Introduction
Hepatocellular carcinoma (HCC) is a prevalent
malignancy and the third leading cause of tumor-associated
mortality worldwide (1). The
mainstay of treatment for patients with HCC at early stage is
curative resection. However, despite significant progression in
diagnosis of HCC, most HCC patients are diagnosed at an advanced
tumor stage when resection is not applicable. Owing to high tumor
aggressiveness and poor susceptibility to standard chemotherapy
strategy, the overall dismal outcome of HCC has not changed
satisfactorily, and the 5-year survival rate is only 7% (2–4).
Hence, further insights into the underlying mechanisms of HCC
pathogenesis and identification of potential molecules for HCC
treatment are urgently needed.
Early growth response (EGR) proteins are a family of
zinc finger transcription factors and induced in various cell types
in response to a wide range of internal and external stimuli, such
as growth factors, cytokines, hypoxia, injury and stress. The EGR
family comprises four members (EGR1, EGR2, EGR3 and EGR4), which
share a high degree of homology at their DNA-binding zinc finger
domains recognizing a GC-rich fragment in the promoter region of
multiple target genes (5–7). EGR proteins are involved in various
physiologic and pathologic processes. EGR1 is the most intensively
studied protein in EGR family, and implicated in ischemic injury,
inflammation, atherosclerosis, and cardiovascular pathogenesis
(8). Additionally, EGR1 also
exhibits tumor-suppressive activity primarily through induction of
tumor cell apoptosis in certain cancer events (9–12).
In contrast to EGR1, the function of EGR3 is poorly understood.
EGR3 has previously been implicated as a regulator in
neuro-development, autoimmunity, inflammation, angiogenesis, and
cancer (13–17). However, both the expression
patterns and functions of EGR3 in human cancers are still
controversial. Suzuki et al (18) reported that ectopic expression of
EGR3 in breast cancer cells caused enhanced cell invasion in
vitro and in vivo. In addition, high expression of EGR3
was observed and associated with poor prognosis in prostate cancer
patients (19). On the contrary, a
recent study showed that high-level EGR3 expression in A549 (lung
adenocarcinoma) cells resulted in strong inhibition of cell growth,
and was prone to show better prognosis in lung adenocarcinoma
(20). Furthermore, EGR3 was a
potent limiting factor for the proliferative potential of
hematopoietic stem cells (HSCs) in leukemia (21). Downregulation of EGR3 was visible
and related to adverse outcome in gastric cancer (22).
The relationship between EGR3 and HCC growth has not
yet been elucidated. To address this question, we observed the
expression pattern in human HCC specimens, human normal hepatic and
HCC cell lines. Furthermore, we evaluated the effect of EGR3 on the
growth of HCC cells and explored the underlying mechanisms.
Fas ligand (FasL), a 40-kDa glycosylated type 2
trans-membrane protein, is a member of the tumor necrosis factor
(TNF) family (23). Binding of
FasL to its surface receptor, Fas, leads to an adaptor molecule,
known as Fas-associated death domain (FADD), recruitment to the
cytoplasmic domain of this death receptor, and activates caspase-8.
Then activated caspase-8 triggers the activation of several
downstream caspase substrates and apoptosis is ultimately induced
in target cells (24,25). In contrast to the ubiquitous Fas
protein, FasL is concentrated in activated T lymphocytes and
natural killer (NK) cells, as well as in some non-lymphoid tissues,
such as eyes, testis, trophoblasts and cancer cells (26–30).
Given its role in apoptosis inhibition, FasL has been studied
extensively in tumor field for the past several years. In a
previous study, high-level FasL expression caused significant
inhibition of cell growth in SGC-7901 cells (human gastric cancer
cells) (31). In addition, FasL
has been reported to play an essential role in gemcitabine-mediated
cell death in non-small cell lung cancer (NSCLC) cells (32). Bianco et al (33) performed intratumoral injection of a
plasmid encoding human FasL into oral malignant melanoma in dogs to
test FasL-based gene therapy. They found that three out of five
dogs displayed tumor volume reduction, providing confirmation of
the pro-apoptotic effect of FasL gene in vivo. These results
suggest therapeutic potential of FasL for certain types of
cancers.
It is well established that the promoter region of
FasL gene possesses multiple sites of DNA-protein interaction,
which can be identified by members of some transcription factor
families, such as nuclear factor in activated T cells (NFAT), EGR
and nuclear factor-kappa B (NF-κB) families, leading to increased
expression of FasL (34). Some
studies have described a key role of EGR3 in triggering
transactivation of the FasL promoter in T cells, HeLa cells and
MCF-7 cells (breast cancer cells) (35,36).
However, little is known about the relationship between EGR3 and
FasL in HCC, and the combined action of EGR3 and FasL on the growth
of cancer cells is even less documented.
In this study, we confirm that EGR3 is frequently
down-regulated in human HCC tissues and cell lines, and high-level
expression of EGR3 exhibits significant growth suppression of HCC
cells both in vitro and in vivo. Furthermore, our
results indicate for the first time that the close cooperation
between EGR3 and FasL is a novel regulator mechanism of the growth
of HCC cells.
Materials and methods
Cell culture and HCC specimens
The human HCC cell lines (PLC/PRF/5, HCC-LM3, Huh7,
HepG2) and human normal hepatic cell line L02 were obtained from
Institute of Liver Diseases, Tongji Hospital of Tongji Medical
College, Huazhong University of Science and Technology (HUST,
Wuhan, China). All the cells were routinely cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island,
NY, USA) and maintained in an incubator with 5% CO2 at
37°C. After approval by the Ethics Committee of Tongji Hospital of
HUST and obtaining the informed consent from all tissue donors,
specimens (including 25 pairs of fresh HCC and adjacent non-tumor
tissues) were collected at Tongji Hospital and stored in liquid
nitrogen immediately after surgery until RNA and protein
extraction.
RNA isolation and quantitative real-time
PCR (qRT-PCR)
Total RNA was extracted from tissues or cells using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. After quantification by NanoDrop 2000
(Thermo Scientific, Wilmington, DE, USA), 500 ng of total RNA was
reversely transcribed into cDNA by a PrimeScript RT Reagent kit
(Takara, Tokyo, Japan). Briefly, each RNA sample was added to the
reaction mixture comprising 5X PrimeScript RT Master Mix and
nuclease-free water, followed by incubation at 37°C for 15 min and
85°C for 5 sec. Then, the target genes were amplified in 10
µl of PCR reaction solution composed of cDNA, primers,
nuclease-free water and SYBR Premix Ex Taq (Takara) in a StepOne
Real-Time PCR system (Applied Biosystem, Carlsbad, CA, USA). The
PCR amplification was conducted at 95°C for 30 sec followed by 40
cycles of 5 sec at 95°C and 30 sec at 60°C. GAPDH was used as an
internal control to normalize gene expression, and relative
quantification was performed using the 2−ΔΔCT
method.
Primers used in this study were as follows: EGR3
sense, 5′-GACATCGGTCTGACCAACGAG-3′; antisense,
5′-GGCGAACTTTCCCAAGTAGGT-3′. FasL sense, 5′-TGCCTTGGTAGGATTGGGC-3′,
antisense, 5′-GCTGGTAGACTCTCGGAGTTC-3′. Bak sense,
5′-ATGGTCACCTTACCTCTGCAA-3′; antisense,
5′-TCATAGCGTCGGTTGATGTCG-3′. p21 sense, 5′-TGTCCGTCAGAACCCATGC-3′;
antisense, 5′-AAAGTCGAAGTTCCATCGCTC-3′. GAPDH sense,
5′-GGGAAGCTTGTCATCAATGG-3′; antisense,
5′-CATCGCCCCACTTGATTTTG-3′.
Western blot analysis
Tissues or cells were lysed in ice-cold RIPA lysis
buffer (Beyotime Biotechnology, Shanghai, China) with a protease
inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA) for
30 min on ice. After centrifugation at 12,000 × g for 10 min at
4°C, the supernatant of lysis was collected and quantified by a BCA
Protein Assay kit (Pierce Biotechnology, Rockford, IL, USA)
according to the manufacturer's instructions. Equal amounts of
protein (40 µg) were separated by 10 or 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then
transferred onto polyvinylidene difluoride (PVDF) membranes
(Millipore, Bedford, MA, USA). After blocking with 5% bovine serum
albumin (BSA; Sigma, St. Louis, MO, USA) for 1 h at room
temperature, membranes were probed with primary antibodies against
EGR3 (sc-191, Santa Cruz, CA, USA; 1:500 dilution), FasL (ab68338,
Abcam, Cambridge, UK; 1:500 dilution), Bak (#6947, Cell Signaling
Technology, Danvers, MA, USA; 1:1,000 dilution), p21 (#2947, Cell
Signaling Technology; 1:1,000 dilution) and GAPDH (10494-1-AP,
Proteintech Group, Chicago, IL, USA; 1:5,000 dilution) at 4°C
overnight. The membranes were subsequently rinsed and further
incubated with a horseradish peroxidase (HRP)-conjugated goat
anti-rabbit secondary antibody (A21020, Abbkine, Redlands, CA, USA;
1:4,000 dilution) for 2 h, followed by visualization with enhanced
chemiluminescence reagent (Pierce Biotechnology). GAPDH served as
an endogenous control to confirm equal loading of proteins.
Plasmid, small interfering RNA (siRNA)
and transfection
The human overexpression plasmid pGV219-EGR3 and its
control plasmid pGV219-Vector were purchased from Genechem Co.,
Ltd. (Shanghai, China). The FasL siRNA (si-FasL) and non-specific
siRNA (si-NC) were designed and synthesized by RiboBio Co., Ltd.
(Guangzhou, China). The effective sequence of siRNA targeting FasL
gene was 5′-GCAAGTCCAACTCAAGGTC-3′. Transient transfection was
performed using the Lipofectamine 2000 reagent (Invitrogen)
according to the manufacturer's instructions. At the indicated
times after transfection, cells were subjected to further
experiments.
CCK-8 assay
Cell proliferation was assessed using Cell Counting
Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan). After
trypsinization and resuspension in culture medium, Huh7 and HCC-LM3
cells were seeded into 96-well plates at 8×103
cells/well. At 80–90% confluency, cells were subjected to transient
transfection, and subsequently cultured for 24, 48 or 72 h. Then,
10 µl of CCK-8 solution was added to each well followed by
incubation at 37°C for 2 h, and optical density (OD) values were
detected at a wavelength of 450 nm with a BioTek ELX800 microplate
reader (BioTek, Vermont, NE, USA). At least six wells were used for
each group, and wells with cell culture medium of the same volume
only served as blanks.
Colony formation assay
At 24 h post-transfection, cells were harvested and
seeded into fresh 6-well plates (1×103 cells/well).
After culture for 2 weeks, the formed colonies were fixed with 4%
paraformaldehyde and stained using 0.1% crystal violet to visualize
colonies for quantification. Percentage of colony formation was
calculated by the following formula: colony formation rate (%) =
numbers of colonies/numbers of seeded cells × 100%.
Apoptosis analysis
Cell apoptosis was quantitatively determined by flow
cytometry with Annexin V-PE/7-AAD Apoptosis Detection kit (BD
Biosciences, San Diego, CA, USA) according to the manufacturer's
instructions. Briefly, at 48 h post-transfection, cells were
collected, washed twice with cold PBS, and resuspended in 1X
binding buffer (1×106 cells/ml). PE-conjugated Annexin V
(5 µl) and 5 µl of 7-amino-actinomycin D (7-AAD) were
subsequently added to 100 µl of cell preparations
(1×105 cells) and incubated for 15 min at room
temperature in the dark. After supplementation with further 400
µl of 1X binding buffer to each sample, the cells were
analyzed by a FACSCalibur instrument (BD Biosciences) within 1
h.
Lentivirus-based EGR3 overexpression
To facilitate in vivo observation of EGR3
function, we commissioned Genechem Co., Ltd. to constructed a
recombinant lentivirus LV-EGR3 and its control LV-Vector, and
transduced them to Huh7 cells according to the manufacturer's
instructions. Briefly, Huh7 cells in good condition were seeded
into 10-cm culture dishes. When reaching 30–40% confluency, cells
were transduced with LV-EGR3 or LV-Vector, respectively.
Twenty-four hours later, cells were washed and covered by fresh
DMEM with 10% FBS. After 48 h, 2 µg/ml puromycin (Sigma) was
applied to screen out the uninfected cells. Once LV-EGR3 or
LV-Vector transfected Huh7 cells were established successfully,
they were used for in vivo tumor xenograft experiment.
Xenograft tumor models
Twelve BALB/c nude mice (female, 4–6-week-old, 16–20
g) were purchased from HFK Bioscience Co., Ltd. (Beijing, China).
The mice were randomly divided into two groups (n=6 in each group)
and bred in the SPF Animal Institute of Tongji Medical College. All
animal experiments were carried out according to the Tongji Medical
College Institutional Animal Care and Use Committee Guidelines. The
LV-EGR3 or LV-Vector transfected Huh7 cells were harvested from
tissue culture dishes and washed twice with PBS. Then cells
(2×107 cells/ml) were resuspended in Matrigel (BD
Biosciences) diluted with serum-free DMEM at the ratio of 1:1, and
injected subcutaneously (2×106 cells per mouse) into the
flank of each mouse. After 10 days, the length (L) and width (W) of
the tumors were measured externally using a digital vernier caliper
every 4 days. Tumor volume was determined according to the formula:
V = (L × W2)/2. At the termination of the experiment,
mice were sacrificed and tumor tissue from each mouse was excised,
photographed and weighed.
Statistics
Statistical analysis was carried out by SPSS 21.0
software (SPSS Inc., Chicago, IL, USA). All the data are presented
as mean ± standard deviation (SD). Statistical differences between
groups and among groups were compared using Student's t-test and
ANOVA, respectively. A value of P<0.05 was considered to
indicate a statistically significant difference. Each experiment
was independently performed at least three times.
Results
Expression of EGR3 is frequently
downregulated in human HCC specimens and cell lines
To evaluate the possible relationship between EGR3
and HCC, EGR3 expression pattern was analyzed in a set of 25 human
HCC specimens, four different human HCC cell lines (PLC/PRF/5,
HCC-LM3, Huh7 and HepG2), and human normal hepatic cell line L02.
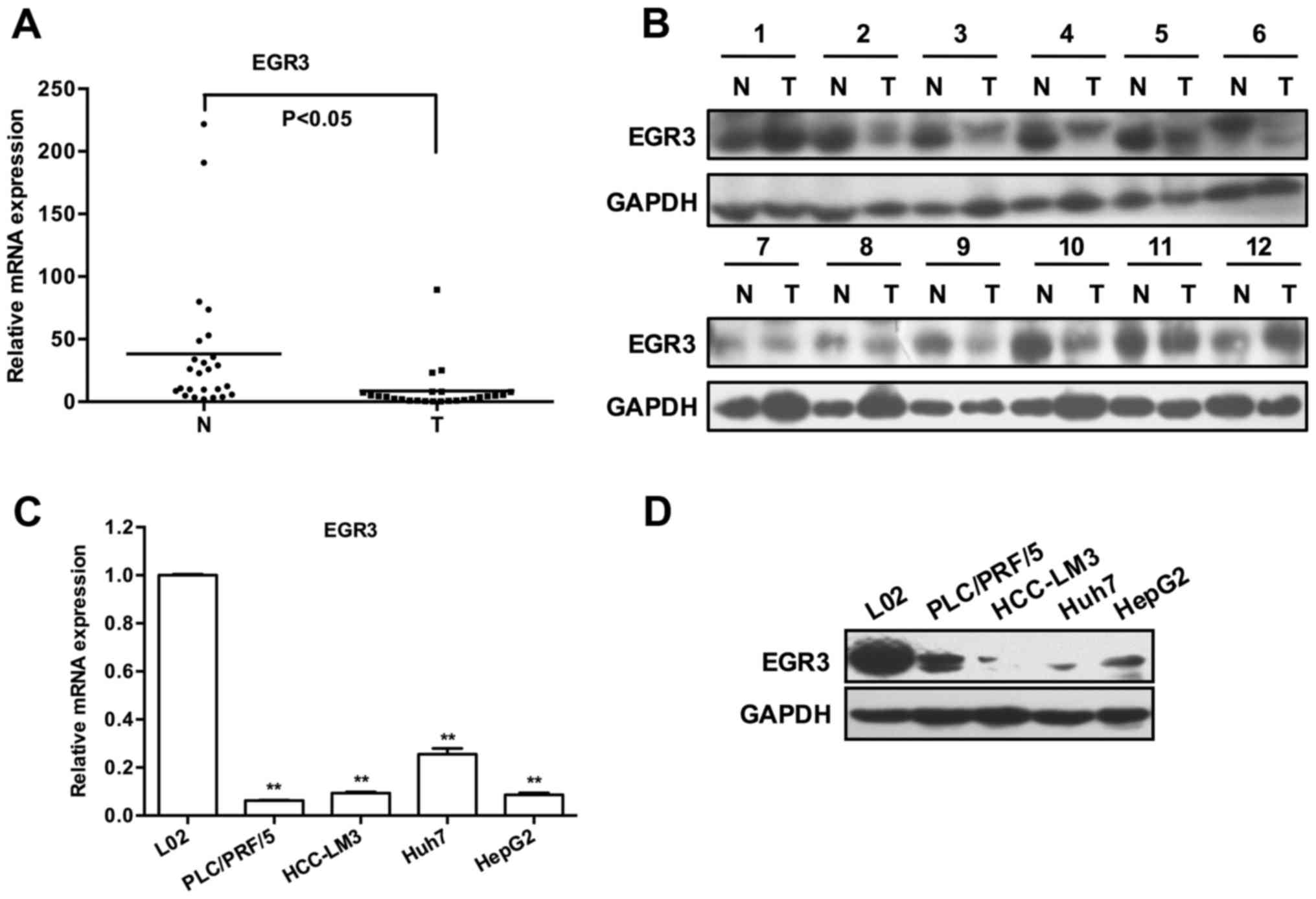
As shown in Fig. 1A, 23 out of 25
cases exhibited lower EGR3 transcripts in HCC tissues than that in
matched adjacent non-tumor tissues (P<0.05). Subsequently, the
protein levels of EGR3 were determined by western blot analysis,
and they were frequently downregulated in HCC tissues as we
expected (Fig. 1B showed protein
bands of EGR3 of partial specimens). Similarly, a remarkable
decrease in the expression of EGR3 mRNA and protein was also
observed in four HCC cell lines compared with the normal hepatic
cell line L02 [Fig. 1C and D;
P<0.01]. These findings revealed a potential association between
low EGR3 expression and HCC pathogenesis.
EGR3 inhibits HCC cell proliferation
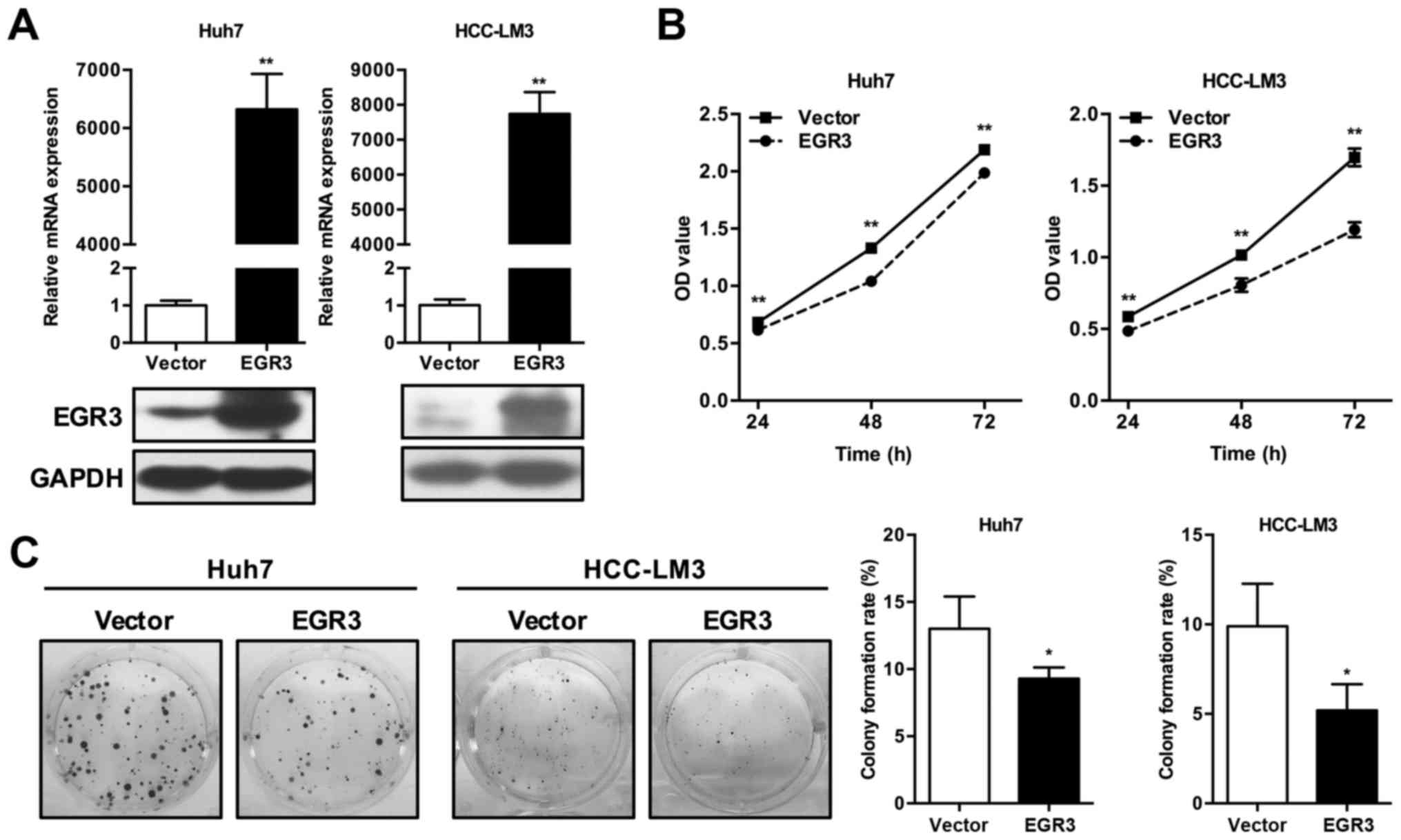
To explore the potential role of EGR3 in HCC,
successful overexpression of EGR3 in Huh7 and HCC-LM3 cells were
first assessed by qRT-PCR and western blot analysis (Fig. 2A). CCK-8 and colony formation
assays were subsequently performed to assess the effect of ectopic
EGR3 expression on proliferation in Huh7 and HCC-LM3 cells. As
demonstrated in Fig. 2B, CCK-8
analysis in two HCC cell lines showed that OD values of EGR3
overexpressing cells at various time-points (24, 48 and 72 h) were
remarkably lower than that of the control cells (P<0.01). In
colony formation assay, high-level expression of EGR3 caused
significant decline of colony-forming ability as evidenced by the
obvious decrease of colony formation rates compared with the
control cells. The colony formation rates of Huh7 cells in vector
control group and EGR3 group were 13.0±2.41 and 9.3±0.82%,
respectively (Fig. 2C; P<0.05).
Similar result of colony formation assay was also observed in
HCC-LM3 cells, as summarized in Fig.
2C (vector control group and EGR3 group, 9.9±2.36 and
5.2±1.46%, respectively; P<0.05). These data indicated the
effect of EGR3 on proliferation inhibition in HCC cells.
EGR3 induces HCC cell apoptosis
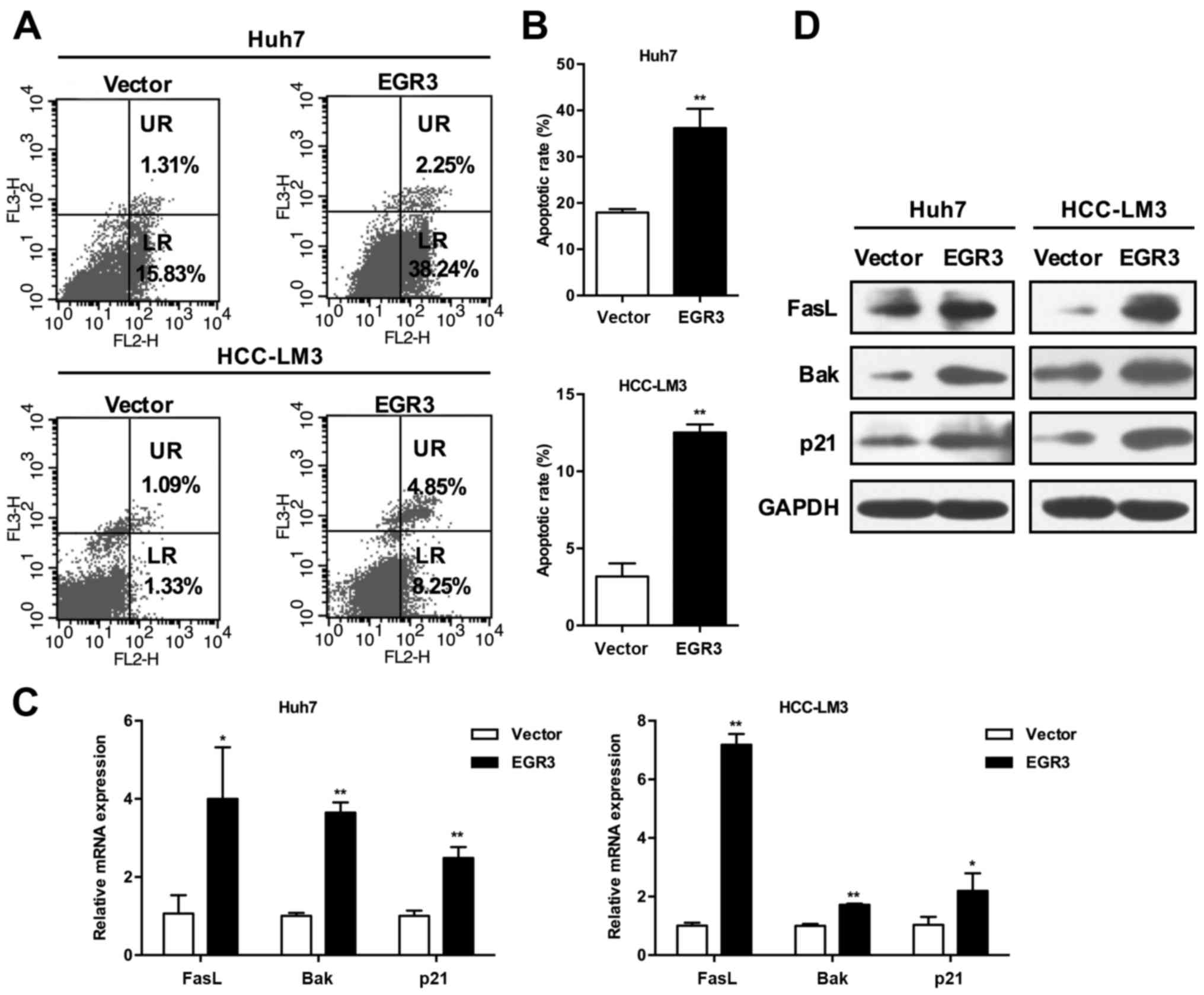
The flow cytometry was performed to evaluate the
involvement of EGR3 in cell apoptosis. Apoptotic cell death was
determined by detecting the cells in the lower right (LR) and upper
right (UR) quadrants of the graphs (Fig. 3A), which are regarded as
early-stage and late-stage apoptotic cells, respectively. As shown
in Fig. 3B, EGR3 significantly
induced apoptosis in the two HCC cell sections. The percentages of
total apoptotic cells (LR+UR quadrants) in vector control group and
EGR3 group were as follows: 17.93±0.78 and 36.21±4.14% in Huh7
cells; 3.18±0.85 and 12.52±0.53% in HCC-LM3 cells (P<0.01).
These results indicated that EGR3 inhibited growth of HCC cells
partially through the induction of apoptosis.
EGR3 upregulates expression of FasL, Bak,
and p21 in HCC cells
Given the previous studies that EGR3 promoted
trans-activation of FasL gene in certain cancer cells, we
speculated that EGR3 also enhanced FasL expression in HCC cells.
Indeed, we found that FasL mRNA and protein expression was elevated
simultaneously with increased expression of EGR3 when EGR3 was
overexpressed in Huh7 and HCC-LM3 cells (Fig. 3C and D; P<0.05, P<0.01).
Furthermore, the expression of pro-apoptotic Bak and cell cycle
inhibitor p21 was also detected. Consistent with the changes of
FasL expression, both mRNA and protein levels of Bak and p21 were
significantly promoted in EGR3 overexpressing cells compared with
the control cells (Fig. 3C and D;
P<0.05, P<0.01). These results suggested that cell growth
inhibition induced by EGR3 was associated with upregulation of
FasL, Bak and p21.
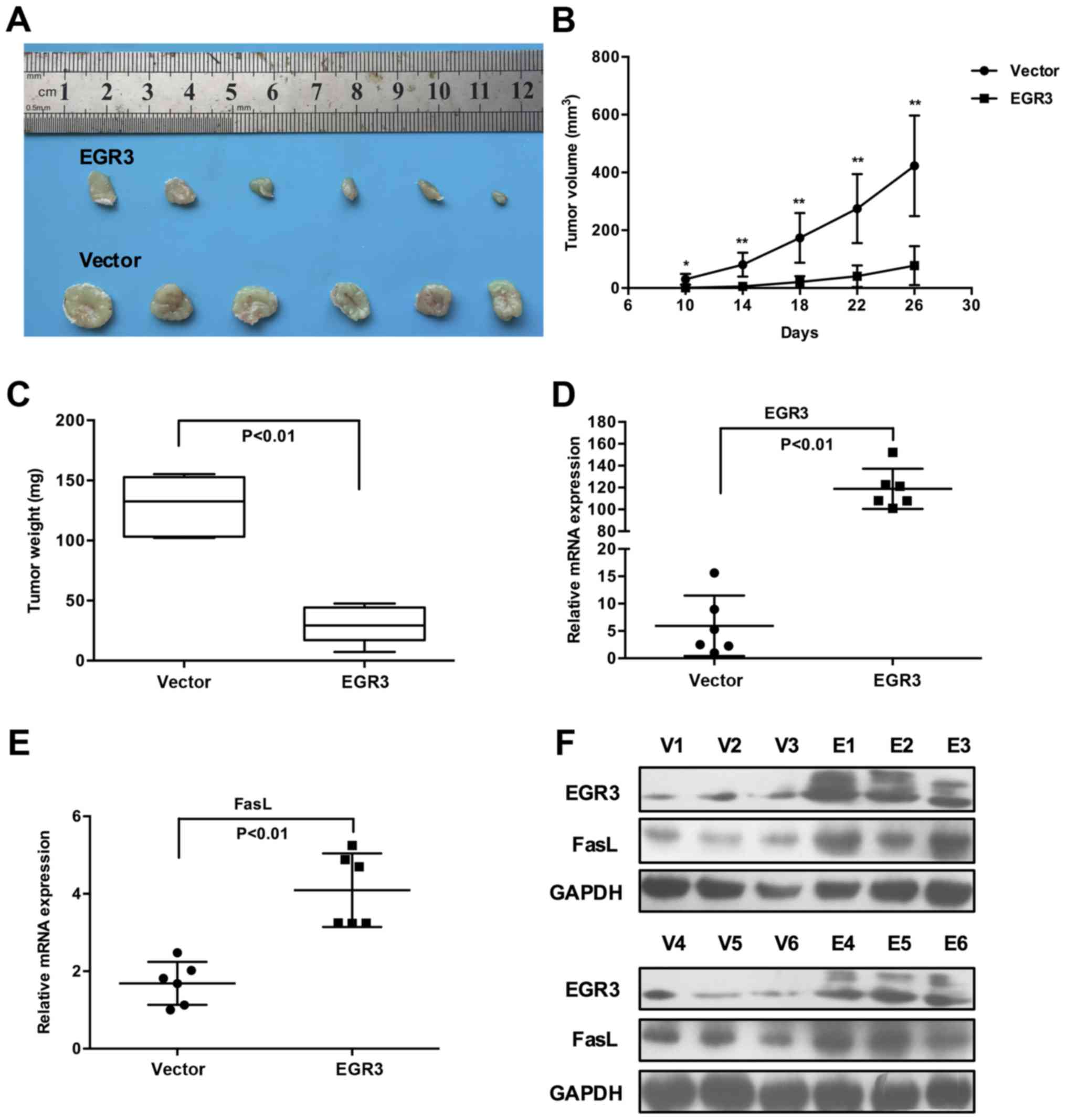
EGR3 restricts tumor growth and increases
FasL expression in xenograft mouse models of HCC
Following in vitro studies, we sought to
determine whether EGR3 restricts in vivo tumor growth by
utilizing xenograft mouse models. In view of the identical
inhibitory effects of EGR3 on Huh7 and HCC-LM3 cell growth, we
selected Huh7 cells for further in vivo studies. At the
outset of the experiment, a recombinant lentivirus LV-EGR3 and its
control LV-Vector were constructed to transduce Huh7 cells. Once
LV-EGR3-Huh7 and LV-Vector-Huh7 cells were established
successfully, they were subcutaneously injected into nude mice, and
tumor growth was monitored regularly. Consistent with our in
vitro results, ectopic expression of EGR3 in Huh7 cells
displayed significantly lower tumorigenicity as evidenced by the
much smaller volumes of tumor masses isolated from nude mice
compared with the control cells (Fig.
4A). Furthermore, a significant retardation of tumor growth and
decreased tumor weights were observed in the LV-EGR3-Huh7 group
when compared with the LV-Vector-Huh7 group (Fig. 4B and C; P<0.05, P<0.01).
Additionally, we also confirmed that the expression of EGR3 was
indeed upregulated in the LV-EGR3-Huh7 group compared to that in
the LV-Vector-Huh7 group, and the expression of FasL was markedly
increased in the xenograft tumor tissues which exhibited high EGR3
expression (Fig. 4D–F; P<0.01).
These data provided strong evidence that EGR3 restricted tumor
growth and promoted FasL expression in vivo.
FasL is essential for the growth
suppression induced by EGR3 in HCC cells
To assess the role of FasL in EGR3-mediated growth
suppression of HCC cells, we established a co-transfection in Huh7
and HCC-LM3 cells with EGR3 overexpression plasmid and FasL siRNA.
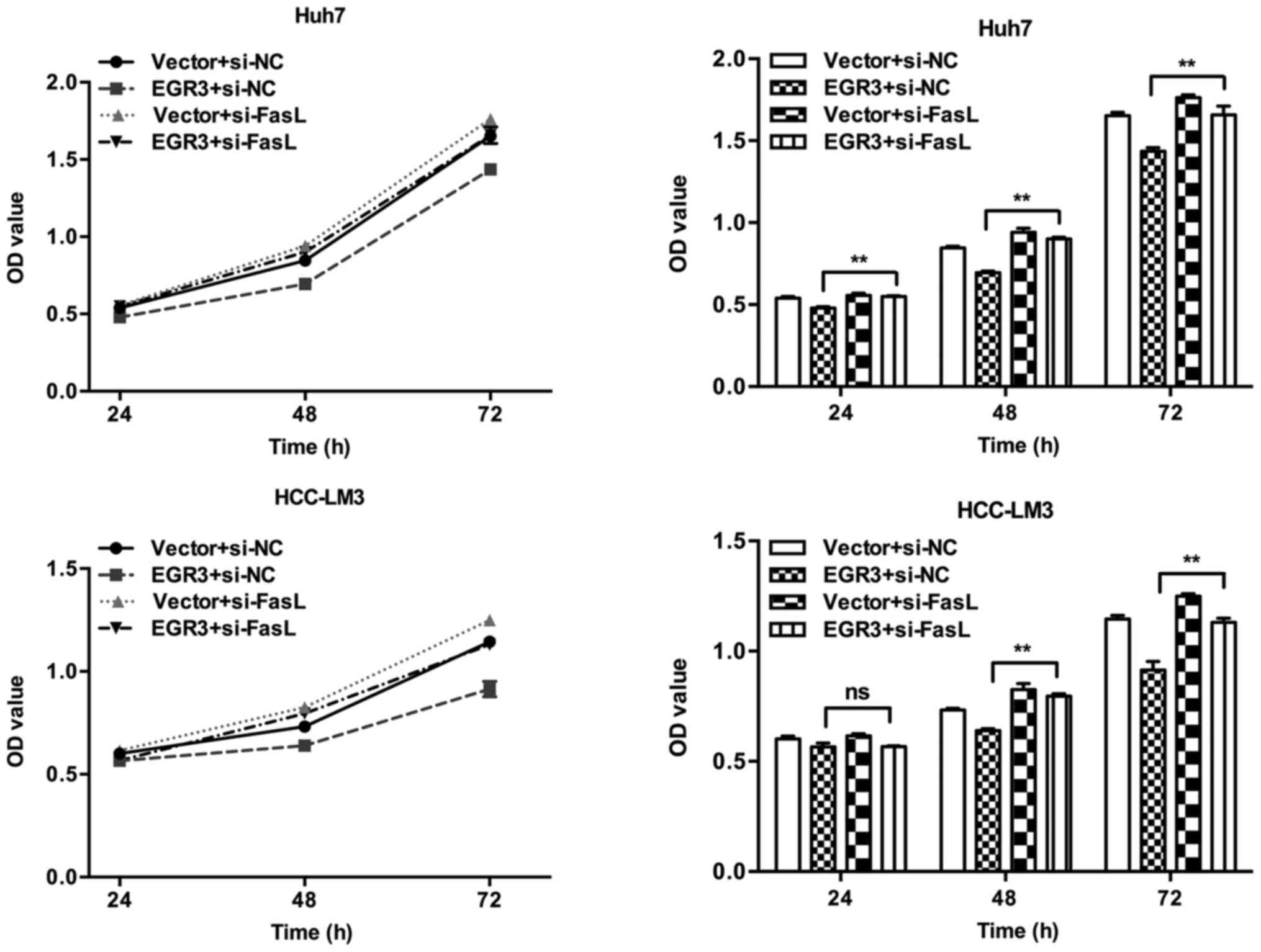
CCK8 analysis showed that siRNA-mediated silencing of FasL gene
significantly attenuated the anti-proliferation effect conferred by
EGR3 overexpression in Huh7 and HCC-LM3 cells (Fig. 5; P<0.01). In addition,
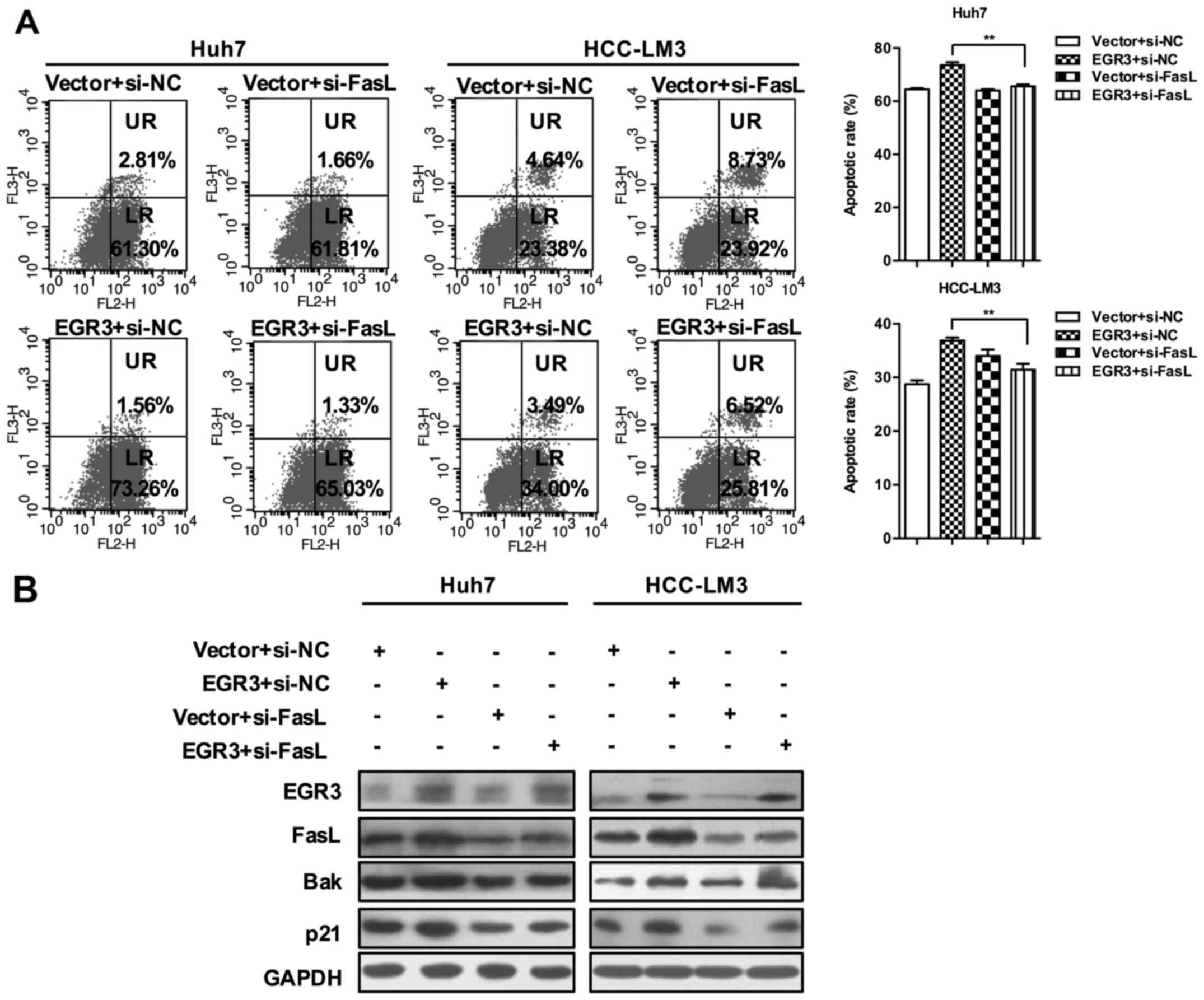
EGR3-induced cell apoptosis was also hampered after silencing of
FasL expression (65.61±0.72 and 73.66±1.02% in Huh7 cells;
31.46±1.13 and 36.88±0.58% in HCC-LM3 cells, Fig. 6A; P<0.01). Finally, we found
that knockdown of FasL gene prevented the increase of Bak and p21
expression induced by EGR3 overexpression as well (Fig. 6B). Taken together, these results
suggested the essential role of FasL in cell growth inhibition
mediated by EGR3 in HCC.
Discussion
HCC is one of the most common malignancies worldwide
and highly prevalent in China. In addition to chronic viral
infections and hepatotoxic agents, the pathogenesis of HCC is
associated with abnormalities of several oncogenes and tumor
suppressor genes (37,38). A series of studies have been
carried out to identify potential molecules for treatment of HCC to
date.
EGR3 is a zinc finger transcription factor, which
was previously reported to be implicated in neurodevelopment,
autoimmunity, inflammation and angiogenesis (13–16).
Furthermore, the effects of EGR3 on tumor initiation and
progression in certain cancers have also been evaluated, and some
studies have described novel role of EGR3 gene as a tumor
suppressor in lung carcinoma, leukemia and gastric cancer (20–22).
However, the relationship between EGR3 and HCC has not yet been
elucidated. In this study, we assessed the effect of EGR3 on HCC
cell growth and investigated the possible molecular mechanisms. We
examined the expression pattern of EGR3 in human HCC specimens,
human normal hepatic and HCC cell lines. We observed that
expression of EGR3 was frequently downregulated in human HCC
tissues and cell lines, suggesting a potential association between
the low expression of EGR3 and HCC pathogenesis. Subsequently, we
utilized gain-of-function approach to determine the effect of EGR3
on cell growth in two HCC cell lines, Huh7 and HCC-LM3 cells. We
demonstrated that ectopic expression of EGR3 inhibited
proliferation and induced apoptosis, leading to cell growth
suppression in Huh7 and HCC-LM3 cells in vitro. Our in
vivo results were further confirmed by xenograft mouse models.
Lentivirus-mediated EGR3 overexpression in Huh7 cells exhibited
obviously lower tumorigenicity as evidenced by the significant
retardation of tumor growth, reduced tumor volumes and decreased
tumor weights. As a whole, these results provided strong evidence
that EGR3 inhibited the growth of HCC cells both in vitro
and in vivo, suggesting the potential of EGR3 as a tumor
suppressor gene for prevention or treatment of HCC.
Apoptosis is an essential cellular activity involved
in multiple physiological and pathological processes. Failure of
apoptosis could allow the survival of abnormal cells, which are
closely related to tumorigenesis. It is well established that
apoptosis can be triggered via two distinct but convergent pathways
known as Fas receptor-mediated pathway and mitochondrial pathway,
respectively (39). The Fas/FasL
apoptotic pathway has been considered a critical mechanism for
eliminating tumor cells. It was previously demonstrated that FasL
was required for curcumin-induced apoptosis in Huh7 cells (40), and the Fas/FasL pathway contributed
to the antitumor effects of a combination therapy of interferon
(IFN)α and 5-fluorouracil (5-FU) against HCC cells (41), and intratumoral injections of an
adenovirus expressing FasL gene into the HNSCC (head and neck
squamous cell carcinoma) cell xenografts induced significant growth
suppression of tumors (42).
Combining these reports and our experimental results, as well as
the role of EGR3 in transactivation of the FasL promoter in certain
cell types (35,36), we speculate that EGR3 may also
promote FasL expression, and FasL is related to the EGR3-mediated
cell growth inhibition in HCC cells. Indeed, we observed a
remarkable increase of FasL mRNA and protein expression in EGR3
overexpressed Huh7 and HCC-LM3 cells. Furthermore, the phenomenon
of high-level FasL expression also existed in xenograft tumor
tissues which exhibited high EGR3 expression. In addition, our
results revealed that the expression of Bak and p21 was
significantly enhanced following upregulation of EGR3 in HCC cells.
p21 is a potent inhibitor of cyclin-dependent kinases (CDKs) and
inhibits the activity of CDKs following binding to them, leading to
cell growth arrest at specific stages of the cell cycle (43). Bak is a pro-apoptotic and acts as a
critical downstream effector in apoptosis process to permeabilize
the outer mitochondrial membrane, leading to the release of
apoptogenic factors including cytochrome c to the cytoplasm
(44). These results indicated
that Bak and p21 were also involved in EGR3-induced cell growth
suppression.
Finally, we sought to determine whether FasL is
essential for the role of EGR3 in HCC cells in vitro. We
established a co-transfection in Huh7 and HCC-LM3 cells with EGR3
overexpression plasmid and FasL siRNA. We found that siRNA-mediated
silencing of FasL gene significantly attenuated the effects of
anti-proliferation and apoptosis conferred by EGR3 overexpression
in Huh7 and HCC-LM3 cells, suggesting an essential role of FasL in
EGR3-mediated growth suppression in HCC cells as expect.
Furthermore, knockdown of FasL also impeded the increase of Bak and
p21 expression induced by EGR3 overexpression. We asked why this
phenomenon appeared. Through consulting literature materials, we
found that binding of FasL with Fas could activate
mitogen-activated protein kinase kinase kinase 5 (ASK1) via death
domain-associated protein Daxx (45). The activated ASK1 itself triggers
the activation of JNK and p38 by phosphorylation cascades involving
a series of protein kinases (46,47).
JNK and p38 pathways are key mechanisms involved in stability and
induction of p53, and induced apoptosis by activation of p53 in
lung carcinoma cells, hepatoma cells, and leukemia cells (48). The p53 tumor suppressor is a potent
transcription factor that activates multiple genes in charge of
apoptosis, cell cycle arrest, and autophagy (49). Bak and p21 are common
transcriptional targets of p53 (50,51).
Based on the above studies, we speculate that overexpression of
EGR3 can upregulate FasL expression followed by activation of JNK
and p38 pathways. Then the tumor suppressor p53 is activated to
transactivate promoters of Bak and p21, and the cell growth is
ultimately inhibited. However, this conjecture needs to be further
verified.
Taken together, in this study, we demonstrate a
novel molecular mechanism through which EGR3 inhibits HCC cell
growth. Our study provides new insight into the close cooperation
between EGR3 and FasL in tumor regulation, suggesting that EGR3 may
be a candidate gene for therapy of HCC.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81302112 and no. 81670554) and
Wuhan application basis research project (no.
2014060101010052).
References
|
1
|
Yang JD and Roberts LR: Hepatocellular
carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 7:448–458.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huo X, Zhang Q, Liu AM, Tang C, Gong Y,
Bian J, Luk JM, Xu Z and Chen J: Overexpression of Yes-associated
protein confers doxorubicin resistance in hepatocellullar
carcinoma. Oncol Rep. 29:840–846. 2013.
|
|
3
|
Tang ZY: Hepatocellular carcinoma - cause,
treatment and metastasis. World J Gastroenterol. 7:445–454. 2001.
View Article : Google Scholar
|
|
4
|
Wu CS, Yen CJ, Chou RH, Li ST, Huang WC,
Ren CT, Wu CY and Yu YL: Cancer-associated carbohydrate antigens as
potential biomarkers for hepatocellular carcinoma. PLoS One.
7:e394662012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gómez-Martín D, Díaz-Zamudio M,
Galindo-Campos M and Alcocer-Varela J: Early growth response
transcription factors and the modulation of immune response:
Implications towards autoimmunity. Autoimmun Rev. 9:454–458. 2010.
View Article : Google Scholar
|
|
6
|
Thiel G and Cibelli G: Regulation of life
and death by the zinc finger transcription factor Egr-1. J Cell
Physiol. 193:287–292. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim HJ, Hong JM, Yoon KA, Kim N, Cho DW,
Choi JY, Lee IK and Kim SY: Early growth response 2 negatively
modulates osteoclast differentiation through upregulation of Id
helix-loop-helix proteins. Bone. 51:643–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bhattacharyya S, Fang F, Tourtellotte W
and Varga J: Egr-1: New conductor for the tissue repair orchestra
directs harmony (regeneration) or cacophony (fibrosis). J Pathol.
229:286–297. 2013. View Article : Google Scholar
|
|
9
|
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo
P, Alexandrov LB, Martincorena I, Dawson KJ, Iorio F, Nik-Zainal S,
Bignell GR, et al: Heterogeneity of genomic evolution and
mutational profiles in multiple myeloma. Nat Commun. 5:29972014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stoddart A, Fernald AA, Wang J, Davis EM,
Karrison T, Anastasi J and Le Beau MM: Haploinsufficiency of
del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce
acute myeloid leukemia in mice. Blood. 123:1069–1078. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boone DN, Qi Y, Li Z and Hann SR: Egr1
mediates p53-independent c-Myc-induced apoptosis via a noncanonical
ARF-dependent transcriptional mechanism. Proc Natl Acad Sci USA.
108:632–637. 2011. View Article : Google Scholar
|
|
12
|
Wirth M, Stojanovic N, Christian J, Paul
MC, Stauber RH, Schmid RM, Häcker G, Krämer OH, Saur D and
Schneider G: MYC and EGR1 synergize to trigger tumor cell death by
controlling NOXA and BIM transcription upon treatment with the
proteasome inhibitor bortezomib. Nucleic Acids Res. 42:10433–10447.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nishimura Y, Takizawa R, Koike S,
Kinoshita A, Satomura Y, Kawasaki S, Yamasue H, Tochigi M, Kakiuchi
C, Sasaki T, et al: Association of decreased prefrontal hemodynamic
response during a verbal fluency task with EGR3 gene polymorphism
in patients with schizophrenia and in healthy individuals.
Neuroimage. 85:527–534. 2014. View Article : Google Scholar
|
|
14
|
Safford M, Collins S, Lutz MA, Allen A,
Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH,
et al: Egr-2 and Egr-3 are negative regulators of T cell
activation. Nat Immunol. 6:472–480. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li S, Miao T, Sebastian M, Bhullar P,
Ghaffari E, Liu M, Symonds AL and Wang P: The transcription factors
Egr2 and Egr3 are essential for the control of inflammation and
antigen-induced proliferation of B and T cells. Immunity.
37:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu D, Evans I, Britton G and Zachary I:
The zinc-finger transcription factor, early growth response 3,
mediates VEGF-induced angiogenesis. Oncogene. 27:2989–2998. 2008.
View Article : Google Scholar
|
|
17
|
Baron VT, Pio R, Jia Z and Mercola D:
Early growth response 3 regulates genes of inflammation and
directly activates IL6 and IL8 expression in prostate cancer. Br J
Cancer. 112:755–764. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suzuki T, Inoue A, Miki Y, Moriya T,
Akahira J, Ishida T, Hirakawa H, Yamaguchi Y, Hayashi S and Sasano
H: Early growth responsive gene 3 in human breast carcinoma: A
regulator of estrogen-meditated invasion and a potent prognostic
factor. Endocr Relat Cancer. 14:279–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pio R, Jia Z, Baron VT and Mercola D; UCI
NCI SPECS Consortium of the Strategic Partners for the Evaluation
of Cancer Signatures-Prostate Cancer: Early growth response 3
(Egr3) is highly overexpressed in non-relapsing prostate cancer but
not in relapsing prostate cancer. PLoS One. 8:e540962013.
View Article : Google Scholar
|
|
20
|
Salotti J, Sakchaisri K, Tourtellotte WG
and Johnson PF: An Arf-Egr-C/EBPβ pathway linked to ras-induced
senescence and cancer. Mol Cell Biol. 35:866–883. 2015. View Article : Google Scholar :
|
|
21
|
Cheng H, Hao S, Liu Y, Pang Y, Ma S, Dong
F, Xu J, Zheng G, Li S, Yuan W, et al: Leukemic marrow infiltration
reveals a novel role for Egr3 as a potent inhibitor of normal
hematopoietic stem cell proliferation. Blood. 126:1302–1313. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liao F, Ji MY, Shen L, Qiu S, Guo XF and
Dong WG: Decreased EGR3 expression is related to poor prognosis in
patients with gastric cancer. J Mol Histol. 44:463–468. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suda T, Takahashi T, Golstein P and Nagata
S: Molecular cloning and expression of the Fas ligand, a novel
member of the tumor necrosis factor family. Cell. 75:1169–1178.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lavrik IN and Krammer PH: Regulation of
CD95/Fas signaling at the DISC. Cell Death Differ. 19:36–41. 2012.
View Article : Google Scholar :
|
|
25
|
Kober AM, Legewie S, Pforr C, Fricker N,
Eils R, Krammer PH and Lavrik IN: Caspase-8 activity has an
essential role in CD95/Fas-mediated MAPK activation. Cell Death
Dis. 2:e2122011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Suda T, Okazaki T, Naito Y, Yokota T, Arai
N, Ozaki S, Nakao K and Nagata S: Expression of the Fas ligand in
cells of T cell lineage. J Immunol. 154:3806–3813. 1995.PubMed/NCBI
|
|
27
|
Montel AH, Bochan MR, Hobbs JA, Lynch DH
and Brahmi Z: Fas involvement in cytotoxicity mediated by human NK
cells. Cell Immunol. 166:236–246. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Griffith TS, Brunner T, Fletcher SM, Green
DR and Ferguson TA: Fas ligand-induced apoptosis as a mechanism of
immune privilege. Science. 270:1189–1192. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Uckan D, Steele A, Cherry, Wang BY,
Chamizo W, Koutsonikolis A, Gilbert-Barness E and Good RA:
Trophoblasts express Fas ligand: A proposed mechanism for immune
privilege in placenta and maternal invasion. Mol Hum Reprod.
3:655–662. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reichmann E: The biological role of the
Fas/FasL system during tumor formation and progression. Semin
Cancer Biol. 12:309–315. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng SY, Li DC, Zhang ZD, Zhao J and Ge
JF: Adenovirus-mediated FasL gene transfer into human gastric
carcinoma. World J Gastroenterol. 11:3446–3450. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siena L, Pace E, Ferraro M, Di Sano C,
Melis M, Profita M, Spatafora M and Gjomarkaj M: Gemcitabine
sensitizes lung cancer cells to Fas/FasL system-mediated killing.
Immunology. 141:242–255. 2014. View Article : Google Scholar :
|
|
33
|
Bianco SR, Sun J, Fosmire SP, Hance K,
Padilla ML, Ritt MG, Getzy DM, Duke RC, Withrow SJ, Lana S, et al:
Enhancing anti-melanoma immune responses through apoptosis. Cancer
Gene Ther. 10:726–736. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kavurma MM and Khachigian LM: Signaling
and transcriptional control of Fas ligand gene expression. Cell
Death Differ. 10:36–44. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mittelstadt PR and Ashwell JD: Cyclosporin
A-sensitive transcription factor Egr-3 regulates Fas ligand
expression. Mol Cell Biol. 18:3744–3751. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Inoue A, Omoto Y, Yamaguchi Y, Kiyama R
and Hayashi SI: Transcription factor EGR3 is involved in the
estrogen-signaling pathway in breast cancer cells. J Mol
Endocrinol. 32:649–661. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee JS and Thorgeirsson SS: Genome-scale
profiling of gene expression in hepatocellular carcinoma:
Classification, survival prediction, and identification of
therapeutic targets. Gastroenterology. 127(Suppl 1): S51–S55. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Thorgeirsson SS, Lee JS and Grisham JW:
Molecular prognostication of liver cancer: End of the beginning. J
Hepatol. 44:798–805. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tang D, Lotze MT, Kang R and Zeh HJ:
Apoptosis promotes early tumorigenesis. Oncogene. 30:1851–1854.
2011. View Article : Google Scholar
|
|
40
|
Wang WZ, Li L, Liu MY, Jin XB, Mao JW, Pu
QH, Meng MJ, Chen XG and Zhu JY: Curcumin induces FasL-related
apoptosis through p38 activation in human hepatocellular carcinoma
Huh7 cells. Life Sci. 92:352–358. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakamura M, Nagano H, Sakon M, Yamamoto T,
Ota H, Wada H, Damdinsuren B, Noda T, Marubashi S, Miyamoto A, et
al: Role of the Fas/FasL pathway in combination therapy with
interferon-alpha and fluorouracil against hepatocellular carcinoma
in vitro. J Hepatol. 46:77–88. 2007. View Article : Google Scholar
|
|
42
|
El Ojeimy S, McKillop JC, El-Zawahry AM,
Holman DH, Liu X, Schwartz DA, Day TA, Dong JY and Norris JS: FasL
gene therapy: A new therapeutic modality for head and neck cancer.
Cancer Gene Ther. 13:739–745. 2006. View Article : Google Scholar
|
|
43
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Westphal D, Dewson G, Czabotar PE and
Kluck RM: Molecular biology of Bax and Bak activation and action.
Biochim Biophys Acta. 1813:521–531. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Swindall AF and Bellis SL: Sialylation of
the Fas death receptor by ST6Gal-I provides protection against
Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem.
286:22982–22990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang X, Khosravi-Far R, Chang HY and
Baltimore D: Daxx, a novel Fas-binding protein that activates JNK
and apoptosis. Cell. 89:1067–1076. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Juo P, Kuo CJ, Reynolds SE, Konz RF,
Raingeaud J, Davis RJ, Biemann HP and Blenis J: Fas activation of
the p38 mitogen-activated protein kinase signalling pathway
requires ICE/CED-3 family proteases. Mol Cell Biol. 17:24–35. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee KB, Kim KR, Huh TL and Lee YM: Proton
induces apoptosis of hypoxic tumor cells by the p53-dependent and
p38/JNK MAPK signaling pathways. Int J Oncol. 33:1247–1256.
2008.PubMed/NCBI
|
|
49
|
Maiuri MC, Galluzzi L, Morselli E, Kepp O,
Malik SA and Kroemer G: Autophagy regulation by p53. Curr Opin Cell
Biol. 22:181–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
51
|
Graupner V, Alexander E, Overkamp T,
Rothfuss O, De Laurenzi V, Gillissen BF, Daniel PT, Schulze-Osthoff
K and Essmann F: Differential regulation of the proapoptotic
multi-domain protein Bak by p53 and p73 at the promoter level Cell
Death Differ. 18:1130–1139. 2011.
|