Introduction
Cervical cancer is the most common carcinoma caused
by oncogenic human papillomavirus (HPV). Globally, it accounted for
an estimated 528,000 new cancer cases worldwide and 266,000 deaths
in 2012 (1) even though HPV
vaccination programs have been implemented worldwide. Conventional
therapy for advanced cervical cancer is radio-therapy,
chemotherapy, or both. However, some populations with
chemo-resistance have poor prognoses. Alternative therapeutics
including molecule-targeting agents have not yet been developed for
cervical cancer.
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is a member of the TNF superfamily and has received
attention for its role as an antitumor agent that has few cytotoxic
effects on normal cells (2–4). Two
major pathways exist downstream of TRAIL signaling: pro-apoptosis
and pro-survival signaling (5).
The apoptosis pathway directly induces the TRAIL-R1/2-DISC
(death-inducing signaling complex)-induced activation of caspase-8
or triggers apoptosis indirectly via a mitochondrial pathway. On
the other hand, the pro-survival pathway induces the activation of
NF-κB or JNK/p38 signaling and increases the expression of
anti-apoptotic or proliferation stimulatory proteins (5). The fate of TRAIL signaling is
controlled by the balance between pro-apoptotic and pro-survival
signaling, which varies among tumor cell types.
In cervical cancer cells, p53-induced apoptosis is
abrogated through the degradation of p53 by the HPV E6 oncoprotein
(6) and thus, the other types of
apoptosis pathway such as TRAIL-induced apoptosis pathway appear to
play a major role in cell programmed death. Overcoming TRAIL
resistance has a potential to improve outcome in treating cervical
cancer. A previous study reported that sensitivity to TRAIL-induced
apoptosis differed among cell lines and that the proteasome
inhibitor MG132 sensitized TRAIL-induced apoptosis in cervical
cancer cells by upregulating death receptor (DR) 4/5 and
inactivating X-linked inhibitor of apoptosis (XIAP) (7). However, the biological mechanisms
preventing TRAIL-induced apoptosis in several cell lines have not
yet been elucidated in detail.
Signal transducer and activator of transcription 3
(STAT3) is activated by tyrosine phosphorylation in response to
various cytokine stimuli (8–10).
STAT3 is induced by various types of cellular stresses such as
hypoxia, reperfusion, and ultraviolet (UV) (11), which often occur in the tumor
microenvironment (TME). STAT3 and its downstream p38
mitogen-activated protein kinase (MAPK) signaling pathway are
well-known regulators of apoptosis or survival of damaged cells
(11–13). The constitutive activation of STAT3
has been reported in various types of malignancies, including
cervical cancer (14–18). Notably, the inhibition of STAT3 has
been shown to shift some apoptosis-resistant cells to TRAIL-induced
apoptosis (19,20). Numerous reports have demonstrated
that several components with the potency to suppress STAT3
activation such as histone deacetylase (HDAC) inhibitors (21–25),
resveratrol (26–30), and curcumin (31–35)
enhanced TRAIL-induced apoptosis in TRAIL-resistant cells.
Due to the importance of the STAT3 pathway
inhibition and TRAIL-induced apoptosis especially in cervical
cancer therapy, we attempted to elucidate the mechanisms
responsible for TRAIL-induced apoptosis with a focus on STAT3
activity.
Materials and methods
Antibodies and reagents
The following antibodies were used at the dilutions
indicated. In western blotting, mouse anti-human α-Tubulin sc-8035
(1:500) purchased from Santa Cruz Biotechnology (TX, USA), mouse
anti-human total STAT3 (124H6) CS#9139 (1:1,000), rabbit anti-human
phospho-STAT3 (Tyr705) (D3A7) CS#9145 (1:1,000), purchased from
Cell Signaling Technologies (MA, USA). The STAT3 inhibitor S3I-201
was purchased from Santa Cruz Biotechnology. Recombinant human
TRAIL was purchased from R&D Systems (Minnesota, MN, USA).
Cell cultures
The cervical cancer cell lines SiHa and CaSki were
maintained in Dulbecco's modified Eagle's medium (Wako, Osaka
Japan) with 10% fetal bovine serum (FBS, Life Technologies, CA,
USA) and antibiotics (antibiotic-antimycotic mixed stock solution,
Nacalai Tesque, Kyoto, Japan). Cells were grown in a humidified
tissue culture incubator at 37°C in 5% CO2.
Cell proliferation assay
Cell proliferation assays were performed to analyze
the effects of TRAIL, the STAT3 inhibitor S3I-201, tunicamycin on
cell proliferation. Five thousand cells were seeded on 96-well
plates. Cell Counting Kit-8 (CCK-8) with the tetrazolium salt WST-8
[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt] (Dojindo, Osaka, Japan) was used and quantified by
monitoring changes in absorbance at 450 nm, which were normalized
relative to the absorbance of control cells. The lengths of the
treatments are described with each result.
Detection of apoptosis by staining with
Annexin V-FITC
Cells (4×105) were cultured in 6-well
plates for 24 h before being treated. SiHa was also pretreated with
S3I-201 or tunicamycin for 24 h with an additional 15–18 h of the
TRAIL treatment. Cells were trypsinized, washed with PBS, and then
analyzed after double staining with an Annexin V Apoptosis
Detection kit (Abcam, MA, USA). The apoptotic cell population was
assessed using flow cytometry.
Immunoblotting
Total cellular extracts were prepared by lysing
cells from dishes in lysis buffer (#9803, Cell Signaling
Technologies) containing protease inhibitor cocktail (Nacalai
Tesque) and phosphatase inhibitor cocktail (Roche, Mannheim,
Germany) on ice for 5 min, and then sonicated briefly. Cells were
centrifuged at 14,000 rpm at 4°C for 10 min. The supernatant was
used in subsequent analyses. In SDS-PAGE, 20 μg of the protein
lysate with 6X sample buffer (Nacalai Tesque) was loaded into each
well. In immunoblotting, 0.45-μm PVDF membranes (Merck Millipore,
Darmstadt, Germany) were used. The membranes were blocked in 5%
milk/TBS-T (TBS containing 0.1% Tween-20) at room temperature for 1
h followed by an incubation with the primary antibodies diluted in
5% milk/TBS-T or 5% BSA/TBS-T for the appropriate time period
indicated in the manufacturer's instructions. After several washes
with TBS-T, the membranes were incubated with secondary antibodies
conjugated with HRP in 5% milk/TBS-T at room temperature for 1 h.
Blots were developed using Immobilon Western Chemiluminescent HRP
substrate (Merck Millipore) according to the manufacturer's
instructions.
Transfections
Small interfering RNA transfections were performed
using Stealth RNAi against STAT3 (HSS186130, HSS186131, and
HSS110279) and non-targeting siRNA (Stealth RNAi™ siRNA Negative
Control, Med GC, Life Technologies) as a control. When cells were
60–70% confluent, transfections were performed using Lipofectamine
RNAiMAX (Life Technologies), Opti-MEM reduced serum medium (Life
Technologies), and a final concentration of 20 nmol/l of siRNAs,
according to the manufacturer's instructions. After 5 h of
incubation, the transfection medium was changed to normal culture
medium without antibiotics. Cells were incubated for 48 h and then
analyzed for each experiment. The transfection sequence was
repeated at least 3 times.
RT-quantitative PCR
Total RNA was extracted from cells using
Blood/Cultured Cell Total RNA Mini kit (Favorgen, Ping Tung,
Taiwan), followed by reverse transcription. cDNA was amplified for
40 cycles in a Light Cycler 480 (Roshe). Expression of CHOP was
normalized by GAPDH mRNA as an internal standard, and spliced XBP1
(sXBP1) was normalized by total XBP1 (tXBP1) calculated by the ΔΔCq
method. The primer pairs were as follows: human total XBP1 (tXBP1)
5′-GGCATCCTTGGCTTGCCTCCA-3′, and 5′-GCCCCCTCAGCAGGTGTTCC-3′, human
spliced XBP1 (sXBP1) 5′-CGCTTGGGGATGGATGCCCTG-3′ and
5′-CCTGCACCTGCTGCGGACT-3′, human CHOP 5′-GGAGCATCAGTCCCCCACTT-3′
and 5′-TGTGGGATTGAGGGTCACATC-3′, and human GAPDH
5′-GAAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′.
Statistical analysis
Data are presented as means ± standard error of the
mean (SEM). Statistical analyses were carried out using Student's
t-test or Dunett's analysis using JMP software. A value of
P<0.05 was considered significant. In the figure legends,
asterisks indicate comparisons with significant difference
(P<0.05).
Results
STAT3 activation is suppressed in CaSki
by the TRAIL stimulation
Hougardy et al previously reported that
HPV16-positive cervical cell line CaSki is the most sensitive cell
line to TRAIL-induced apoptosis while HPV16-positive SiHa is the
most resistant one among cervical cancer cell lines that were
tested (7).
In order to investigate the cause of the difference
in TRAIL sensitivity, we first performed a comprehensive analysis
on apoptotic signaling in both cell types under physiological
conditions using an apoptosis array. However, no marked differences
were observed in the expression levels of apoptosis-related
molecules including pro-apoptotic protein survivin or TRAIL
receptors: DR4 and DR5 (data not shown).
STAT3 has been reported to be involved in apoptosis
resistance in various cancers (36–38).
We investigated the involvement of STAT3 in TRAIL sensitivity
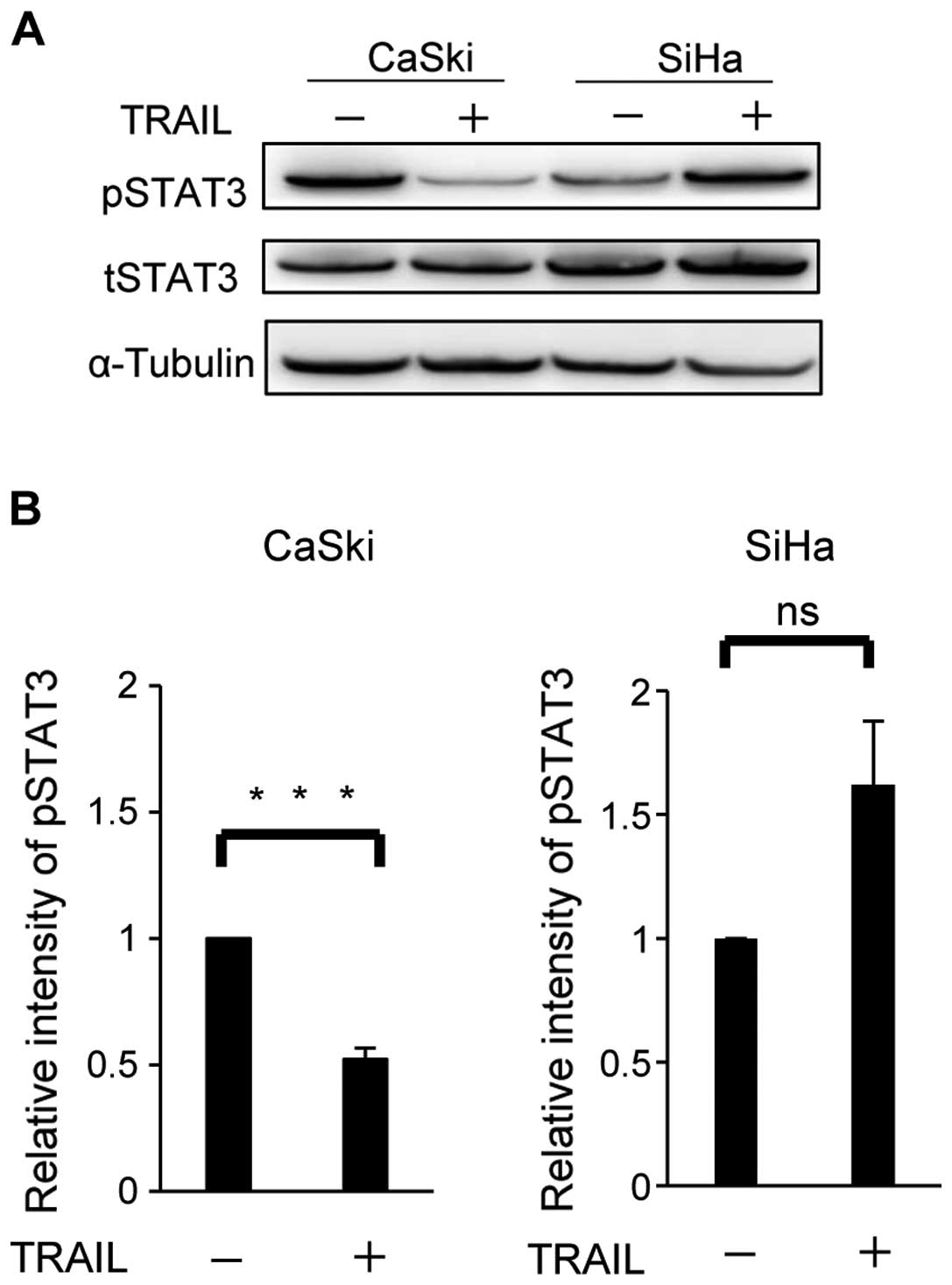
between CaSki and SiHa (Fig. 1).
Western blotting revealed that the expression levels of total STAT3
(tSTAT3) were similar in both cell types regardless of the TRAIL
stimulation. The phosphorylation of STAT3 (pSTAT3) was suppressed
in CaSki by TRAIL, but tend to be upregulated or remained at the
same level in SiHa (Fig. 1, upper
panels). Six independent experiments revealed that the protein
level of pSTAT3 decreased significantly (P=0.0003) by TRAIL in
CaSki, but not in SiHa (P=0.0957) (Fig. 1, lower panels). These results
indicated that the modulation of STAT3 activity by TRAIL might be
involved in the different responses to the TRAIL stimulation.
siSTAT3 enhances TRAIL-induced
apoptosis
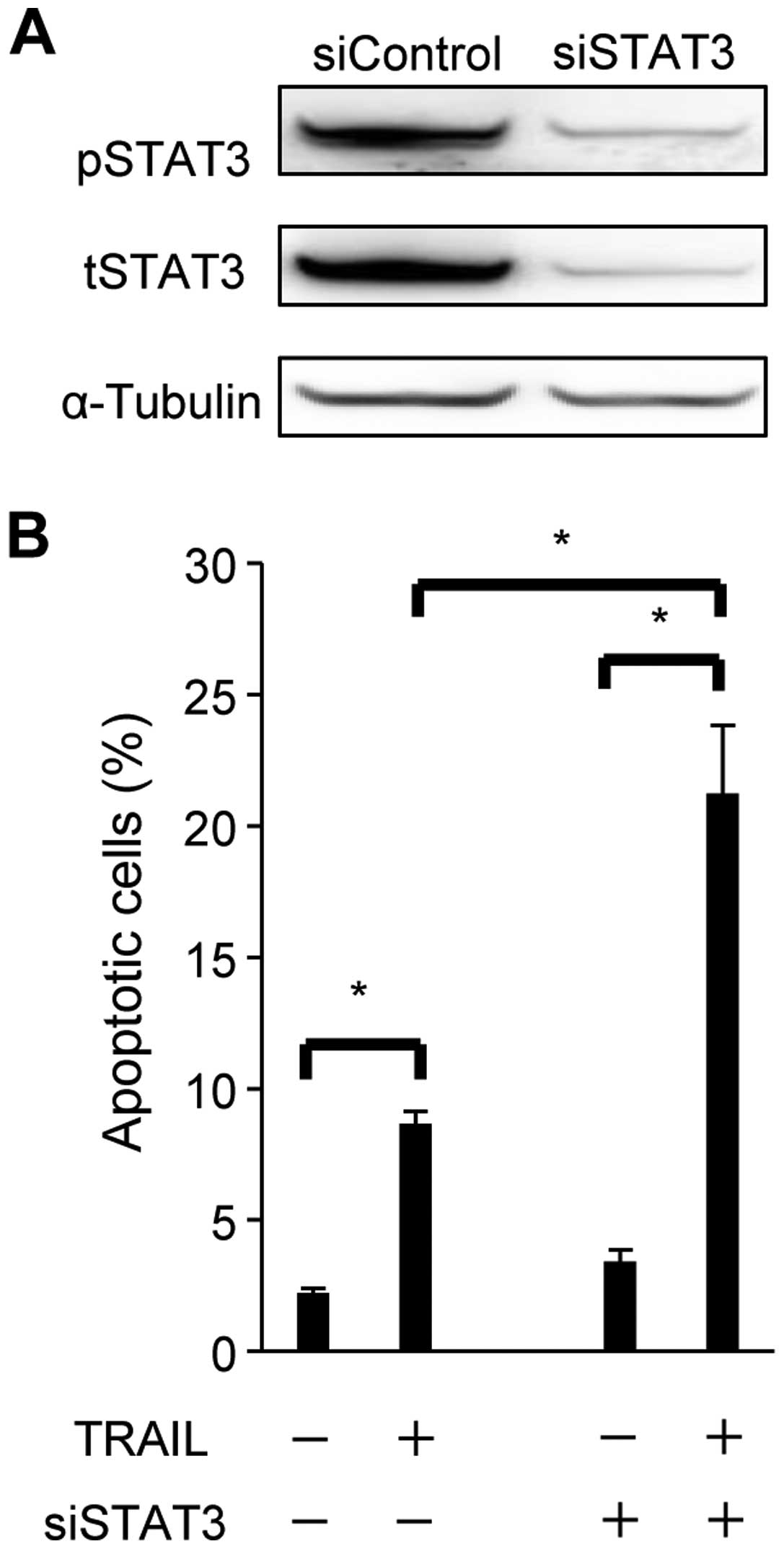
In order to examine the role of STAT3 in resistance
to TRAIL-induced apoptosis, optimized siRNA for STAT3 (siSTAT3) or
control siRNA was transduced into SiHa (Fig. 2). Total STAT3 (tSTAT3) as well as
phosphorylation of STAT3 (pSTAT3) expression were sufficiently
knocked-down at the protein level by siSTAT3 in SiHa (Fig. 2A). STAT3 knockdown (siSTAT3) and
control cells were exposed to TRAIL and then assessed for
apoptosis. Although the TRAIL stimulation induced apoptosis in
control and siSTAT3 cells, the extent of apoptosis observed was
markedly higher in siSTAT3 cells [21.2(±2.6)%] than in control
cells [8.7(±0.44)%] (Fig. 2B). The
increase induced in apoptosis by the knockdown of STAT3 did not
occur in cells not exposed to TRAIL. These results indicated that
the knockdown of STAT3 enhanced sensitivity to TRAIL-induced
apoptosis in SiHa.
Next, we investigated the effect of STAT3
suppression on cisplatin (CDDP)-induced apoptosis in the SiHa
cells, as CDDP is one of the standard chemotherapeutics for
advanced or recurrent cervical cancer (39). Unlike TRAIL-induced apoptosis,
STAT3 inhibition did not enhance CDDP-induced apoptosis in the SiHa
cells (data not shown).
The STAT3 inhibitor enhances sensitivity
to TRAIL-induced apoptosis
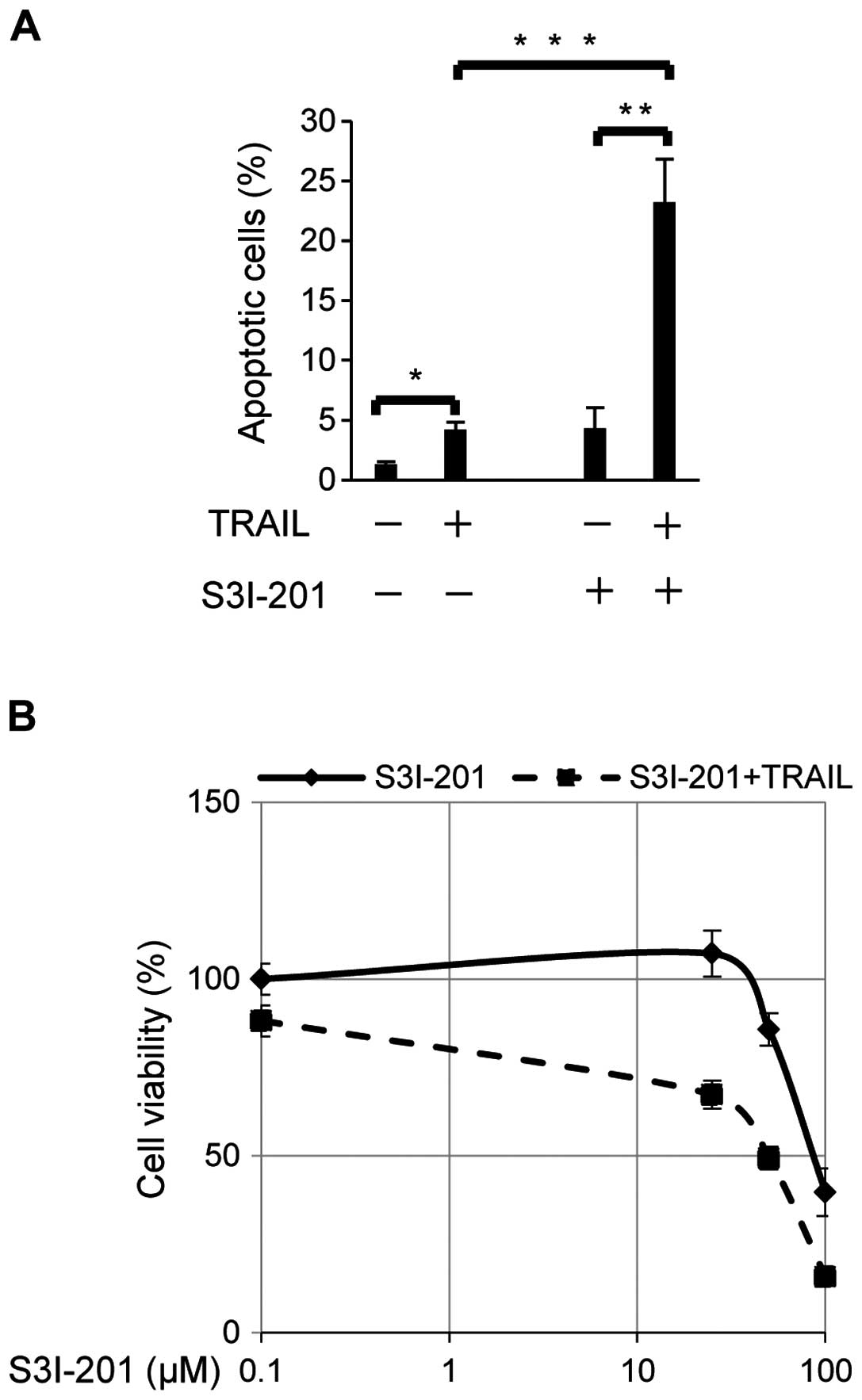
As a molecular-targeting agent in a clinical
setting, S3I-201 is an attractive STAT3 inhibitor which inhibits
STAT3 dimerization, DNA biding and transcriptional activities
(40). We hypothesized that
S3I-201 may enhance sensitivity to TRAIL-induced apoptosis in cells
resistant to TRAIL. Apoptosis was examined in TRAIL-stimulated SiHa
cells treated with or without the STAT3 inhibitor S3I-201 (Fig. 3A). Increases of less than 5% were
observed in the proportion of apoptotic cells by the TRAIL
stimulation without exposure to S3I-201. TRAIL-stimulated SiHa
showed a five-fold increase [4.3(±0.97)% to 23(±2.1)%] in the
proportion of apoptotic cells following exposure to 100 μM of
S3I-201 (Fig. 3A). Furthermore,
SiHa cells were exposed to S3I-201 at different doses (25, 50 and
100 μM) with or without a sequential 100 ng/ml of TRAIL
stimulation, and cell viability at each dose was blotted on the
curve (Fig. 3B). Although S3I-201
alone suppressed cell viability at doses >50 μM, the combination
of S3I-201 and TRAIL more effectively suppressed cell viability in
SiHa. These results indicated that the STAT3 inhibitor, synergistic
with TRAIL, enhanced sensitivity to apoptosis in TRAIL-resistant
SiHa cell line.
Tunicamycin sensitized TRAIL-induced
apoptosis by suppressing STAT3 activation in SiHa
Previous studies demonstrated that several ER stress
inducers suppressed STAT3 phosphorylation (41,42).
We hypothesized that ER stress inducers may sensitize cells to
TRAIL-induced apoptosis through the inactivation of STAT3. The
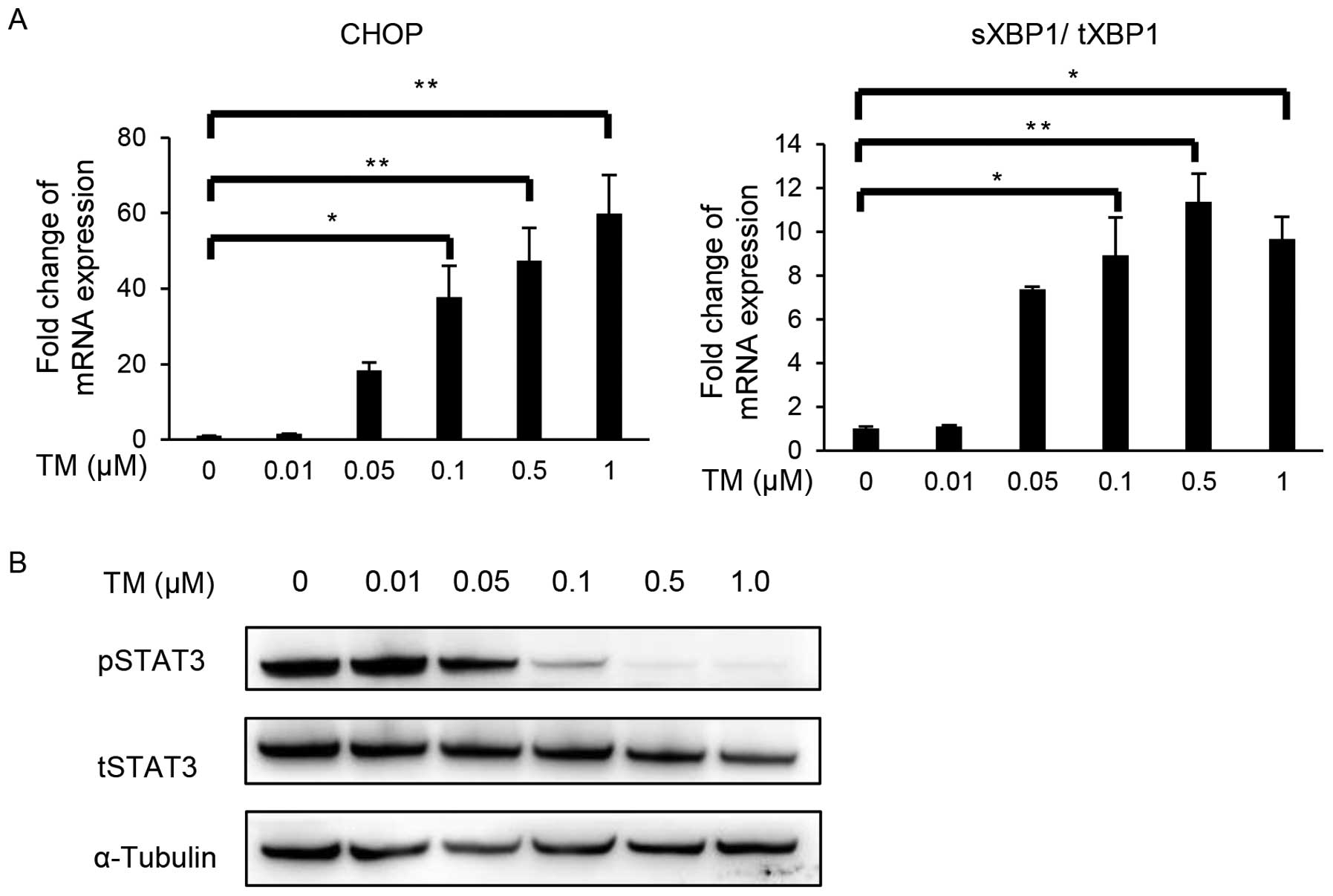
TRAIL-resistant cell line, SiHa, was exposed to different doses of
the ER stress inducer, tunicamycin (TM) (0.01, 0.05, 0.1, 0.5 and
1.0 μM) to examine the phosphorylation of STAT3 and TRAIL-induced
apoptosis. We confirmed that TM upregulated C/EBP homologous
protein (CHOP) and X-box-binding protein-1 (XBP1) splicing levels
in a dose-dependent manner, indicating that TM activated an
unfolded protein reaction (UPR): ER stress branches (Fig. 4A). Western blotting for pSTAT3 and
tSTAT3 demonstrated that TM successfully suppressed the activation
(phosphorylation) of STAT3 in a dose-dependent manner (Fig. 4B).
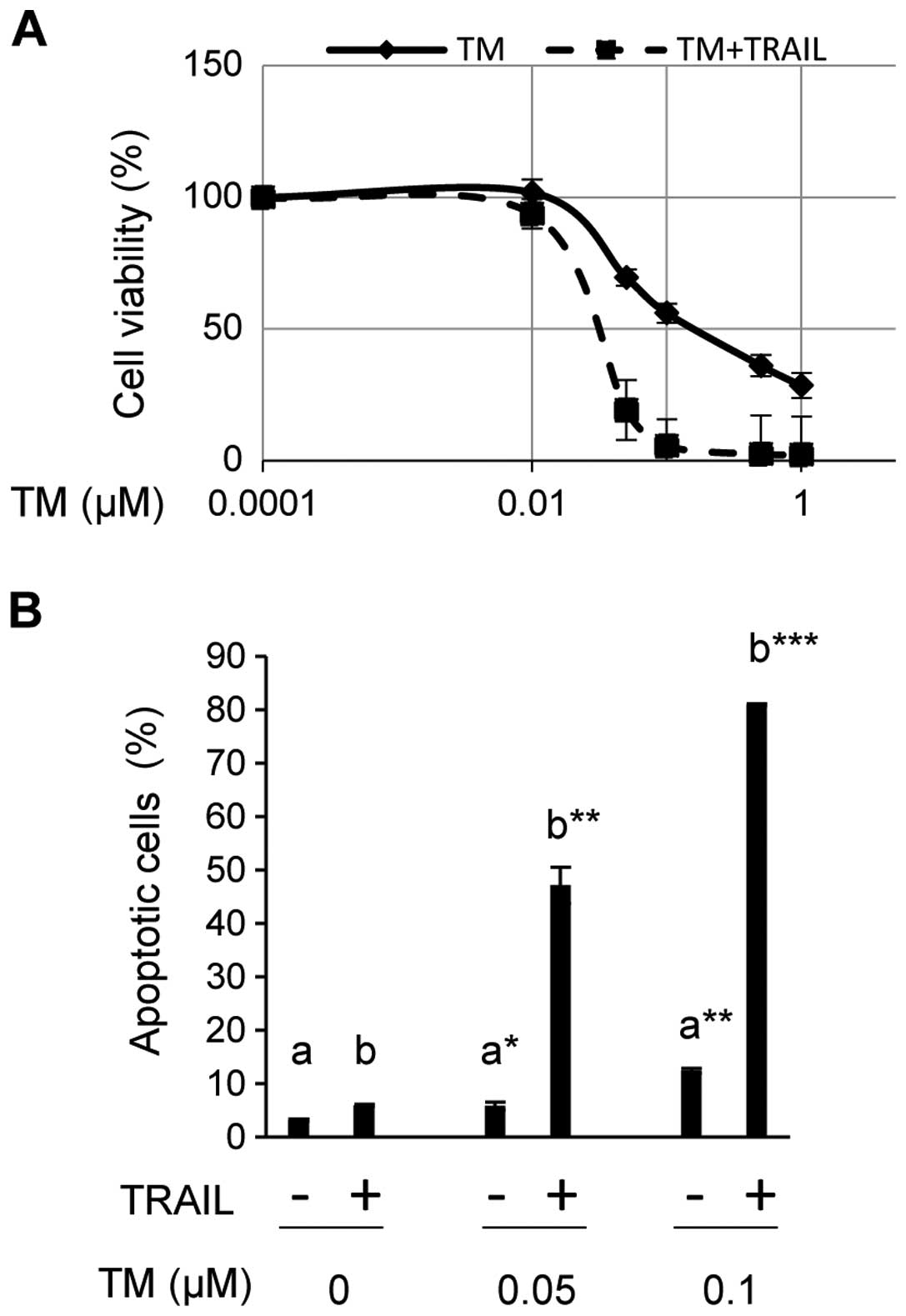
SiHa was then exposed to TM at different doses
(0.01, 0.05, 0.1, 0.5 and 1.0 μM) for 24 h with or without a
sequential 100 ng/ml of TRAIL stimulation, and cell viability was
assessed using CCK-8 (Fig. 5A). TM
reduced cell viability to less than 50% at doses greater than 0.1
μM in the absence of TRAIL (Fig.
5A). Since TM increased spliced XBP1 and CHOP, which play
central roles in ER stress-mediated apoptosis (43) (Fig.
4A), decreases in cell viability appeared to reflect TM-induced
ER stress-mediated apoptosis. However, cell viability curves
revealed that TM in combination with TRAIL had more suppressive
effect on the cell viability compared with TM alone. In the
presence of TRAIL, viable cells were barely detected at the same
doses of TM (Fig. 5A). We also
examined the proportion of apoptotic cells among SiHa exposed to
TRAIL and/or TM at different doses (Fig. 5B). The proportion of apoptotic
cells was only 5.9(±0.22)% in SiHa exposed to TRAIL alone, but
increased to 47.1(±3.4)% and 81.0(±0.2)% in SiHa exposed to TRAIL
in combination with TM at 0.05 and 0.1 μM, respectively. Exposure
to TM alone induced apoptosis in 5.8(±0.68)% and 12.4(±0.48)% of
cells at these doses. Therefore, the combination of TM and TRAIL
had a synergistic effect on the induction of apoptosis.
Discussion
In this study, we showed that STAT3 activation was
suppressed by TRAIL in the TRAIL-sensitive cell line CaSki, but not
in the TRAIL-resistant SiHa cell line. The inhibition of STAT3
expression using siRNA technology and the suppression of STAT3
activity using a STAT3 inhibitor increased the sensitivity of the
SiHa cells to TRAIL-induced apoptosis. Furthermore, an ER stress
inducer (TM) also effectively increased their sensitivity to
TRAIL-induced apoptosis accompanied by STAT3 inactivation. These
results indicated that STAT3 regulated the TRAIL sensitivity in the
SiHa cells.
We first examined the differences in the basal
expression of apoptosis-related molecules including
apoptosis-related receptors. However, no marked differences were
observed in the expression levels of apoptosis-related molecules or
TRAIL receptors (data not shown). In contrast, western blotting
revealed a difference in the phosphorylation of STAT3 with TRAIL
stimulation (Fig. 1), whereas
other major pro-apoptotic signaling pathways remained unchanged
(data not shown). These results indicate that STAT3 activity might
be involved in the difference in the responses of the two cell
lines to TRAIL stimulation, which is consistent with previous
findings showing that the inhibition of STAT3 signals sensitizes
TRAIL-induced apoptosis (41,42).
Previous studies reported that several components that have the
potency to suppress STAT3 shift the fate of some
apoptosis-resistant malignant cells to TRAIL-induced apoptosis. In
these studies, a JAK2 inhibitor, histone deacetylase (HDAC)
inhibitor, resveratrol, and curcumin were used as inducers of
TRAIL-induced apoptosis (21–35).
Taken together, these data suggested that STAT3 might be the
central molecule in the resistance of the SiHa cells to TRAIL. In
this study, we further confirmed that both suppression of STAT3
expression using siRNA technology and inhibition of STAT3
activation using the STAT3 inhibitor S3I-201 increased the
TRAIL-induced apoptosis in the SiHa cells (Figs. 2 and 3). Oncogenes and p53 are the central
regulators of the apoptosis induction in cervical cancer. A
previous report demonstrated that knockdown of STAT3 suppressed the
expression of viral E6 and E7 oncoproteins as well as upregulated
p53 expression (44). Upregulation
of p53 might also enhanced the apoptosis-inducing function of the
STAT3 knockdown in our model. These results indicated that
targeting STAT3 expression or activity combined with TRAIL
stimulation could be a strategy for cervical cancer treatment.
ER stress-mediated apoptosis occurs following the
disruption of the UPR balance under prolonged ER stress. UPR
branches (PERK, IRE1a, and ATF6) are well known to cross-talk with
each other and various cellular signaling pathways (45). However, it currently remains
unclear whether STAT3 preferentially involves UPR or ER
stress-mediated apoptosis. Several studies have shown that some ER
stress inducers suppress the phosphorylation of STAT3 (41,42).
The results of this study showed that 0.1 μM of tunicamycin (TM)
upregulated CHOP, which plays a central role in ER stress-mediated
apoptosis (43), although it
induced apoptosis in only 10% of the treated cells (Fig. 5B). This result indicated that TM
activated UPR, but did not induce ER stress-mediated apoptosis at
this dose. In contrast, the combination of TRAIL and 0.1 μM of TM
induced apoptosis in 80% of the treated cells (Fig. 5B), which suggested that the
combination of TRAIL and TM had a synergistic effect on the
induction of apoptosis. Although TM is known to itself upregulate
DR4 or DR5 and sensitize cells to TRAIL-induced apoptosis (46–49),
only a slight increase in DR5 mRNA level in the SiHa cells was
observed after TM treatment in our experiments (data not shown).
The inactivation of STAT3 by TM may be one of the key mechanisms to
increase the sensitivity of SiHa cells to TRAIL-induced apoptosis,
which possibly resulted in the synergistic effect observed.
In addition to the TRAIL-induced apoptosis pathway,
we also investigated the effect of STAT3 suppression on the
CDDP-induced apoptosis pathway. Although previous reports
demonstrated that the inhibition of STAT3 enhanced the CDDP-induced
apoptosis in various cancers (50,51),
in our model, the inhibition of STAT3 did not enhance the
CDDP-induced apoptosis. In the CDDP-induced apoptosis, DNA damage
is induced by intra-strand or inter-strand cross-link between two
adjacent G residues (52). This
DNA damage induces p53-dependent apoptosis. However, HPV-positive
cervical cancer lacks p53 expression and the DNA damage-induced
apoptosis pathway is considered to be different from other types of
cancer. The result suggested that STAT3 inhibition could be an
effective therapeutic modality for cervical cancer when combined
with TRAIL rather than with CDDP.
In conclusion, in this study, we showed that
different TRAIL sensitivity among cell lines might be regulated by
STAT3 activity and that the inactivation of STAT3 enhances the
sensitivity of the cells to TRAIL-induced apoptosis, even in a
TRAIL-resistant cancer cell line. Our results suggest the potential
of STAT3 inhibition in combination with TRAIL-based therapy for
cervical cancer.
Acknowledgements
We would like to thank Dr Terufumi Yokoyama for
expert advice on experimental methodologies. We thank Editage
(www.editage.com) for English language editing.
Abbreviations:
|
TRAIL
|
tumor necrosis factor-related
apoptosis inducing ligand
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
References
|
1
|
Ferlay JSI, Ervik M, Dikshit R, Eser S,
Mathers C, Rebelo M, Parkin DM, Forman D and Bray F:
Incidence/mortality data. GLOBOCAN 2012 1.0. 2012, https://www.iarc.fr.
|
|
2
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang L and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar
|
|
4
|
Wiley SR, Schooley K, Smolak PJ, Din WS,
Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA,
et al: Identification and characterization of a new member of the
TNF family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kruyt FA: TRAIL and cancer therapy. Cancer
Lett. 263:14–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moody CA and Laimins LA: Human
papillomavirus oncoproteins: Pathways to transformation. Nat Rev
Cancer. 10:550–560. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hougardy BM, Maduro JH, van der Zee AG, de
Groot DJ, van den Heuvel FA, de Vries EG and de Jong S: Proteasome
inhibitor MG132 sensitizes HPV-positive human cervical cancer cells
to rhTRAIL-induced apoptosis. Int J Cancer. 118:1892–1900. 2006.
View Article : Google Scholar
|
|
8
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Akira S: Roles of STAT3 defined by
tissue-specific gene targeting. Oncogene. 19:2607–2611. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levy DE and Lee CK: What does Stat3 do? J
Clin Invest. 109:1143–1148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dudley AC, Thomas D, Best J and Jenkins A:
The STATs in cell stress-type responses. Cell Commun Signal.
2:82004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramírez de Arellano A, Lopez-Pulido EI,
Martínez-Neri PA, Estrada Chávez C, González Lucano R,
Fafutis-Morris M, Aguilar-Lemarroy A, Muñoz-Valle JF and
Pereira-Suárez AL: STAT3 activation is required for the
antiapoptotic effects of prolactin in cervical cancer cells. Cancer
Cell Int. 15:832015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Banerjee K and Resat H: Constitutive
activation of STAT3 in breast cancer cells: A review. Int J Cancer.
138:2570–2578. 2016. View Article : Google Scholar
|
|
15
|
Benekli M, Xia Z, Donohue KA, Ford LA,
Pixley LA, Baer MR, Baumann H and Wetzler M: Constitutive activity
of signal transducer and activator of transcription 3 protein in
acute myeloid leukemia blasts is associated with short disease-free
survival. Blood. 99:252–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takemoto S, Ushijima K, Kawano K,
Yamaguchi T, Terada A, Fujiyoshi N, Nishio S, Tsuda N, Ijichi M,
Kakuma T, et al: Expression of activated signal transducer and
activator of transcription-3 predicts poor prognosis in cervical
squamous-cell carcinoma. Br J Cancer. 101:967–972. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schoppmann SF, Jesch B, Friedrich J,
Jomrich G, Maroske F and Birner P: Phosphorylation of signal
transducer and activator of transcription 3 (STAT3) correlates with
Her-2 status, carbonic anhydrase 9 expression and prognosis in
esophageal cancer. Clin Exp Metastasis. 29:615–624. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mali SB: Review of STAT3 (Signal
Transducers and Activators of Transcription) in head and neck
cancer. Oral Oncol. 51:565–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kusaba M, Nakao K, Goto T, Nishimura D,
Kawashimo H, Shibata H, Motoyoshi Y, Taura N, Ichikawa T, Hamasaki
K, et al: Abrogation of constitutive STAT3 activity sensitizes
human hepatoma cells to TRAIL-mediated apoptosis. J Hepatol.
47:546–555. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lanuti P, Bertagnolo V, Pierdomenico L,
Bascelli A, Santavenere E, Alinari L, Capitani S, Miscia S and
Marchisio M: Enhancement of TRAIL cytotoxicity by AG-490 in human
ALL cells is characterized by downregulation of cIAP-1 and cIAP-2
through inhibition of Jak2/Stat3. Cell Res. 19:1079–1089. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh TR, Shankar S and Srivastava RK:
HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL
in breast carcinoma. Oncogene. 24:4609–4623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fulda S: Histone deacetylase (HDAC)
inhibitors and regulation of TRAIL-induced apoptosis. Exp Cell Res.
318:1208–1212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Earel JK Jr, VanOosten RL and Griffith TS:
Histone deacetylase inhibitors modulate the sensitivity of tumor
necrosis factor-related apoptosis-inducing ligand-resistant bladder
tumor cells. Cancer Res. 66:499–507. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fandy TE, Shankar S, Ross DD, Sausville E
and Srivastava RK: Interactive effects of HDAC inhibitors and TRAIL
on apoptosis are associated with changes in mitochondrial functions
and expressions of cell cycle regulatory genes in multiple myeloma.
Neoplasia. 7:646–657. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gupta M, Han JJ, Stenson M, Wellik L and
Witzig TE: Regulation of STAT3 by histone deacetylase-3 in diffuse
large B-cell lymphoma: Implications for therapy. Leukemia.
26:1356–1364. 2012. View Article : Google Scholar
|
|
26
|
Ganapathy S, Chen Q, Singh KP, Shankar S
and Srivastava RK: Resveratrol enhances antitumor activity of TRAIL
in prostate cancer xenografts through activation of FOXO
transcription factor. PLoS One. 5:e156272010. View Article : Google Scholar
|
|
27
|
Taguchi A, Koga K, Kawana K, Makabe T, Sue
F, Miyashita M, Yoshida M, Urata Y, Izumi G, Tkamura M, et al:
Resveratrol enhances apoptosis in endometriotic stromal cells. Am J
Reprod Immunol. 75:486–492. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Quoc Trung L, Espinoza JL, Takami A and
Nakao S: Resveratrol induces cell cycle arrest and apoptosis in
malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS One.
8:e551832013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kotha A, Sekharam M, Cilenti L, Siddiquee
K, Khaled A, Zervos AS, Carter B, Turkson J and Jove R: Resveratrol
inhibits Src and Stat3 signaling and induces the apoptosis of
malignant cells containing activated Stat3 protein. Mol Cancer
Ther. 5:621–629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tameda M, Sugimoto K, Shiraki K, Inagaki
Y, Ogura S, Kasai C, Yoneda M, Okamoto R, Yamamoto N, Takei Y, et
al: Resveratrol sensitizes HepG2 cells to TRAIL-induced apoptosis.
Anticancer Drugs. 25:1028–1034. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bharti AC, Donato N and Aggarwal BB:
Curcumin (diferu-loylmethane) inhibits constitutive and
IL-6-inducible STAT3 phosphorylation in human multiple myeloma
cells. J Immunol. 171:3863–3871. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alexandrow MG, Song LJ, Altiok S, Gray J,
Haura EB and Kumar NB: Curcumin: A novel Stat3 pathway inhibitor
for chemoprevention of lung cancer. Eur J Cancer Prev. 21:407–412.
2012. View Article : Google Scholar :
|
|
33
|
Shankar S, Chen Q, Sarva K, Siddiqui I and
Srivastava RK: Curcumin enhances the apoptosis-inducing potential
of TRAIL in prostate cancer cells: Molecular mechanisms of
apoptosis, migration and angiogenesis. J Mol Signal. 2:102007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park S, Cho DH, Andera L, Suh N and Kim I:
Curcumin enhances TRAIL-induced apoptosis of breast cancer cells by
regulating apoptosis-related proteins. Mol Cell Biochem. 383:39–48.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Glienke W, Maute L, Wicht J and Bergmann
L: Curcumin inhibits constitutive STAT3 phosphorylation in human
pancreatic cancer cell lines and downregulation of survivin/BIRC5
gene expression. Cancer Invest. 28:166–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu H, Kortylewski M and Pardoll D:
Crosstalk between cancer and immune cells: Role of STAT3 in the
tumour microenvironment. Nat Rev Immunol. 7:41–51. 2007. View Article : Google Scholar
|
|
37
|
Zhang HF and Lai R: STAT3 in Cancer -
Friend or Foe? Cancers (Basel). 6:1408–1440. 2014. View Article : Google Scholar
|
|
38
|
Pensa S, Regis G, Boselli D, Novelli F and
Poli V: STAT1 and STAT3 in tumorigenesis: Two sides of the same
coin. Madame Curie Bioscience Database. Landes Bioscience; Austin,
TX: 2009
|
|
39
|
Pectasides D, Kamposioras K, Papaxoinis G
and Pectasides E: Chemotherapy for recurrent cervical cancer.
Cancer Treat Rev. 34:603–613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Siddiquee K, Zhang S, Guida WC, Blaskovich
MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence
NJ, et al: Selective chemical probe inhibitor of Stat3, identified
through structure-based virtual screening, induces antitumor
activity. Proc Natl Acad Sci USA. 104:7391–7396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kimura K, Yamada T, Matsumoto M, Kido Y,
Hosooka T, Asahara S, Matsuda T, Ota T, Watanabe H, Sai Y, et al:
Endoplasmic reticulum stress inhibits STAT3-dependent suppression
of hepatic gluconeogenesis via dephosphorylation and deacetylation.
Diabetes. 61:61–73. 2012. View Article : Google Scholar :
|
|
42
|
Yang Z, Liu Y, Liao J, Gong C, Sun C, Zhou
X, Wei X, Zhang T, Gao Q, Ma D, et al: Quercetin induces
endoplasmic reticulum stress to enhance cDDP cytotoxicity in
ovarian cancer: Involvement of STAT3 signaling. FEBS J.
282:1111–1125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
44
|
Shukla S, Mahata S, Shishodia G, Pandey A,
Tyagi A, Vishnoi K, Basir SF, Das BC and Bharti AC: Functional
regulatory role of STAT3 in HPV16-mediated cervical carcinogenesis.
PLoS One. 8:e678492013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
46
|
Jiang CC, Chen LH, Gillespie S, Kiejda KA,
Mhaidat N, Wang YF, Thorne R, Zhang XD and Hersey P: Tunicamycin
sensitizes human melanoma cells to tumor necrosis factor-related
apoptosis-inducing ligand-induced apoptosis by up-regulation of
TRAIL-R2 via the unfolded protein response. Cancer Res.
67:5880–5888. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jung YH, Lim EJ, Heo J, Kwon TK and Kim
YH: Tunicamycin sensitizes human prostate cells to TRAIL-induced
apoptosis by upregulation of TRAIL receptors and downregulation of
cIAP2. Int J Oncol. 40:1941–1948. 2012.PubMed/NCBI
|
|
48
|
Hasegawa A, Osuga Y, Hirota Y, Hamasaki K,
Kodama A, Harada M, Tajima T, Takemura Y, Hirata T, Yoshino O, et
al: Tunicamycin enhances the apoptosis induced by tumor necrosis
factor-related apoptosis-inducing ligand in endometriotic stromal
cells. Hum Reprod. 24:408–414. 2009. View Article : Google Scholar
|
|
49
|
Shiraishi T, Yoshida T, Nakata S, Horinaka
M, Wakada M, Mizutani Y, Miki T and Sakai T: Tunicamycin enhances
tumor necrosis factor-related apoptosis-inducing ligand-induced
apoptosis in human prostate cancer cells. Cancer Res. 65:6364–6370.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hu Y, Hong Y, Xu Y, Liu P, Guo DH and Chen
Y: Inhibition of the JAK/STAT pathway with ruxolitinib overcomes
cisplatin resistance in non-small-cell lung cancer NSCLC.
Apoptosis. 19:1627–1636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Selvendiran K, Bratasz A, Kuppusamy ML,
Tazi MF, Rivera BK and Kuppusamy P: Hypoxia induces chemoresistance
in ovarian cancer cells by activation of signal transducer and
activator of transcription 3. Int J Cancer. 125:2198–2204. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|