Introduction
The tumor protein D52 (TPD52) protein family
consists of TPD52 (1), -53
(1–4), -54 (2,4), and
-55 (5). The first identified
protein of this family, TPD52, was found to be overexpressed in
breast and lung cancers (6,7).
Other family members have also been reported to be highly expressed
in colon (8,9), ovary (10–12),
testis (5,13), prostate (14), and breast (15–17)
cancers. Previous reports showed that overexpression of
tpd52 in non-malignant 3T3 fibroblasts induces malignant
transformation and increases cell proliferation and
anchorage-independent growth (18,19).
Moreover, overexpression of tpd52 led to an increase in cell
proliferation as well as phosphorylation of Akt/protein kinase B
(PKB) in prostate cancer (20–22)
and also protected the cells from apoptosis induced by androgen
deprivation via activation of the Stat3/Bcl-2 pathway
(21). In addition, TPD52
regulates cell migration and invasion (23) and inhibits DNA damage repair
(24). TPD53 regulates the cell
cycle and is highly upregulated at the G2-M phase transition
(25). TPD52, -53, and -54 also
interact with each other through their coiled-coil domains
(4). They also have numerous other
binding partners, such as MAL2 (26), the phospholipid binding protein
Annexin VI (27), the SNARE
protein (Synaptobrevin 2), a main components of the SNARE complex
(28), 14–3–3 (29), and adipose differentiation-related
protein (ADRP) (30). These
reports strongly suggest that TPD52 family protein members are
important candidate targets of molecular therapy (31).
Signaling pathways such as mitogen-activated protein
kinase (MAPK), Akt/PKB, and integrin signaling regulate cell
proliferation (32), survival
(33), and metastasis (34). Moreover, cross-talk of integrin
signaling with MAPK signaling (35) and Akt/PKB signaling (36,37)
has been widely reported. Despite these findings, the direct
effects of TPD52 family protein members on cell proliferation,
migration, invasion, and metastasis in oral squamous cell carcinoma
cell (OSCC) are still under investigation.
We recently reported (38) that TPD54 is highly expressed in
both OSCC and in hyperplastic epidermal cells around cancer tissue.
We also showed that TPD54 is a negative regulator of extracellular
matrix (ECM)-dependent attachment and migration in OSCC (39). However, little is known concerning
the detailed physiological and pathological functions of TPD54.
Herein, as an extension of our previous study, we investigated in
greater detail the expression of D52 proteins in OSCC tissue, and
the effects of TPD52, -53 and -54 that underlie cell growth,
migration, and invasion of OSCC cells. The results showed that
TPD54 was overexpressed in both highly and poorly differentiated
OSCC tissue, while the expression of TPD52 was not as high as that
of TPD54. TPD53 was moderately expressed. Furthermore, TPD54
inhibited the colony formation and migration. These results
indicated that overexpression of TPD54 negatively regulates tumor
growth in vivo.
Materials and methods
Co-immunoprecipitation (Co-IP)
analysis
Co-immunoprecipitation analysis was carried out
using a commercial kit [Immunoprecipitation kit (Protein G),
Sigma-Aldrich, St. Louis, MO, USA]. Subconfluent SAS cells in a
10-cm culture dish were lysed with the lysis buffer, and the total
extract was immunoprecipitated with 5 µg of anti-TPD52,
-TPD53, -TPD54 or -GAPDH antibodies, or 5 µg of pre-immune
mouse IgG (described above), according to the manufacturer's
protocol. Subsequently, the precipitated proteins were eluted in 1X
SDS sample buffer (Bio-Rad, Hercules, CA, USA) containing 5% of
2-mercaptoethanol, and co-precipitating proteins were then analyzed
by western blotting as described in another subsection.
Protein preparation and western blot
analysis
Total cellular proteins were collected with Triton
X-100 lysis buffer [50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 0.5%
Triton X-100, 5 mM ethrylendiaminetetraacetic acid (EDTA), 1 mM
sodium o-vanadate] supplemented with Complete Mini protease
inhibitor tablet (Roche Diagnostics, Mannheim, Germany) 48 h after
gene-transfection. Protein concentrations were measured using the
Quick Start Bradford reagent (Bio-Rad) with bovine serum albumin
(BSA) as a standard. For western blot analysis, 20 µg of
total cellular protein was analyzed by sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) using a 4–20%
gradient gel (Bio-Rad) and blotted onto a polyvinylidene difluoride
membrane using iBlot 2 (Thermo Fisher Scientific, Waltham, MA,
USA). After blocking of non-specific binding with 0.2% non-fat dry
milk (Cell Signaling Technology, Danvers, MA, USA) in Tris-buffered
saline (Takara Bio, Shiga, Japan), the membrane was incubated with
the following primary antibodies: anti-TPD52 [1/1,000 dilution;
rabbit monoclonal antibody, Abcam, Branford, CT, USA (ab182578)];
anti-TPD53 [1/1,000 dilution; rabbit polyclonal antibody,
Proteintech, Rosemont, IL, USA (14732-1-AP)]; anti-TPD54 [1/1,000
dilution; rabbit polyclonal antibody, Proteintech (11795-1-AP)];
anti-hemagglutinin (HA) [1/1,000 dilution; rabbit polyclonal
antibody, Clontech, Mountain View, CA, USA (631207)]; anti-green
fluorescent protein (GFP) [1/1,000 dilution; rabbit polyclonal
antibody, Santa Cruz, Dallas, TX, USA (sc-8334)]; or
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [1/10,000
dilution; mouse monoclonal antibody, Sigma-Aldrich (G9295)]
antibody and horseradish-peroxidase-conjugated secondary antibody
(for rabbit, donkey polyclonal antibody (NA934V); for mouse, sheep
polyclonal antibody (NA931V) (GE Healthcare UK Ltd.,
Buckinghamshire, UK) as described previously (39). Subsequent washings were conducted
using TBS-T. The protein bands were visualized using Amersham ECL
Western Blotting Detection reagents (GE Healthcare UK Ltd.) and a
ChemiDoc XRS Plus ImageLab System (Bio-Rad).
Clinical samples
All samples were acquired from patients who
underwent treatment for squamous cell carcinoma in Showa University
Dental Hospital from January 2001 through March 2015. All patients
provided informed consent before enrollment in the study, in
accordance with the protocol approved by the Institutional Review
Board at Showa University Dental Hospital (approval no.
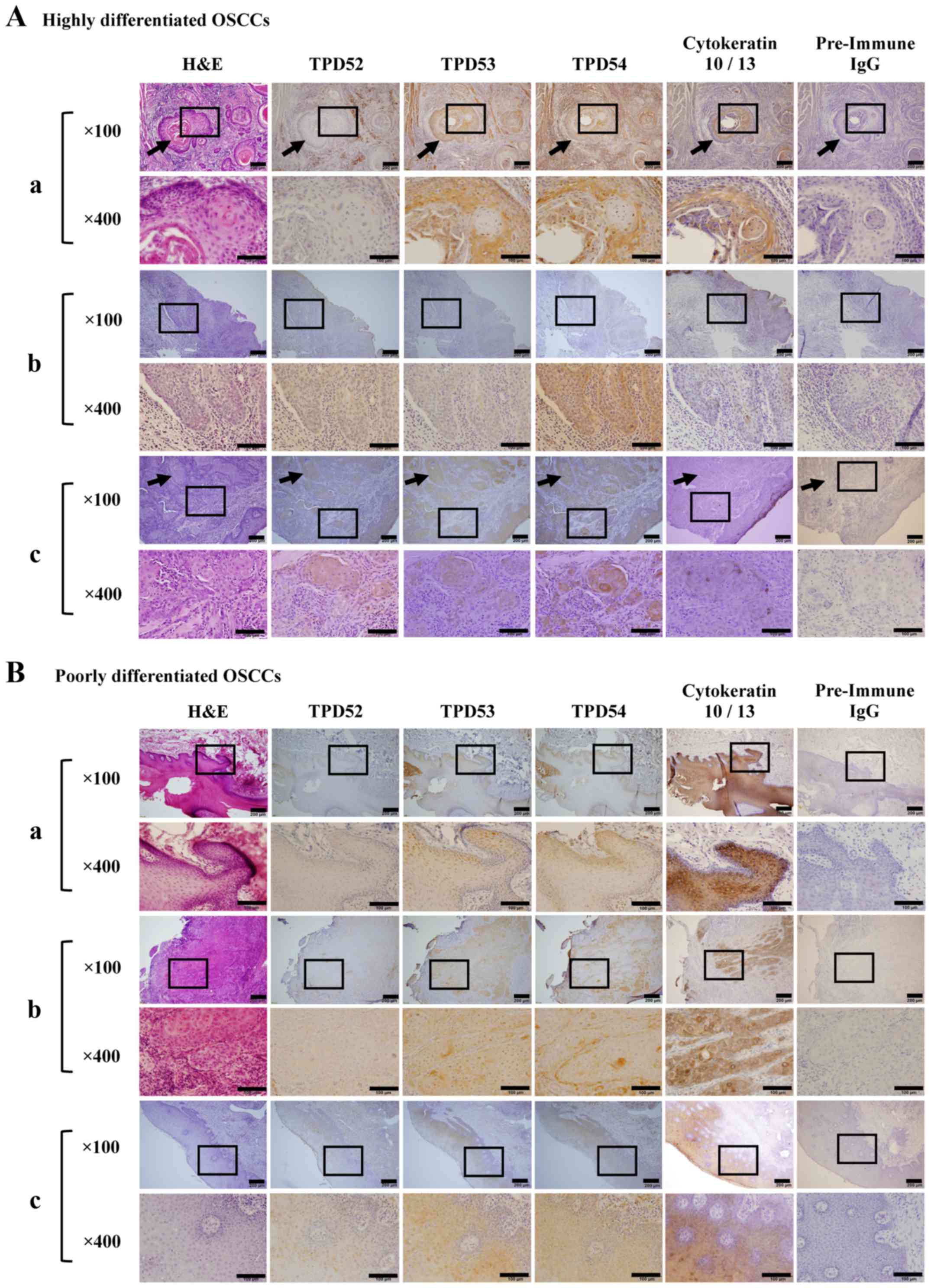
DH2015-013). Primary lesions were resected from tongue (Fig. 2A-a-c and B-a, -b) and gingiva
(Fig. 2B-c). Differentiation grade
was determined according to the pathological reports submitted from
the Division of Pathology, Department of Oral Diagnostic Sciences,
School of Dentistry, Showa University.
Cell culture
SAS (40), HSC2,
and HSC3 cells (41) (human oral
squamous cell carcinoma-derived cell lines) were grown in
high-glucose Dulbecco's modified Eagle's medium (HDMEM) (Wako,
Osaka, Japan) supplemented with 10% fetal bovine serum (FBS), 100
U/ml penicillin, and 100 mg/ml streptomycin (Thermo Fisher
Scientific) at 37°C in an atmosphere containing 5%
CO2.
Molecular constructs, small interfering
RNA (siRNA) and gene transfection
The coding regions of human tpd52 and
-53 cDNAs were amplified by an RT-PCR technique using
single-stranded cDNA reverse-transcribed from total RNA of SAS
cells that was used as a template. All of the primer sequences used
are shown in Table I. Final
concentrations of the primers were 10 µM, respectively. PCR
cycles were 95°C for 3 min; then, 94°C for 30 sec, 60°C for 30 sec,
72°C for 1 min (×40 cycles); and followed by 72°C for 5 min. The
amplified products were subcloned into a pGEM T-Easy vector
(Promega, Madison, WI, USA) using a TA-cloning technique. For
construction of expression vectors of hemagglutinin (HA)-tagged
human tpd52 and -53, cDNA of the coding region was
amplified by PCR employing a sense-BglII-adaptor primer and
an antisense KpnI-adaptor primer. The amplicons were
double-digested with BglII and KpnI and inserted into
the corresponding site of the pCMV-HA vector (Clontech
Laboratories) as described previously (39). The resulting expression vectors
were confirmed by sequencing using an ABI PRISM 310 Genetic
Analyzer (Thermo Fisher Scientific) and the BigDye Terminator v3.1
Cycle Sequencing kit (Thermo Fisher Scientific). The expression
vector for human tpd54 was that used in a previously
described experiment (40). siRNA
against human tpd52 and -54 was purchased from
Sigma-Aldrich and Thermo Fisher Scientific, respectively. Control
siRNA was purchased from Thermo Fisher Scientific. Expression
vectors and siRNAs were transfected with Lipofectamine 2000 (Thermo
Fisher Scientific) according to the manufacturer's protocol.
 | Table IPrimer sequences for molecular
constructs. |
Table I
Primer sequences for molecular
constructs.
| Primer
sequences |
|---|
| TPD52 | |
| Sense |
5′-GTCTGCTTATCAGGAGGGGC-3′ |
| Antisense |
5′-GGCAGTGGGTAGCAGAACAA-3′ |
| TPD53 | |
| Sense |
5′-GAGGTAACCAGAAGCGGCTA-3′ |
| Antisense |
5′-ACAATGTCAAGGCCTGGGTT-3′ |
| HA-tagged | |
| TPD52 | |
| Sense |
5′-GAAGATCTACATGGATTGTAGAGAGATGGA-3′ |
| Antisense |
5′-GGGGTACCTCACAGGCTCTCCTGTGTCTTT-3′ |
| HA-tagged | |
| TPD53 | |
| Sense |
5′-GAAGATCTACATGGAGGCGCAGGCACAAGG-3′ |
| Antisense |
5′-GGGGTACCTTAGCACTGCAGCTCCTCCTCC-3′ |
Generation of stable clones of cell
lines
tpd52 and tpd54 overexpressing (OE)
stable clones were obtained by using GFP Fusion TOPO TA Expression
kits (Thermo Fisher Scientific). Single-stranded cDNAs were
reverse-transcribed from total RNA of SAS cells that was used as a
template. The amplified products were subcloned into a
pcDNA3.1/NT-GFP-TOPO vector using TOPO cloning technology (Thermo
Fisher Scientific). pcDNA3.1/NT-GFP (Thermo Fisher Scientific) was
used as a control vector for OE experiments. TPD54 knocked-down
(KD) stable clones were obtained by using BLOCK-iT Pol miR RNAi
Expression Vector kits (Thermo Fisher Scientific). A
double-stranded oligo targeting tpd54 was prepared by
annealing two single-stranded oligos. The amplified products were
subcloned into a pcDNA6.2-GW/Em-GFP-miR vector using Gateway
cloning technology. pcDNA6.2-GW/Em-GFP-miR-neg (Thermo Fisher
Scientific) was used as a negative control miRNA vector. All of the
primer sequences used are shown in Table II. The resulting expression
vectors were verified by sequencing and were transfected with
Lipofectamine 2000 (Thermo Fisher Scientific). To obtain
tpd52 and tpd54 OE stable clones, transfected SAS
cells were cultivated in the presence of 0.5 mg/ml of Geneticin
(Thermo Fisher Scientific). tpd54 knock-down stable clones
were obtained by cultivation in the presence of 50 µg/ml of
Blasticidin S HCl (Thermo Fisher Scientific), according to the
manufacturer's protocol.
| Table IIPrimer sequences for the generation
of stable clones. |
Table II
Primer sequences for the generation
of stable clones.
| Primer
sequences |
|---|
| TPD52 OE | |
| Sense |
5′-ATGGATTGTAGAGAGATGGA-3′ |
| Antisense |
5′-TCACAGGCTCTCCTGTGTCTTT-3′ |
| TPD54 OE | |
| Sense |
5′-ATGGACTCCGCCGGCCAAGATATCAACCTG-3′ |
| Antisense |
5′-TTAGAAAGGTGCGGGATCCGACAGGGGCTT-3′ |
| TPD54 KD | |
| Sense |
5′-TGCTGTTCAAATTCATGCAAACGCGGGTTTTGGCCACTGACTGACCCGCGTTTATGAATTTGAA-3′ |
| Antisense | 5′-CCTGTTCAAATTCATAAACGCGGGTCAGTCAGTGGCCAAAACCCGCGTTTGCATGAATTTGAAC-3′ |
Cell growth assay
One thousand cells were seeded into a 96-well tissue
culture plate in triplicate. After 48 h, cell growth was assayed
using the tetrazolium salt
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay, as described previously (42). The experiment was performed in
triplicate.
Anchorage-independent growth assay
The anchorage-independent growth assay was carried
out by using a commercial kit (CytoSelect 96-Well In Vitro
Tumor Sensitivity assay, Cell Biolabs, San Diego, CA, USA)
according to the manufacturer's protocol. In total, 2500
transfected cells were grown in soft agar. After a week, the cells
were examined under a microscope (Eclipse TS100/TS100-F, Nikon,
Tokyo, Japan), photographed with a digital CCD camera (DS-Fil,
Nikon), and formed colonies were counted (colonies/field). A colony
was defined as a cell cluster that was >50 µm in
diameter. Thereafter, cell growth was assayed using the MTT assay.
The experiment was performed in triplicate.
Cell migration and invasion assays
Chemotaxis, haptotaxis, and invasion assays were
carried out using a commercially available Boyden chamber kit
(Chemotaxicell; Kurabo, Osaka, Japan). For the chemotaxis assay,
the cells were starved in FBS-free medium for 24 h and then 10,000
of the transfected cells were seeded into a chamber inserted in a
24-well culture plate containing 10% FBS as a chemotactic agent.
After 48 h, the cells were fixed with 10% formalin (Wako) and
stained using crystal violet (43). Migrated cells were photographed,
and the numbers of migrated cells were counted (cells/field). For
the haptotaxis assay, the membranes were pre-coated with type I
collagen (Nitta Gelatin Inc., Osaka, Japan), according to the
manufacturer's protocol. Approximately 100,000 transfected cells
were seeded into a chamber inserted in a 24-well culture plate.
After 48 h, the cells were fixed and stained using crystal violet.
Migrated cells were photographed, and were then counted, as
described for the chemotaxis assay. The wound healing assay was
carried out as described previously (44). The transfected cells (100,000) were
seeded into a 24-well culture plate. Once the cells were confluent,
a wound was made by scratching (0.9 mm width). After wound closure
was first observed, the cells were fixed, stained using crystal
violet and photographed. The wound width was then measured. For the
invasion assay, the cells were starved in FBS-free medium for 24 h.
Similar to the chemotaxis assay, 10,000 of the transfected cells
were seeded into a chamber, which was pre-coated with type I
collagen (Nitta Gelatin Inc.), and was then inserted in a 24-well
culture plate. After 48 h, the cells were fixed and stained using
crystal violet. Migrated cells were photographed and were counted
in a manner similar to the chemotaxis and haptotaxis assays. Each
experiment was performed in triplicate.
Mice
This study was approved by the Institutional Animal
Care and Use Committee (approval no. 15024) and was carried out
according to the Showa University Guidelines for Animal
Experiments. Four-week-old female balb/c nu/nu mice (n=3 for each
experimental group) were purchased from Clea Japan, Inc. (Tokyo,
Japan) and were maintained under pathogen-free conditions.
Approximately 1.0×107 cells in 100 µl of
phosphate-buffered saline (PBS) were injected subcutaneously into a
unilateral flank. Tumor-bearing mice were treated with 1 nmol of
siRNAs (Koken, Tokyo, Japan) in 100 µl of AteloGene (Koken),
which were injected into subcutaneous spaces around tumor sections
once a week for 7 weeks from day 7. The AteloGene and siRNA mixture
was generated according to the manufacturer's protocol. At the end
of the experiment, mice were sacrificed by CO2
asphyxiation, and resected tumor, liver, and lung were fixed in
formalin (Wako) and stained with hematoxylin and eosin (H&E)
(Sakura Finetek Japan, Tokyo, Japan), as described previously
(45). Tumor volume was determined
by direct measurement, and was calculated using the formula π/6 ×
(large diameter) × (small diameter)2 (45). The Kaplan-Meier method was used for
survival analysis (46).
Immunohistochemistry
Resected specimens were fixed with 10% formalin,
embedded in paraffin, stained with H&E, and then
immunohistochemically stained for TPD52 family proteins as
described previously (46).
Antigen retrieval was carried out with citrate-phosphate buffer
(0.01 M, pH 6.0) at 121°C for 20 min. Endogenous peroxidases were
blocked by incubating the sample with 10%
H2O2 (Wako) for 10 min. Proteins were blocked
using a commercial kit (Dako, Carpinteria, CA, USA) according to
the manufacturer's protocol. The sections were incubated at 4°C
overnight with primary antibodies (anti-TPD52 antibody, 1:50
dilution, rabbit polyclonal antibody; Biorbyt, Cambridgeshire, UK
(orb100564); anti-TPD53 antibody, 1:200 dilution, rabbit polyclonal
antibody, Proteintech (14732-1-AP); anti-TPD54 antibody, 1:200
dilution, rabbit polyclonal antibody, Proteintech (11795-1-AP);
anti-cytokeratin 10/13, 1/200 dilution, mouse monoclonal antibody,
Santa Cruz (DE-K13); control pre-immune IgG, 1/200 dilution, mouse
IgG, Santa Cruz (sc-2762). The next day, the sections were
incubated with secondary antibodies (EnVision+
system-HRP labelled polymer anti-rabbit/anti-mouse; Dako). Finally,
sections were reacted with a 3, 3′-diaminobenzidine (DAB)
peroxidase substrate kit (Dako) for color development and were
examined under a microscope.
Statistical analysis
All values are expressed as means ± standard
deviation of triplicate data sets. The statistical significance of
differences between groups was analyzed using a paired Student's
t-test. A p-value of <0.05 was considered statistically
significant.
Compliance with ethical standards
Ethical approval
All applicable international, national, and/or
institutional guidelines for the care and use of animals were
followed. All procedures performed in studies involving animals
were in accordance with the ethical standards of Showa University
at which the studies were conducted. Animal experiments were
approved by the Institutional Animal Care and Use Committee and
were carried out according to the Showa University Guidelines for
Animal Experiments (approval no. 15024). Regarding the management
of specimens, before enrollment in the study, informed consent was
obtained from all individual participants included in the study, in
accordance with the protocol approved by the Institutional Review
Board at Showa University Dental Hospital (approval no.
DH2015-013). The experiments complied with the current laws of
Japan.
Results
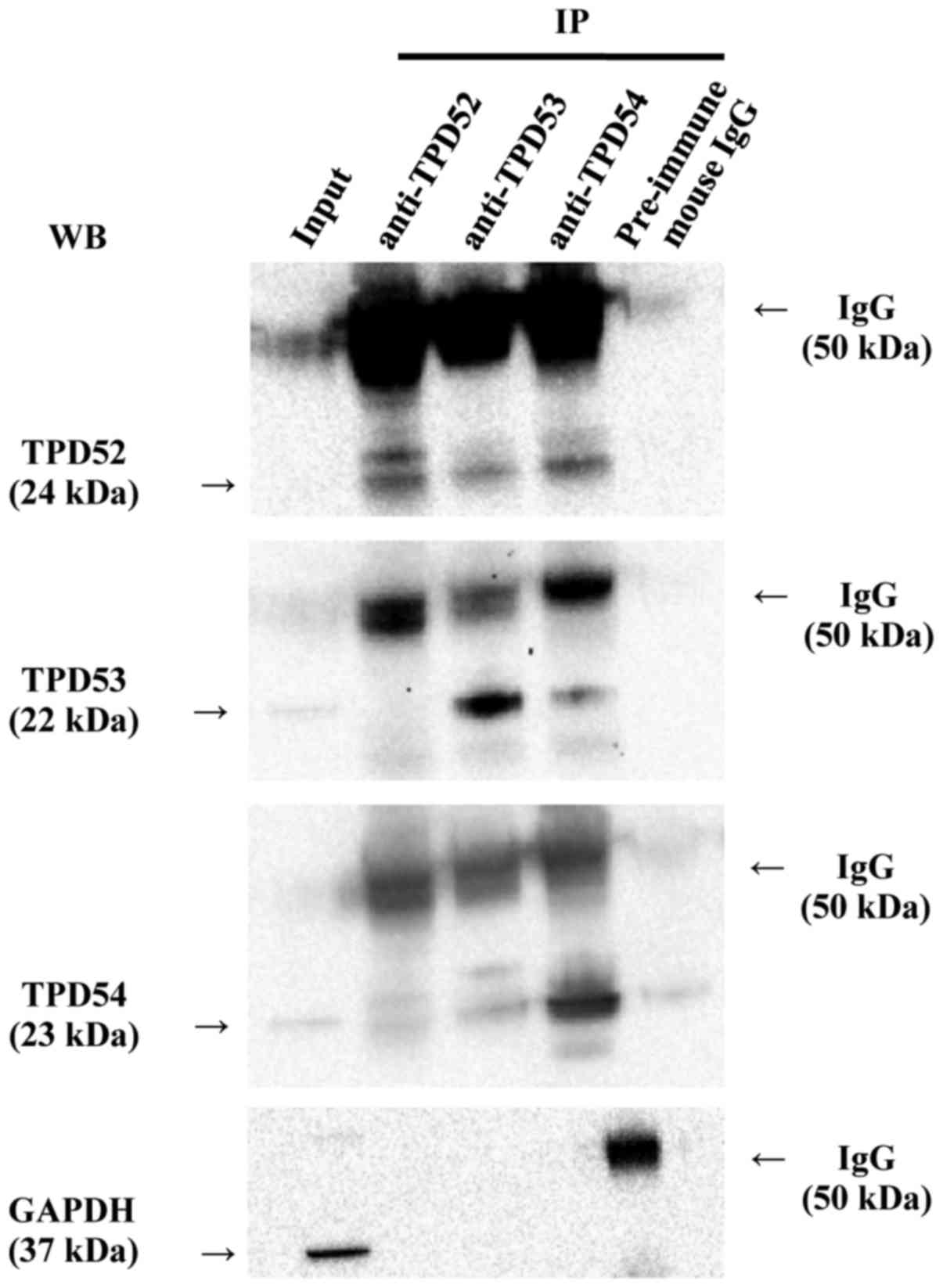
TPD52 family proteins form
hetero-complexes
Since previous studies (2,4)
showed that TPD52 family proteins form homo-and hetero-complexes in
various cells, we investigated whether such molecular interactions
were observed in SAS cells by a co-immunoprecipitation (co-IP)
assay (Fig. 1). In western
blotting of the total cellular protein (input), the blotted bands
of TPD54, TPD53, and TPD52 gave strong, weak, and faint signals,
respectively, in agreement with our previous study (39). Of more importance, TPD52, -53, and
-54 were found to co-immunoprecipitate with each other. Co-IP with
control pre-immune mouse IgG indicated the specificity of the
binding, despite the presence of a very faint pseudo-positive
signal that was probably due to insufficient washing of the
blots.
TPD54 is highly expressed in OSCC
We recently showed that TPD54 is highly expressed in
oral squamous cell carcinomas (38). However, the expression of TPD52 and
-53 in OSCC, and the differences in TPD52 family protein expression
in highly and poorly differentiated tumors are still unclear. We
therefore analyzed the detailed expression patterns of TPD52 family
proteins in OSCC using immunohistochemical staining (Fig. 2). Three cases each of highly and
poorly differentiated tumors were randomly chosen from tongue
(Fig. 2A-a-c and B-a, -b) and
gingiva (Fig. 2B-c).
Differentiation grade was determined according to the pathological
reports. Immunohistochemical staining showed that cytokeratin 10/13
[a marker of SCC cells (47)] was
highly expressed in all of these OSCCs. A cancer 'pearl' structure
was observed in some of the highly differentiated tissues (arrows
in Fig. 2 indicate a typical
cancer pearl structure), but was not observed in poorly
differentiated tissue. Additionally, staining with pre-immune IgG
(negative control) indicated the specificity of TPD52 family immune
staining. In agreement with our previous study (38), TPD54 was highly expressed in the
cancer region and the surrounding connective tissue, regardless of
the tumor differentiation level. In contrast, the expression of
TPD52 in either highly or poorly differentiated OSCC was lower than
that of TPD54, and TPD52 was barely expressed in normal tissue.
TPD53 was moderately expressed in the cancer region.
TPD54 inhibited colony formation in an
anchorage-independent manner
The higher expression level of TPD54 compared to
TPD52 and -53 suggested that TPD54 might play a more important role
in OSCCs. Furthermore, our previous study (39) showed that TPD54 affects cell
attachment to the ECM and cell migration of OSCC cells. Based on
those findings, we hypothesized that TPD54 might be a key protein
in OSCC cells, and might negatively affect tumor progression
mediated by TPD52 and -53. To further assess this possibility,
tpd52, -53, and -54 were overexpressed in SAS
cells. Additionally, in this experiment, HA-tagged tpd52 or
-53 expression vectors were transfected into SAS cell lines
that stably expressed GFP-tpd54 or GFP-miRNA
against-tpd54. Subsequent western blot analysis (Fig. 3) showed that protein expression
resulting from transient overexpression of tpd52 or
-53 and stable overexpression or knock-down of tpd54
was observed as expected. Expression marker tags (HA and GFP) and
internal control (GAPDH) proteins were also detected. Since
expression profiles of control miRNA cell lines were similar to
those of control cell lines (Fig.
3), protein expression in the control miRNA cell lines in
further experiments are not shown to simplify the experiments.
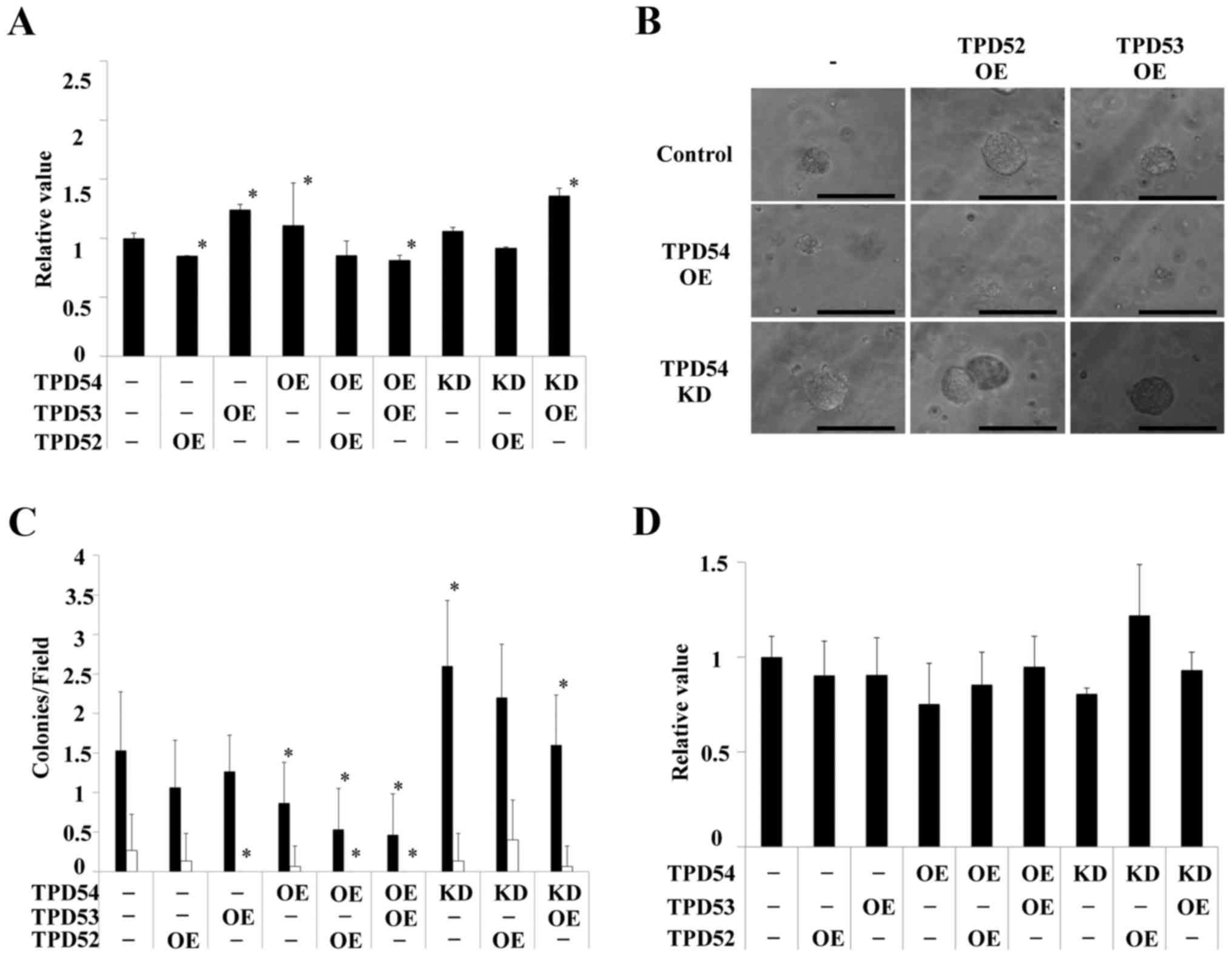
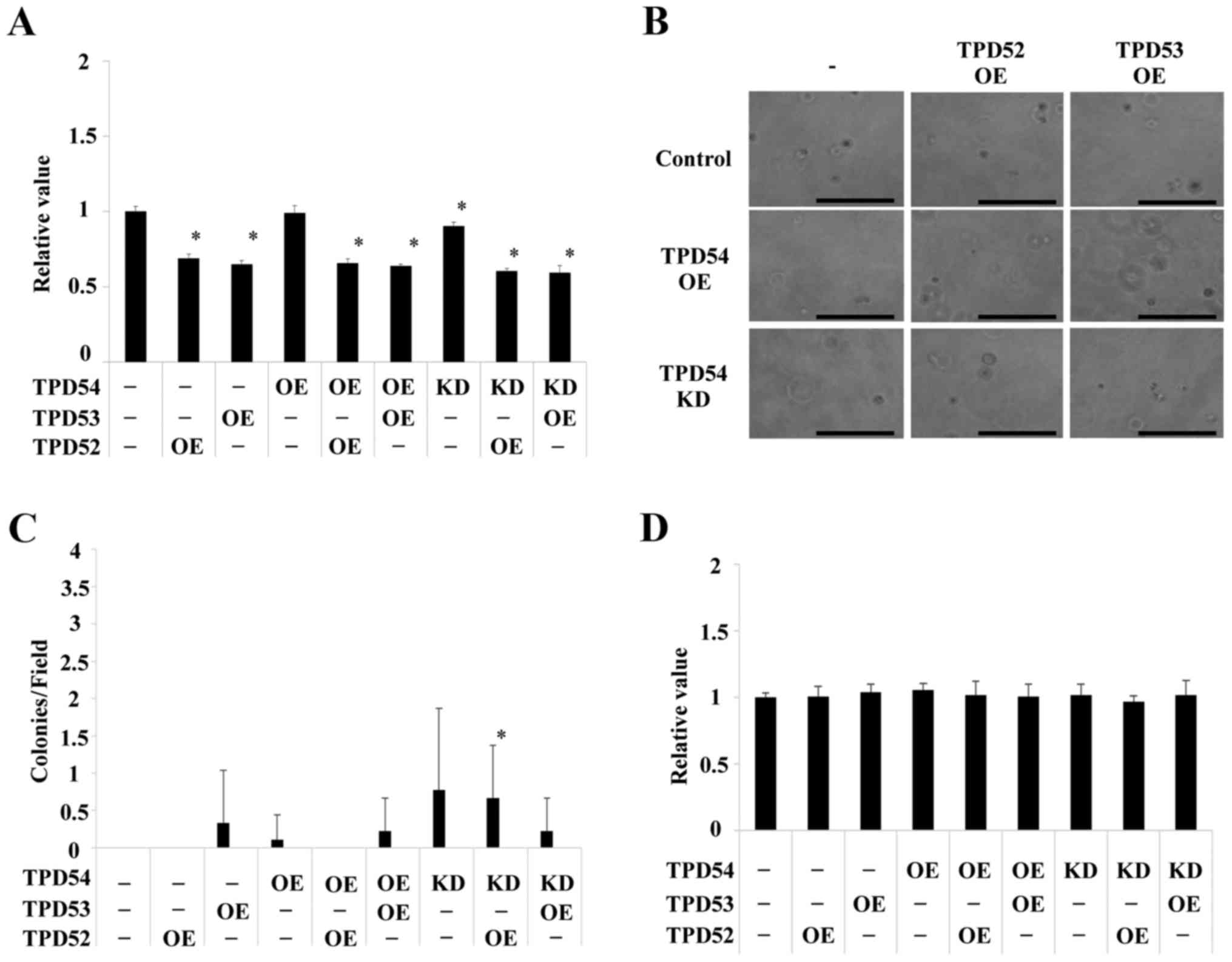
These cells were then assayed using an MTT assay of the monolayer
cultured cells (Fig. 4A), a soft
agar colony formation assay (Fig. 4B
and C) and an MTT assay of the colonies formed in soft agar
(Fig. 4D). Significant differences
in cell growth between the control and several of the differently
transfected cells were observed in the MTT assay of monolayer
cultures (*p<0.05 versus control cells) (Fig. 4A), but the differences were smaller
than those between the cells in colony formation assays (Fig. 4B–D). Colony formation of the
tpd54-overexpressing cells was greatly limited in an
anchorage-independent manner versus control cells, regardless of
the co-expression of tpd52 or tpd53 (Fig. 4B). Conversely, knock-down of
tpd54 enhanced the number of colonies formed (Fig. 4C). MTT assay of the cells in this
colony forming experiment showed no differences in cell growth
(Fig. 4D), indicating that cell
viability was not an important factor even in an
anchorage-dependent culture. These findings suggested that TPD54
might be capable of attenuating the tumorigenicity of other TPD52
family proteins, and that TPD54 and other TPD52 family proteins
might have opposite effects on primary tumor formation. Next, it
was examined whether these effects on cell growth are reproduced in
other OSCC cell lines, or not. The same experiments were carried
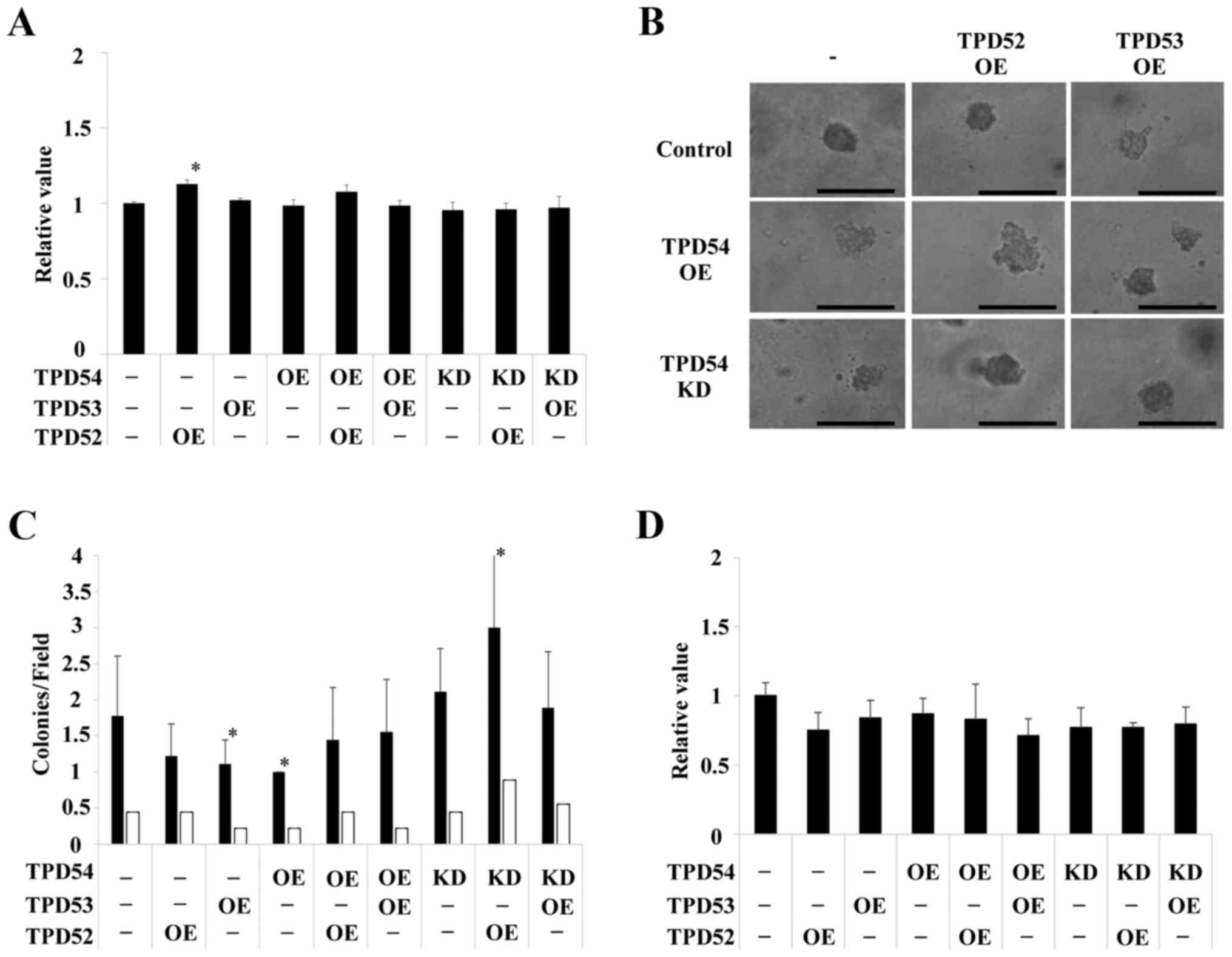
out in HSC2 (Fig. 5) and HSC3
(Fig. 6) cells. HSC2 and HSC3
cells also showed significant differences between transfected cells
versus control cells in an anchorage-independent manner in colony
formation (*p<0.05 versus control cells) as well as
in cell growth in a monolayer culture. However, the effects
observed in HSC2 and HSC3 cells were slightly different than those
observed in SAS cells. Therefore, SAS cells, in which OE and KD of
TPD52 proteins were the most effective were used as representative
OSCC cells in the following experiments.
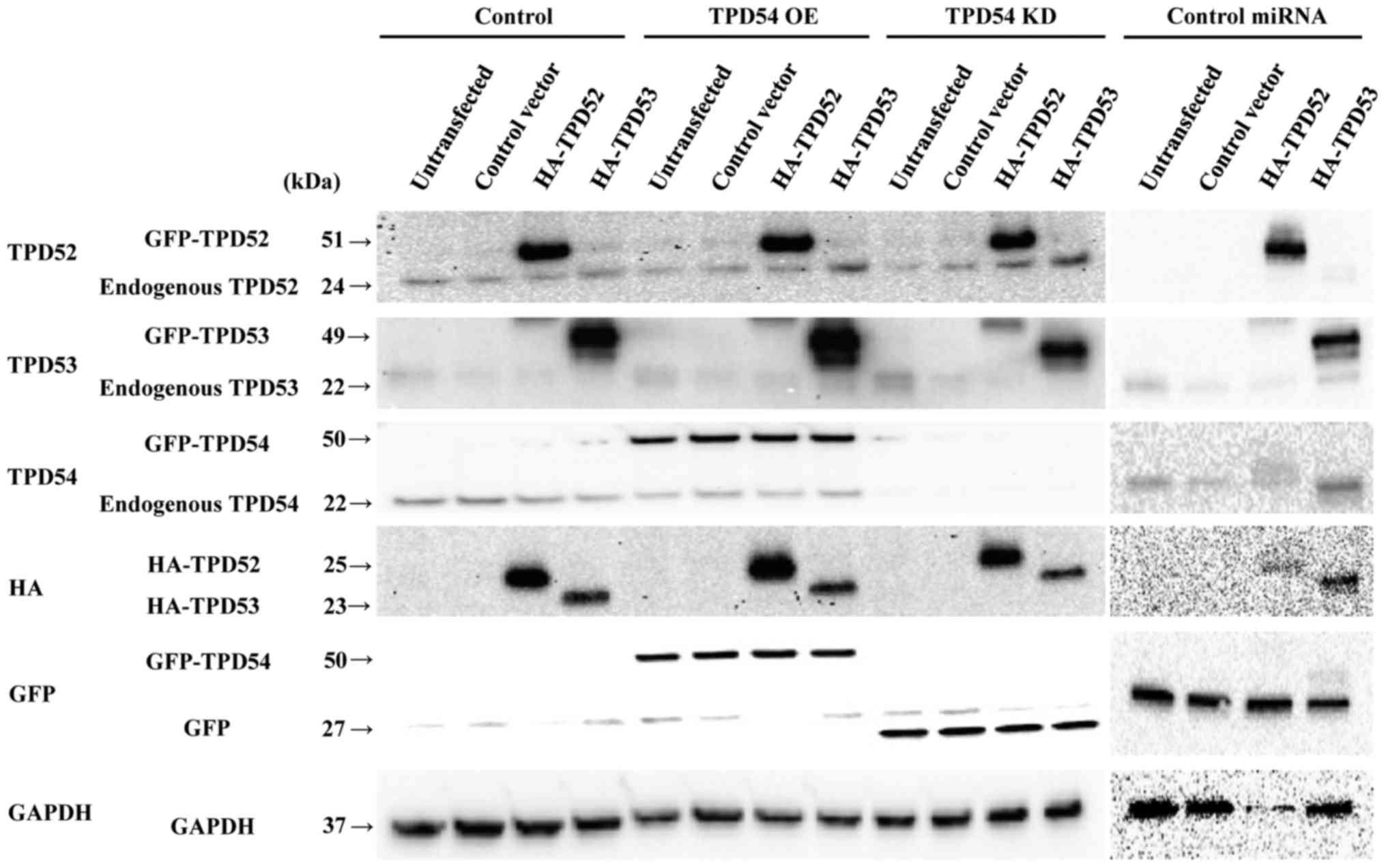 | Figure 3Western blot analysis of SAS cells
with overexpression or knock-down of tpd54 and/or
overexpression of tpd52 or -53. Control-HA and
HA-tpd52 and -53 expression vectors were transfected
into cells of the SAS cell line in which GFP-empty (control),
GFP-tpd54 (TPD54 OE) genes, GFP-tpd54miRNA (TPD54
KD), or control miRNA were stably expressed, or not
(untransfected). After 48 h, total cellular proteins were collected
and analyzed by western blotting for TPD52, -53, and -54, HA, GFP,
and GAPDH (an internal control) expression. |
TPD54 downregulates the cell migration of
OSCC-derived cells
Our previous report (39) showed that TPD54 plays a crucial
role in the cell migration and invasion of OSCC cells. To further
assess the effects of TPD52, -53, and -54 on the cell migration of
OSCC cells, we performed chemotaxis, haptotaxis and wound healing
assays of the transfected cells (Fig.
7). tpd54 overexpression reduced cell migration versus
control cells in a chemotaxis assay when expressed either alone or
in combination with overexpression of tpd52 or -53
(Fig. 7A). However, little effects
on haptotaxis were observed following overexpression or knock-down
of tpd54 alone or combined with overexpression of
tpd52 or -53 (Fig.
7B). Overexpression of tpd52 or -53 alone did not
affect either chemotaxis or haptotaxis. tpd54-overexpressing
cells (tpd54+, 53−, 52−)
showed decreased wound closure compared to control in a wound
healing assay (*p<0.05, Fig. 7C). No differences versus control
were observed for the other experimental groups (Fig. 7C). We additionally investigated the
effect of the different cell transfection combinations on cell
invasion (Fig. 7D). However, cell
invasion was barely modulated by any of these combinations. These
results indicated that TPD54 might regulate cell migration through
the modulation of molecular events such as integrin signaling.
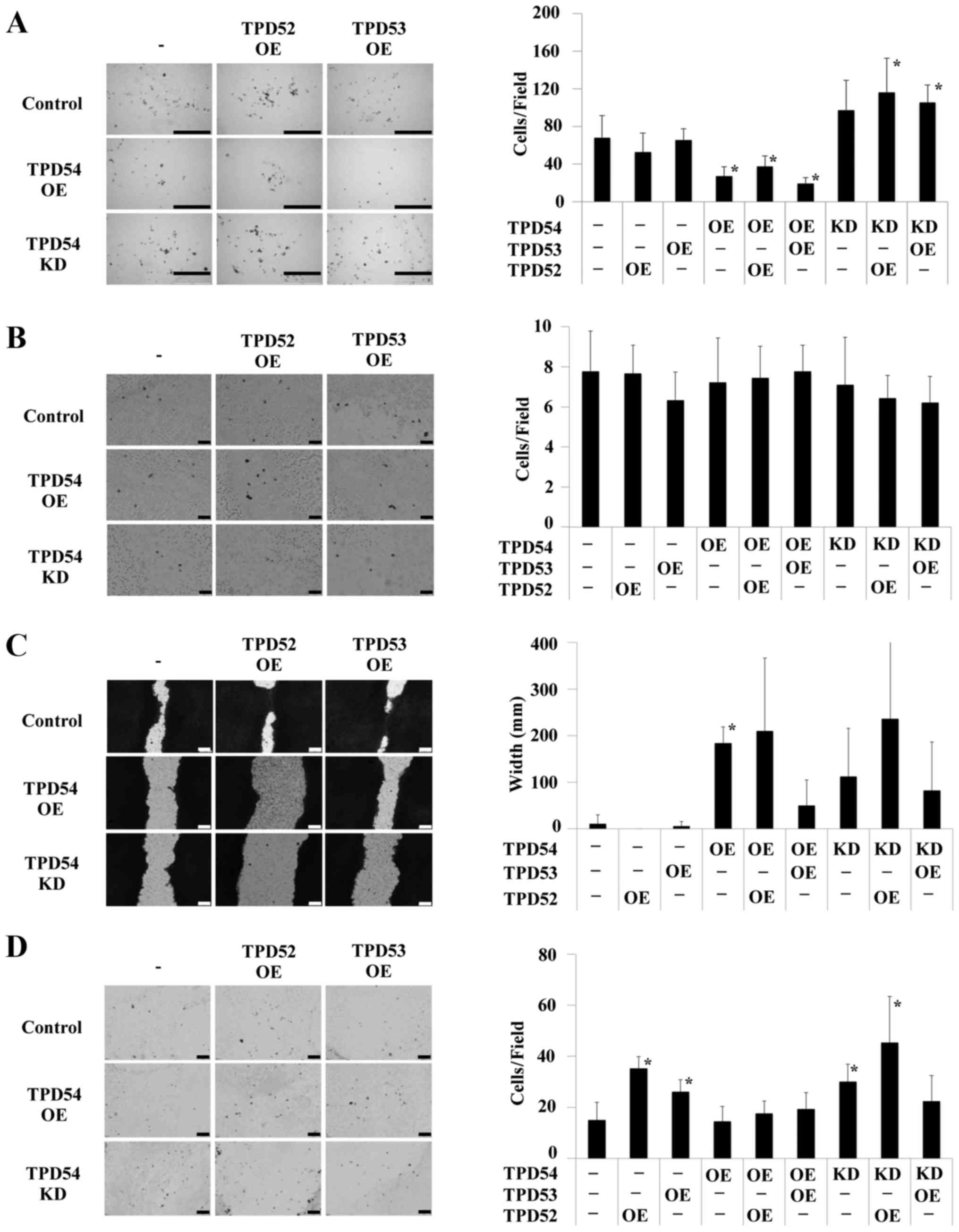 | Figure 7Effects of overexpression or
knocking-down of tpd54, and/or overexpression of
tpd52 and -53 on cell migration and invasion.
Control-HA (−), or HA-tpd52 (TPD52 OE), -53 (TPD53
OE) expression vectors were transfected into SAS cells in which
GFP-empty (control), GFP-tpd54 (TPD54 OE) genes, or
GFP-tpd54shRNA (TPD54 KD) was stably expressed. One hundred
thousand transfected cells were analyzed in chemotaxis (A),
haptotaxis (B), wound healing (C), and cell invasion (D) assays.
(A) Chemotaxis assay. Left and right panels show optical
microscopic images and a graph of the number of migrated cells,
respectively. Bar, 500 µm. *p<0.05 versus
control. (B) Haptotaxis assay. Left and right panels show optical
microscopic images and a graph of the number of migrated cells,
respectively. Bar, 100 µm. (C) Wound healing assay. Left and
right panels show optical microscopic images and a graph of wound
widths, respectively. Bar, 100 µm. *p<0.05
versus control. (D) Cell invasion assay. Left and right panels show
optical microscopic images and a graph of the number of migrated
cells, respectively. Bar, 200 µm. *p<0.05
versus control. Details of each assay can be found in Materials and
methods. |
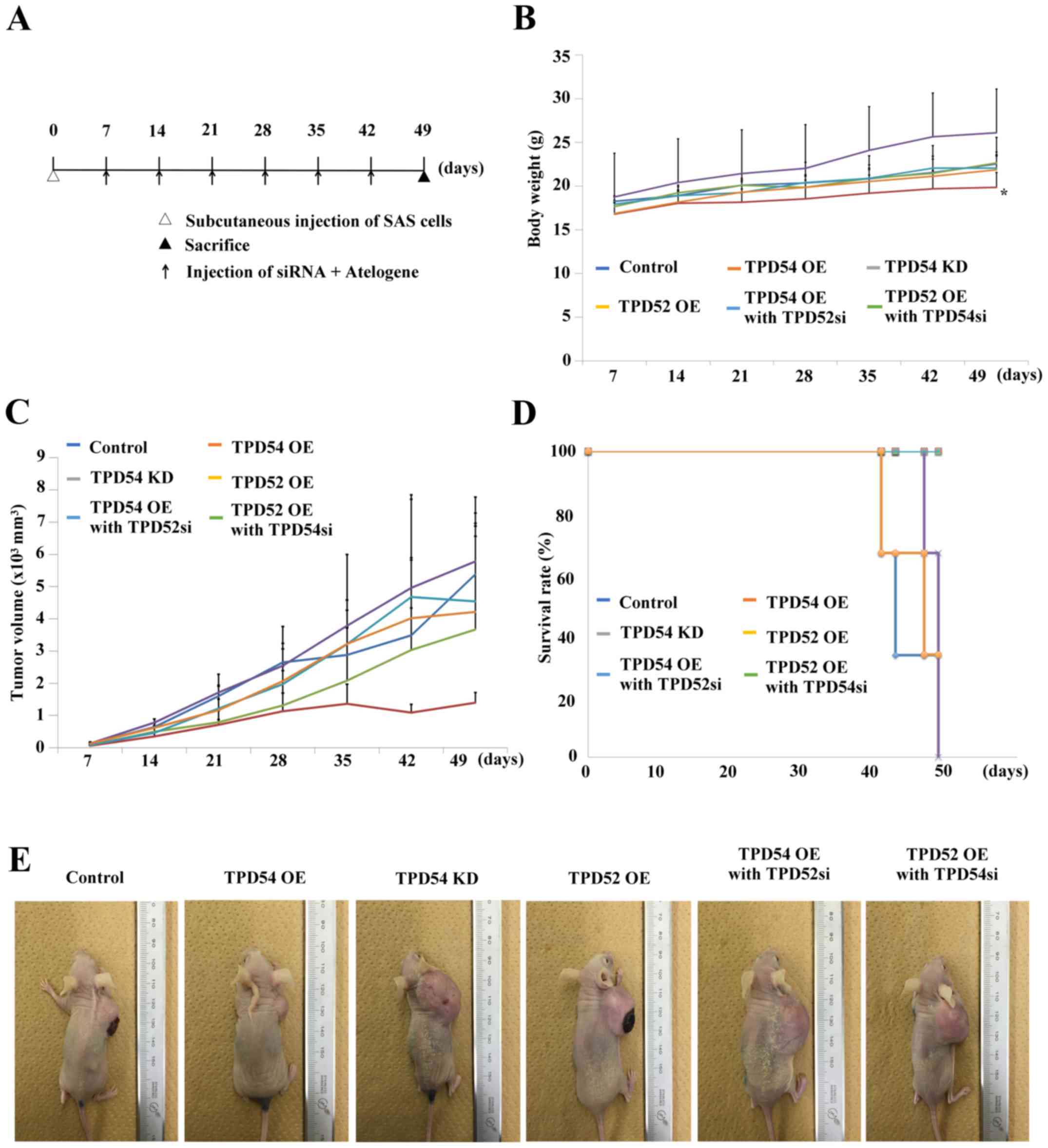
tpd54 overexpression attenuated tumor
growth in vivo
Since our previous in vitro study suggested
that TPD54 has negative effects on cell proliferation and migration
in OSCC cells, we further examined the ability of TPD54 to modulate
tumor growth and/or metastasis in an in vivo study. The
scheme of this study is outlined in Fig. 8A. In these experiments, one
nanomole of siRNAs in Atelogene was injected once a week into each
mouse (n=3 per group) with a xenografted tumor of OSCC cells, in
order to obtain sustainable knock-down of tpd52 or
-54. A significant difference in body weight between the
tpd54 overexpressing group and control was seen
(*p<0.05) (Fig. 8B)
although no significant tumor suppression was observed in the
tpd54 overexpressing group (Fig. 8C and E). However, contrary to our
expectations, knock-down of tpd52 in tpd54
overexpressing cells showed little effect on tumor growth. No
significant differences in survival rate between the different
groups of mice, as calculated by Kaplan-Meier's method, were
observed (Fig. 8D). At the
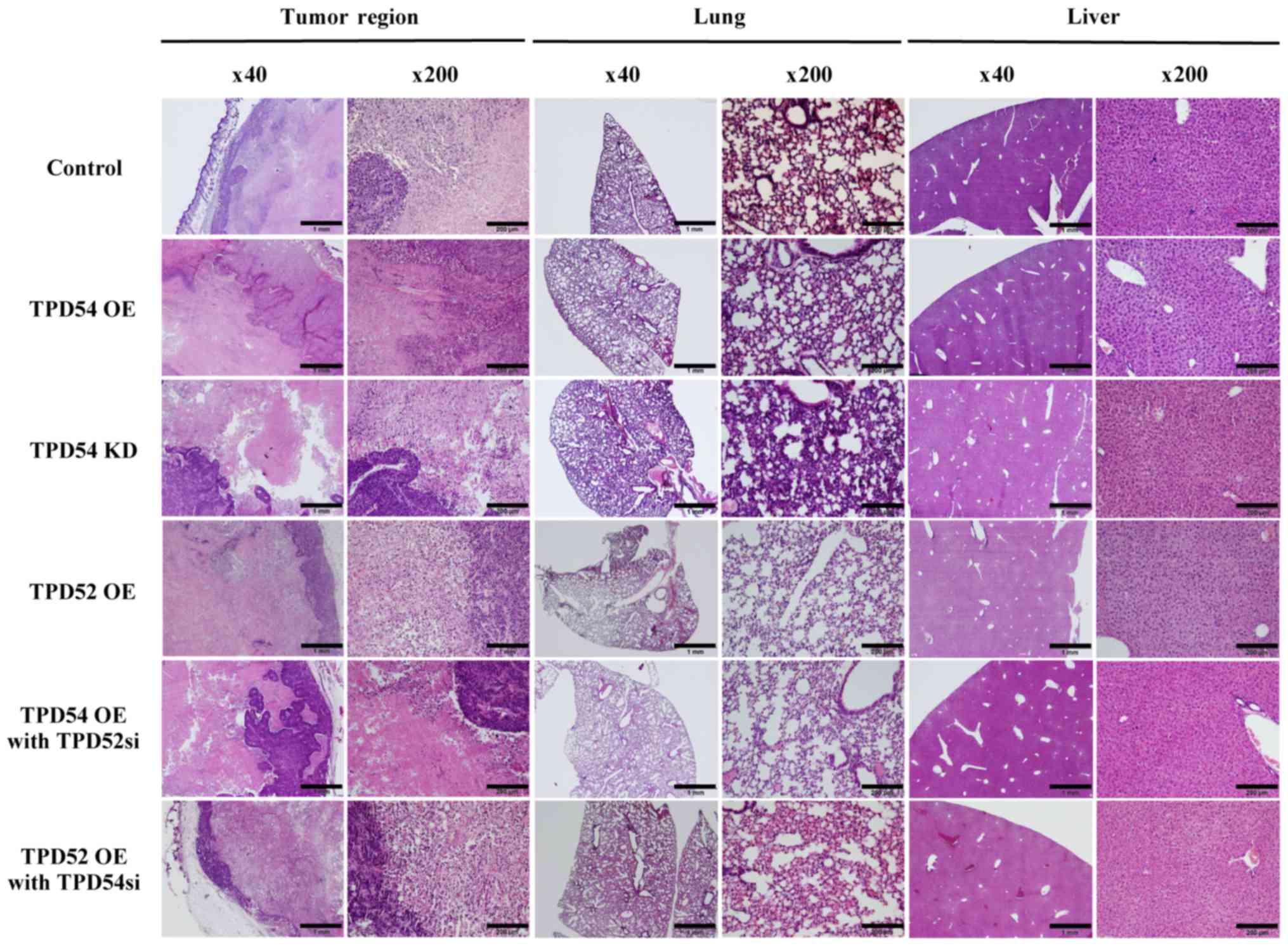
endpoint, specimens of the original tumor, and of lung and liver
tissue were H&E stained for confirmation of metastasis
(Fig. 9). However, there were few
histological differences between the groups. Metastasis was not
observed in any group by microscopic examination. These results
indicated that TPD54 might function as a negative regulator of
tumor growth, suggesting that interactions between TPD52 family
protein members might be involved in the growth of OSCC.
Discussion
Tumor protein D52 (TPD52) family members are
expressed in several types of cancers, including OSCC (5–17).
Additionally, the cellular functions of this family have been
widely studied. Previous reports indicated that overexpression of
tpd52 in nonmalignant 3T3 fibroblasts induced an increase in
the growth rate and also enabled the cells to exist in an
anchorage-independent manner in vitro and to undergo
metastatic growth in vivo (18). Moreover, overexpression of
tpd52 led to increased tumor growth in cancer cells
(48). Downregulation of
tpd52 led to a decrease in cell migration, invasion
(23), and apoptosis (20). TPD53 regulates the cell cycle and
is highly upregulated at the G2-M phase transition (25). However, little is known about the
cellular functions of TPD54, particularly in OSCCs. We recently
showed that TPD54 is expressed in OSCCs and in nearby hypertrophic
squamous cells, and we suggested that this protein might be a
candidate marker of oral epithelial carcinogenesis (38). The expression of TPD52 and -53 in
OSCC and differences in their expression in high and poor
differentiation stages of OSCC are still unclear. We therefore
determined the detailed expression patterns of TPD52 family
proteins in OSCC (Fig. 1). Indeed,
TPD54 was highly expressed in the cancer region, regardless of the
differentiation stage. However, TPD52 was expressed at a lower
level than TPD54 in either highly or poorly differentiated OSCC.
TPD53 was moderately expressed in the cancer region. A previous
report (4) indicated homo- or
heteromeric interactions between TPD52, -53, and -54 and suggested
that these proteins might play several kinds of roles in cancer
progression. These combined data suggested that TPD52 and TPD54
interact with each other and that these proteins, in particular
TPD54, play important roles in cancer progression in OSCC. In the
present study, co-IP experiments (Fig.
1) confirmed interactions between those TPD family proteins,
suggesting that they do form hetero-protein complexes on OSCC
cells. Furthermore, in our previous experiments involving
tpd54 overexpression or knock-down in OSCC-derived SAS
cells, we observed little effects on cell proliferation, caspase
activity, MMP activity or cell invasion in monolayer cultures
(39). However, tpd54
overexpression led to a decrease in anchorage-independent
proliferation, regardless of apoptosis (39). In the present study, we
investigated in more detail the cellular effects of expression
levels of TPD52 family proteins by using gene co-expression. These
studies indicated little differences in cell proliferation between
several differently transfected cells in a monolayer culture.
However, in anchorage-independent culture, strong suppression of
colony formation was observed in tpd54 overexpressing cells,
regardless of the co-expression of tpd52 or tpd53. In
addition, knock-down of tpd54 enhanced colony formation. The
correlation between anchorage-independent proliferation and the
in vivo behavior of cells is well established (49,50).
The combination of these data with our previous results suggested a
significant involvement of TPD54 in tumor growth in the modulation
of cancer metastasis. These results prompted us to further assess
the combinatorial effects of TPD52 family proteins on cell
migration and invasion. These assays showed that TPD54 negatively
controls cell migration, leading us to hypothesize that TPD52
proteins might modulate the affinity of integrins for the ECM
(39).
Since these in vitro results suggested that
TPD54 has negative effects on cell proliferation and migration in
OSCC cells, we examined the involvement of TPD54 in tumor growth
in vivo. In those experiments, a significant difference in
body weight between tpd54 overexpressing mice and controls
was seen, although no significant tumor suppressive effect was
observed in this group. However, tumor growth in several other
experimental groups with different combinations of TPD-family
proteins did not agree with our expectations. We therefore consider
that several as yet unknown molecular pathways might be involved,
and/or that homo- and heteromeric interactions of TPD52 family
proteins through their coiled-coil motif with each other or with
other heteromeric partners (4,26,28,29,51–53)
are likely to participate in tumor growth regulation by these
proteins. Li et al (54)
reported that prostate leucine zipper (PrLZ), which is a TPD52
isoform, is highly expressed in prostate cancers and interacts with
the androgen receptor. We consider that this interaction may also
occur in OSCC cells. Analysis of the effects of such an interaction
on consequent interactions of TPD52 family proteins might be
essential in order to solve this issue. The details of TPD52 family
interactions are still unclear, particularly in OSCC cells. We are
currently in the process of investigating the cellular growth,
invasion and metastasis mediated by the interaction of TPD52 family
protein members. He et al recently reported that TPD54 has a
role in the promotion of cancer progression in OSCC cell lines
(55), which is the opposite
result to the present study. This discrepancy may be due to the
different cells used in the two studies. Thus, the CAL27 and KB
cell lines that they employed in their experiments are an oral
adenosquamous carcinoma cell line (56) and a HeLa contaminant-epidermal
carcinoma cell line (57),
respectively. However, the OSCC cells that we employed are SAS
cells, which were isolated from tongue OSCC (40). These cells might harbor cells with
more of the original phenotypes of OSCC cells. In any case, more
detailed investigations are required in the future in order to
address this discrepancy.
For tumor therapy, molecular targeted therapies are
currently in the spotlight, next to surgery, radiation, and
chemotherapy. TPD52 family proteins are also considered as novel
candidate target proteins since these proteins are expressed in
many types of cancers (31). In
conclusion, our study showed that TPD54 acts as a negative
regulator of anchorage-independent proliferation and cell migration
of OSCC cells in vitro. Moreover, TPD54 decreased body
weight gain and tended to attenuate tumor growth in vivo.
These combined data suggest that an increase in the expression of
tpd54 might improve outcomes in OSCC patients. As yet, only
a few studies of TPD54 have been reported. Further investigation of
TPD54 is still required.
Acknowledgments
This study was supported by Grants-in-Aid for
Scientific Research (KAKENHI) from the Japan Society for the
Promotion of Science (JSPS) [KAKENHI C to Y.M. (15K11301), S.K.
(24593060) and T.S. (15K11269)]. The whole manuscript was proofread
by FOLTE (Tokyo, Japan). The authors wish to thank all of the staff
at the Department of Oral and Maxillofacial Surgery, School of
Dentistry, Showa University for their helpful suggestions; Dr Kenji
Mishima of the Division of Pathology, Department of Oral Diagnostic
Sciences, School of Dentistry, Showa University for pathological
diagnostic advice; and Ms. Miho Yoshihara for secretarial
assistance.
References
|
1
|
Byrne JA, Mattei MG and Basset P:
Definition of the tumor protein D52 (TPD52) gene family through
cloning of D52 homologues in human (hD53) and mouse (mD52).
Genomics. 35:523–532. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nourse CR, Mattei MG, Gunning P and Byrne
JA: Cloning of a third member of the D52 gene family indicates
alternative coding sequence usage in D52-like transcripts. Biochim
Biophys Acta. 1443:155–168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Byrne JA, Mattei MG, Basset P and Gunning
P: Identification and in situ hybridization mapping of a mouse
Tpd52l1 (D53) orthologue to chromosome 10A4-B2. Cytogenet Cell
Genet. 81:199–201. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Byrne JA, Nourse CR, Basset P and Gunning
P: Identification of homo- and heteromeric interactions between
members of the breast carcinoma-associated D52 protein family using
the yeast two-hybrid system. Oncogene. 16:873–881. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cao Q, Chen J, Zhu L, Liu Y, Zhou Z, Sha
J, Wang S and Li J: A testis-specific and testis developmentally
regulated tumor protein D52 (TPD52)-like protein TPD52L3/hD55
interacts with TPD52 family proteins. Biochem Biophys Res Commun.
344:798–806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Byrne JA, Tomasetto C, Garnier JM, Rouyer
N, Mattei MG, Bellocq JP, Rio MC and Basset P: A screening method
to identify genes commonly overexpressed in carcinomas and the
identification of a novel complementary DNA sequence. Cancer Res.
55:2896–2903. 1995.PubMed/NCBI
|
|
7
|
Chen SL, Maroulakou IG, Green JE,
Romano-Spica V, Modi W, Lautenberger J and Bhat NK: Isolation and
characterization of a novel gene expressed in multiple cancers.
Oncogene. 12:741–751. 1996.PubMed/NCBI
|
|
8
|
Malek RL, Irby RB, Guo QM, Lee K, Wong S,
He M, Tsai J, Frank B, Liu ET, Quackenbush J, et al: Identification
of Src transformation fingerprint in human colon cancer. Oncogene.
21:7256–7265. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Petrova DT, Asif AR, Armstrong VW, Dimova
I, Toshev S, Yaramov N, Oellerich M and Toncheva D: Expression of
chloride intracellular channel protein 1 (CLIC1) and tumor protein
D52 (TPD52) as potential biomarkers for colorectal cancer. Clin
Biochem. 41:1224–1236. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Byrne JA, Balleine RL, Schoenberg Fejzo M,
Mercieca J, Chiew YE, Livnat Y, St Heaps L, Peters GB, Byth K,
Karlan BY, et al: Tumor protein D52 (TPD52) is overexpressed and a
gene amplification target in ovarian cancer. Int J Cancer.
117:1049–1054. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Byrne JA, Maleki S, Hardy JR, Gloss BS,
Murali R, Scurry JP, Fanayan S, Emmanuel C, Hacker NF, Sutherland
RL, et al: MAL2 and tumor protein D52 (TPD52) are frequently
overexpressed in ovarian carcinoma, but differentially associated
with histological subtype and patient outcome. BMC Cancer.
10:4972010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fejzo MS, Dering J, Ginther C, Anderson L,
Ramos L, Walsh C, Karlan B and Slamon DJ: Comprehensive analysis of
20q13 genes in ovarian cancer identifies ADRM1 as amplification
target. Genes Chromosomes Cancer. 47:873–883. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Willems A, De Gendt K, Allemeersch J,
Smith LB, Welsh M, Swinnen JV and Verhoeven G: Early effects of
Sertoli cell-selective androgen receptor ablation on testicular
gene expression. Int J Androl. 33:507–517. 2010. View Article : Google Scholar
|
|
14
|
Rubin MA, Varambally S, Beroukhim R,
Tomlins SA, Rhodes DR, Paris PL, Hofer MD, Storz-Schweizer M,
Kuefer R, Fletcher JA, et al: Overexpression, amplification, and
androgen regulation of TPD52 in prostate cancer. Cancer Res.
64:3814–3822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen H, Pimienta G, Gu Y, Sun X, Hu J, Kim
MS, Chaerkady R, Gucek M, Cole RN, Sukumar S, et al: Proteomic
characterization of Her2/neu-overexpressing breast cancer cells.
Proteomics. 10:3800–3810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crugliano T, Quaresima B, Gaspari M,
Faniello MC, Romeo F, Baudi F, Cuda G, Costanzo F and Venuta S:
Specific changes in the proteomic pattern produced by the
BRCA1-Ser1841Asn missense mutation. Int J Biochem Cell Biol.
39:220–226. 2007. View Article : Google Scholar
|
|
17
|
Scanlan MJ, Gout I, Gordon CM, Williamson
B, Stockert E, Gure AO, Jäger D, Chen YT, Mackay A, O'Hare MJ, et
al: Humoral immunity to human breast cancer: Antigen definition and
quantitative analysis of mRNA expression. Cancer Immun.
1:42001.
|
|
18
|
Lewis JD, Payton LA, Whitford JG, Byrne
JA, Smith DI, Yang L and Bright RK: Induction of tumorigenesis and
metastasis by the murine orthologue of tumor protein D52. Mol
Cancer Res. 5:133–144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shehata M, Bièche I, Boutros R,
Weidenhofer J, Fanayan S, Spalding L, Zeps N, Byth K, Bright RK,
Lidereau R, et al: Nonredundant functions for tumor protein
D52-like proteins support specific targeting of TPD52. Clin Cancer
Res. 14:5050–5060. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ummanni R, Teller S, Junker H, Zimmermann
U, Venz S, Scharf C, Giebel J and Walther R: Altered expression of
tumor protein D52 regulates apoptosis and migration of prostate
cancer cells. FEBS J. 275:5703–5713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang D, He D, Xue Y, Wang R, Wu K, Xie H,
Zeng J, Wang X, Zhau HE, Chung LW, et al: PrLZ protects prostate
cancer cells from apoptosis induced by androgen deprivation via the
activation of Stat3/Bcl-2 pathway. Cancer Res. 71:2193–2202. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang H, Wang J, Pang B, Liang RX, Li S,
Huang PT, Wang R, Chung LW, Zhau HE, Huang C, et al: PC-1/PrLZ
contributes to malignant progression in prostate cancer. Cancer
Res. 67:8906–8913. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao P, Zhong W, Ying X, Yao B, Yuan Z, Fu
J and Zhou Z: Comparative proteomic analysis of
anti-benzo(a)pyrene-7,8-dihydrodiol-9,10-epoxide-transformed and
normal human bronchial epithelial G0/G1 cells. Chem Biol Interact.
186:166–173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sims AH, Finnon P, Miller CJ, Bouffler SD,
Howell A, Scott D and Clarke RB: TPD52 and NFKB1 gene expression
levels correlate with G2 chromosomal radiosensitivity in
lymphocytes of women with and at risk of hereditary breast cancer.
Int J Radiat Biol. 83:409–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boutros R, Fanayan S, Shehata M and Byrne
JA: The tumor protein D52 family: Many pieces, many puzzles.
Biochem Biophys Res Commun. 325:1115–1121. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wilson SH, Bailey AM, Nourse CR, Mattei MG
and Byrne JA: Identification of MAL2, a novel member of the mal
proteolipid family, though interactions with TPD52-like proteins in
the yeast two-hybrid system. Genomics. 76:81–88. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thomas DD, Kaspar KM, Taft WB, Weng N,
Rodenkirch LA and Groblewski GE: Identification of Annexin VI as a
Ca2+-sensitive CRHSP-28-binding protein in pancreatic
acinar cells. J Biol Chem. 277:35496–35502. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Proux-Gillardeaux V, Galli T, Callebaut I,
Mikhailik A, Calothy G and Marx M: D53 is a novel endosomal
SNARE-binding protein that enhances interaction of syntaxin 1 with
the synaptobrevin 2 complex in vitro. Biochem J. 370:213–221. 2003.
View Article : Google Scholar
|
|
29
|
Boutros R, Bailey AM, Wilson SHD and Byrne
JA: Alternative splicing as a mechanism for regulating 14-3-3
binding: Interactions between hD53 (TPD52L1) and 14-3-3 proteins. J
Mol Biol. 332:675–687. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kamili A, Roslan N, Frost S, Cantrill LC,
Wang D, Della-Franca A, Bright RK, Groblewski GE, Straub BK, Hoy
AJ, et al: TPD52 expression increases neutral lipid storage within
cultured cells. J Cell Sci. 128:3223–3238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Byrne JA, Frost S, Chen Y and Bright RK:
Tumor protein D52 (TPD52) and cancer-oncogene understudy or
understudied oncogene? Tumour Biol. 35:7369–7382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fornaro M, Manes T and Languino LR:
Integrins and prostate cancer metastases. Cancer Metastasis Rev.
20:321–331. 2001. View Article : Google Scholar
|
|
35
|
Wu WS: The signaling mechanism of ROS in
tumor progression. Cancer Metastasis Rev. 25:695–705. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bhaskar PT and Hay N: The two TORCs and
Akt. Dev Cell. 12:487–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Legate KR, Wickström SA and Fässler R:
Genetic and cell biological analysis of integrin outside-in
signaling. Genes Dev. 23:397–418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fujita A and Kondo S: Identification of
TPD54 as a candidate marker of oral epithelial carcinogenesis. J
Oral Maxillofac Surg Med Pathol. 27:770–774. 2015. View Article : Google Scholar
|
|
39
|
Mukudai Y, Kondo S, Fujita A, Yoshihama Y,
Shirota T and Shintani S: Tumor protein D54 is a negative regulator
of extracellular matrix-dependent migration and attachment in oral
squamous cell carcinoma-derived cell lines. Cell Oncol (Dordr).
36:233–245. 2013. View Article : Google Scholar
|
|
40
|
Takahashi K: Establishment and
characterization of a cell line(SAS) from poorly differentiated
human squamous cell carcinoma of the tongue. Jpn Stomatological
Soc. 38:20–28. 1989.
|
|
41
|
Momose F, Araida T, Negishi A, Ichijo H,
Shioda S and Sasaki S: Variant sublines with different metastatic
potentials selected in nude mice from human oral squamous cell
carcinomas. J Oral Pathol Med. 18:391–395. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tsukamoto H, Kondo S, Mukudai Y, Nagumo T,
Yasuda A, Kurihara Y, Kamatani T and Shintani S: Evaluation of
anticancer activities of benzo[c]phenanthridine alkaloid
sanguinarine in oral squamous cell carcinoma cell line. Anticancer
Res. 31:2841–2846. 2011.PubMed/NCBI
|
|
43
|
Enomoto-Iwamoto M, Iwamoto M, Mukudai Y,
Kawakami Y, Nohno T, Higuchi Y, Takemoto S, Ohuchi H, Noji S and
Kurisu K: Bone morphogenetic protein signaling is required for
maintenance of differentiated phenotype, control of proliferation,
and hypertrophy in chondrocytes. J Cell Biol. 140:409–418. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen Y, Lu B, Yang Q, Fearns C, Yates JR
III and Lee JD: Combined integrin phosphoproteomic analyses and
small interfering RNA - based functional screening identify key
regulators for cancer cell adhesion and migration. Cancer Res.
69:3713–3720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yasuda A, Kondo S, Nagumo T, Tsukamoto H,
Mukudai Y, Umezawa K and Shintani S: Anti-tumor activity of
dehydroxymethylepoxyquinomicin against human oral squamous cell
carcinoma cell lines in vitro and in vivo. Oral Oncol. 47:334–339.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shiogama S, Yoshiba S, Soga D, Motohashi H
and Shintani S: Aberrant expression of EZH2 is associated with
pathological findings and P53 alteration. Anticancer Res.
33:4309–4317. 2013.PubMed/NCBI
|
|
47
|
Mukudai Y, Zhang M, Shiogama S, Kondo S,
Ito C, Motohashi H, Kato K, Fujii M, Shintani S, Shigemori H, et
al: Methanol and butanol extracts of Paeonia lutea leaves repress
metastasis of squamous cell carcinoma. Evid Based Complement
Alternat Med. 2016:60872132016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang R, Xu J, Mabjeesh N, Zhu G, Zhou J,
Amin M, He D, Marshall FF, Zhau HE and Chung LW: PrLZ is expressed
in normal prostate development and in human prostate cancer
progression. Clin Cancer Res. 13:6040–6048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cifone MA and Fidler IJ: Correlation of
patterns of anchorage-independent growth with in vivo behavior of
cells from a murine fibrosarcoma. Proc Natl Acad Sci USA.
77:1039–1043. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hahn WC, Counter CM, Lundberg AS,
Beijersbergen RL, Brooks MW and Weinberg RA: Creation of human
tumour cells with defined genetic elements. Nature. 400:464–468.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen SL, Zhang XK, Halverson DO, Byeon MK,
Schweinfest CW, Ferris DK and Bhat NK: Characterization of human N8
protein. Oncogene. 15:2577–2588. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Proux V, Provot S, Felder-Schmittbuhl MP,
Laugier D, Calothy G and Marx M: Characterization of a leucine
zipper-containing protein identified by retroviral insertion in
avian neuroretina cells. J Biol Chem. 271:30790–30797. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sathasivam P, Bailey AM, Crossley M and
Byrne JA: The role of the coiled-coil motif in interactions
mediated by TPD52. Biochem Biophys Res Commun. 288:56–61. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li L, Xie H, Liang L, Gao Y, Zhang D, Fang
L, Lee SO, Luo J, Chen X, Wang X, et al: Increased PrLZ-mediated
androgen receptor transactivation promotes prostate cancer growth
at castration-resistant stage. Carcinogenesis. 34:257–267. 2013.
View Article : Google Scholar :
|
|
55
|
He Y, Chen F, Cai Y and Chen S: Knockdown
of tumor protein D52-like 2 induces cell growth inhibition and
apoptosis in oral squamous cell carcinoma. Cell Biol Int.
39:264–271. 2015. View Article : Google Scholar
|
|
56
|
Jiang L, Ji N, Zhou Y, Li J, Liu X, Wang
Z, Chen Q and Zeng X: CAL 27 is an oral adenosquamous carcinoma
cell line. Oral Oncol. 45:e204–e207. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Eagle H: Propagation in a fluid medium of
a human epidermoid carcinoma, strain KB. Proc Soc Exp Biol Med.
89:362–364. 1955. View Article : Google Scholar : PubMed/NCBI
|