat were not found to be
caused by an enhancement of viral replication by the HDACi.
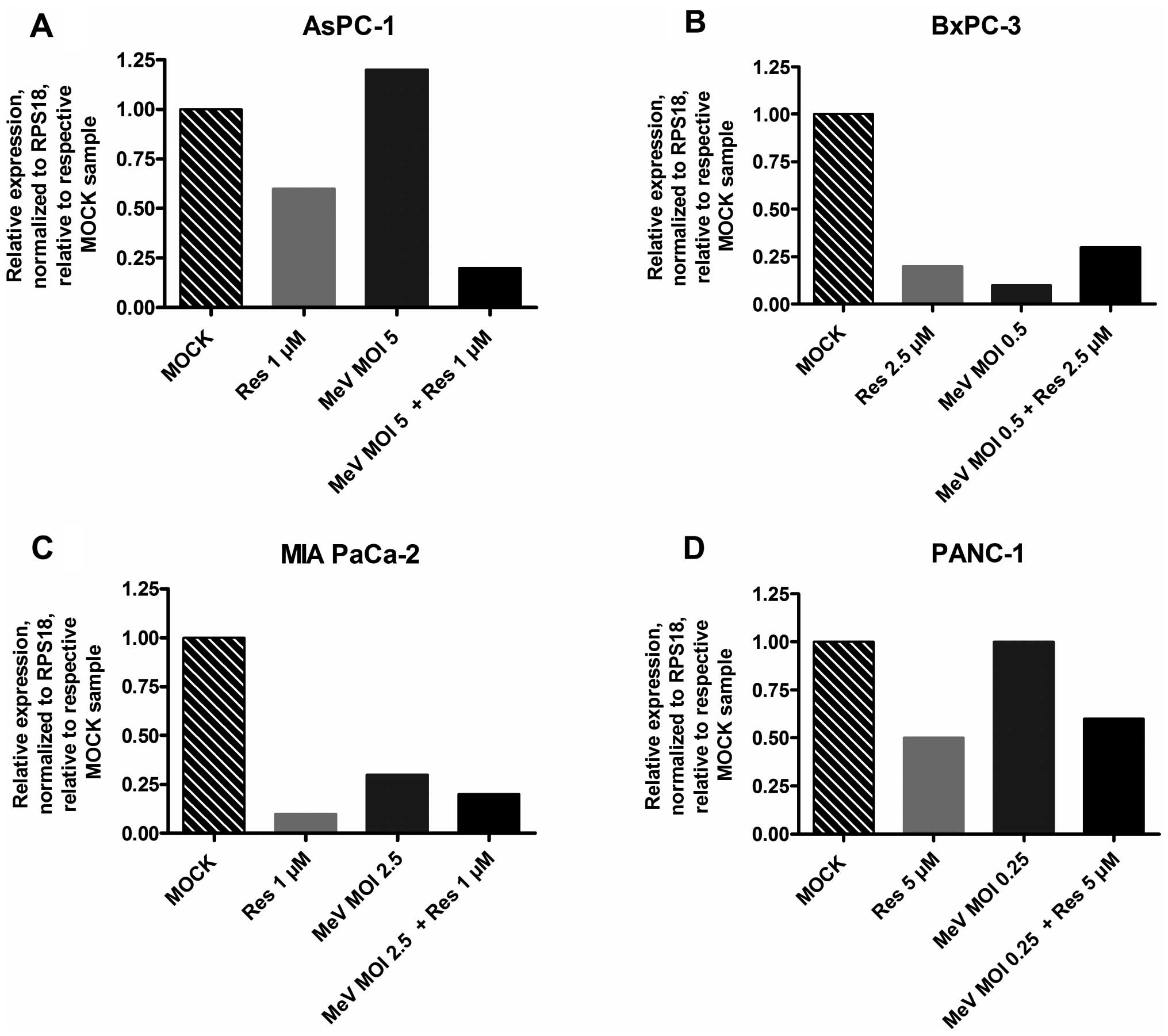
Expression of surrogate parameter zinc
finger protein 64 decreased in the course of resminostat treatment
of pancreatic cancer cells
Decrease in the expression of zinc finger protein 64
(zfp64) has been revealed to be a good surrogate parameter for the
pharmacological activity of resminostat. Therefore, we examined
mRNA expression of zfp64 after monotreatment with either
resminostat or MeV-GFP and after combination treatment (resminostat
plus MeV-GFP) using the same resminostat concentrations and MOIs as
in all prior experiments (Fig. 7).
In the presence of resminostat, zfp64 expression was found to be
down regulated in each tumor cell line as early as after five hours
of treatment initiation. Under epi-virotherapeutic co-treatment
with resminostat and MeV-GFP, we still observed a lower expression
of zfp64 as compared to the mock-treated control (with AsPC-1 tumor
cells showing an even lower expression under co-treatment as
compared to resminostat treatment alone; Fig. 7A). In contrast, different
expression patterns of zfp64 were found when tumor cells had only
been infected with MeV-GFP; in these cases, zfp64 was only
downregulated in BxPC-3 and MIA PaCa-2 tumor cells (Fig. 7B and C), but there was no
detectable regulation in AsPC-1 and PANC-1 tumor cells (Fig. 7A and D).
In conclusion, our experiments provide evidence that
the pharmacodynamic function of resminostat did not seem to be
impaired in MeV-GFP-infected pancreatic cancer cell lines.
Resminostat did not impair activation of
IFN signaling
In most studies investigating epi-virotherapeutic
approaches so far, damping of the anti-viral response by HDACi was
highlighted as a potential explanation for underlying synergistic
antitumoral effects of this combined treatment approach.
Accordingly, we were interested in the functionality of
IFN-signaling of pancreatic cancer cells in the presence and
absence of resminostat during infections with MeV-GFP.
Many tumor cells are known to exhibit defects in IFN
signaling and are therefore considered to be susceptible to
OV-mediated oncolysis (53). In
this context, we first examined whether pancreatic cancer cells
have the ability of initiating an IFN response in the course of an
infection by MeV. For this purpose, pancreatic cancer cells were
infected with MeV-GFP at standard MOIs (0.25 for PANC-1, 0.5 for
BxPC-3, 2.5 for MIA PaCa-2, and 5 for AsPC-1, respectively).
Furthermore, control samples were generated by stimulating cells
with IFN-β for 24 h. Samples were taken at 24, 48, 72, and 96 hpi.
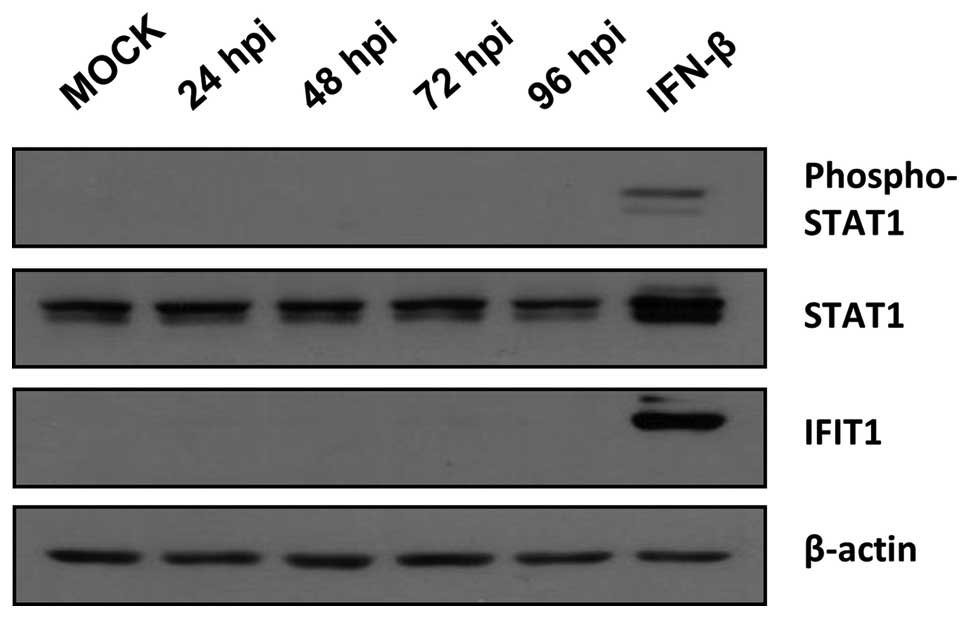
In AsPC-1, BxPC-3, and PANC-1 tumor cells phosphorylation of STAT1
and expression of IFN-induced protein with tetratricopeptide
repeats 1 (IFIT1) were observed at the latest at 72 hpi indicating
an unaltered activation of IFN signaling (data not shown). In
contrast, in MIA PaCa-2 cells neither phosphorylation of STAT1 nor
expression of IFIT1 was detected after MeV-GFP infection being
indicative of a severe defect in IFN signaling in this distinct
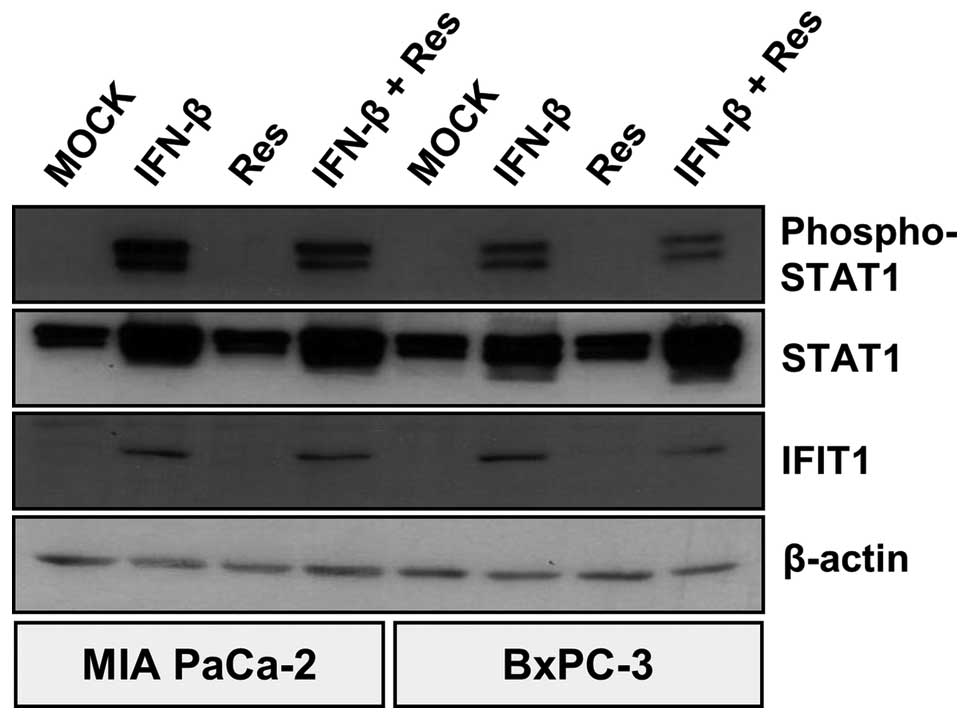
tumor cell line (Fig. 8).
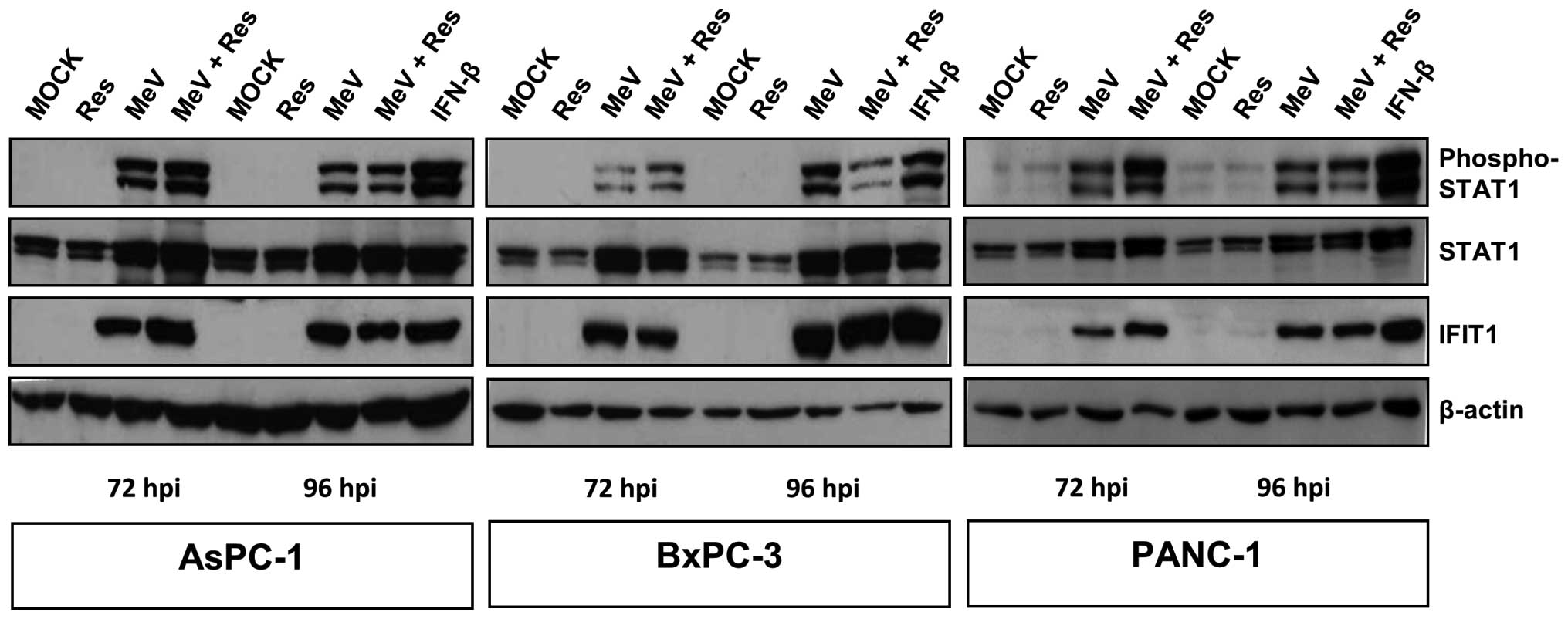
We then investigated the impact of resminostat on
MeV-GFP-induced activation of IFN signaling in AsPC-1, BxPC-3, and
PANC-1 cells. As a result, resminostat monotreatment did neither
result in phosphorylation of STAT1 nor in expression of IFIT1.
However, both MeV-GFP infection alone as well as the
epi-virotherapeutic combination resminostat plus MeV-GFP were found
to activate IFN signaling at both 72 and 96 hpi, indicated by
phosphorylation of STAT1 and expression of IFIT1 (Fig. 9). As MIA PaCa-2 cells did not
initiate IFN signaling after MeV-GFP infection, we stimulated these
tumor cells with IFN-β (please note: BxPC-3 cells were used as a
control in this experiment). Some of these were additionally
treated with resminostat. As a result, IFN-β treatment was found to
induce IFN signaling; but similar to all prior results, resminostat
was unable to inhibit phosphorylation of STAT1 and expression of
IFIT1 (Fig. 10). These results
clearly imply that resminostat does not impair the IFN response of
pancreatic cancer cells that had been initiated by infection with
MeV-GFP. Consequently, resminostat does not elicit synergistic
effects due to an impairment of the anti-viral response.
Discussion
Oncolytic viruses have recently made a major move
toward their full establishment in clinical practice by approval of
Imlygic® both by the American Food and Drug
Administration (FDA) and by the European Medicines Agency (EMA)
(7).
In our study, an epi-virotherapeutic approach was
pursued, augmenting oncolytic MeV with the oral HDACi resminostat.
Both agents already have been evaluated independently as well as
recently in combination for the treatment of different solid tumors
with encouraging results (1,14,24,45,54–57).
Here, we tested a series of four human pancreatic cancer cell
lines: i) for their sensitivity to both agents in monotreatment and
subsequently, ii) toward the effect of epi-virotherapeutic
co-treatment.
At the outset, monotreatment experiments revealed
that both agents, oncolytic MeV-GFP as well as resminostat, caused
dose- and time-dependent tumor cell killing in all tested human
pancreatic cancer cell lines. Strikingly, the cytotoxic effect of
resminostat on a specific cancer cell line could not be predicted
from the results obtained in OV cytotoxicity assays and vice versa.
This is most clearly visible when comparing the virotherapeutic
with the epigenetic results obtained with PANC-1 cells emphasizing
that there are no cross-resistances between OV and other cytotoxic
drugs such as HDACi.
Subsequently, cooperative effects were evaluated by
performing SRB cell viability assays and afterwards confirmed
utilizing the xCELLigence system. The results showed that the
epi-virotherapeutic approach elicited beneficial cytotoxic effects
in all four pancreatic cancer cell lines. Regarding MIA PaCa-2
tumor cells, considerable synergistic results were observed:
virus-mediated reduction in the tumor cell masses was found to be
improved in the presence of resminostat from 35 to 81% (at MOI 2.5)
as well as from 55 to 92% (at MOI 5) (Fig. 4). Similarly, epi-virotherapeutic
treatment of the other three cancer cell lines exhibited stronger
effects than obtained in monotreatment. In further experiments we
found that virus growth curves revealed no significant differences
in the presence or absence of resminostat, suggesting that
resminostat neither facilitated virus entry nor enhanced virus
replication.
With regard to studies that have already
investigated the therapeutic potential of epi-virotherapeutic
treatment of different tumor entities, the most frequently examined
and highlighted molecular mechanism of synergism is the ability of
HDACi to impair the anti-viral immune response of host tumor cells,
thereby facilitating virus replication and spread. Many underlying
mechanisms have been revealed, describing involvement of HDAC
activity in almost each step of IFN signaling. Virus infection
leads to phosphorylation of IFN-regulatory factors (IRFs), homo- or
heterodimerization and translocation into the nucleus where IFN-β
expression is induced (58).
Trichostatin A (TSA) was shown to prevent proper IRF-3 function,
thereby hindering cells to produce IFN-β (25). Downstream signaling of the IFN-β
receptor likewise requires HDAC activity, enabling proper receptor
activation, STAT dimerization, and IRF-9 function as well as the
formation of the IFN-stimulated gene factor-3 (ISGF3) (26–28).
Also, HDAC are involved in the expression of IFN-stimulated-genes
(ISGs) (29). Accordingly, HDAC
inhibitors were proven to impair the expression of ISGs when tumor
cells were coincidently infected with oncolytic viruses (30,36,59).
Due to these findings, the enhanced oncolytic effect was
retrospectively assigned to the interference with IFN
signaling.
In contrast to these observations, the present
epi-virotherapeutic approach did not modulate IFN signaling as
indicated by an unaltered phosphorylation of STAT1 and expression
of the ISG IFIT1 in any of the tested pancreatic cancer cell lines.
Moreover, no obvious alteration in virus growth kinetics could be
observed. For these reasons, our experiments do not support the
prevailing opinion of HDACi damping the IFN-response thus enhancing
OV-mediated oncolysis. In respect of implementing our
epi-virotherapeutic approach into clinical practice, it is
potentially not preferable that type I IFN production is impaired.
Since especially IFN-α and IFN-β are essential cytokines that
attract and prime cytotoxic and T helper cells by causing
expression of important receptors on cancer cells (such as MHC I),
type I IFN secretion from tumor sites might amplify an antitumor
immune response (60,61).
Other studies having examined the potential of HDACi
to enhance different virotherapeutics obtained similar findings.
After having infected different infection-resistant cancer cells
with vaccinia virus (VV) that had retained their B18R gene,
functioning as an IFN antagonist, the HDACi TSA was still capable
of amplifying OV-mediated oncolysis, suggesting that its antitumor
effect was not based on an immunosuppressive function (19). In our study, MIA PaCa-2 was the
only pancreatic cancer cell line which did not exhibit an
activation of the IFN signaling pathway after MeV infection.
Despite this lack of establishing a proper anti-viral state, it was
not the most susceptible cell line to MeV-mediated oncolysis and
more noteworthy, epi-virotherapeutic treatment showed the most
pronounced effect in this cell line, stressing that HDACi seem to
enhance virus-mediated oncolysis by eliciting other effects than
damping the IFN response. This raises the question which additional
mechanisms could explain the enhancement of virus-mediated cell
death by epi-virotherapeutic co-treatment.
Explanations, amongst others, were provided by Liu
et al (31). Using an
epi-virotherapeutic approach consisting of oncolytic
herpes-simplex-virus (HSV) and TSA in a panel of tumor and normal
quiescent cells, they obtained beneficial cytoreductive effects
compared to monotreatment. These effects could be attributed
neither to the dosing schedule nor to enhanced infectivity or virus
replication. The authors rather ascribed the results to a decrease
in expression of cyclin D1, mediating cell cycle arrest, and VEGF,
reinforcing the hypothesis of vascular shutdown induced by OV
(5).
Beyond the above, further replication-independent
mechanisms have been illustrated, highlighting the impact of HDACi
on cell signaling. Thus, HDACi cause hyperacetylation of NF-κB,
thereby increasing its nuclear retention and DNA binding capacity.
Due to its promotion of HSV gene expression, this HDACi-mediated
effect elicited synergistic tumor killing in oral squamous cell
carcinoma (SCC) cells (62).
Furthermore, combined treatment was shown to increase the
expression of p21 which mediates cell cycle arrest, consequently
slowing down tumor progression and resulting in the induction of
tumor cell apoptosis.
Recently, Shulak et al found a mechanism
explaining NF-κB activity accompanied by an enhanced OV-mediated
oncolysis. They pointed out that hyperacetylation and nuclear
retention of NF-κB induced the expression of several
autophagy-related genes. They argued that the induction of
autophagy led to an impairment of IFN signaling but also to
vesicular stomatitis virus (VSV)-mediated apoptosis in prostate
cancer cells (36). Autophagy is a
process that is per se frequently enhanced in tumor cells
since it serves as a stress response to oxidative stress, lack of
nutrients, and hypoxia as it is commonly present in the
microenvironment of solid tumors (63). Interestingly, pancreatic cancer
cells even require this catabolic process in order to prevent
accumulation of ROS, thereby contributing to tumor growth as well
as establishing the basis for drug resistance (64,65).
Despite these pro-survival aspects, some viruses are notably
capable of exploiting the autophagic machinery for the purpose of
efficient replication (38).
Attenuated MeV derived from the Edmonston strain actually
induce and require autophagy for efficient replication (37). Since hydroxamic acid based HDACi
equally increase autophagic activity (66), it is tempting to speculate that the
effect elicited by resminostat in combination with oncolytic MeV is
caused by an enhanced self-digestion and subsequently enhanced
tumor cell death.
Physiologically, cell signaling often requires
protein modifications such as phosphorylation or acetylation but
beyond targeting cell proteins, even pathogenic proteins can serve
as substrates for those modifications, resulting either in enhanced
or impaired activity. In this context, it was revealed that a
portion of the NS-1 protein, representing the major pathogenic and
most important protein for replication of the rat parvovirus H-1PV,
gets acetylated during virus infection (67). Noteworthy, treatment with VPA
caused hyperacetylation of NS-1 resulting in an accumulation of ROS
and an enhanced transcriptional activity. Ultimately, DNA damage in
cancer cells was observed consequently inducing apoptosis. Those
findings were confirmed later in vivo, resulting in complete
disappearance of implanted tumors in mice that had undergone
co-treatment with H-1PV and VPA (18). Likewise, HDACi-related
hyperacetylation of microtubules accelerated nuclear translocation
of oncolytic HSV-genomes, thereby enhancing the antitumor effect in
glioma stem-like cells (39).
In conclusion, our results provide evidence that the
epi-virotherapeutic combination of oncolytic MeV and the HDACi
resminostat constitutes a beneficial option in the treatment of
advanced pancreatic ductal adenocarcinoma. We revealed an
augmentation of MeV-mediated oncolysis by resminostat. Treatment of
MIA PaCa-2 cells resulted even in a synergistic enhancement of the
tumor-killing potential when compared to the monotherapies.
Molecular mechanisms underlying the synergistic effects and the
potential of our epi-virotherapeutic approach in vivo have
to be elucidated in animal models in the future.
Acknowledgements
For the statistical analysis the methodical advice
from the Institute of Clinical Epidemiology and Applied Biometrics
of the University Hospital Tuebingen were utilized. We wish to
thank Professor Martin Eichner for his excellent support, and we
further wish to thank Hannes Schramm (University Hospital
Tuebingen) who helped us with imaging and the augmentation of
oncolytic viruses by histone deacetylase inhibitors. Dr Tanja Wulff
is an employee of 4SC AG. T.P.E. was funded by the intramural
IZKF-scholarship of the Faculty of Medicine, University of
Tuebingen. S.B. was supported by the German Childhood Cancer
Foundation (DKS) and M.B. by grants of the European Foundation for
Alcohol Research (ERAB). We further acknowledge the support by the
German Research Foundation (DFG) and the Open Access Publishing
Fund of the Eberhard Karls University of Tuebingen.
References
|
1
|
Russell SJ, Peng KW and Bell JC: Oncolytic
virotherapy. Nat Biotechnol. 30:658–670. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hammill AM, Conner J and Cripe TP:
Oncolytic virotherapy reaches adolescence. Pediatr Blood Cancer.
55:1253–1263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bourke MG, Salwa S, Harrington KJ,
Kucharczyk MJ, Forde PF, de Kruijf M, Soden D, Tangney M, Collins
JK and O'Sullivan GC: The emerging role of viruses in the treatment
of solid tumours. Cancer Treat Rev. 37:618–632. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith TT, Roth JC, Friedman GK and
Gillespie GY: Oncolytic viral therapy: Targeting cancer stem cells.
Oncolytic Virother. 2014:21–33. 2014.PubMed/NCBI
|
|
5
|
Breitbach CJ, De Silva NS, Falls TJ, Aladl
U, Evgin L, Paterson J, Sun YY, Roy DG, Rintoul JL, Daneshmand M,
et al: Targeting tumor vasculature with an oncolytic virus. Mol
Ther. 19:886–894. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andtbacka RH, Kaufman HL, Collichio F,
Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I,
Agarwala SS, et al: Talimogene laherparepvec improves durable
response rate in patients with advanced melanoma. J Clin Oncol.
33:2780–2788. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ledford H: Cancer-fighting viruses win
approval. Nature. 526:622–623. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Russell SJ, Federspiel MJ, Peng KW, Tong
C, Dingli D, Morice WG, Lowe V, O'Connor MK, Kyle RA, Leung N, et
al: Remission of disseminated cancer after systemic oncolytic
virotherapy. Mayo Clin Proc. 89:926–933. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiocca EA: The host response to cancer
virotherapy. Curr Opin Mol Ther. 10:38–45. 2008.PubMed/NCBI
|
|
10
|
Kim M, Zinn KR, Barnett BG, Sumerel LA,
Krasnykh V, Curiel DT and Douglas JT: The therapeutic efficacy of
adenoviral vectors for cancer gene therapy is limited by a low
level of primary adenovirus receptors on tumour cells. Eur J
Cancer. 38:1917–1926. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berchtold S, Lampe J, Weiland T, Smirnow
I, Schleicher S, Handgretinger R, Kopp HG, Reiser J, Stubenrauch F,
Mayer N, et al: Innate immune defense defines susceptibility of
sarcoma cells to measles vaccine virus-based oncolysis. J Virol.
87:3484–3501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Escobar-Zarate D, Liu YP, Suksanpaisan L,
Russell SJ and Peng KW: Overcoming cancer cell resistance to VSV
oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 20:582–589.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Noll M, Berchtold S, Lampe J, Malek NP,
Bitzer M and Lauer UM: Primary resistance phenomena to oncolytic
measles vaccine viruses. Int J Oncol. 43:103–112. 2013.PubMed/NCBI
|
|
14
|
Nguyen TL, Wilson MG and Hiscott J:
Oncolytic viruses and histone deacetylase inhibitors - a
multi-pronged strategy to target tumor cells. Cytokine Growth
Factor Rev. 21:153–159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakashima H, Nguyen T and Chiocca EA:
Combining HDAC inhibitors with oncolytic virotherapy for cancer
therapy. Dovepress. 2015:183–191. 2015.
|
|
16
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: Emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar
|
|
17
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li J, Bonifati S, Hristov G, Marttila T,
Valmary-Degano S, Stanzel S, Schnölzer M, Mougin C, Aprahamian M,
Grekova SP, et al: Synergistic combination of valproic acid and
oncolytic parvovirus H-1PV as a potential therapy against cervical
and pancreatic carcinomas. EMBO Mol Med. 5:1537–1555. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
MacTavish H, Diallo JS, Huang B, Stanford
M, Le Boeuf F, De Silva N, Cox J, Simmons JG, Guimond T, Falls T,
et al: Enhancement of vaccinia virus based oncolysis with histone
deacetylase inhibitors. PLoS One. 5:e144622010. View Article : Google Scholar
|
|
20
|
White MC and Frampton AR Jr: The histone
deacetylase inhibitor valproic acid enhances equine herpesvirus
type 1 (EHV-1)-mediated oncolysis of human glioma cells. Cancer
Gene Ther. 20:88–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bridle BW, Chen L, Lemay CG, Diallo JS,
Pol J, Nguyen A, Capretta A, He R, Bramson JL, Bell JC, et al: HDAC
inhibition suppresses primary immune responses, enhances secondary
immune responses, and abrogates autoimmunity during tumor
immunotherapy. Mol Ther. 21:887–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cody JJ, Markert JM and Hurst DR: Histone
deacetylase inhibitors improve the replication of oncolytic herpes
simplex virus in breast cancer cells. PLoS One. 9:e929192014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alvarez-Breckenridge CA, Yu J, Price R,
Wei M, Wang Y, Nowicki MO, Ha YP, Bergin S, Hwang C, Fernandez SA,
et al: The histone deacetylase inhibitor valproic acid lessens NK
cell action against oncolytic virus-infected glioblastoma cells by
inhibition of STAT5/T-BET signaling and generation of gamma
interferon. J Virol. 86:4566–4577. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ruf B, Berchtold S, Venturelli S, Burkard
M, Smirnow I, Prenzel T, Henning SW and Lauer UM: Combination of
the oral histone deacetylase inhibitor resminostat with oncolytic
measles vaccine virus as a new option for epi-virotherapeutic
treatment of hepatocellular carcinoma. Mol Ther Oncolytics.
2:150192015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nusinzon I and Horvath CM: Positive and
negative regulation of the innate antiviral response and beta
interferon gene expression by deacetylation. Mol Cell Biol.
26:3106–3113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Génin P, Morin P and Civas A: Impairment
of interferon-induced IRF-7 gene expression due to inhibition of
ISGF3 formation by trichostatin A. J Virol. 77:7113–7119. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang X, Gao JS, Guan YJ, McLane KE, Yuan
ZL, Ramratnam B and Chin YE: Acetylation-dependent signal
transduction for type I interferon receptor. Cell. 131:93–105.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuan ZL, Guan YJ, Chatterjee D and Chin
YE: Stat3 dimerization regulated by reversible acetylation of a
single lysine residue. Science. 307:269–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang HM, Paulson M, Holko M, Rice CM,
Williams BR, Marié I and Levy DE: Induction of
interferon-stimulated gene expression and antiviral responses
require protein deacetylase activity. Proc Natl Acad Sci USA.
101:9578–9583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Otsuki A, Patel A, Kasai K, Suzuki M,
Kurozumi K, Chiocca EA and Saeki Y: Histone deacetylase inhibitors
augment antitumor efficacy of herpes-based oncolytic viruses. Mol
Ther. 16:1546–1555. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu TC, Castelo-Branco P, Rabkin SD and
Martuza RL: Trichostatin A and oncolytic HSV combination therapy
shows enhanced antitumoral and antiangiogenic effects. Mol Ther.
16:1041–1047. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wunder T, Schmid K, Wicklein D, Groitl P,
Dobner T, Lange T, Anders M and Schumacher U: Expression of the
coxsackie adenovirus receptor in neuroendocrine lung cancers and
its implications for oncolytic adenoviral infection. Cancer Gene
Ther. 20:25–32. 2013. View Article : Google Scholar
|
|
33
|
Kasman L, Onicescu G and Voelkel-Johnson
C: Histone deacetylase inhibitors restore cell surface expression
of the coxsackie adenovirus receptor and enhance CMV promoter
activity in castration-resistant prostate cancer cells. Prostate
Cancer. 2012:1371632012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stiff A, Caserta E, Sborov DW, Nuovo GJ,
Mo X, Schlotter SY, Canella A, Smith E, Badway J, Old M, et al:
Histone deacetylase inhibitors enhance the therapeutic potential of
reovirus in multiple myeloma. Mol Cancer Ther. 15:830–841. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meng S, Xu J, Wu Y and Ding C: Targeting
autophagy to enhance oncolytic virus-based cancer therapy. Expert
Opin Biol Ther. 13:863–873. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shulak L, Beljanski V, Chiang C, Dutta SM,
Van Grevenynghe J, Belgnaoui SM, Nguyên TL, Di Lenardo T, Semmes
OJ, Lin R, et al: Histone deacetylase inhibitors potentiate
vesicular stomatitis virus oncolysis in prostate cancer cells by
modulating NF-κB-dependent autophagy. J Virol. 88:2927–2940. 2014.
View Article : Google Scholar :
|
|
37
|
Richetta C, Grégoire IP, Verlhac P, Azocar
O, Baguet J, Flacher M, Tangy F, Rabourdin-Combe C and Faure M:
Sustained autophagy contributes to measles virus infectivity. PLoS
Pathog. 9:e10035992013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dreux M and Chisari FV: Viruses and the
autophagy machinery. Cell Cycle. 9:1295–1307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakashima H, Kaufmann JK, Wang PY, Nguyen
T, Speranza MC, Kasai K, Okemoto K, Otsuki A, Nakano I, Fernandez
S, et al: Histone deacetylase 6 inhibition enhances oncolytic viral
replication in glioma. J Clin Invest. 125:4269–4280. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guillerme JB, Gregoire M, Tangy F and
Fonteneau JF: Antitumor virotherapy by attenuated measles virus
(MV). Biology (Basel). 2:587–602. 2013.
|
|
41
|
Hutzen BRC and Studebaker AW: Advances in
the design and development of oncolytic measles viruses. Dovepress.
4:109–118. 2015.
|
|
42
|
Bitzer M, Horger M, Giannini EG, Ganten
TM, Wörns MA, Siveke JT, Dollinger mM, Gerken G, Scheulen ME, Wege
H, et al: Resminostat plus sorafenib as second-line therapy of
advanced hepatocellular carcinoma - The SHELTER Study. J Hepatol.
65:280–288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Russell SJ and Peng KW: Measles virus for
cancer therapy. Curr Top Microbiol Immunol. 330:213–241.
2009.PubMed/NCBI
|
|
44
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brunetto AT, Ang JE, Lal R, Olmos D,
Molife LR, Kristeleit R, Parker A, Casamayor I, Olaleye M, Mais A,
et al: First-inhuman, pharmacokinetic and pharmacodynamic phase I
study of Resminostat, an oral histone deacetylase inhibitor, in
patients with advanced solid tumors. Clin Cancer Res. 19:5494–5504.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kitazono S, Fujiwara Y, Nakamichi S,
Mizugaki H, Nokihara H, Yamamoto N, Yamada Y, Inukai E, Nakamura O
and Tamura T: A phase I study of resminostat in Japanese patients
with advanced solid tumors. Cancer Chemother Pharmacol.
75:1155–1161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Weiland T, Lampe J, Essmann F, Venturelli
S, Berger A, Bossow S, Berchtold S, Schulze-Osthoff K, Lauer UM and
Bitzer M: Enhanced killing of therapy-induced senescent tumor cells
by oncolytic measles vaccine viruses. Int J Cancer. 134:235–243.
2014. View Article : Google Scholar
|
|
48
|
Spearman C: The method of ‘right and wrong
cases’ (‘constant stimuli’) without gauss's formulae. Br J Psychol.
2:227–242. 1908.
|
|
49
|
Kärber G: Beitrag zur kollektiven
behandlung pharmakologischer reihenversuche. Naunyn Schmiedebergs
Arch Exp Pathol Pharmakol. 162:480–483. 1931.(In German).
View Article : Google Scholar
|
|
50
|
Walewski J, Pszkiewicz-Kozik E, Warzewska
A, Borsaru G, Moicean A, Hellmann A, Mayer J, Hauns B, Mais A,
Henning SW, et al: Final results of the phase II SAPHIRE trial of
resminostat (4SC-201) in patients with relapsed/refractory Hodgkin
lymphoma. Presented at 53rd ASH Annual Meeting and Exposition.
(abstract 2675); 2011, http://www.4sc.de/wp-content/uploads/SAPHIRE-Poster-ASH-San-Diego-2011.pdf.
|
|
51
|
Abassi YA, Xi B, Zhang W, Ye P, Kirstein
SL, Gaylord MR, Feinstein SC, Wang X and Xu X: Kinetic cell-based
morphological screening: Prediction of mechanism of compound action
and off-target effects. Chem Biol. 16:712–723. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Weiland T, Berger A, Essmann F, Lauer UM,
Bitzer M and Venturelli S: Kinetic tracking of therapy-induced
senescence using the real-time cell analyzer single plate system.
Assay Drug Dev Technol. 10:289–295. 2012. View Article : Google Scholar
|
|
53
|
Stojdl DF, Lichty BD, tenOever BR,
Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L,
Atkins H, et al: VSV strains with defects in their ability to
shutdown innate immunity are potent systemic anti-cancer agents.
Cancer Cell. 4:263–275. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Abend A and Kehat I: Histone deacetylases
as therapeutic targets - from cancer to cardiac disease. Pharmacol
Ther. 147:55–62. 2015. View Article : Google Scholar
|
|
55
|
Mottamal M, Zheng S, Huang TL and Wang G:
Histone deacetylase inhibitors in clinical studies as templates for
new anticancer agents. Molecules. 20:3898–3941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Feng W, Zhang B, Cai D and Zou X:
Therapeutic potential of histone deacetylase inhibitors in
pancreatic cancer. Cancer Lett. 347:183–190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xu C, Li H, Su C and Li Z: Viral therapy
for pancreatic cancer: Tackle the bad guys with poison. Cancer
Lett. 333:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Honda K, Takaoka A and Taniguchi T: Type I
interferon [corrected] gene induction by the interferon regulatory
factor family of transcription factors. Immunity. 25:349–360. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nguyên TL, Abdelbary H, Arguello M,
Breitbach C, Leveille S, Diallo JS, Yasmeen A, Bismar TA, Kirn D,
Falls T, et al: Chemical targeting of the innate antiviral response
by histone deacetylase inhibitors renders refractory cancers
sensitive to viral oncolysis. Proc Natl Acad Sci USA.
105:14981–14986. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fuertes MB, Kacha AK, Kline J, Woo SR,
Kranz DM, Murphy KM and Gajewski TF: Host type I IFN signals are
required for antitumor CD8+ T cell responses through
CD8{alpha}+ dendritic cells. J Exp Med. 208:2005–2016.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Prestwich RJ, Errington F, Diaz RM, Pandha
HS, Harrington KJ, Melcher AA and Vile RG: The case of oncolytic
viruses versus the immune system: Waiting on the judgment of
Solomon. Hum Gene Ther. 20:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Katsura T, Iwai S, Ota Y, Shimizu H, Ikuta
K and Yura Y: The effects of trichostatin A on the oncolytic
ability of herpes simplex virus for oral squamous cell carcinoma
cells. Cancer Gene Ther. 16:237–245. 2009.
|
|
63
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: Implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar
|
|
64
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gammoh N, Lam D, Puente C, Ganley I, Marks
PA and Jiang X: Role of autophagy in histone deacetylase
inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl
Acad Sci USA. 109:6561–6565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hristov G, Krämer M, Li J, El-Andaloussi
N, Mora R, Daeffler L, Zentgraf H, Rommelaere J and Marchini A:
Through its nonstructural protein NS1, parvovirus H-1 induces
apoptosis via accumulation of reactive oxygen species. J Virol.
84:5909–5922. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kaufman HL, Kohlhapp FJ and Zloza A:
Oncolytic viruses: A new class of immunotherapy drugs. Nat Rev Drug
Discov. 14:642–662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gujar S, Dielschneider R, Clements D,
Helson E, Shmulevitz M, Marcato P, Pan D, Pan L, Ahn D-G, Alawadhi
A, et al: Multifaceted therapeutic targeting of ovarian peritoneal
carcinomatosis through virus-induced immunomodulation. Mol Ther.
21:338–347. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Moehler MH, Zeidler M, Wilsberg V,
Cornelis JJ, Woelfel T, Rommelaere J, Galle PR and Heike M:
Parvovirus H-1-induced tumor cell death enhances human immune
response in vitro via increased phagocytosis, maturation, and
cross-presentation by dendritic cells. Hum Gene Ther. 16:996–1005.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kroesen M, Gielen P, Brok IC, Armandari I,
Hoogerbrugge PM and Adema GJ: HDAC inhibitors and immunotherapy; a
double edged sword? Oncotarget. 5:6558–6572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Setiadi AF, Omilusik K, David MD, Seipp
RP, Hartikainen J, Gopaul R, Choi KB and Jefferies WA: Epigenetic
enhancement of antigen processing and presentation promotes immune
recognition of tumors. Cancer Res. 68:9601–9607. 2008. View Article : Google Scholar : PubMed/NCBI
|