Introduction
The small cell carcinoma of the ovary hypercalcemic
type (SCCOHT) represents a rare form of an aggressive ovarian tumor
with poor prognosis and is predominantly diagnosed in young women
associated in most cases with paraendocrine hypercalcemia (1–3).
Previous research has identified specific genetic alterations in
SCCOHT including loss of SMARCA4 (BRG1) and SMARCA2 (BRM) functions
by both, somatic and germline mutations (4–7).
These SWI/SNF family proteins display ATPase and helicase
activities and act as transcriptional activators by altering
chromatin structures in the vicinity of target genes whereas
functional loss of these proteins is associated with tumorigenic
development (8). Related mutations
including SMARCA4 and SMARCB1 were also observed in certain brain
tumors, particularly in atypical teratoid/rhabdoid tumors (AT/RTs)
and further genotypic similarities acknowledged SCCOHT as ovarian
tumors with AT/RT properties (9).
Accordingly, SCCOHT were suggested as malignant rhabdoid tumor of
the ovary (10) whereby AT/RTs are
composed of several entities requiring further discrimination
(11).
Effective and standardized therapeutic approaches
remain unclear for SCCOHT cancer whereby different treatment
modalities such as surgery, chemotherapy, radiotherapy, and/or
high-dose chemotherapy with autologous stem cell rescue revealed a
heterogeneous outcome for the patients (12). The two cellular models for this
disease, SCCOHT-1 and BIN-67 have demonstrated multiple
chemotherapeutic resistances by continued tumor growth of
appropriate xenotransplants (13,14).
Moreover, in vitro and in vivo studies with these
cell lines exhibited various metabolic and functional alterations
during interaction with other cell populations of the tumor stroma,
in particular mesenchymal stroma/stem cells (MSC).
In general, MSC represent a heterogeneous cell
population of multipotent progenitor cells which participate in
structural and functional organization of connective tissue and are
involved in the maintenance of tissue homeostasis. Various debates
about developmental and functional properties are accompanying MSC
which are almost ubiquitously present in most tissues. MSC are
attracted to damaged tissue sites and wounds to modulate initial
immune cell activities, to enable a neo-vascularization process,
and to promote tissue repair (15–18).
Likewise, aggressive tumor growth such as SCCOHT creates invasive
tissue damage and an inflammatory environment whereby recruited MSC
develop their natural functionality and produce certain
extracellular matrix proteins to support regeneration of the
damaged tissue and contribute to angiogenesis and
neo-vascularization by stimulation of endothelial cells. This
functionality of MSC is exploited by the tumor cells to take
advantage of these conditions for further enhanced tumor growth
(19–21). Indeed, previous in vitro and
in vivo studies with breast cancer cells substantiate a
tumor- and metastasis-promoting role of MSC within the tumor stroma
by direct cell-cell interactions and/or indirectly via the release
of cytokines/chemokines or microvesicles including exosomes
(22–24). This is also supported by effects
during culture of SCCOHT-1 cells in the presence of MSC-derived
exosomes which enabled new metabolic activities in these tumor
cells such as acquisition of MMP-2 and ecto-5′-nucleotidase
activities (25). Whereas
interaction of MSC with SCCOHT-1 cells is associated with certain
alteration of tumor cell properties, these MSC-mediated activities
also provide the capability to re-organize the tumor
microenvironment.
Therefore, in this study, we tested therapeutic
compounds to treat SCCOHT-1 and BIN-67 cells and found a
combination with substantial in vitro tumor cell killing.
Subsequent in vivo tests of this combination in
SCCOHT-1-induced mouse tumors, however, demonstrated surprising
effects of tumor cell protection and accordingly, we investigated
in further experiments the reasons for this divergence by focusing
on distinct extracellular matrix-associated proteins and the role
of MSC within the tumor microenvironment.
Materials and methods
Cell culture of tumor cells
SCCOHT-1 cells represent a spontaneously
proliferating population derived from a patient with recurrent
small cell carcinoma of the ovary hypercalcemic type (SCCOHT) and
were maintained in RPMI-1640 with medium supplements as described
previously (13).
BIN-67 cells were kindly provided by Dr Barbara
Vanderhyden (University of Ottawa, Canada) and cultured with
RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented
with 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 U/ml
penicillin and 100 μg/ml streptomycin (26).
Cells were cultivated at 37°C in a humidified
atmosphere containing 5% CO2 and tested for mycoplasma
by the luminometric MycoAlert Plus mycoplasma detection kit (Lonza
Inc., Rockland, ME, USA) according to the manufacturer’s
instructions.
Labeling of SCCOHT cells by lentiviral
transduction
Wild-type SCCOHT-1 (SCCOHT-1wt) cells and
BIN-67 cells were transduced with a 3rd generation lentiviral SIN
vector containing the GFP gene to generate SCCOHT-1GFP
cells and BIN-67GFP cells as previously described
(23). These labeled populations
were used for chemosensitivity assays and for discrimination of the
SCCOHT cells in co-cultures and in the mouse tissue after forming
tumors and metastases.
Cell culture of primary human mesenchymal
stroma/stem cells (MSC)
The use of primary MSC following tissue explant
culture was approved by the Ethics Committee of Hannover Medical
School, Project #443 on February 26th, 2009, and informed written
consent was obtained from each patient.
MSC-like cells were isolated from explant cultures
of human umbilical cords as reported previously (27) and cultured in α-MEM medium (Sigma
Chemie GmbH, Steinheim, Germany) supplemented with 10% of
allogeneic human AB-serum (commercially obtained from blood bank,
University Campus Lübeck, Germany), 100 U/ml penicillin, 100 μg/ml
streptomycin and 2 mM L-glutamine (Sigma). For subculture, MSC were
harvested by accutase (Sigma) treatment for 3 min at 37°C.
Co-culture of MSC and SCCOHT-1GFP cells was performed in
MSC culture medium at the indicated cell ratios for ≤72 h.
Cell line authentication
Authentication of SCCOHT-1 and BIN-67 cell lines was
performed by short tandem repeat (STR) fragment analysis using the
GenomeLab human STR primer set (Beckman Coulter Inc., Fullerton,
CA, USA). The fragment analysis demonstrated a similar STR pattern
according to the STR patterns in a previous report (28).
Cytotoxicity measurements by fluoroscan
assay
The proliferative capacity of SCCOHT-1GFP
cells and BIN-67GFP cells in the presence and absence of
different chemotherapeutics and corresponding IC50
measurement was performed as described previously (29). Briefly, the two tumor cell lines
were incubated with different concentrations of talazoparib
(BMN-673), vorinostat (SAHA), and romidepsin (FK228) (all from
Selleck Chemicals LLC, Houston, TX, USA), respectively. For
fluorescence measurement 3,000 cells/well were seeded with standard
culture medium (100 μl/well) in flat bottom 96-well plates
(Nunc/Thermo Fischer Scientific, Roskilde, Denmark) and incubated
overnight to allow attachment. Thereafter, 100 μl of culture medium
was added to the cells as control and in further wells 100 μl of
culture medium containing the maximal solvent concentration was
added to the cells as solvent control, respectively. Moreover, 100
μl of the three chemotherapeutics were added to the cells and dosed
in a 3-fold serial dilution, respectively. Following incubation of
the cells with these chemotherapeutic compounds at different time
points up to 72 h, the medium was removed and the cells were lysed
with 5% (w/v) SDS. Afterwards, the fluorescence intensities of GFP
in the cell homogenate which corresponded to the appropriate cell
number of ovarian cancer cells was measured at excitation 485
nm/emission 520 nm using the Fluoroscan Ascent Fl(Thermo Fisher
Scientific).
Cell cycle analysis
Measurement of cell cycle distribution was performed
in 5×105 SCCOHT-1 cells and BIN-67 cells after culture
in the absence or presence of either 0.5 μM foretinib (GSK1363089;
PF-02341066) (Selleck Chemicals LLC), 20 nM FK228, or both drugs
together (0.5 μM foretinib + 20 nM FK228), respectively. Following
treatment of SCCOHT-1 cells and BIN-67 for different time
intervals, the cells were fixed in 70% (v/v) ice-cold ethanol at
4°C for 24 h. Thereafter, the fixed cells were stained with CyStain
DNA 2 step kit (Sysmex Europe GmbH, Norderstedt, Germany) and
filtered through a 50-μm filter. Flow cytometry analysis was
performed in a Galaxy FACSan (Partec) using FloMax analysis
software (Partec).
Immunohistochemical analysis of SCCOHT
mouse tumors
Immunohistochemistry was performed on an automated
staining instrument (Benchmark Ultra; Ventana, Tuscon, AZ, USA)
using the CC1 mild antigen retrieval procedure.
Paraformaldehyde-fixed thin tissue sections were stained for HE,
Ki67, and Elastin-van-Gieson.
Transcript analysis by RT-PCR
Total RNA was isolated from 3 control and 3 treated
SCCOHT-1 tumor tissues using RNeasy Mini kit (Qiagen, Hilden,
Germany) according to the manufacturer’s instructions. RNA (1 μg)
was reverse transcribed into cDNA using 500 μM of dNTP (R0193), 5
μM Oligo(dT)18 primer (S0132), 5 μM Random Hexan primer (S0142), 1
U RiboLock™ RNase Inhibitor (E00381) and 5 U RevertAid™ M-MuLV
Reverse Transcriptase (EP0441) in the supplied reaction buffer (all
reagents from Thermo Scientific, Schwerte, Germany). The cDNA
reactions were performed for 10 min/25°C, 1 h/37°C and stopped at
72°C for 10 min. As a template 2.5 μl of cDNA was used with
following specific primers (all primers customized by Eurofins, MWG
GmbH, Ebersberg, Germany): CD73 (sense, 5′-CGC AAC AAT GGC ACA ATT
AC-3′; antisense, 5′-CTC GAC ACT TGG TGC AAA GA-3′; amplification
product 241 bp) (25); CD90
(sense, 5′-GGA CTG AGA TCC CAG AAC CA-3′; antisense, 5′-ACG AAG GCT
CTG GTC CAC TA-3′; amplification product 124 bp) (25); human VEGF-A (sense, 5′-CCT CAG TGG
GCA CAC ACT CC-3′; antisense, 5′-CGA AAC CAT GAA CTT TCT GC-3′
amplification product 302 bp) (28); human cyclin D1 (sense, 5′-CTT CCT
CTC CAA AAT GCC AG-3′); antisense, 5′-AGA GAT GGA AGG GGG AAA GA-3′
amplification product 568 bp) (30); mouse VEGF-R2 (sense, 5′-ATA ACC TGG
CTG ACC CGA TTC-3′; antisense, 5′-TCG GTG ATG TAC ACG ATG CC-3′
amplification product 614 bp) (28); mouse laminin B2 (sense, 5′-AGA AGG
CAG AGA CAG TCC AAG C-3′; antisense, 5′-GTA TTG GTC ACC TAC TTG TTC
C-3′ amplification product 328 bp (31); mouse elastin (sense, 5′-ACT AAG CTG
GCT GGG CAT AC-3′; antisense, 5′-CCA AAG AGC ACA CCA ACA ATC-3′
amplification product 282 bp; mouse fibronectin-1 (sense, 5′-CAG
TGT TGG GCA ACA AAT GA-3′; antisense, 5′-GGCATGTGAGCTTAAAGCCA-3′
amplifications product 569 bp) (32); GAPDH as a control PCR (sense,
5′-ACC ACA GTC CAT GCC ATC AC-3′; antisense, 5′-TCC ACC ACC CTG TTG
CTG TA-3′; amplification product 452 bp).
PCR reactions included 0.2 μM of each primer, 200 μM
of dNTP (R0193, Thermo Scientific) and 0.05 U Taq DNA
polymerase (EPO402, Thermo Scientific) in the supplied reaction
buffer. PCR cycling conditions were performed 30 sec at 94°C, 1 min
at 60°C and 72°C for 1 min, respectively, including an initial 30
sec denaturation step at 94°C and a final 10-min extension step at
72°C (35 cycles). Aliquots of 25 μl of each RT-PCR product were
separated on a 2% agarose gel including the standard GeneRuler 100
bp DNA Ladder (Thermo Scientific) and visualized by GelRed™
(Biotium Inc., Hayward, CA, USA) staining.
In vivo experiments
Animal research using NODscid mice was
carried out by following internationally recognized guidelines on
animal welfare and was approved by the institutional licensing
committee ref. #33.14-42502-04-12/0814 on June 26th, 2012.
Approximately 3×106
SCCOHT-1GFP cells were injected subcutaneously into ~5
week-old-female NODscid mice. Within 10 days, all SCCOHT-1-injected
mice had developed small subcutaneous tumors. A therapeutic
approach was performed with a combined therapy of foretinib and
FK228. In detail, a systemic therapy was performed by oral
application of 200 μl foretinib (25 mg/kg) (Selleck Chemicals LLC)
dissolved in 30% (v/v) propylene glycol, 5% (v/v) Tween-80, and 65%
(v/v) of a 5% (w/v) dextrose solution in H2O. Oral
application was performed using plastic feeding tubes (18 ga × 30
mm) (Instech Laboratories, Plymouth, PA, USA) whereby the amount of
applied foretinib was half of that used in previous experiments
(28). Other in vivo mouse
experiments in recent studies were performed even at 3- to 4-fold
higher foretinib concentrations of 60–100 mg/kg (33) whereas this compound is administered
to patients between 3.6 and 4.5 mg/kg in clinical studies. In
addition to a daily foretinib treatment, the mice received an
intraperitoneal application of 20 μg FK228 dissolved in 500 μl of
0.9% NaCl from a DMSO stock solution with a final concentration of
0.74% (v/v) DMSO every 72 h. Control tumor-carrying mice received
200 μl of a daily oral application of 30% (v/v) propylene glycol,
5% (v/v) Tween-80, and 65% (v/v) of a 5% (w/v) dextrose solution in
H2O and an intraperitoneal application of 500 μl with
0.74% (v/v) DMSO in 0.9% NaCl every 72 h.
Following 10 days of therapy, 4 mice of the treated
tumors and 4 mice with a control tumor were sacrificed by cervical
dislocation. The GFP-positive tumors were dissected under UV light,
washed in PBS, weighed and either cultured in a primary explant
culture for chemosensitivity testing, or RNA extracted for
subsequent PCR analysis, or fixed in 4% glutardialdehyde solution
for histopathological evaluations.
Results
SCCOHT-1wt cells were developed in our
lab from a recurrent SCCOHT tumor biopsy and SCCOHT-1GFP
cells were generated from SCCOHT-1wt cells after
lentiviral GFP transduction and clonal selection. Functional
differences revealed an enhanced in vitro proliferative
capacity of SCCOHT-1GFP cells compared to
SCCOHT-1wt cells and a significantly accelerated in
vivo tumor development in NODscid mice (tumor within ~10
days/2×106 subcutaneously injected
SCCOHT-1GFP cells) compared to the corresponding
wild-type population (tumor after >8 weeks/2×106
subcutaneously injected SCCOHT-1wt cells). Since
reference cell lines are not available, an extended STR analysis
was performed to substantiate the fragment patterns of these unique
SCCOHT-1 populations. Therefore, SCCOHT-1wt cells were
tested together with SCCOHT-1GFP cells and various
tissues including PBMC, normal ovarian tissue and SCCOHT tumor
tissue from the originating patient. All 11 gene fragments and the
gender analysis revealed similar STR pattern in both cell
populations and the three tissues, respectively, with the exception
that one fragment of D13S317 remained undetectable in transfected
SCCOHT-1GFP cells (Table
I).
 | Table IComparative STR fragment analysis was
performed in SCCOHT-1 cells, SCCOHT-1GFP cells, and
different patient-originating tissues including PBMC, patient
normal tissue and patient SCCOHT tumor tissue. |
Table I
Comparative STR fragment analysis was
performed in SCCOHT-1 cells, SCCOHT-1GFP cells, and
different patient-originating tissues including PBMC, patient
normal tissue and patient SCCOHT tumor tissue.
| STR fragment |
SCCOHTwt |
SCCOHTGFP | Patient PBMC | Patient normal
tissue | Patient tumor
tissue |
|---|
| Penta D A1 | 12 | 12 | 12 | 12 | 12 |
| Penta D A2 | 15 | 15 | 15 | 15 | 15 |
| Penta E A1 | 10 | 10 | 10 | 10 | 10 |
| Penta E A2 | 13 | 13 | 13 | 13 | 13 |
| AMEL A1 | X-chr | X-chr | X-chr | X-chr | X-chr |
| AMEL A2 | X-chr | X-chr | X-chr | X-chr | X-chr |
| CSF1PO A1 | 10 | 10 | 10 | 10 | 10 |
| CSF1PO A2 | 11 | 11 | 11 | 11 | 11 |
| D13S317 A1 | 8 | | 8 | 8 | 8 |
| D13S317 A2 | 12 | 12 | 12 | 12 | 12 |
| D16S539 A1 | 9 | 9 | 9 | 9 | 9 |
| D16S539 A2 | 13 | 13 | 13 | 13 | 13 |
| D18S51 A1 | 13 | 13 | 13 | 13 | 13 |
| D18S51 A2 | 13 | 13 | 13 | 13 | 13 |
| D3S1358 A1 | 15 | 15 | 15 | 15 | 15 |
| D3S1358 A2 | 17 | 17 | 17 | 17 | 17 |
| D7S820 A1 | 10 | 10 | 10 | 10 | 10 |
| D7S820 A2 | 11 | 11 | 11 | 11 | 11 |
| D8S1179 A1 | 13 | 13 | 13 | 13 | 13 |
| D8S1179 A2 | 14 | 14 | 14 | 14 | 14 |
| TH01 A1 | 7 | 7 | 7 | 7 | 7 |
| TH01 A2 | 8 | 8 | 8 | 8 | 8 |
| TPOX A1 | 8 | 8 | 8 | 8 | 8 |
| TPOX A2 | 9 | 9 | 9 | 9 | 9 |
Loss of SMARCA4 (BRG1) and SMARCA2 (BRM) in SCCOHT
contributes to altered chromatin remodeling capabilities and tumor
growth which is also property of the corresponding cellular models.
Therefore, certain DNA-associated proteins were targeted by
appropriate inhibitors in the two only available cell lines for
this disease to date, known as SCCOHT-1 and BIN-67. Following
lentiviral GFP-labeling, the SCCOHT-1GFP and
BIN-67GFP cells were used in a fluorescence-based assay
and treated with different chemotherapeutic compounds including the
poly-ADP-ribose polymerase (PARP) inhibitor BMN-673 which prevents
PARP-mediated DNA repair of single strand DNA breaks via the
base-excision repair pathway and thereby enhances the accumulation
of DNA damage and promotes genomic instability. Moreover,
deacetylated histones are involved in reduced transcriptional
activities associated with enhanced DNA protection and
heterochromatin remodeling which can be blocked by appropriate
histon deacetylase (HDAC) inhibitors such as SAHA and FK228,
respectively. Both tumor cell populations demonstrated low
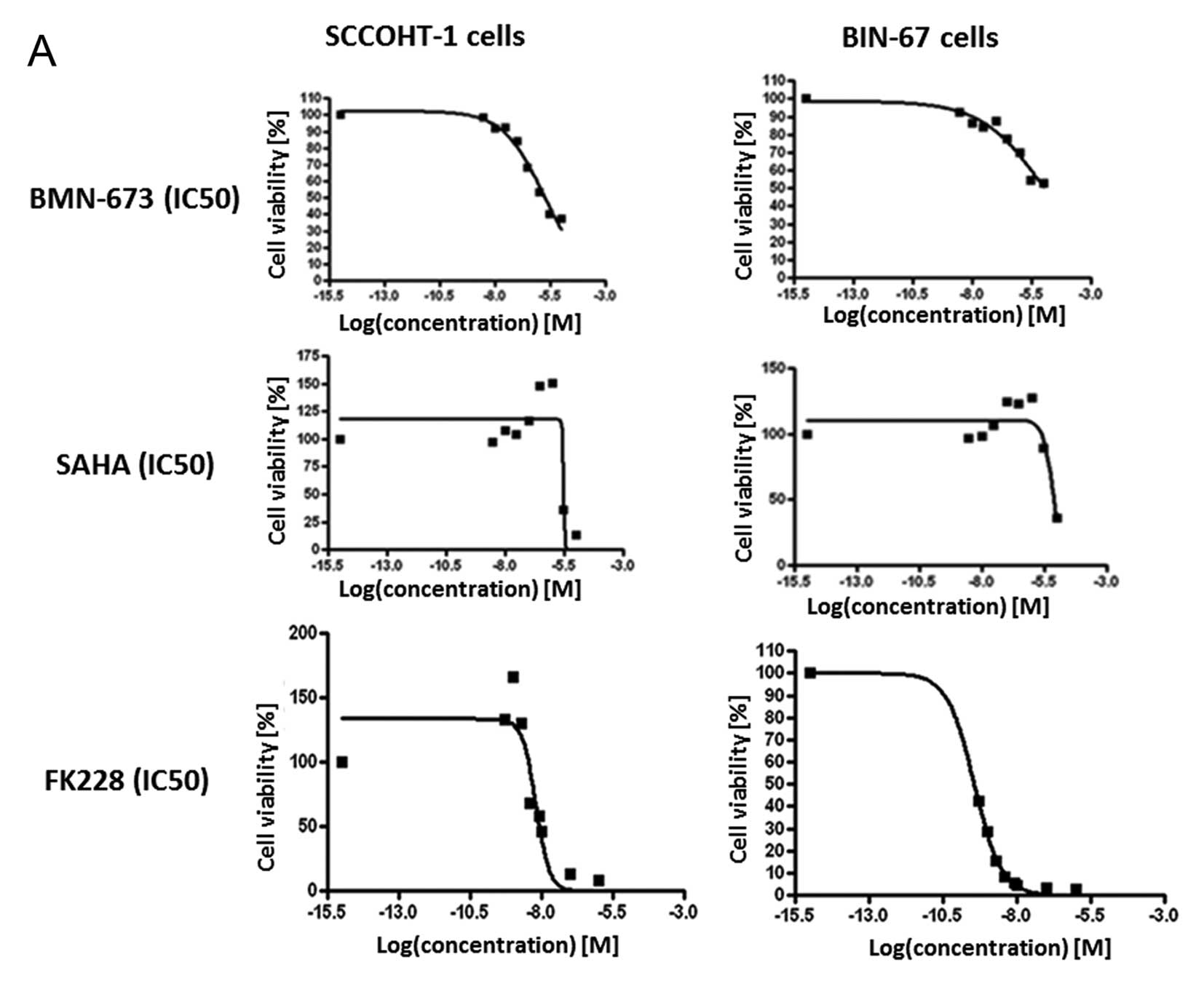
responsiveness to BMN-673 with an IC50 of
1.65×10−6 M for SCCOHT-1GFP cells (Fig. 1A, upper left panel) and an
IC50 of 1.16×10−5 M for BIN-67GFP
cells (Fig. 1A, upper right panel)
after 72 h. Likewise, high IC50 values were obtained for
SAHA with 2.86×10−6 M for SCCOHT-1GFP cells
(Fig. 1A, middle left panel) and
6.9×10−6 M for BIN-67GFP cells (Fig. 1A, middle right panel) after 72 h.
In contrast, a markedly elevated sensitivity of the tumor cells was
observed for the HDAC inhibitor FK228 with an IC50 of
6.29×10−9 M for SCCOHT-1GFP cells after 72 h
(Fig. 1A, lower left panel) and an
IC50 of 3.63×10−10 M for BIN-67GFP
cells after 144 h (Fig. 1A, lower
right panel).
Previous research has demonstrated effective tumor
regression of SCCOHT-induced mouse tumors by high calcium together
with the microtubule inhibitory epothilone B (29) or by the c-Met inhibitor foretinib
(28). Considering the promising
in vitro results for the HDAC inhibitor FK228 (Fig. 1A, lower panels), a combination of
these differentially functional antitumor agents was tested in a
fluorescence-based proliferation assay using the two GFP-labeled
SCCOHT cell lines. In these experiments, 0.5 μM foretinib was
applied with epothilone B (2 and 10 nM, respectively) and FK228 (20
and 100 nM, respectively) either alone, or in a two drug or three
drug combination for 24, 72 and 120 h, respectively, in
SCCOHT-1GFP cells and BIN-67GFP cells. One of
the most effective growth inhibition in both cell lines with low
drug concentration was observed with a combination of 0.5 μM
foretinib and 20 nM FK228 demonstrating synergistic effects as
compared to both compounds alone and revealed only 1.31±0.01% (n=3)
of proliferating SCCOHT-1GFP cells and 5.44±0.2% (n=3)
of proliferating BIN-67GFP cells after 120 h. This
growth inhibition of ~95% in BIN-67GFP cells and ~98% in
SCCOHT-1GFP cells was also confirmed by cell cycle
analysis. Stimulation of SCCOHT-1 cells with 0.5 μM foretinib for
≥72 h was associated with a G2/M accumulation and populations with
even more DNA content (>G2/M) compared to controls. In contrast,
SCCOHT-1 treatment with 20 nM FK228 or a combination of 0.5 μM
foretinib and 20 nM FK228 demonstrated an initial G2/M arrest with
subsequent accumulation of dead or dying cells in a sub-G1 phase
after 72 h (Fig. 1B, left
histograms). The slower proliferating BIN-67 cells were observed
for ≥144 h and exhibited an initial G2/M accumulation with 0.5 μM
foretinib after 48 h followed by G1 arrest and some cells in
sub-G1. Incubation of BIN-67 cells with 20 nM FK228 or a
combination of 0.5 μM foretinib and 20 nM FK228, however, was
accompanied by little if any detectable cells in the proliferative
cell cycle and an accumulation of dead or dying cells in a sub-G1
phase after 144 h (Fig. 1B, right
histograms). Together, these data demonstrated an efficient in
vitro tumor cell killing of >95% by a combination of 0.5 μM
foretinib with 20 nM FK228.
To confirm these findings in vivo,
SCCOHT-1GFP-induced tumors in NODscid mice were treated
with a combination of an intraperitoneal FK228 application every 72
h and a daily oral foretinib application over a period of 10 days.
Simultaneously, control mice were treated with the appropriate
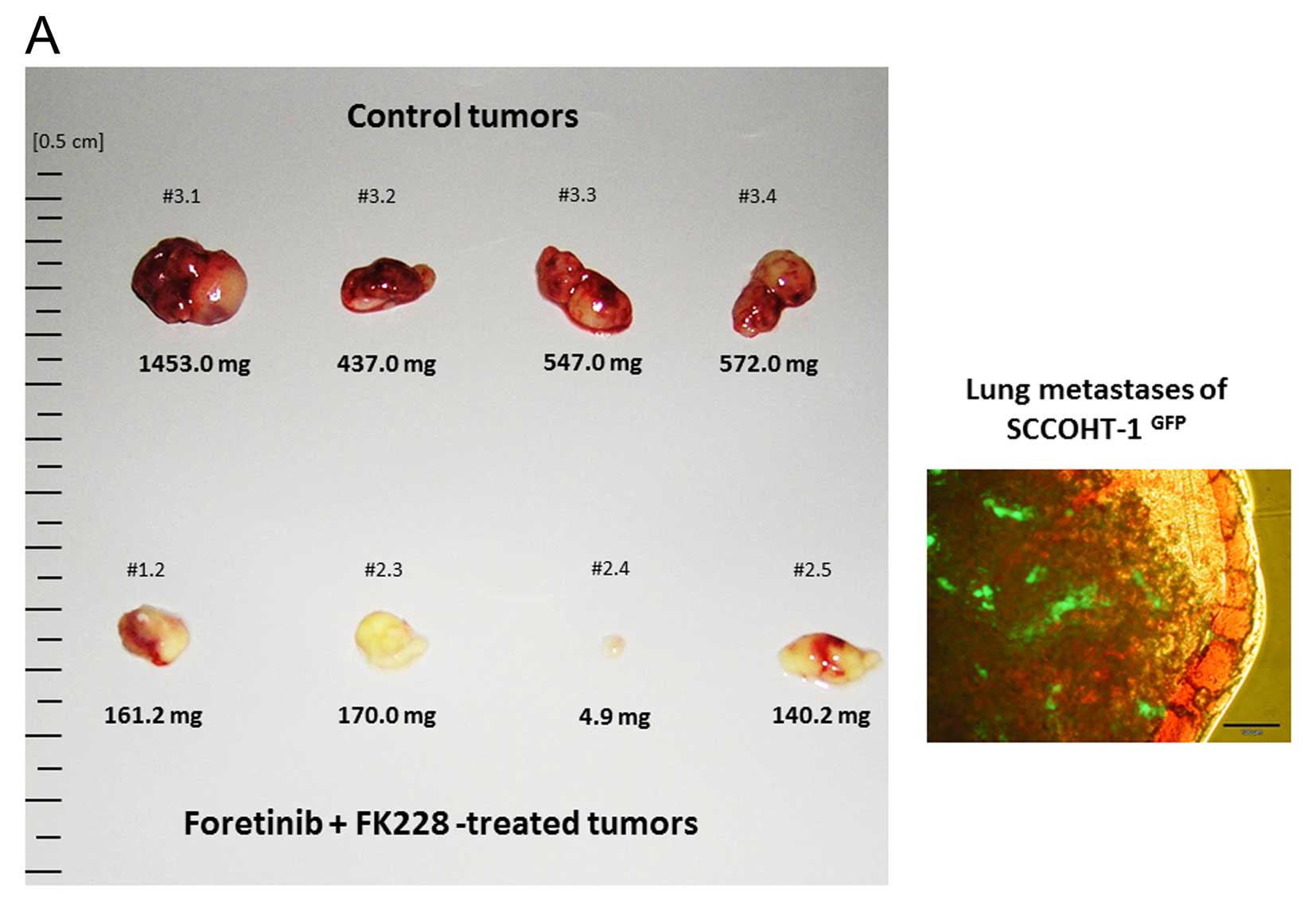
solvents. Although tumor size was significantly reduced in the
combined treatment (Fig. 2A, left
panel), distant organ metastasis could be observed as documented by
the distribution of green fluorescence from SCCOHT-1GFP
tumor cells within lung tissue (Fig.
2A, right panel). Whereas five mice died in the course of the
experiment for unknown reasons, there was little if any change in
the body weight of the control mice or during chemotherapeutic
application within the 10 days of treatment. However, a 6.3-fold
reduction in tumor weight was measured after treatment with
~0.752±0.408 g (n=4) in control tumors compared to 0.119±0.067 g
(n=4) in foretinib/FK228-exposed tumors (Fig. 2B). Likewise, the relation of tumor
weight to mouse weight revealed 3.763±1.727% (n=4) in control
tumors compared to 0.675±0.386% (n=4) in foretinib/FK228-treated
tumors which displayed a 5.6-fold tumor reduction (Fig. 2C).
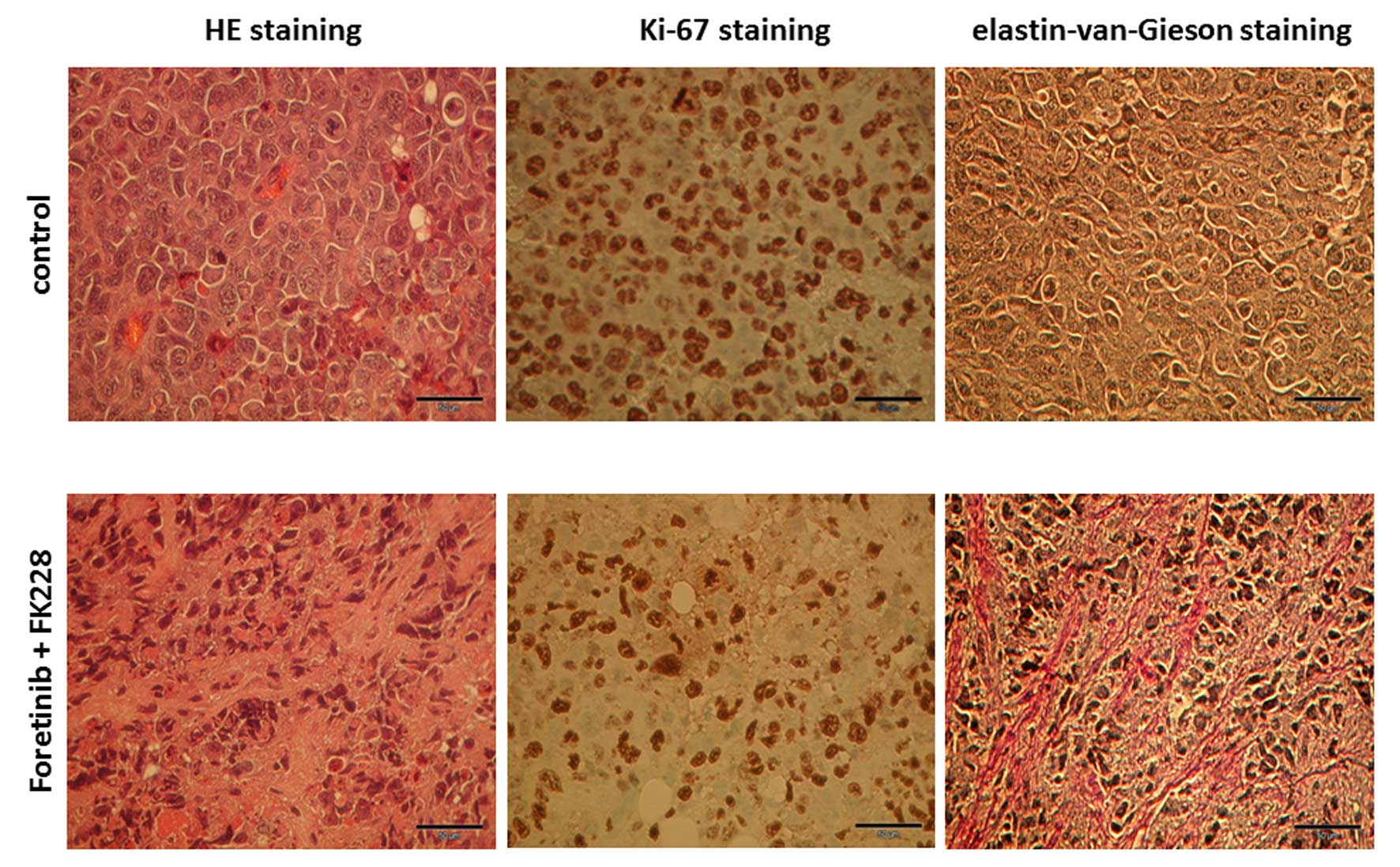
Histopathological evaluation of the mouse tumors by
hematoxylin/eosin (HE) revealed an increased vascularization of
control tumors compared to foretinib/FK228 treatment (Fig. 3, left panels). Likewise, expression
of the proliferation marker Ki-67 was markedly enhanced in control
tumors (Fig. 3, middle panels). In
contrast, a significant amount of extracellular matrix structures
with positive staining for elastin-van-Gieson appeared
predominantly in the foretinib/FK228-treated tumor (Fig. 3, right panels).
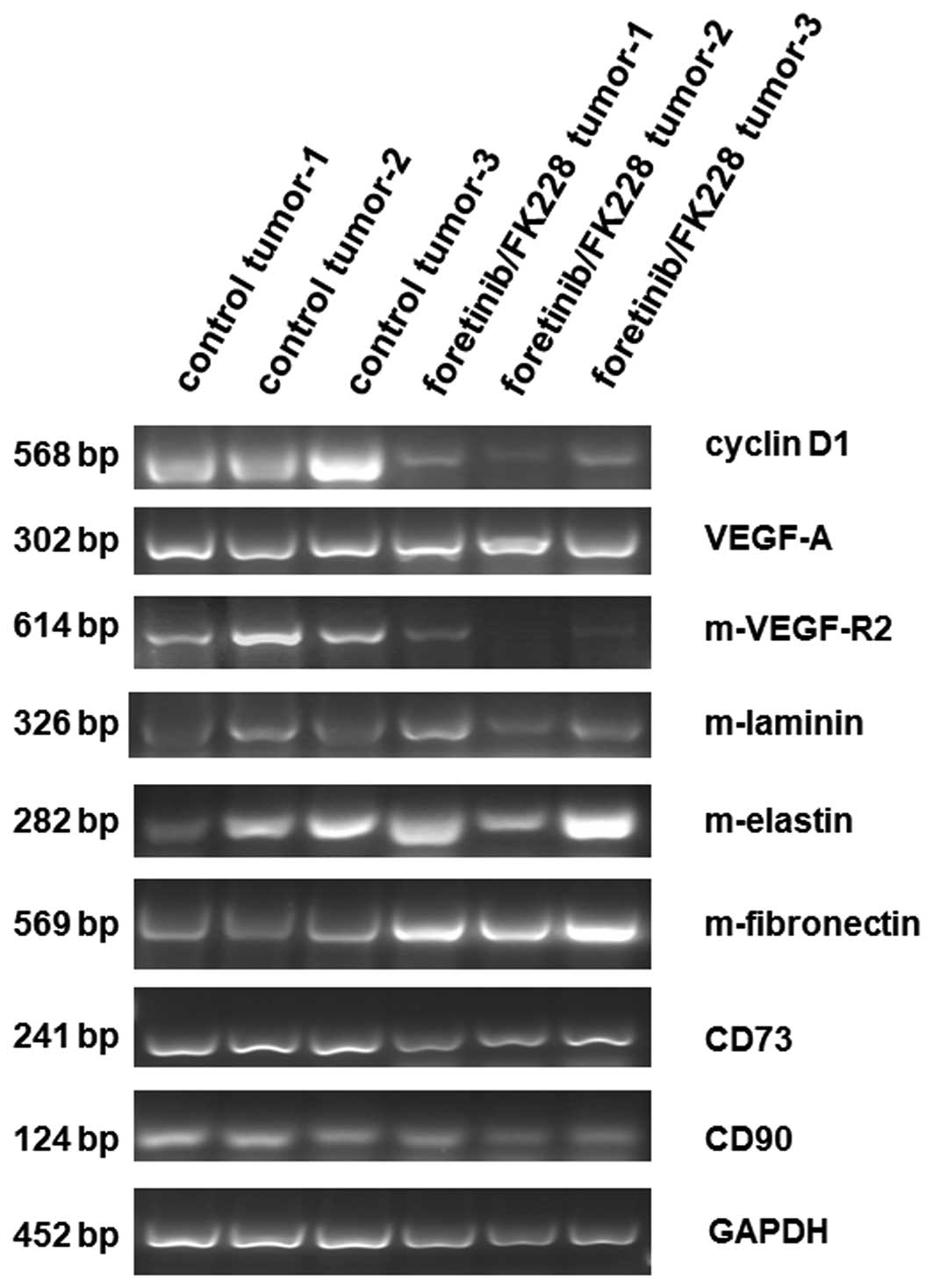
Transcripts of three different control tumors were
compared to three foretinib/FK228-treated mouse tumors (Fig. 4). Whereas the cell cycle associated
cyclin D1 was down-modulated in each of the three treated tumors,
there was little if any difference in the mRNA levels of the
vascular endothelial growth factor-A (VEGF-A). With respect to
mouse-specific gene expression derived from appropriate mouse MSC
and further cell populations within the tumor microenvironment, a
significant down-modulation of the murine vascular endothelial
growth factor receptor-2 (m-VEGF-R2) was detectable in the treated
mouse tumors. Vice versa, mRNAs of various extracellular matrix
proteins including laminin, elastin, and fibronectin were
upregulated in the mouse tissue of foretinib/FK228-treated tumors
suggesting enhanced expression of extracellular matrix structures
by cells of the tumor microenvironment where MSC play a major role.
Indeed, similar levels of the MSC-specific markers CD73 and CD90
were detectable in all tumor specimens. Unaltered expression levels
of GAPDH served as a loading control (Fig. 4).
Based upon these data, a potential direct
contribution of MSC to changes in extracellular matrix protein
expression and a possible role of MSC to protect the tumor cells
were investigated by appropriate western blots and co-culture
experiments.
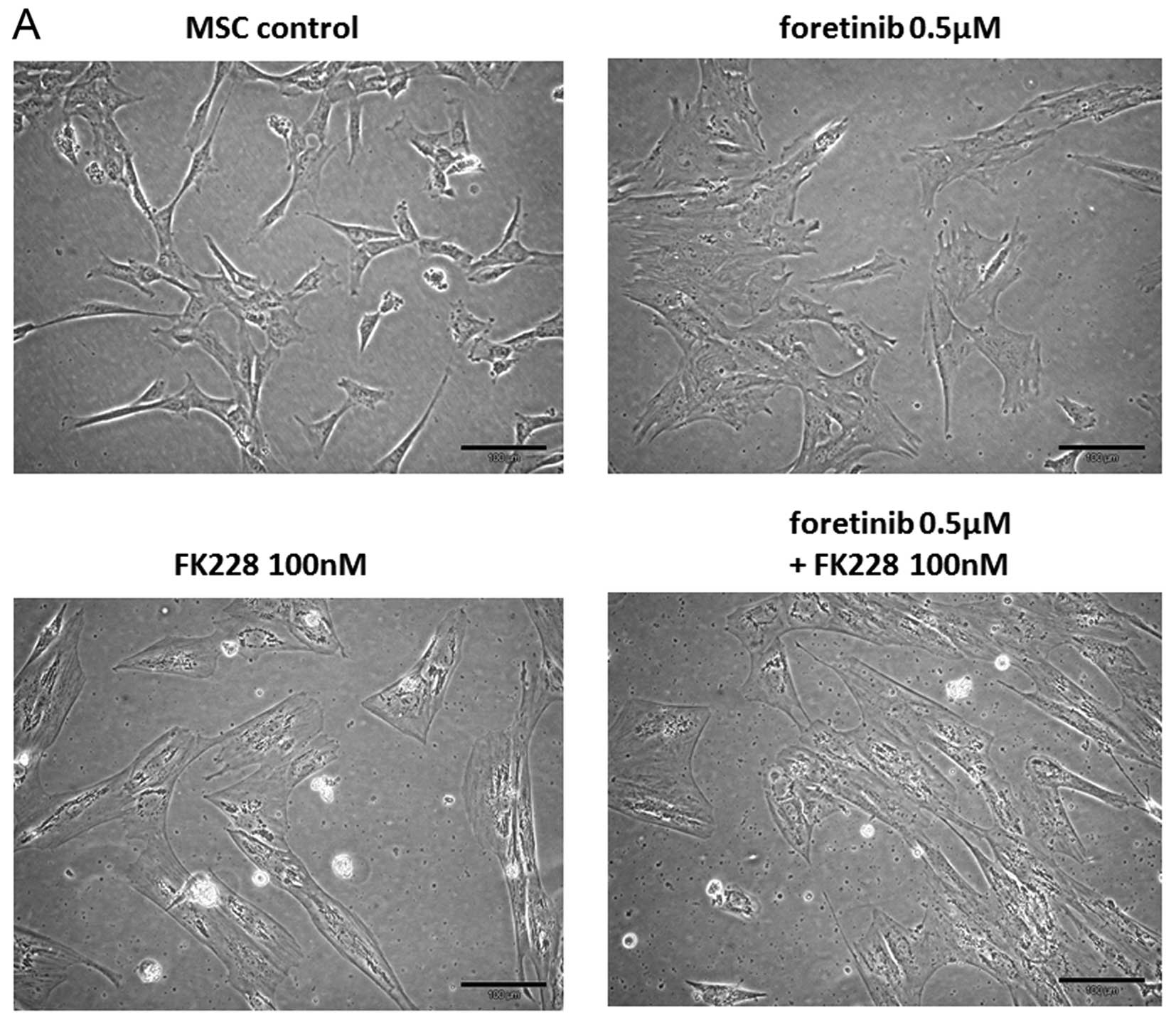
Exposure of MSC cultures to 0.5 μM foretinib for 72
h revealed a similar morphology of a singularized and differently
shaped cell culture compared to untreated control cells. In
contrast, treatment of MSC with either 100 nM FK228 alone or a
combination of both drugs foretinib/FK228 was associated with a
formation of paralleled cell aggregates displaying fibroblast-like
extensions and a marked accumulation of cell-associated filament
structures (Fig. 5A).
Western blot analysis demonstrated a reduced
expression of the matrix-restructuring enzyme MMP-9 after treatment
with 0.5 μM foretinib, 100 nM FK228 or a combination of both.
Little if any change was observed in the expression of structural
proteins of the connective tissue within the extracellular matrix
including fibronectin and laminin in MSC and foretinib-exposed MSC.
However, treatment of MSC with FK228 or a combination of foretinib
with FK228 was associated with enhanced expression of fibronectin
and laminin. Unaltered levels of GAPDH served as loading control
(Fig. 5B).
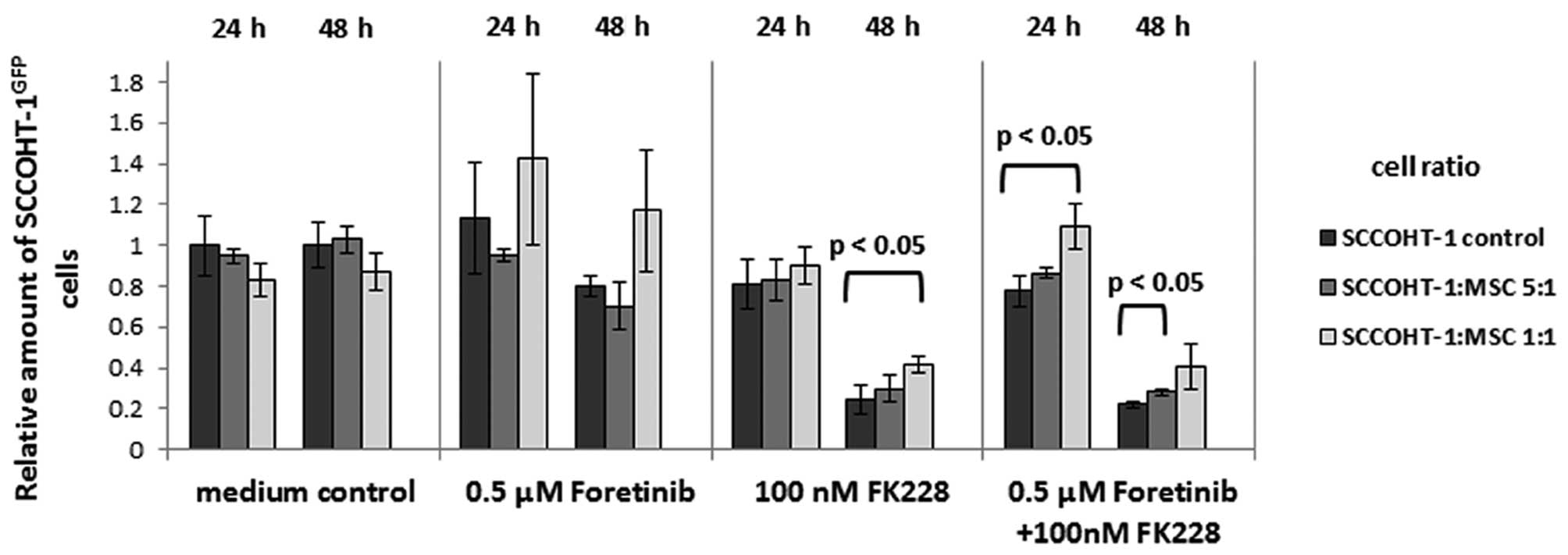
A co-culture of MSC with SCCOHT-1GFP
cells at a cell ratio of 1:5 revealed a marked increase in the
amount of tumor cells compared to a corresponding
SCCOHT-1GFP monoculture in the presence of 100 nM FK228
after 24 and 48 h. Similar results were obtained after exposure of
the mono- and co-cultures to a combination of 0.5 μM foretinib +
100 nM FK228 (Fig. 6). These
effects of an elevated amount of tumor cells in the
chemotherapeutics-treated co-culture compared to the
SCCOHT-1GFP mono-culture was even more pronounced in the
presence of more MSC with a cell ratio of 1:1 (Fig. 6). Together, these data demonstrated
that FK228- or foretinib/FK228-treated MSC undergo morphological
changes which was associated with alterations of the
microenvironment by enhanced expression of structural proteins such
as laminin and fibronectin. Moreover, MSC provide a certain
protection for the tumor cells against cytotoxic effects of these
chemotherapeutics.
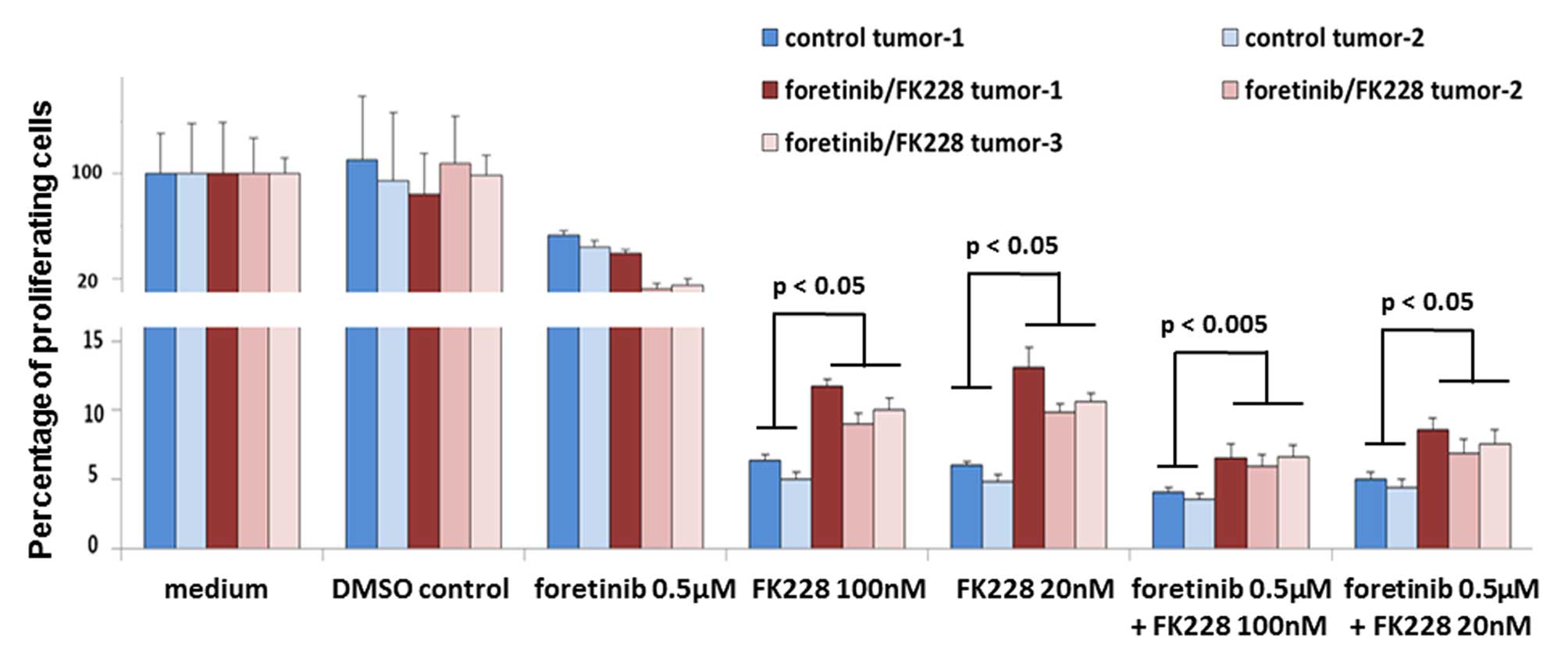
To further substantiate these effects of the tumor
microenvironment in altering the chemoreactivity of SCCOHT-1 cells
during tumor development, explant cultures were performed from two
control tumors and three foretinib/FK228-treated mouse tumors and
the derived cells were exposed in vitro to foretinib and
FK228 alone or in combination. Thus, the sensitivity to foretinib
alone was increased in two of the three treated tumors. In
contrast, incubation of explant cells from all three previously
treated tumors with 20 nM FK228, 100 nM FK228, or corresponding
combinations with 0.5 μM foretinib was always associated with a
significantly elevated chemoresistance by ~100% compared to explant
cells from previously untreated mouse tumors (Fig. 7).
Discussion
To date, little knowledge is available on a
successful therapy of rare tumors such as SCCOHT which is very
unsatisfactory for the respective patients. Previous research has
demonstrated some favorable results with epothilone B and higher
calcium concentrations (29) as
well as with foretinib as a multi-kinase inhibitor of c-Met and
VEGF-Rs (28,33).
A more promising approach by in vitro testing
of various chemotherapeutic compounds alone and in combination
revealed an efficient tumor cell killing of >95% in SCCOHT-1 and
BIN-67 cells following treatment with a combination of foretinib
and FK228. Both compounds are part of clinical studies,
respectively, whereby a combination of these drugs displays
multiple effects. Foretinib inhibits c-Met signaling in SCCOHT-1
cells (28) whilst overexpression
of c-Met and a paralleled hyperactivation in ovarian cancers
correlates with poor prognosis by triggering tumor growth,
metastasis and angiogenesis (34).
In addition, FK228 predominantly targets tumor cells by interacting
with the binding pocket of Zn-dependent histone deacetylase to
block this enzyme activity and accordingly heterochromatin
formation. In vivo treatment with foretinib/FK228 reduced
angiogenesis and cell growth of SCCOHT-1 tumor xenografts by
diminished detection of capillaries and Ki-67 positive cells which
was also substantiated by down-modulation of the
proliferation-associated cyclin D1 and the mVEGF-R2 involved in
neo-vascularization. Inhibition of VEGF-R2 signaling by foretinib
has been reported (33) and
constitutive VEGF expression by SCCOHT-1-induced tumor xenografts
paralleled by a down-modulation of the murine VEGF-R2 in
foretinib/FK228-treated mice also indicated selective signaling
blockage of cells within the tumor micro-environment rather than
the tumor cells themselves.
Unfortunately however, little if any synergistic
effects were observed with the tumor cell targeting compound FK228
in vivo, which left the combination of foretinib/FK228 below
expectations by demonstrating much less effective tumor mass
reduction as compared to previous studies with the c-Met inhibitor
alone and by the simultaneous detection of lung metastases. In
fact, the combination of foretinib and FK228 with an ~6-fold
reduction in tumor mass was less efficient compared to foretinib
alone in previous studies which displayed a >10-fold reduction
of SCCOHT-1-induced tumor xenografts (28) suggesting certain resistance and/or
some protective mechanisms induced during in vivo
application.
Supportive evidence to follow this hypothesis was
presented by the altered histopathology of foretinib/FK228-treated
tumors with elevated expression of elastin van Gieson-positive
structures in the tumor stroma. Moreover, analysis of the tumor
tissue revealed enhanced expression of structural and connective
tissue protein products such as laminin, elastin, and fibronectin.
Whereas the majority of these extracellular matrix proteins and
predominantly fibronectin is thought to be produced by MSC and
stromal fibroblasts in the tumor microenvironment, analysis of the
mouse tumor tissue also confirmed the expression of MSC-like
markers including CD73 and CD90 which suggests an important role of
MSC during foretinib/FK228-mediated SCCOHT-1 tumor cell protection.
Indeed, potential protection of SCCOHT-1 cells was substantiated
in vitro by MSC-induced expression of laminin and
fibronectin after incubation with foretinib/FK228. In this context,
previous research has demonstrated a protective role of MSC for
cervical tumor cells (35).
Interaction of MSC with ovarian cancer cells also contributed to
increased metastatic ability (36). More importantly, co-cultures of MSC
with SCCOHT-1GFP cells revealed a significantly elevated
resistance of the tumor cells against the cytotoxic action of FK228
alone or a combination of foretinib/FK228 when compared to a
SCCOHT-1GFP mono-culture which further confirmed
tumor-protective mechanisms of MSC. In addition, this MSC-mediated
protection of SCCOHT-1GFP cells was also observed in
co-cultures of MSC populations with the tumor cells, whereby
increasing protective effects correlated with a corresponding
elevated cell ratio of MSC. Finally, increased drug resistance was
detectable in previously foretinib/FK228-treated compared to
untreated SCCOHT-1 explant tumor xenografts after repeated exposure
to FK228 alone or a combination of foretinib/FK228.
Summary and outlook
Taken together, these findings demonstrated a
tumor-protective role of MSC in SCCOHT tumors which would be in
agreement with the conceptually natural functionality of MSC to
generally support tissue repair and regeneration (37).
In contrast to very promising data of substantial
tumor cell killing in SCCOHT tumor cell mono-culture assays in
vitro, a markedly reduced effect was achieved by the
chemotherapeutics in a multi-cell population microenvironment in
vitro and in vivo. These discrepancies are linked to
tumor cell protection by concomitant activation of particularly MSC
within the tumor stroma. Consequently, a more effective therapeutic
approach necessitates the application of an increased drug dosage
in vivo to overcome the MSC-mediated protection of tumor
cells. However, this also includes the risk of simultaneously
increased side effects in treated patients. Therefore,
drug-mediated alterations of tumor microenvironment-associated
aberrant MSC with subsequent tumor cell protection may represent a
more general phenomenon which exposes the tumor stroma as
additional target for more focused chemo-therapeutic
effectiveness.
Acknowledgements
We thank Dr Petar Jelinic for providing initial
substances. Yuanyuan Yang is supported by a postdoctoral fellowship
from the DAAD and CSC. This study was supported by a grant from the
Erich und Gertrud Roggenbuck-Stiftung for Cancer Research to Ralf
Hass.
References
|
1
|
Dickersin GR, Kline IW and Scully RE:
Small cell carcinoma of the ovary with hypercalcemia: A report of
eleven cases. Cancer. 49:188–197. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ulbright TM, Roth LM, Stehman FB, Talerman
A and Senekjian EK: Poorly differentiated (small cell) carcinoma of
the ovary in young women: Evidence supporting a germ cell origin.
Hum Pathol. 18:175–184. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McCluggage WG, Oliva E, Connolly LE,
McBride HA and Young RH: An immunohistochemical analysis of ovarian
small cell carcinoma of hypercalcemic type. Int J Gynecol Pathol.
23:330–336. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jelinic P, Mueller JJ, Olvera N, Dao F,
Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA, et al:
Recurrent SMARCA4 mutations in small cell carcinoma of the ovary.
Nat Genet. 46:424–426. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Witkowski L, Carrot-Zhang J, Albrecht S,
Fahiminiya S, Hamel N, Tomiak E, Grynspan D, Saloustros E, Nadaf J,
Rivera B, et al: Germline and somatic SMARCA4 mutations
characterize small cell carcinoma of the ovary, hypercalcemic type.
Nat Genet. 46:438–443. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramos P, Karnezis AN, Craig DW, Sekulic A,
Russell ML, Hendricks WP, Corneveaux JJ, Barrett MT, Shumansky K,
Yang Y, et al: Small cell carcinoma of the ovary, hypercalcemic
type, displays frequent inactivating germline and somatic mutations
in SMARCA4. Nat Genet. 46:427–429. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karnezis AN, Wang Y, Ramos P, Hendricks
WP, Oliva E, D’Angelo E, Prat J, Nucci MR, Nielsen TO, Chow C, et
al: Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and
SMARCA2/BRM is highly sensitive and specific for small cell
carcinoma of the ovary, hypercalcaemic type. J Pathol. 238:389–400.
2016. View Article : Google Scholar :
|
|
8
|
Kadoch C, Hargreaves DC, Hodges C, Elias
L, Ho L, Ranish J and Crabtree GR: Proteomic and bioinformatic
analysis of mammalian SWI/SNF complexes identifies extensive roles
in human malignancy. Nat Genet. 45:592–601. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Foulkes WD, Clarke BA, Hasselblatt M,
Majewski J, Albrecht S and McCluggage WG: No small surprise - small
cell carcinoma of the ovary, hypercalcaemic type, is a malignant
rhabdoid tumour. J Pathol. 233:209–214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Witkowski L, Goudie C, Foulkes WD and
McCluggage WG: Small-cell carcinoma of the ovary of hypercalcemic
type (malignant rhabdoid tumor of the ovary): A review with recent
developments on pathogenesis. Surg Pathol Clin. 9:215–226. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johann PD, Erkek S, Zapatka M, Kerl K,
Buchhalter I, Hovestadt V, Jones DT, Sturm D, Hermann C, Segura
Wang M, et al: Atypical teratoid/rhabdoid tumors are comprised of
three epigenetic subgroups with distinct enhancer landscapes.
Cancer Cell. 29:379–393. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Witkowski L, Goudie C, Ramos P, Boshari T,
Brunet JS, Karnezis AN, Longy M, Knost JA, Saloustros E, McCluggage
WG, et al: The influence of clinical and genetic factors on patient
outcome in small cell carcinoma of the ovary, hypercalcemic type.
Gynecol Oncol. 141:454–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Otte A, Göhring G, Steinemann D,
Schlegelberger B, Groos S, Länger F, Kreipe HH, Schambach A,
Neumann T, Hillemanns P, et al: A tumor-derived population
(SCCOHT-1) as cellular model for a small cell ovarian carcinoma of
the hypercalcemic type. Int J Oncol. 41:765–775. 2012.PubMed/NCBI
|
|
14
|
Gamwell LF, Gambaro K, Merziotis M, Crane
C, Arcand SL, Bourada V, Davis C, Squire JA, Huntsman DG, Tonin PN,
et al: Small cell ovarian carcinoma: Genomic stability and
responsiveness to therapeutics. Orphanet J Rare Dis. 8:332013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pittenger MF, Mackay AM, Beck SC, Jaiswal
RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S and
Marshak DR: Multilineage potential of adult human mesenchymal stem
cells. Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caplan AI: Why are MSCs therapeutic? New
data: New insight. J Pathol. 217:318–324. 2009. View Article : Google Scholar
|
|
17
|
Hass R, Kasper C, Böhm S and Jacobs R:
Different populations and sources of human mesenchymal stem cells
(MSC): A comparison of adult and neonatal tissue-derived MSC. Cell
Commun Signal. 9:122011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bianco P: ‘Mesenchymal’ stem cells. Annu
Rev Cell Dev Biol. 30:677–704. 2014. View Article : Google Scholar
|
|
19
|
Friedl P and Alexander S: Cancer invasion
and the microenvironment: Plasticity and reciprocity. Cell.
147:992–1009. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ungefroren H, Sebens S, Seidl D, Lehnert H
and Hass R: Interaction of tumor cells with the microenvironment.
Cell Commun Signal. 9:182011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hass R and Otte A: Mesenchymal stem cells
as all-round supporters in a normal and neoplastic
microenvironment. Cell Commun Signal. 10:262012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karnoub AE, Dash AB, Vo AP, Sullivan A,
Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R and Weinberg
RA: Mesenchymal stem cells within tumour stroma promote breast
cancer metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mandel K, Yang Y, Schambach A, Glage S,
Otte A and Hass R: Mesenchymal stem cells directly interact with
breast cancer cells and promote tumor cell growth in vitro and in
vivo. Stem Cells Dev. 22:3114–3127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang Y, Otte A and Hass R: Human
mesenchymal stroma/stem cells exchange membrane proteins and alter
functionality during interaction with different tumor cell lines.
Stem Cells Dev. 24:1205–1222. 2015. View Article : Google Scholar :
|
|
25
|
Yang Y, Bucan V, Baehre H, von der Ohe J,
Otte A and Hass R: Acquisition of new tumor cell properties by
MSC-derived exosomes. Int J Oncol. 47:244–252. 2015.PubMed/NCBI
|
|
26
|
Upchurch KS, Parker LM, Scully RE and
Krane SM: Differential cyclic AMP responses to calcitonin among
human ovarian carcinoma cell lines: a calcitonin-responsive line
derived from a rare tumor type. J Bone Miner Res. 1:299–304. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Y, Melzer C, Bucan V, von der Ohe J,
Otte A and Hass R: Conditioned umbilical cord tissue provides a
natural three-dimensional storage compartment as in vitro stem cell
niche for human mesenchymal stroma/stem cells. Stem Cell Res Ther.
7:282016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Otte A, Rauprich F, von der Ohe J, Yang Y,
Kommoss F, Feuerhake F, Hillemanns P and Hass R: c-Met inhibitors
attenuate tumor growth of small cell hypercalcemic ovarian
carcinoma (SCCOHT) populations. Oncotarget. 6:31640–31658.
2015.PubMed/NCBI
|
|
29
|
Otte A, Rauprich F, Hillemanns P,
Park-Simon TW, von der Ohe J and Hass R: In vitro and in vivo
therapeutic approach for a small cell carcinoma of the ovary
hypercalcaemic type using a SCCOHT-1 cellular model. Orphanet J
Rare Dis. 9:1262014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsikitis M, Zhang Z, Edelman W, Zagzag D
and Kalpana GV: Genetic ablation of Cyclin D1 abrogates genesis of
rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci USA.
102:12129–12134. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mazaud Guittot S, Vérot A, Odet F, Chauvin
MA and le Magueresse-Battistoni B: A comprehensive survey of the
laminins and collagens type IV expressed in mouse Leydig cells and
their regulation by LH/hCG. Reproduction. 135:479–488. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu F, Ma FF, Zhang W, Li Y, Wei FY and
Zhou L: Qualitative research of alternatively splice variants of
fibronectin in different development stage of mice heart. J Thorac
Dis. 7:2307–2312. 2015.
|
|
33
|
Zillhardt M, Park SM, Romero IL, Sawada K,
Montag A, Krausz T, Yamada SD, Peter ME and Lengyel E: Foretinib
(GSK1363089), an orally available multikinase inhibitor of c-Met
and VEGFR-2, blocks proliferation, induces anoikis, and impairs
ovarian cancer metastasis. Clin Cancer Res. 17:4042–4051. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Parr C and Jiang WG: Expression of
hepatocyte growth factor/scatter factor, its activator, inhibitors
and the c-Met receptor in human cancer cells. Int J Oncol.
19:857–863. 2001.PubMed/NCBI
|
|
35
|
Montesinos JJ, Mora-García ML, Mayani H,
Flores-Figueroa E, García-Rocha R, Fajardo-Orduña GR,
Castro-Manrreza ME, Weiss-Steider B and Monroy-García A: In vitro
evidence of the presence of mesenchymal stromal cells in cervical
cancer and their role in protecting cancer cells from cytotoxic T
cell activity. Stem Cells Dev. 22:2508–2519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lis R, Touboul C, Halabi NM, Madduri AS,
Querleu D, Mezey J, Malek JA, Suhre K and Rafii A: Mesenchymal cell
interaction with ovarian cancer cells induces a background
dependent prometastatic transcriptomic profile. J Transl Med.
12:592014. View Article : Google Scholar
|
|
37
|
Melzer C, Yang Y and Hass R: Interaction
of MSC with tumor cells. Cell Commun Signal. 14:202016. View Article : Google Scholar : PubMed/NCBI
|