Introduction
Chondrosarcoma is the second most commonly occurring
primary bone tumor (1–3). At present, wide-margin surgical
resection remains the main curative method of treatment for
chondrosarcoma, which continues to have poor prognosis due to
resistance to conventional chemo- and radiotherapy (4–6).
Consequently, there exists a critical clinical need to explore
novel molecular-targeted therapies for this disease.
The transcription factor signal transducer and
activator of transcription 3 (Stat3) is a point of convergence for
numerous oncogenic and inflammatory signaling pathways, including
cytokines, growth factors and oncogenes (7–9).
Stat3 participates in cell growth and survival regulating cell
proliferation, apoptosis and autophagy (9,10).
Constitutive activation of Stat3 represents a key player in tumor
angiogenesis and metastasis and has been observed in other cancers
of the bone including osteosarcoma (10–15).
As such, Stat3 has emerged as a promising molecular target for
cancer therapy.
Bortezomib, formerly known as PS341, is a selective
proteasome inhibitor, which has been approved by the US Food and
Drug Administration for the treatment of patients with multiple
myeloma and mantle cell lymphoma (16,17).
Recently it was indicated that bortezomib also has crucial
antineoplastic activity against some solid tumors, including
ovarian and prostate cancers as well as squamous cell cancer of the
head and neck, Ewing's sarcoma and osteosarcoma. Preclinical data
have suggested that bortezomib's antineoplastic effect occurs via
diverse mechanisms, exerting significantly different roles in a
tissue and cancer-specific context (18–21).
Moreover, it has been shown that inactivation of Stat3 plays an
important role in bortezomib-mediated treatment of myeloma
(22). However, the effects of
bortezomib on human chondrosarcoma have not been adequately studied
and, in particular, the mechanisms by which bortezomib exerts its
antitumor functions in human chondrosarcoma have not been
previously investigated.
While the majority of published studies on the
antineoplastic actions of bortezomib have focused on suppression of
proliferation and promotion of apoptosis (17,19),
little is known about the effects of bortezomib on modulating the
cell cycle progression and metastasis in cancer.
In the present study, we investigated the in
vitro and in vivo effects of bortezomib on human
chondrosarcoma. In particular, we demonstrated the importance of
Stat3 signaling for the antitumor actions of this agent, including
apoptosis promotion, cell cycle arrest, inhibition of migration and
invasion. Thus, we report on the therapeutic potential of
bortezomib on human chondrosarcoma, based on our improved
understanding of this agent.
Materials and methods
Animals and ethical statement
Six-week-old BALB/c female athymic nude mice were
obtained (Vital River Laboratories Co., Ltd., Beijing, China). The
present study was carried out in accordance with the
recommendations in the Guide for the Chinese Ethics Review
Committees. The protocol was approved by the Ethics Committee of
Peking University People's Hospital. All experiments was carried
out under the ethics approval of Peking University People's
Hospital and was conducted according to the National Institutes of
Health guidelines. Nude mice were maintained under specific
pathogen-free (SPF) conditions, and bedding, water and daily
rations were sterilized.
Tissue specimen
The paraffin-embedded pathological specimens from 12
patients with chondrosarcoma and adjacent non-tumor tissues were
obtained from the Department of Pathology and the Musculoskeletal
Tumor Center, Peking University Peoples Hospital (Beijing, China).
Informed consents (written in the light of the ethical guidelines)
were obtained from all the patients. All human specimens were
approved by the Research Ethics Committee of Peking University
Peoples Hospital (Beijing, China).
Cell culture, cell viability assay and
colony formation assay
The human articular chondrocyte cell line HC-a
(Sciencell Research Laboratories, Carlsbad, CA, USA) was maintained
in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15%
fetal bovine serum (FBS), plus antibiotics. SW1353 cells were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) and were maintained in L-15 medium (Gibco, Grand Island,
NY, USA). OUMS-27 cells, HCS-2/8 cells and JJ012 cells were kindly
gifted from Dr J. Block (Rush Medical College, Chicago, IL, USA)
and were cultured in DMEM (HyClone Laboratories, Inc., Logan, UT,
USA) supplemented with 10% fetal calf serum (FCS; Gibco) at 37°C in
a humidified atmosphere with 5% CO2. All experiments
were performed during the exponential phase of cell growth. Cell
viability was assessed using the CCK-8 assay as previously
described (23). Cells were plated
in 96-well plates at a density of 5,000 cells in 100 µl
medium/well one day before the experiment. The cells were treated
with bortizomib (Sigma-Aldrich Chemical, Co., St. Louis, MO, USA)
with indicated condition, which included gene knockdown. The cell
viability was examined by CCK-8 assay. For colony formation assay,
following treatment, adherent cells were trypsinized and 1,000
viable cells were subcultured in 6-well plates (in triplicate).
Cells were allowed to adhere and colonize for 14 days. Media were
removed and cells were fixed in methyl alcohol for 15 min and
stained with crystal violet staining solution for visualizing
colonies, as previously described (23).
Western blot analysis
Equal amounts of proteins collected from cell
lysates were loaded on 10–15% SDS-PAGE gels using a NuPAGE system
(Invitrogen, Carlsbad, CA, USA) and then transferred onto PVDF
membranes as previously described (24). The following antibodies were used
in the experiments: anti-p-STAT3, anti-STAT3, anti-c-Myc,
anti-E-cadherin, anti-vimentin, anti-cyclin D1, anti-Bax,
anti-Bcl-2, and anti-GAPDH were all from Cell Signaling Technology
(Beverly, MA, USA). Anti-N-cadherin, anti-Slug, anti-MMP9 and
anti-CD44 were from Abcam (Cambridge, MA, USA).
Immunohistochemistry
Paraffin sections were reacted with rabbit
polyclonal anti-p-Stat3, anti-vimentin, anti-N-cadherin and
anti-E-cadherin antibodies (1:200 dilution). Sections stained with
non-immune rabbit serum (1:200 dilution) in phosphate-buffered
saline (PBS) instead of primary antibody served as negative
controls. Cells exhibiting positive staining on cell membranes and
in the cytoplasm and nucleus were counted in at least 10
representative fields (×400 magnification) and the mean percentage
of positive cells was calculated. Immunostaining was assessed by
two independent pathologists blinded to clinical characteristics
and outcomes.
Cell cycle and apoptosis analysis by flow
cytometry
Cells for cell cycle analysis were fixed in 70%
ethanol, digested with RNaseA and labeled with propidium iodide
(PI). Apoptotic cells were analyzed with Annexin V/FITC kit (BD
Biosciences, San Jose, CA, USA) according to the manufacturer's
instructions and were analyzed by flow cytometry after compound
treatment as previously described (25).
Wound healing assay
Cells (2×105) OUMS-27, HCS-2/8 and SW1353
were seeded into a 24-well plate. The tumor cells grew to
confluence 24 h later. An artificial wound was introduced with a
P10 pipette tip/well. Data of the wounded area were taken at 0 and
24 h with a microscope (Olympus Corp). The assay was repeated three
times.
Transwell assay
HCS-2/8, OUMS-27 and SW1353 cells were respectively
harvested, washed and suspended with DMEM (Gibcο/Life Technologies,
11965-092), L-15 medium (Gibco) and seeded to the upper chambers of
Transwell inserts (8 µm pore size; Corning Incorporated,
Corning, NY, USA) with/without low concentration bortezomib (10 nM)
in the migration assay. The upper chambers were coated with
Matrigel (BD Biosciences, 354234) prior to the inoculation of the
cancer cells and bortezomib in the invasion assay. The lower
compartments were filled with DMEM or L-15 medium supplemented with
5% FBS. The cells in the upper chamber were removed with a swab
after incubation for 12 h in the migration assay or 24 h in the
invasion assay. The cells that migrated to the lower layer and
attached to the membrane were stained with crystal violet and were
numbered in five fields per well under a microscope. The assay was
repeated three times.
Mesenchymal-epithelial transition (MET)
induction
OUMS-27 and HCS-2/8 cells were cultured as adherent
cells in the complete medium overnight. The cells were maintained
in either medium alone or medium supplemented with 2.5 ng/ml TGF-β1
(R&D Systems, Minneapolis, MN, USA), with or without 20 nM
bortezomib in a humidified 5% CO2 incubator at 37°C. The
morphological images of the cells were taken seven days after the
incubation. The experiments were repeated three times.
Gene knockdown using siRNA
The siRNAs to Stat3 or control siRNA were all
purchased from Suzhou GenePharma, Co., Ltd. (Suzhou, China). Cells
were transfected with siRNA using Lipofectamine 3000, purchased
from Origene Technologies Inc. (Rockville, MD, USA) according to
the manufacturer's instructions. Cells were incubated for 48 h
before further treatment.
Generation of xenografts
Six-week-old BALB/c female athymic nude mice (Vital
River Laboratories) were subcutaneously injected in the right flank
with cells (2×106 in 0.1 ml PBS). Once a palpable tumor
developed, the mice were randomly divided into two groups and
intraperitoneally administered dimethyl sulfoxide (DMSO) or
bortezomib at a dose of 0.5 mg/kg every other day for 30 days. The
volume of xenografts was measured every 5 days (tumor volume =
(length × width2)/2). The mice were sacrificed after 30
days. The tumor samples were processed for routine IHC. The tumor
metastatic ability of HCS-2/8 cells (5×106) was observed
following cell injection intravenously into the tail vein. Four
weeks later, the mice were randomly divided into two groups and
intraperitoneally administered with DMSO or bortezomib at a dose of
0.5 mg/kg every other day for 30 days (n=6 per group), the mice
were sacrificed and the number of metastatic nodules on the lung
surface was counted. Metastatic lungs were fixed with 4%
paraformaldehyde before dehydration and paraffin embedding.
Paraffin sections were stained with hematoxylin and eosin according
to the standard protocols.
Statistical analysis
Kaplan-Meier analysis was used to estimate the
cumulative cause-specific survival rate and the differences in
mouse survival with DMSO/bortezomib (n=5 per group). The influence
of bortezomib on the growth, apoptosis, migration, invation, cell
cycle and tumor formation of chondrosarcoma cells were analyzed by
the Student's t-test. In all statistical analyses, statistical
significance in the two-sided test was indicated with P≤0.05 and
P<0.01 was remarkably significant.
Results
p-Stat3 and EMT-related protein
expression in the normal articular cartilage, in chondrosarcoma and
in their corresponding cell lines HC-a, OUMS-27, HCS-2/8, SW1353
and JJ012
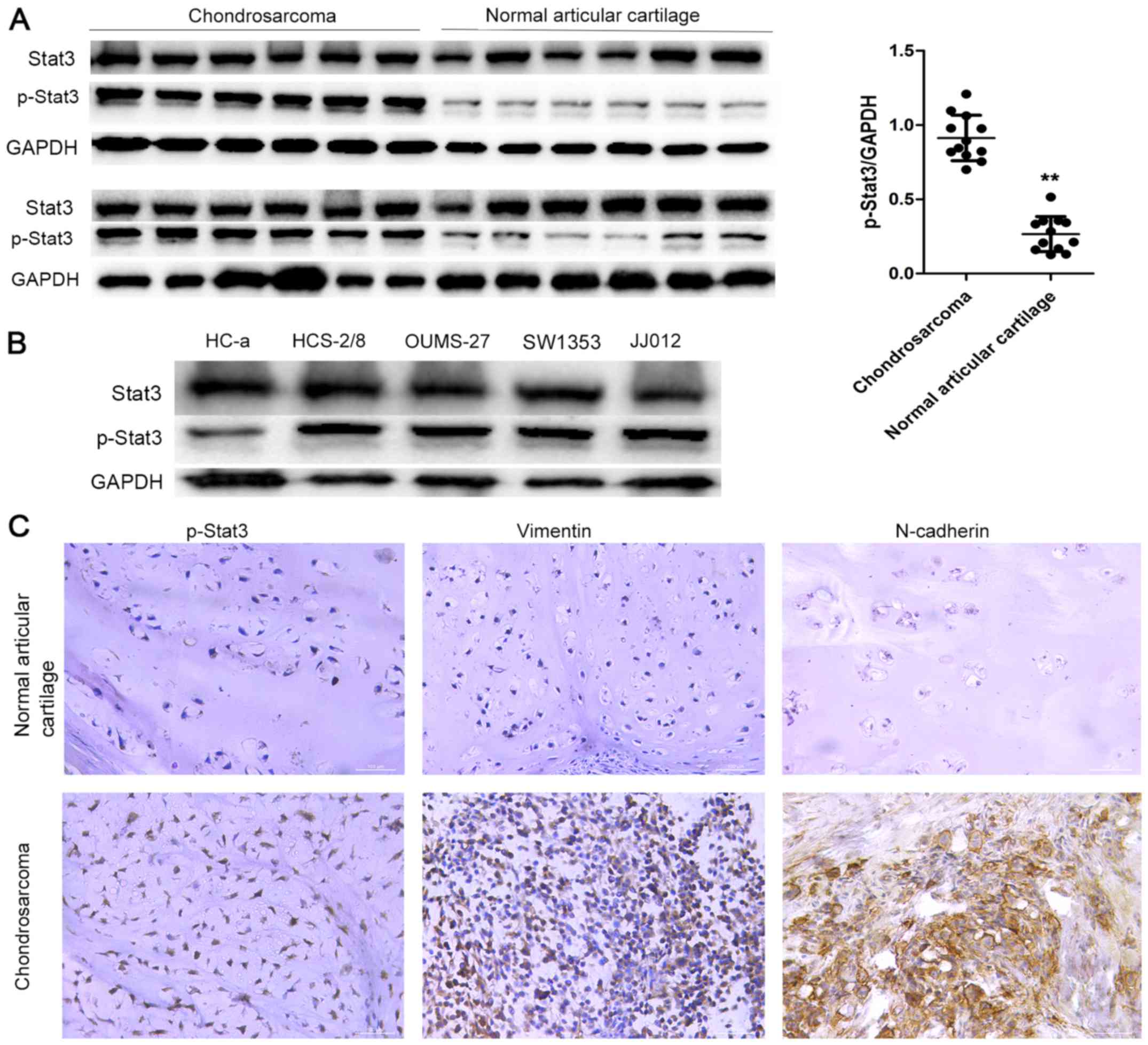
We examined the expression of p-Stat3 using western
blot analysis in 12 samples taken from the normal articular
cartilage and chondrosarcoma tissues, respectively. Western blot
analysis showed that p-Stat3 was highly expressed in
chondrosarcomas and considerably lower in the normal articular
cartilage tissues (Fig. 1A and B).
These results indicated that p-Stat3 might have tumor-promoting
roles in chondrosarcoma. To validate our results, we analyzed the
expression of p-Stat3 in the corresponding cell lines HC-a,
HCS-2/8, OUMS-27, SW1353 and JJ012 by western blotting and observed
similar results (Fig. 1C).
We also assessed the expression of p-Stat3 and
vimentin with immunohistochemistry (IHC). Weak expression of
p-Stat3, N-cadherin and vimentin was observed in the normal
articular cartilage, but strong expression was recorded in the
chondrosarcoma tissues (Fig.
1D).
Bortezomib specifically inhibits the
growth and promotes apoptosis of human chondrosarcoma cells
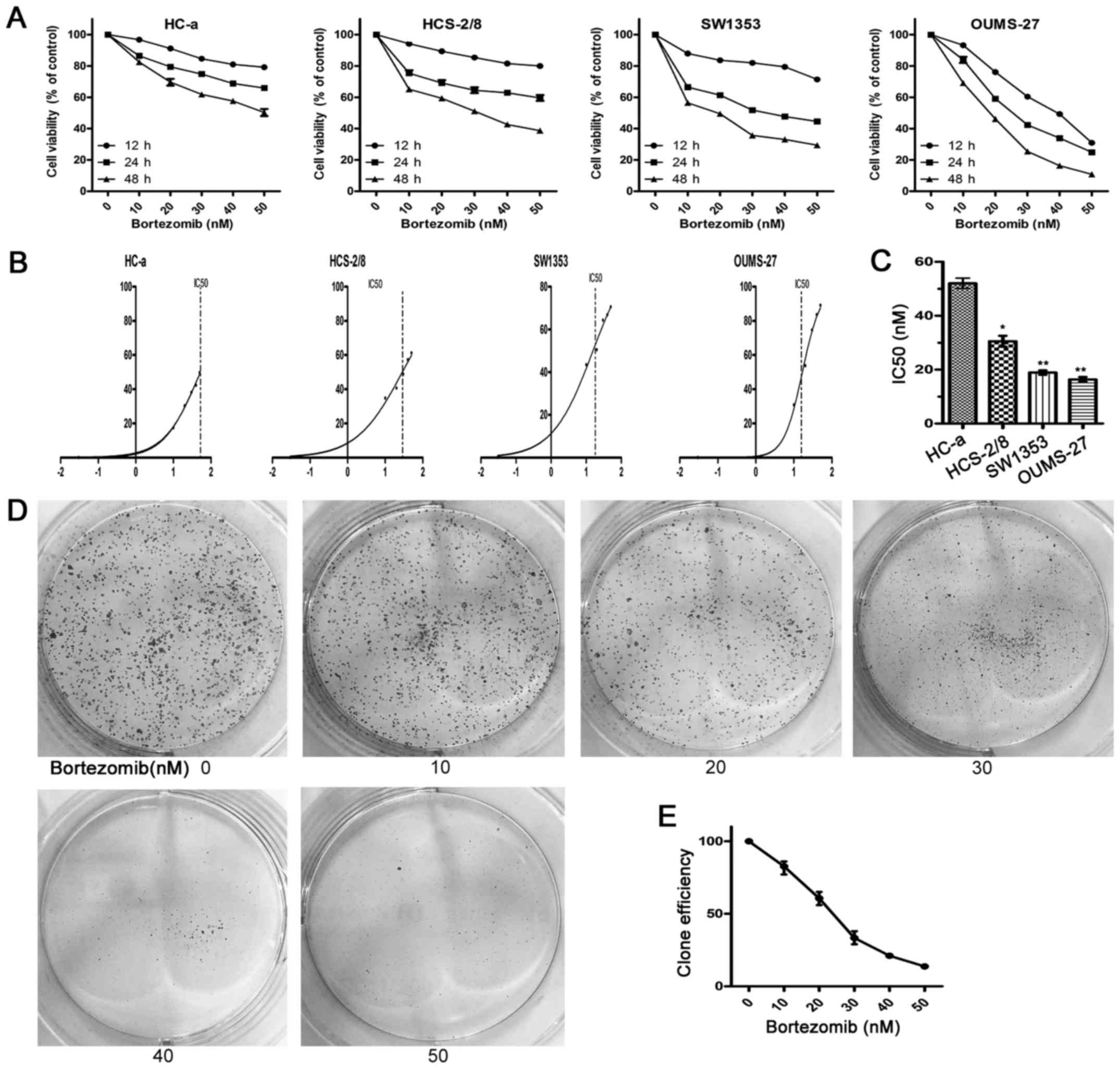
To investigate the effect of bortezomib on growth of
human chondrosarcoma cells, we first examined the function of
bortezomib in three human chondrosarcoma cell lines (OUMS-27,
HCS-2/8 and SW1353) and human articular chondrocyte cells (HC-a),
respectively. We cultured each cell line with different
concentration of bortezomib for 12, 24 or 48 h and analyzed cell
viability. The growth of human chondrosarcoma cells was inhibited
by bortezomib in a dose- and time-dependent manner. By contrast,
the viability of HC-a cells only decreased slightly (Fig. 2A). As shown in Fig. 2B and C, the IC50 of
bortezomib in HC-a cells was higher than that needed for
chondrosarcoma cells (HCS-2/8, OUMS-27 and SW1353). In colony
formation assays, bortezomib decreased colony formation compared
with control (Fig. 2D and E).
These data showed that bortezomib specifically suppressed the
growth of human chondrosarcoma cells but not normal cells in
vitro.
Bortezomib induces apoptotic cell death
and promotes G0/G1 phase arrest
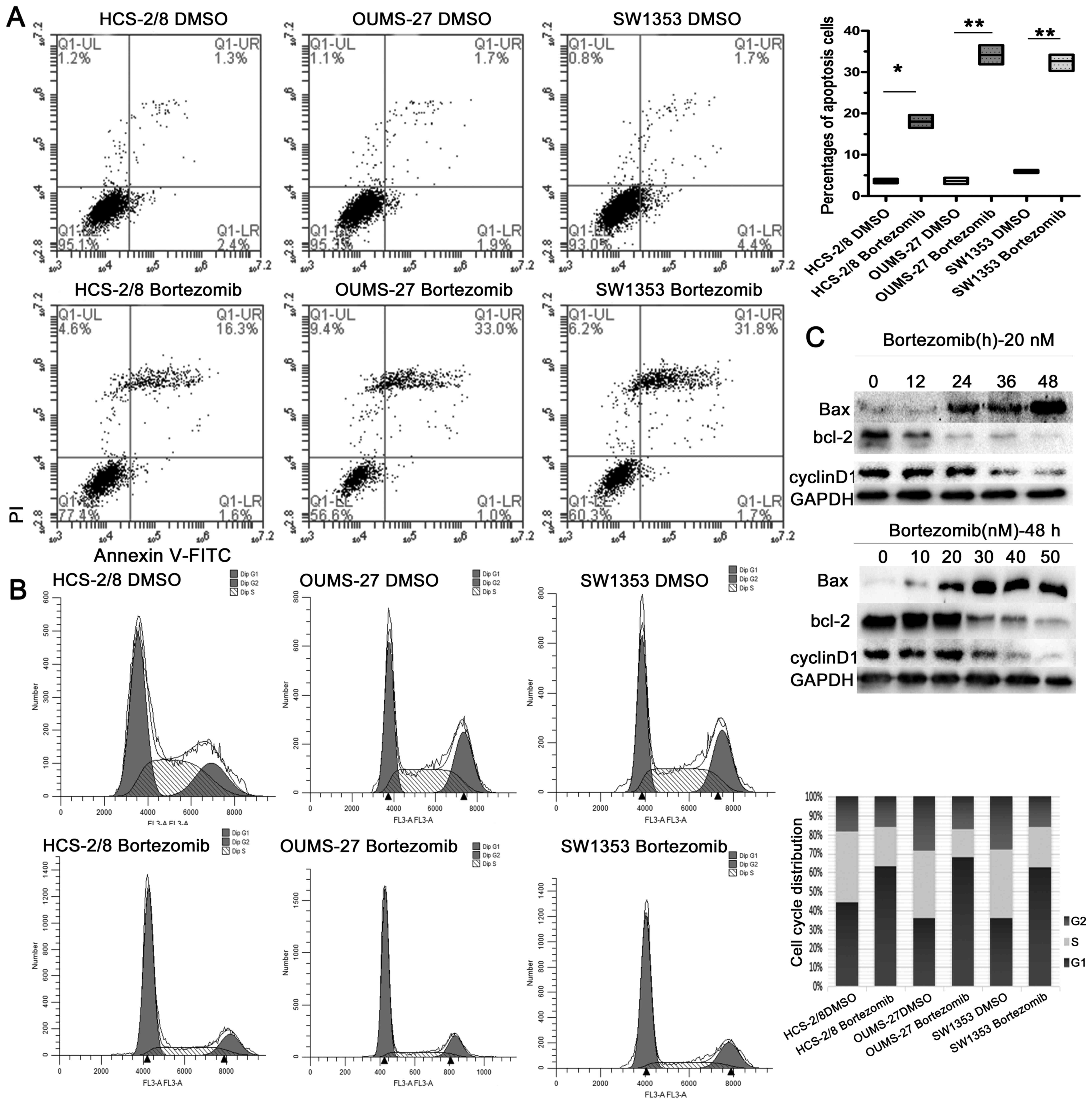
To investigate the role of bortezomib in cell death
of chondrosarcoma cells, chondrosarcoma cells were analyzed by flow
cytometry following Annexin V-FITC and propidium iodide (PI) dual
staining. As shown in Fig. 3A,
bortezomib significantly induced apoptosis. As key executors of
cell apoptosis, the ratio of Bax to Bcl-2 proteins increased
following a 48-h treatment with bortezomib, or with 20 nM
bortezomib for indicated time-points (Fig. 3C).
As previously reported, siRNA-mediated inhibition of
STAT3 promoted G0/G1 cell cycle arrest (26). We evaluated the effects of
bortezomib on cell proliferation and cell cycle arrest by
performing cell cycle analysis. Our analysis demonstrated that
chondrosarcoma cells were mostly arrested within the G0/G1 phase,
indicating there was a reduced number of dividing tumor cells
following bortezomib treatment. There were also fewer cells under S
phase, demonstrating that bortezomib could also inhibit DNA
replication (Fig. 3B). In
addition, cyclin D1, a G0/G1 phase-related protein, was decreased
following treatment, as analyzed by western blotting assay
(Fig. 3C). Together, these results
illustrate that apoptosis, as well as cell G0/G1 phase arrest, are
involved in the response of chondrosarcoma to bortezomib
treatment.
Bortezomib attenuates the migratory and
invasive capacities of chondrosarcoma cells
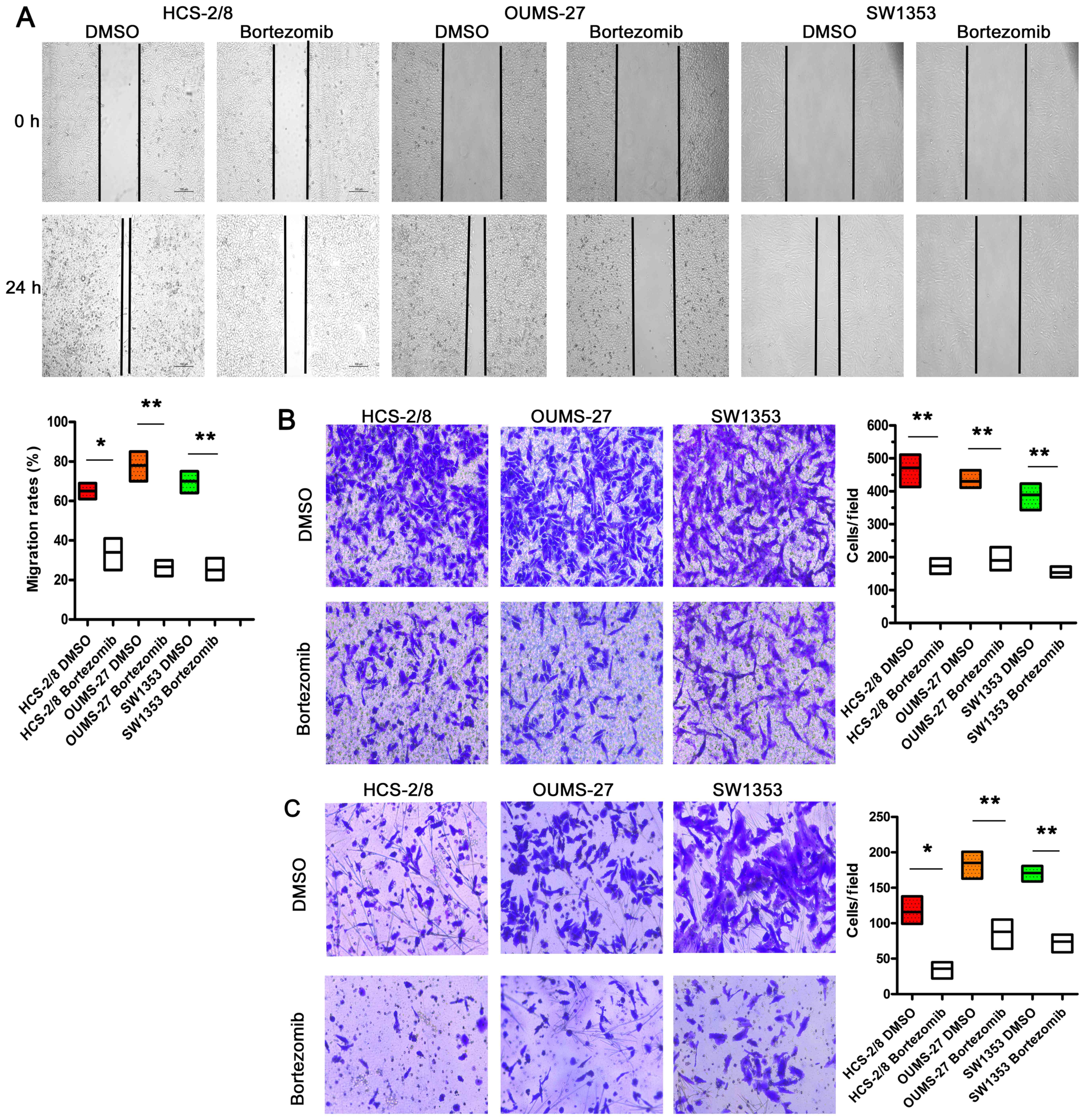
Chondrosarcoma cells display high migration and
invasion capacities, that can be attenuated by bortezomib, as
determined by wound healing assay (Fig. 4A) and transwell assays (Fig. 4B), respectively. In the
wound-healing assay, bortezomib could very effectively suppress the
migration of chondrosarcoma cells. Moreover, treatment with
bortezomib resulted in markedly decreased invasive abilities of
HCS-2/8, OUMS-27 and SW1353 cells in Matrigel (Fig. 4C). These data show that, in human
chondrosarcoma cells, bortezomib effectively attenuates the
migratory and invasive capacities.
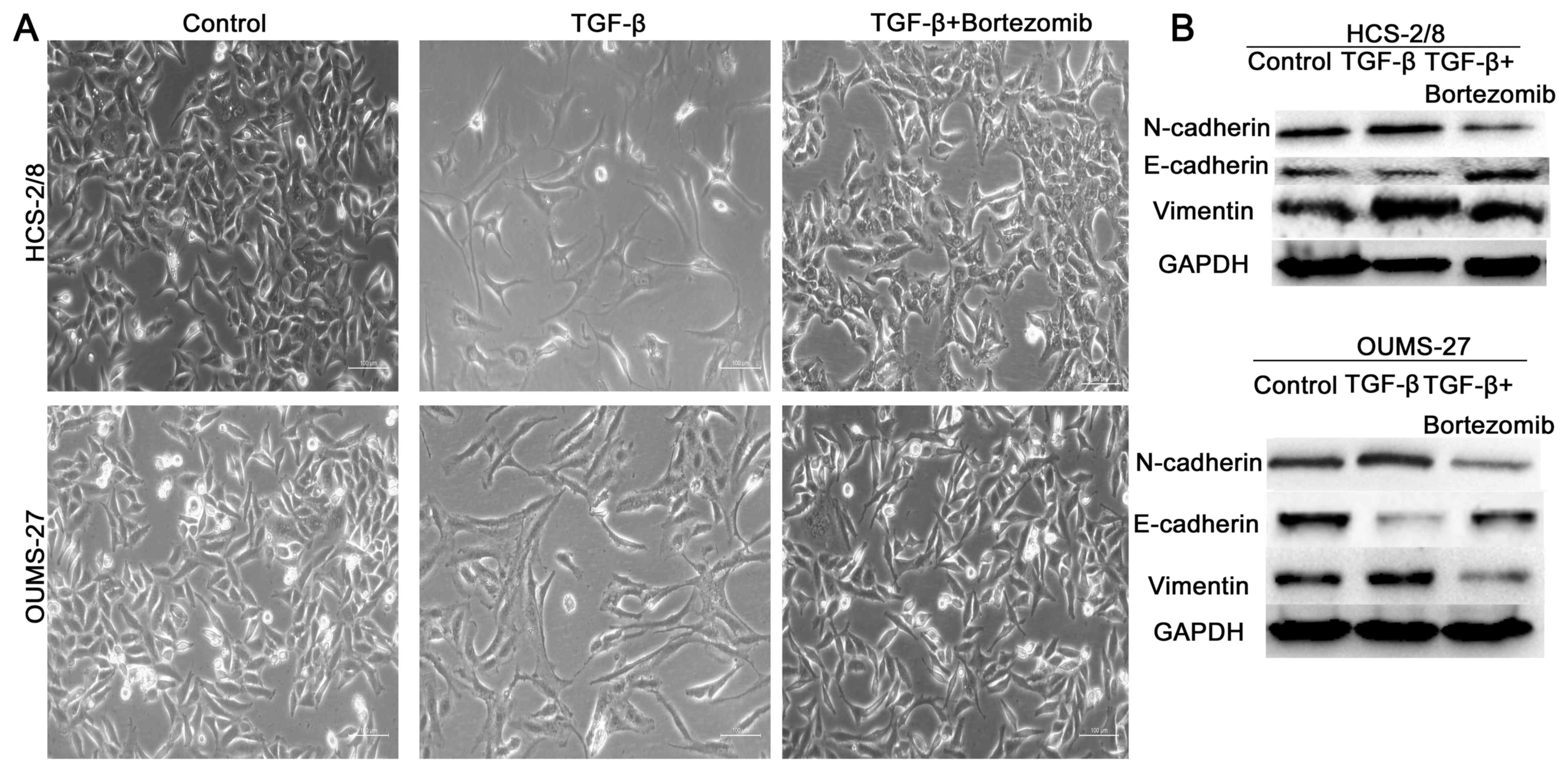
Bortezomib induces the
mesenchymal-epithelial transition of chondrosarcoma cells
Epithelial-mesenchymal transition (EMT) is a
critical process for epithelial cells to harbor mesenchymal
properties and is closely involved in cancer invasion and
metastasis (27). To evaluate the
effect of bortezomib on this process, we enhanced mesenchymal
properties of chondrosarcoma cells with TGF-β1 (27,28)
and found that the cell lines showed mesenchymal appearance, which
was fibroblast-like. When treated with bortezomib, however, the
chondrosarcoma cell lines maintained an epithelial appearance
(Fig. 5A), indicating that
bortezomib efficiently induced the mesenchymal-epithelial
transition. Moreover, bortezomib impaired the expression of
mesenchymal cell markers, including N-cadherin, vimentin and slug.
However, the expression of E-cadherin, an epithelial cell marker,
increased (Fig. 5B).
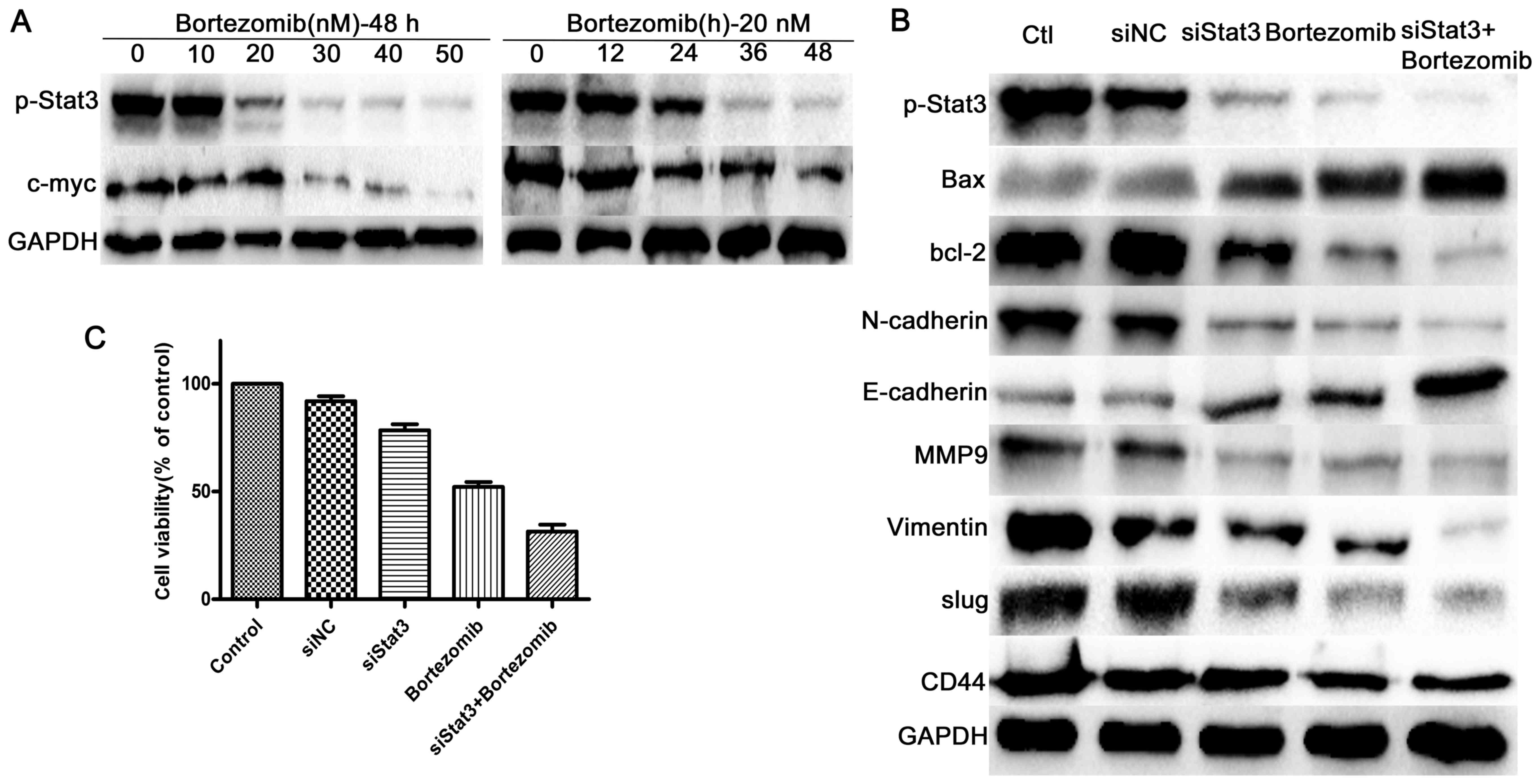
Bortezomib inhibits the Stat3 signalling
pathway
As Stat3 is a well-known cancer therapeutic target,
we studied whether Stat3 is engaged in bortezomib action in
chondrosarcoma cells. Western blotting assay demonstrated that
expression of p-Stat3 and its target c-Myc were decreased in SW1353
cells in dose- and time-dependent manner following bortezomib
treatment (Fig. 6A). As shown in
Fig. 3C, the other two direct
downstream targets of Stat3, cyclin D1 and Bcl-2 decreased in the
same way.
To further confirm the regulatory role of Stat3
signaling in bortezomib-treated chondrosarcoma, we characterized
the effects of bortezomib in SW1353 cells in which Stat3 was
silenced with siRNA. In accordance with bortezomib treatment,
knockdown of Stat3 not only induced apoptosis, cell cycle arrest as
indicated by increased bax and decreased cyclin D1, but also led to
promotion of MET, as evidenced by enhanced E-cadherin expression
and decreased N-cadherin, vimentin, MMP-9, CD44 and slug. The
expression of p-Stat3 was decreased following siStat3 or bortezomib
treatment for 48 h, while the level of p-Stat3 was more
significantly repressed when treated both siStat3 and bortezomib,
analyzed by western blot assay (Fig.
6B).
Moreover, silencing of Stat3 resulted in enhanced
bortezomib-induced decreases in cell viability. As shown in
Fig. 6C, the cell viability of
siStat3 silenced SW1353 cells treated with 20 nM bortezomib for 48
h was decreased more significantly than that treated with siStat3
or only with 20 nM bortezomib. Thus, these data suggest that Stat3
inactivation is involved in botezomib-mediated inhibition of
growth, cell cycle pregression, metastasis and induced
apoptosis.
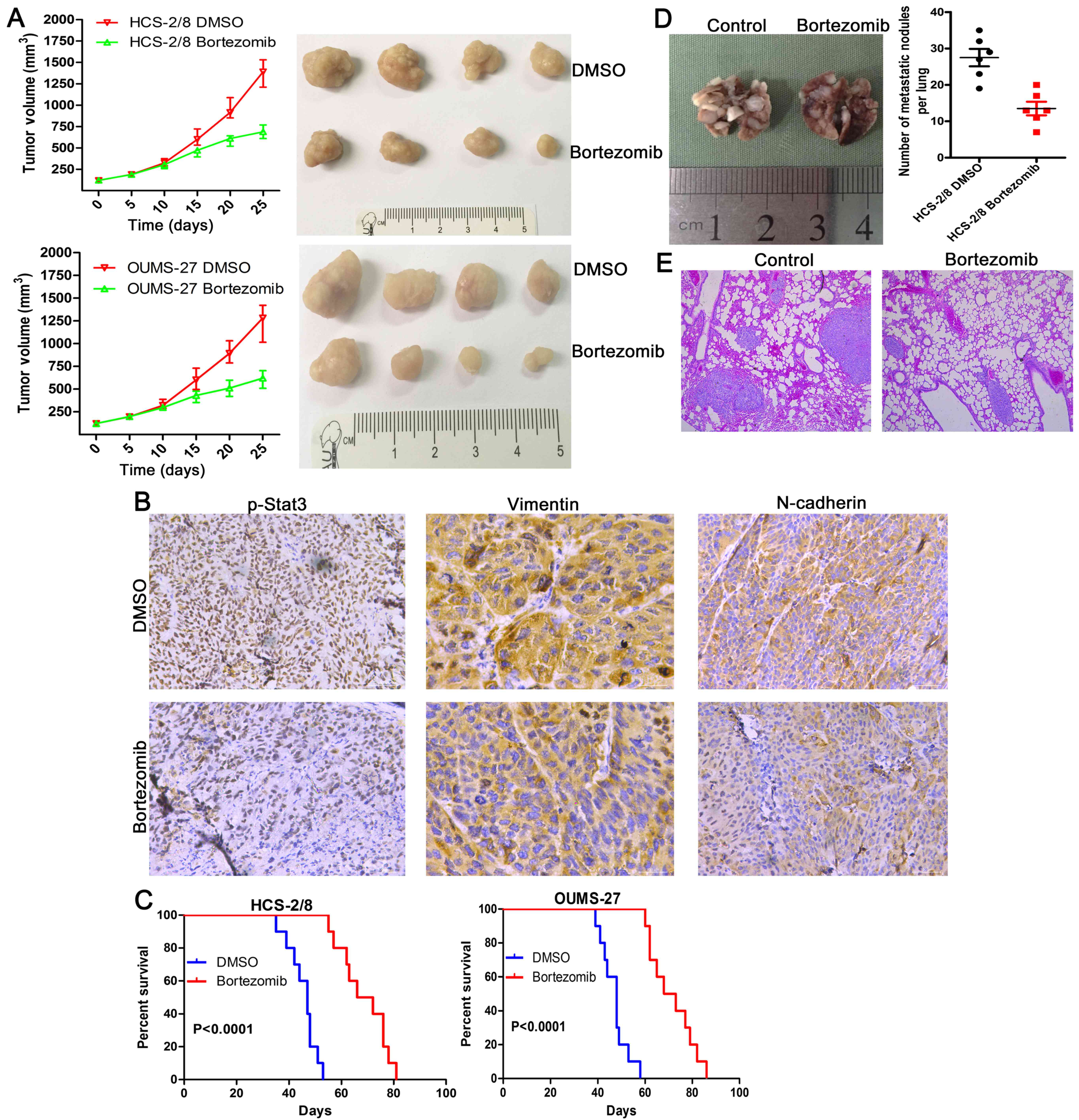
Bortezomib inhibits chondrosarcoma tumor
growth in vivo
Five days after HCS-2/8 and OUMS-27 cells were
injected subcutaneously into the right armpit of nude BALB/c mice,
the mice were randomly divided into two groups and
intraperitoneally administered DMSO or botezomib at a dose of 0.5
mg/kg every other day for 30 days. The volume of xenografts was
measured every five days (tumor volume = (length ×
width2)/2). In line with the in vitro data,
bortezomib administration was very effective in inhibiting tumor
growth in vivo throughout the course of treatment, resulting
in decreased tumor size (Fig. 7A).
These data indicate that bortezomib reduces tumor volume and growth
rate of chondrosarcoma cells in vivo.
The tumors were removed and evaluated by
immunohistochemistry. Bortezomib decreased p-Stat3, vimentin and
N-cadherin expression in tumors formed by HCS-2/8 and OUMS-27 cells
(Fig. 7B).
Furthermore, bortezomib-treated mice had a
significantly longer survival time compared with control mice
(Fig. 7C).
Bortezomib inhibits metastasis in
vivo
To extend our in vitro observations, we
investigated whether bortezomib could regulate tumor metastasis
in vivo. After HCS-2/8 cells were injected intravenously
into the tail vein at 4-weeks, the mice were intraperitoneally
administered DMSO or bortezomib at a dose of 0.5 mg/kg every other
day for another 30 days. Bortezomib-treated mice displayed
statistically significantly lower numbers of lung metastases than
those treated with DMSO (Fig. 7D).
Hematoxylin and eosin staining, of the lungs revealed fewer lung
metastatic nodes in the mice treated with bortezomib (Fig. 7E).
Discussion
Chondrosarcoma is the second most frequent type of
primary bone cancer following osteosarcoma (1–3),
with limited effective treatment options. At present, surgical
resection remains the only effective means of treating
chondrosarcomas, but remains associated with poor prognosis due to
their resistance to adjuvant treatments such as radio- and
chemotherapy. However, molecular targeted therapies have completely
changed the treatment paradigm of some tumors due to its high
efficiency and low toxicity.
Persistent activation of signal transducer and
activator of transcription-3 (Stat3) is found in a wide range of
solid malignancies of tumors, containing musculoskeletal tumor,
such as Ewing's sarcoma and osteosarcoma (13,29–31).
In accordance with our studies, we found that the Stat3 signal
pathway is abnormally activated in chondrosarcoma. In addition,
inhibiting Stat3 by specific siRNA could suppress the growth of
human chondrosarcoma cells.
Bortezomib is a dipeptidyl boronic acid, which is a
selective inhibitor of the proteasome. Many studies have showed
that bortezomib can reversibly suppress the proteasome pathway by
binding with the 20S proteasome complex directly and blocking its
enzymatic activity (17,32). However, the effects of bortezomib
appear to be cell type-specific or context dependent.
In the present study we found that a low
concentration of bortezomib (20 nM) inhibited the growth of
chondrosarcoma cells alone and did not affect normal articular
chondrocyte cells. Mesenchymal-related traits, which is the key to
tumor metastasis, was also efficiently inhibited by bortezomib
treatment. Moreover, bortezomib caused cell cycle arrest and
apoptosis of chondrosarcoma cells at a low concentration (20 nM).
The molecular mechanisms of action of bortezomib included: growth
inhibition, apoptosis promotion, cell cycle arrest, promotion of
MET and the inactivation of Stat3.
Our findings are consistent with studies which have
revealed that bortezomib inhibited tumor growth via inactivation of
the Stat3 pathway (33). While
some studies have suggested that bortezomib facilitated G0/G1
arrest in cancer cells (23,34),
we found that bortezomib markedly induced G0/G1 phase arrest by
downregulating cyclin D1, consequently resulting in chondrosarcoma
cell quiescent and impeding tumor progression.
EMT is thought to play a significant role in
metastasis (35,36). Metastatic relapses are
characterized by rapidly proliferating, drug-resistant tumors that
are associated with a high mortality rate (37). Metastasis is a complicated process,
during which cells undergo phenotypic alteration and migrate away
from the primary tumor, survive in the vasculature or lymphatics,
and colonize metastatic sites (38). Stat3 is activated to trigger the
molecular events in the EMT of various types of human tumors
(39–41).
In the present study, following exposure of
chondrosarcoma cells to bortezomib for 6 days, the cells displayed
an epithelial-like morphology. Furthermore, there were increased
expression of E-cadherin (an epithelial marker) and decreased
expressions of N-cadherin and vimentin (mesenchymal markers). In
tumors formed by HCS-2/8 and OUMS-27 cells injected into nude
BALB/c mice, bortezomib reduced the expression of N-cadherin and
vimentin. These results demonstrate that, exposure to bortezomib
leads to chondrosarcoma cells undergoing MET in vitro and
in vivo.
In conclusion, bortezomib demonstrates an
antineoplastic role on chondrosarcoma both in vitro and
in vivo. These beneficial effects can be explained by
bortezomib-mediated Stat3 suppression. This study suggests a
promising therapeutics target in chondrosarcoma and probably in
other kinds of metastatic malignant tumors.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (nos. 81572633 and
81472509).
Abbreviations:
|
Stat3
|
the transcription factor signal
transducer and activator of transcription 3
|
|
IHC
|
immunohistochemistry
|
|
MET
|
mesenchymal-epithelial transition
|
|
siRNA
|
small interfering RNA
|
|
CCK-8
|
Cell Counting kit-8
|
References
|
1
|
O'Neal LW and Ackerman LV: Chondrosarcoma
of bone. Cancer. 5:551–577. 1952. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee FY, Mankin HJ, Fondren G, Gebhardt MC,
Springfield DS, Rosenberg AE and Jennings LC: Chondrosarcoma of
bone: An assessment of outcome. J Bone Joint Surg Am. 81:326–338.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bauer HC, Brosjo O, Kreicbergs A and
Lindholm J: Low risk of recurrence of enchondroma and low-grade
chondrosarcoma in extremities. 80 patients followed for 2–25 years.
Acta Orthop Scand. 66:283–288. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eriksson AI, Schiller A and Mankin HJ: The
management of chondrosarcoma of bone. Clin Orthop Relat Res. (153):
44–66. 1980.PubMed/NCBI
|
|
5
|
Bovée JV, Cleton-Jansen AM, Taminiau AH
and Hogendoorn PC: Emerging pathways in the development of
chondrosarcoma of bone and implications for targeted treatment.
Lancet Oncol. 6:599–607. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gelderblom H, Hogendoorn PC, Dijkstra SD,
van Rijswijk CS, Krol AD, Taminiau AH and Bovée JV: The clinical
approach towards chondrosarcoma. Oncologist. 13:320–329. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darnell JE Jr: Reflections on STAT3,
STAT5, and STAT6 as fat STATs. Proc Natl Acad Sci USA.
93:6221–6224. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kijima T, Niwa H, Steinman RA, Drenning
SD, Gooding WE, Wentzel AL, Xi S and Grandis JR: STAT3 activation
abrogates growth factor dependence and contributes to head and neck
squamous cell carcinoma tumor growth in vivo. Cell Growth Differ.
13:355–362. 2002.PubMed/NCBI
|
|
10
|
Chen X, Ying Z, Lin X, Lin H, Wu J, Li M
and Song L: Acylglycerol kinase augments JAK2/STAT3 signaling in
esophageal squamous cells. J Clin Invest. 123:2576–2589. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bu LL, Deng WW, Huang CF, Liu B, Zhang WF
and Sun ZJ: Inhibition of STAT3 reduces proliferation and invasion
in salivary gland adenoid cystic carcinoma. Am J Cancer Res.
5:1751–1761. 2015.PubMed/NCBI
|
|
12
|
Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin
W, Ke J, Huang J, Yeung SC and Zhang H: Metformin promotes
autophagy and apoptosis in esophageal squamous cell carcinoma by
downregulating Stat3 signaling. Cell Death Dis. 5:e10882014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anderson JL, Titz B, Akiyama R,
Komisopoulou E, Park A, Tap WD, Graeber TG and Denny CT:
Phosphoproteomic profiling reveals IL6-mediated paracrine signaling
within the Ewing sarcoma family of tumors. Mol Cancer Res.
12:1740–1754. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Ni J, Yi S, Song D and Ding M:
Protein inhibitor of activated STAT xα depresses cyclin D and
cyclin D kinase, and contributes to the inhibition of osteosarcoma
cell progression. Mol Med Rep. 13:1645–1652. 2016.
|
|
15
|
Sandoval-Usme MC, Umaña-Pérez A, Guerra B,
Hernández-Perera O, García-Castellano JM, Fernández-Pérez L and
Sánchez-Gómez M: Simvastatin impairs growth hormone-activated
signal transducer and activator of transcription (STAT) signaling
pathway in UMR-106 osteosarcoma cells. PLoS One. 9:e877692014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kane RC, Farrell AT, Sridhara R and Pazdur
R: United States Food and Drug Administration approval summary:
Bortezomib for the treatment of progressive multiple myeloma after
one prior therapy. Clin Cancer Res. 12:2955–2960. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Dou QP: Bortezomib as the first proteasome inhibitor anticancer
drug: Current status and future perspectives. Curr Cancer Drug
Targets. 11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cavo M: Proteasome inhibitor bortezomib
for the treatment of multiple myeloma. Leukemia. 20:1341–1352.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin DH, Chun YS, Lee DS, Huang LE and
Park JW: Bortezomib inhibits tumor adaptation to hypoxia by
stimulating the FIH-mediated repression of hypoxia-inducible
factor-1. Blood. 111:3131–3136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mackay H, Hedley D, Major P, Townsley C,
Mackenzie M, Vincent M, Degendorfer P, Tsao MS, Nicklee T, Birle D,
et al: A phase II trial with pharmacodynamic endpoints of the
proteasome inhibitor bortezomib in patients with metastatic
colorectal cancer. Clin Cancer Res. 11:5526–5533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim GP, Mahoney MR, Szydlo D, Mok TS,
Marshke R, Holen K, Picus J, Boyer M, Pitot HC, Rubin J, et al: An
international, multi-center phase II trial of bortezomib in
patients with hepatocellular carcinoma. Invest New Drugs.
30:387–394. 2012. View Article : Google Scholar
|
|
22
|
Hideshima T, Chauhan D, Hayashi T, Akiyama
M, Mitsiades N, Mitsiades C, Podar K, Munshi NC, Richardson PG and
Anderson KC: Proteasome inhibitor PS-341 abrogates IL-6 triggered
signaling cascades via caspase-dependent downregulation of gp130 in
multiple myeloma. Oncogene. 22:8386–8393. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Z, Liu S, Zhu M, Zhang H, Wang J, Xu
Q, Lin K, Zhou X, Tao M, Li C and Zhu H: PS341 inhibits
hepatocellular and colorectal cancer cells through the FOXO3/CTNNB1
signaling pathway. Sci Rep. 6:220902016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tingting R, Wei G, Changliang P, Xinchang
L and Yi Y: Arsenic trioxide inhibits osteosarcoma cell
invasiveness via MAPK signaling pathway. Cancer Biol Ther.
10:251–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun Y, Guo W, Ren T, Liang W, Zhou W, Lu
Q, Jiao G and Yan T: Gli1 inhibition suppressed cell growth and
cell cycle progression and induced apoptosis as well as autophagy
depending on ERK1/2 activity in human chondrosarcoma cells. Cell
Death Dis. 5:e9792014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun KX, Xia HW and Xia RL: Anticancer
effect of salidroside on colon cancer through inhibiting JAK2/STAT3
signaling pathway. Int J Clin Exp Pathol. 8:615–621.
2015.PubMed/NCBI
|
|
27
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wake MS and Watson CJ: STAT3 the oncogene
- still eluding therapy? FEBS J. 282:2600–2611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stobbe-Maicherski N, Wolff S, Wolff C,
Abel J, Sydlik U, Frauenstein K and Haarmann-Stemmann T: The
interleukin-6-type cytokine oncostatin M induces aryl hydrocarbon
receptor expression in a STAT3-dependent manner in human HepG2
hepatoma cells. FEBS J. 280:6681–6690. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Salas S, Jiguet-Jiglaire C, Campion L,
Bartoli C, Frassineti F, Deville JL, Maues De Paula A, Forest F,
Jézéquel P, Gentet JC, et al: Correlation between ERK1 and STAT3
expression and chemoresistance in patients with conventional
osteosarcoma. BMC Cancer. 14:6062014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dalla Via L, Nardon C and Fregona D:
Targeting the ubiquitin-proteasome pathway with inorganic compounds
to fight cancer: A challenge for the future. Future Med Chem.
4:525–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee JH, Kim C, Kim SH, Sethi G and Ahn KS:
Farnesol inhibits tumor growth and enhances the anticancer effects
of bortezomib in multiple myeloma xenograft mouse model through the
modulation of STAT3 signaling pathway. Cancer Lett. 360:280–293.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim JE, Lee JI, Jin DH, Lee WJ, Park GB,
Kim S, Kim YS, Wu TC, Hur DY and Kim D: Sequential treatment of HPV
E6 and E7-expressing TC-1 cells with bortezomib and celecoxib
promotes apoptosis through p-p38 MAPK-mediated downregulation of
cyclin D1 and CDK2. Oncol Rep. 31:2429–2437. 2014.PubMed/NCBI
|
|
35
|
Liang YJ, Wang QY, Zhou CX, Yin QQ, He M,
Yu XT, Cao DX, Chen GQ, He JR and Zhao Q: MiR-124 targets Slug to
regulate epithelial-mesenchymal transition and metastasis of breast
cancer. Carcinogenesis. 34:713–722. 2013. View Article : Google Scholar
|
|
36
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial-mesenchymal transition in cancer: Parallels between
normal development and tumor progression. J Mammary Gland Biol
Neoplasia. 15:117–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Romero-Pérez L, López-García MÁ,
Díaz-Martín J, Biscuola M, Castilla MÁ, Tafe LJ, Garg K, Oliva E,
Matias-Guiu X, Soslow RA, et al: ZEB1 overexpression associated
with E-cadherin and microRNA-200 downregulation is characteristic
of undifferentiated endometrial carcinoma. Mod Pathol.
26:1514–1524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lim YY, Wright JA, Attema JL, Gregory PA,
Bert AG, Smith E, Thomas D, Lopez AF, Drew PA, Khew-Goodall Y, et
al: Epigenetic modulation of the miR-200 family is associated with
transition to a breast cancer stem-cell-like state. J Cell Sci.
126:2256–2266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014.PubMed/NCBI
|
|
40
|
Yuan W, Li T, Mo X, Wang X, Liu B, Wang W,
Su Y, Xu L and Han W: Knockdown of CMTM3 promotes metastasis of
gastric cancer via the STAT3/Twist1/EMT signaling pathway.
Oncotarget. 7:29507–29519. 2016.PubMed/NCBI
|
|
41
|
Tania M, Khan MA and Fu J: Epithelial to
mesenchymal transition inducing transcription factors and
metastatic cancer. Tumour Biol. 35:7335–7342. 2014. View Article : Google Scholar : PubMed/NCBI
|