Introduction
Pancreatic cancer (PC) remains a deadly disease.
Gemcitabine has been considered as the standard therapy for
patients with unresectable or metastatic disease for over a decade
(1,2). Recently, overall survival (OS) has
been significantly prolonged using combination therapies, such as
gemcitabine plus erlotinib, a combination of oxaliplatin,
irinotecan, fluorouracil and leucovorin (FOLIFRINOX), a combination
of nab-paclitaxel and gemcitabine, or a combination of
nanoliposomal irinotecan, fluorouracil and leucovorin (NAPOLI-1)
(3–6). However, despite such recent progress,
the OS rate of PC patients is still <5% (1,2).
Erlotinib, an epidermal growth factor receptor (EGFR) tyrosine
kinase inhibitor (TKI), was the first drug approved for the
treatment of PC after showing a survival benefit when combined with
gemcitabine over traditional gemcitabine alone (3). Although PC has been well
characterized at the genetic level, the molecular mechanisms
linking genetic changes to the aggressive nature of this disease
remain unclear (7). Multiple
genetic alterations, such as K-ras mutation or the loss of
p53 and SMAD4, are thought to influence the
progression of PC (8).
Nevertheless, to date, the inhibition of EGFR by erlotinib is the
only targeted approach to demonstrate a survival benefit.
The PI3K/Akt/mTOR pathway, which is located
downstream of the EGFR pathway and regulates cell survival and
apoptosis, is frequently upregulated or altered in many cancers,
and components of the PI3K/Akt/mTOR pathway can also be targeted in
the treatment of many cancers (9).
Among them, the inhibition of mTOR with rapalogs has initially
shown a clinical efficacy in some solid tumors (10). More recently, many agents in
clinical development have been designed to inhibit other components
of this pathway, including Akt, PI3K and PTEN. Akt, the major
downstream component of the PI3K/Akt/mTOR pathway, is a
serine/threonine kinase that plays a critical role in regulating
diverse cellular function including cell gowth, proliferation,
survival, glucose metabolism, genome stability, transcription and
protein synthesis, and neurovascularization (9). The Akt family has three isoforms:
Akt1, Akt2 and Akt3. These isoforms are structually homologous but
exhibit distinct features. Akt1 and Akt2 are ubiquitously
expressed, whereas Akt3 is found predominantly in the brain, heart
and kidneys (11). The Akt
isoforms are known to carry specific genetic alterations in
different tumor types. Akt1 amplification has been detected
in gastric adenocarcinoma, and the selective activation of
Akt3 in combination with a loss of PTEN has been found in
sporadic melanoma (12). In
contrast, amplification or high levels of expression of Akt2
are frequently found in human pancreatic, lung, colorectal,
ovarian, and breast cancers (12–19),
and high Akt2 expression levels are positively correlated
with the aggressiveness of cancer or poor survival rates in
colorectal, ovarian, and breast cancers (16–19).
In PC, the activation of the PI3K/Akt/mTOR pathway is a biological
indicator of aggressiveness (20),
and a recent report has shown that EGFR-TKI resistance in PC is
associated with the upregulation of the PI3K/Akt/mTOR pathway in
vitro (21). Thus, high Akt2
expression levels have been hypothesized to induce resistance to
the EGFR-TKI, erlotinib, in patients with PC. In this study, we
investigated the association between the Akt2 expression level and
the outcome of patients with advanced PC who had received erlotinib
treatment as well as the contribution of Akt2 to resistance to
anti-EGFR therapies in vitro.
Materials and methods
Patients and clinical specimens
Twenty-six patients with advanced PC that received
Tarceva® (erlotinib) treatment in combination with
gemcitabine at Kindai University Hospital between 2010 and 2014
were included. Among them, 22 patients who had been diagnosed based
on the results of endoscopic ultrasonography-guided fine needle
aspiration (EUS-FNA) participated in this study. Progression-free
survival (PFS) was defined as the time from the initiation of
erlotinib treatment until the first observation of disease
progression or death from any cause, while OS was defined as the
time from the initiation of erlotinib treatment until death from
any cause. The stage of disease was classified according to the
clinical TNM staging system. Tumor response was evaluated using
computed tomography (CT) according to the Response Evaluation
Criteria in Solid Tumors. This study was performed retrospectively
and was approved by the ethics committee of the Kindai University
Faculty of Medicine.
Immunohistochemistry
The immunohistochemical method used in this study
has been previously described (22). Briefly, 4-μm tissue sections
from formalin-fixed, paraffin-embedded blocks were sectioned and
placed onto charged slides. The slides were then deparaffinized and
hydrated; endogenous peroxidase activity was blocked using 3%
H2O2 in methanol and normal goat serum. The
slides were incubated in a rabbit polyclonal antibody specific for
Akt2 (1:200; Proteintech Chicago, IL, USA) overnight at 4°C.
Immunohistochemical staining was performed using the rabbit
Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA,
USA), and the slides were developed in diaminobenzidine (DAB kit,
Thermo Fisher Scientific, Waltham, MA, USA) according to the
manufacturer's protocols, then counterstained with hematoxylin. The
staining assessment was performed using ImageJ software (http://imagej.nih.gov/ij/).
Cell culture and reagents
The HCC827, PC-9, Ma-1, and H358 cell lines [human
non-small cell lung cancer (NSCLC) cell lines] and the BxPC-3 and
PANC-1 cell lines (human PC cell lines) were maintained in
RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% fetal
bovine serum (FBS; Sigma-Aldrich). The CCK81 cell line [a human
colorectal cancer (CRC) cell line] and the gpIRES-293 cell line
were maintained in DMEM medium (Nissui Pharmaceutical, Tokyo,
Japan) with 10% FBS. All the cells were maintained in a 5%
CO2-humidified atmosphere at 37°C. Erlotinib and BYL719
were purchased from Selleck Chemicals (Houston, TX, USA).
Copy number assay
The Akt2 copy number was determined using a
commercially available and predesigned TaqMan Copy Number assay
(Applied Biosystems, Foster City, CA, USA), as described previously
(23). Genomic DNA was extracted
from each of the cell lines using the QIAamp DNA Mini kit (Qiagen,
Hilden, Germany), according to the manufacturer's instructions. The
primer ID used for Akt2 was Hs04028824_cn. The TERT
locus was used for the internal reference copy number. Human
genomic DNA (Takara, Shiga, Japan) was used as a normal control. A
PCR analysis was performed using the ABI PRISM 7900HT Sequence
Detection system (Applied Biosystems), and the results were
analyzed using CopyCaller software version 2.0 (Applied
Biosystems). The experiment was performed in triplicate.
Real-time reverse-transcription PCR
(RT-PCR)
A total of 1 μg of RNA was isolated from the
cells using Isogen reagent (Nippon Gene, Tokyo, Japan) and then
converted to cDNA using the Gene Amp RNA-PCR kit (Applied
Biosystems). Real-time PCR was performed using SYBR Premix Ex Taq
and Thermal Cycler Dice (Takara) under the following conditions:
95°C for 5 min, followed by 50 cycles of 95°C for 10 sec and 60°C
for 30 sec, as described previously (23). GAPDH was used to normalize
the expression levels in the subsequent quantitative analyses. The
experiment was performed in triplicate. The primers used for this
study were as follows: Akt2 F, CCGCCTGTGCTTTGTGATGG; R,
TTTCCAGCTTGATGTCGCGG. GAPDH F, GCACCGTCAAGGCTGAGAAC; and R,
ATGGTGGTGAAGACGCCAGT.
In vitro growth inhibition assay
The growth-inhibitory effects of the drugs were
examined using a 3-(4,
5-di-methyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
(MTT; Sigma-Aldrich), as described previously (23). The experiment was performed in
triplicate.
Plasmid construction, viral production,
and stable transfectants
The methods used in this section have been
previously described (24).
Complementary DNA (cDNA) encoding human full length Akt2 was
prepared by PCR using Prime STAR HS DNA polymerase (Takara) and the
following primers: forward,
5′-GGGAATTCGCCGCCATGAATGAGGTGTCTGTCATCAAAG-3′; reverse,
5′-CCCTCGAGGCCCAGTCACTCGCGGATGCTGGC-3′. The full length Akt2
was subcloned into a pCR-Blunt II-TOPO cloning vector (Invitrogen,
Carlsbad, CA, USA) as EcoRI-XhoI fragments.
Akt2 in the TOPO cloning vector was cut out and transferred
to a pQCLIN retroviral vector (Clontech Laboratories, Inc., Palo
Alto, CA, USA) together with the enhanced green fluorescent protein
(EGFP) following the internal ribosome entry site sequence (IRES)
to monitor the expression of the inserts indirectly. The nucleotide
sequence of the construct was verified by DNA sequence analysis. A
pVSV-G vector (Clontech) for the constitution of the envelope and
the pQCLIN-IG constructs were cotransfected into gpIRES-293 cells
using FuGENE6 transfection reagent (Roche Diagnostics, Basel,
Switzerland). After 48 h of transfection, the culture medium was
collected and viral particles were concentrated by centrifugation
at 15,000 × g for 3 h at 4°C. The viral pellet was suspended in
fresh DMEM medium. The titer of the viral vector was calculated
using the EGFP-positive cells that were infected by the serial
dilution of virus-containing media, and the multiplicity of
infection was determined. The vectors and the stable viral
transfectant BxPC-3, Ma-1, and CCK81 cell lines were designated as
pQCLIN-EGFP, pQLCIN-Akt2, BxPC-3/EGFP, BxPC-3/Akt2, Ma-1/EGFP,
Ma-1/Akt2, CCK81/EGFP, and CCK81/Akt2, respectively.
Western blot analysis
The western blot analysis was performed as described
previously (24). Subconfluent
cells were washed with cold phosphate-buffered saline (PBS) and
lysed using lysis A buffer containing 1% Triton X-100, 20 mM
Tris-HCl (pH 7.0), 5 mM EDTA, 50 mM sodium chloride, 50 mM
pyrophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate,
and a protease inhibitor mix, Complete™ (Roche Diagnostics).
Whole-cell lysates were separated using SDS-PAGE and were blotted
onto a polyvinylidene fluoride membrane. The membrane was blocked
for 1 h with 5% skim milk in a TBS buffer (pH 8.0) with 0.1%
Tween-20. The membrane was then washed 3 times with TBS and
incubated overnight with the primary antibody at 4°C. After washing
3 times with TBS, the membrane was incubated with a horseradish
peroxidase-conjugated secondary antibody for 1 h at room
temperature. The membrane was then washed, followed by
visualization using an ECL detection system and LAS-4000 (GE
Healthcare, Buckinghamshire, UK). Rabbit antibodies specific for
EGFR, phospho-EGFR, Akt, phospho-Akt, caspase-3, cleaved caspase-3,
and β-actin were obtained from Cell Signaling (Beverly, MA, USA).
To evaluate the influence of the drugs on phosphorylation and an
apoptosis-related molecule, the cells were stimulated for 1–3 and
24 h, respectively.
Database analysis
To analyze the prevalence of Akt2
amplification and high expression levels, the cBioPortal for Cancer
Genomics database (http://www.cbioportal.org/public-portal/) was searched
(25,26). Within the database, The Cancer
Genome Atlas (TCGA) datasets (http://cancergenome.nih.gov/) of several cancers are
analyzed.
Statistical analysis
Continuous variables were analyzed using Student's
t-test, and the results were expressed as the average and standard
deviations (SD). The univariate relationship between each
independent variable was examined using the Fisher's exact test.
The statistical analyses were two-tailed and were performed using
Microsoft Excel (Microsoft, Redmond, WA, USA) and JMP Pro 11 (SAS
Institute, Cary, NC, USA). P<0.05 was considered statistically
significant.
Results
Akt2 expression and patient
characteristics
A total of 22 patients with advanced PC and a good
performance status (0–1) who had been diagnosed based on the
results of EUS-FNA were enrolled in this study. The patients with
PC ranged in age from 17 to 70 years, with a median age of 61
years, and the male: female ratio was 12:10. To evaluate the
relationship between Akt2 expression and prognosis, an
immunohistochemical analysis was performed using biopsy specimens
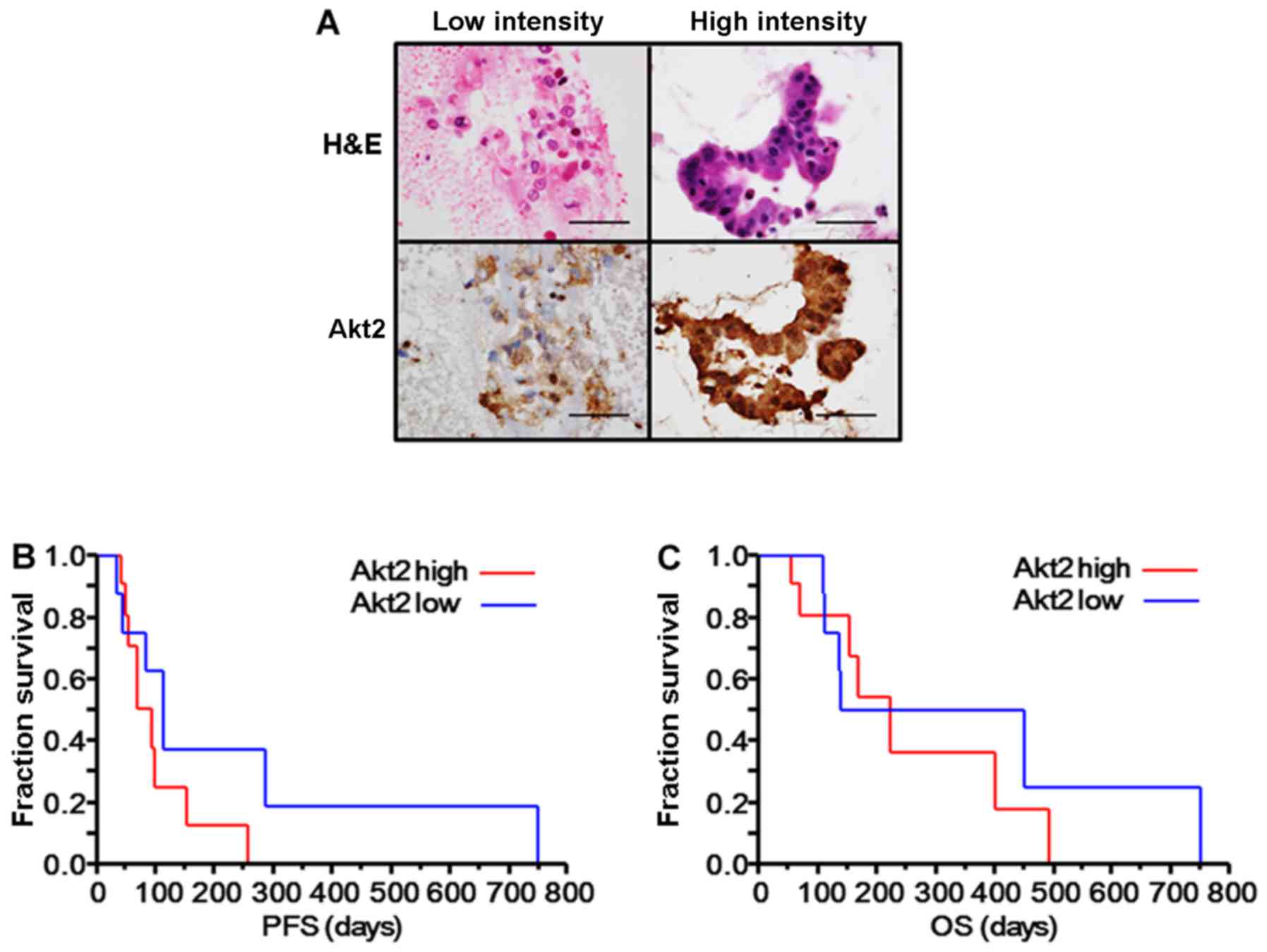
from these patients (Fig. 1A).
Three of the 22 specimens could not be evaluated properly using
immunohistochemistry for Akt2 because of the poor conditions of the
samples and were excluded from additional analyses. The staining
intensity was determined as the relative mean grey value (mean grey
value/maximum grey value) using ImageJ software, and the patients
were divided into two groups: a low intensity group (relative mean
grey value <70%, n=8) and a high intensity group (relative mean
grey value ≥70%, n=11). No significant differences in the patient
characteristics were observed between the two groups, but patients
with a high Akt2 intensity tended to have a poorer response to
erlotinib plus gemcitabine (0/11 vs. 2/8, P=0.16) (Table I). Though the difference was not
significant, patients with a high Akt2 intensity also tended to
have a shorter PFS (median PFS: 92 vs. 113 days, P=0.19) (Fig. 1B) and OS after the initiation of
erlotinib plus gemcitabine (median OS: 224 days vs. 295 days,
P=0.59) (Fig. 1C). These results
suggest that Akt2 might be associated with erlotinib resistance in
advanced PC.
 | Table IPatient characteristics and
associations with Akt2 expression. |
Table I
Patient characteristics and
associations with Akt2 expression.
| Patients
characteristics | Akt2 intensity
| P-valueb |
|---|
Low
(n=8) | High
(n=11) |
|---|
| Age, years | | | |
| <60 | 4 | 5 | 1.0 |
| ≥60 | 4 | 6 | |
| Gender | | | |
| Male | 4 | 7 | 0.66 |
| Female | 4 | 4 | |
| T stage | | | |
| T1-3 | 5 | 5 | 0.65 |
| T4 | 3 | 6 | |
| N stage | | | |
| N0 | 3 | 7 | 0.37 |
| N1-3 | 5 | 4 | |
| M stage | | | |
| M0 | 3 | 2 | 0.60 |
| M1 | 5 | 9 | |
| Treatment line of
erlotinib plus gemcitabine | | | |
| First line | 4 | 5 | 1.0 |
| Second line or
later | 4 | 6 | |
| Response to
erlotinib plus gemcitabine | | | |
| PR | 2 | 0 | 0.16 |
| SD or PD | 6 | 11 | |
| Median PFS
(days)a | 113 | 92 | 0.19 |
| Median OS
(days)a | 295 | 224 | 0.59 |
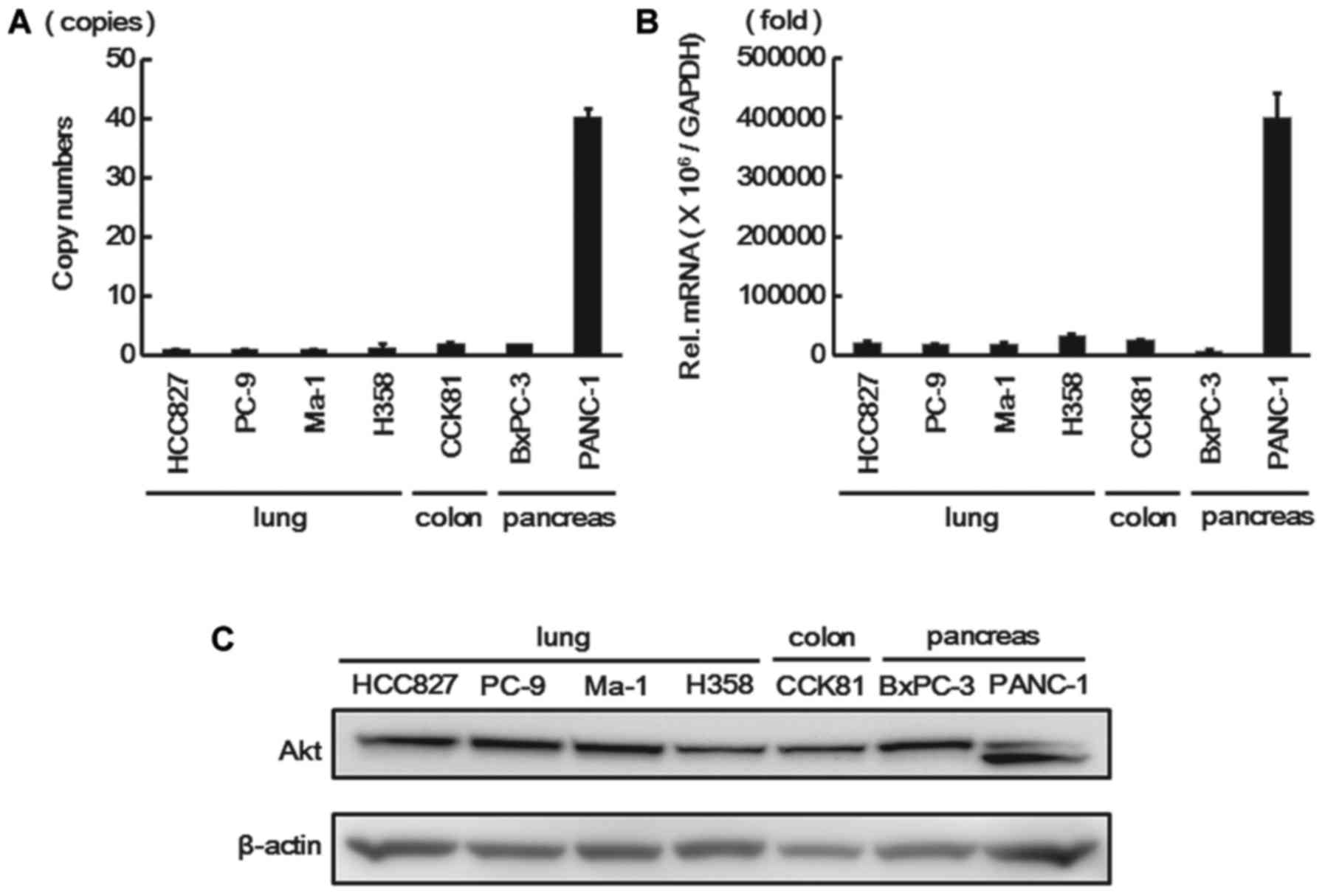
Akt2 copy number, Akt2 gene expression,
and Akt2 protein expression in diverse cancer cell lines
To investigate the relationship with Akt2 expression
and the response to anti-EGFR therapies, we evaluated the Akt2 gene
copy numbers, Akt2 gene expression, and Akt2 protein expression in
several cancer cell lines for which anti-EGFR therapies are
commonly used. Seven diverse cancer cell lines (four NSCLC cell
lines, one CRC cell line, and two PC lines) were used because the
PANC-1 cell line has been reported to exhibit Akt2
amplification and the other cell lines are sensitive to anti-EGFR
therapies. The Akt2 gene copy number, Akt2 gene expression, and
Akt2 protein expression for these cell lines were estimated using a
copy number assay, real-time RT-PCR, and western blotting,
respectively. The PANC-1 cell line had high copy numbers of the
Akt2 gene (40 copies) and the Akt2 gene expression level was also
markedly elevated, whereas the other cell lines had neither a high
Akt2 copy number nor a high expression level (Fig. 2A and B). Western blot analysis also
revealed that Akt2 protein was highly expressed in the PANC-1 cell
line (Fig. 2C).
Combined effect of erlotinib and a PI3K
inhibitor in an Akt2-amplified and highly expressed PANC-1 cell
line
To investigate the influence of Akt2 on the
resistance to anti-EGFR therapies, the effect of erlotinib against
the PANC-1 cell line (an Akt2-amplified and highly expressed
cell line) was tested using an MTT assay. As is shown in Fig. 3A, the PANC-1 cell line was
particularly resistant to erlotinib, compared with the other cell
lines that did not exhibit either amplification or a high
Akt2 expression level. Next, the combined effect of
erlotinib and BYL719, a PI3K inhibitor, was examined in the PANC-1
cell line. The combined treatment of erlotinib and BYL719 inhibited
the cellular growth more intensively than erlotinib monotherapy
(Fig. 3B). This combined effect in
the PANC-1 cell line has already been reported (21), but we also confirmed the efficacy
of combination therapy in this study. Whereas the phosphorylation
of Akt persisted after erlotinib monotherapy, the combined
treatment decreased the phosphorylation of Akt (Fig. 3C). In contrast to erlotinib or
BYL719 monotherapy, the combined treatment of erlotinib and BYL719
increased the expression level of an apoptosis-related molecule,
cleaved caspase-3 (Fig. 3D).
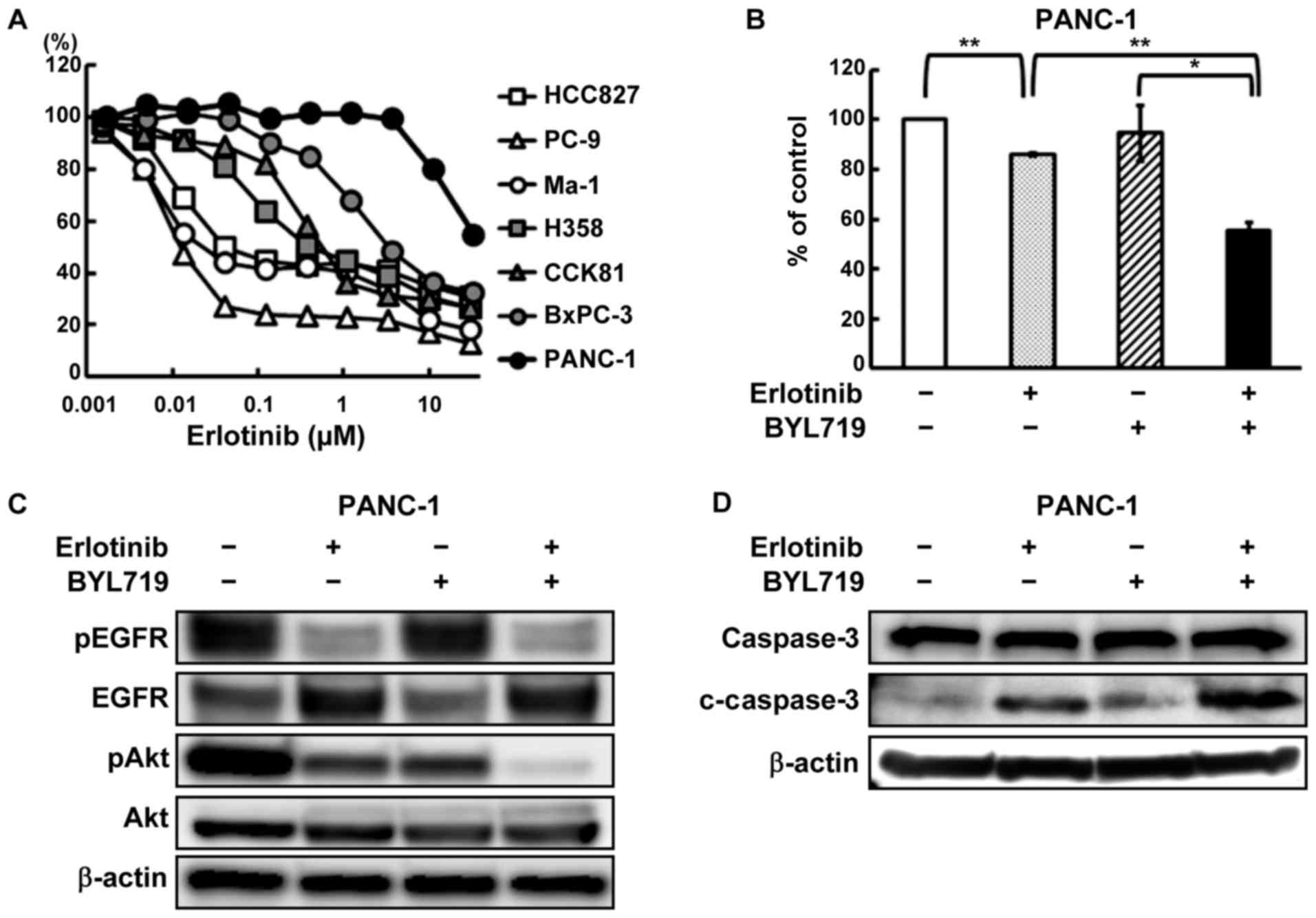 | Figure 3Combined effect of erlotinib and
BYL719 in an Akt2-amplified and highly expressed PANC-1 cell
line. (A) Growth inhibitory effects of erlotinib in diverse cancer
cell lines. The cells were exposed to each concentration of
erlotinib for 72 h, and the growth inhibitory effects were
evaluated using an MTT assay. The HCC827, PC-9, and Ma-1 cell lines
(non-small cell lung cancer cell lines harboring EGFR
mutaions) were hypersensitive to erlotinib. The PANC-1 cell line
was particularly resistant to erlotinib. Lines, mean of independent
triplicate experiments. (B) Growth inhibitory effects of erlotinib
and/or BYL719 in the PANC-1 cell line. The cells were treated with
10 μM of erlotinib and/or 2 μM of BYL719 for 72 h,
and the growth inhibitory effects were evaluated using an MTT
assay. The combined treatment of erlotinib and BYL719 inhibited the
cellular growth more intensively, compared with erlotinib
monotherapy. Columns, mean of independent triplicate experiments;
error bars, SD; *P<0.05; **P<0.01. (C)
Phosphorylation of EGFR and Akt in the PANC-1 cell line. The cells
were treated with 10 μM of erlotinib and/or 2 μM of
BYL719, and the samples were collected 1 h after drug stimulation.
The phophorylation of Akt persisted in the erlotinib monotherapy,
while the combined treatment with BYL719 decreased the
phosphorylation of Akt. β-actin was used as an internal control.
pEGFR, phospho-EGFR; pAkt, phopho-Akt. (D) Expression of an
apoptosis-related molecule in the PANC-1 cell line. Twenty-four
hours after the cells were exposed to the drugs (erlotinib, 10
μM; BYL719, 2 μM), the samples were collected.
Although erlotinib or BYL719 monotherapy did not increase the
expression of cleaved caspase-3, the combined treatment increased
the expression. β-actin was used as an internal control.
c-caspase-3, cleaved caspase-3. |
Akt2 overexpression led to resistance to
anti-EGFR therapies
To evaluate the contribution of Akt2 to the EGFR-TKI
response, a stably Akt2-overexpressed PC cell line was
created. The BxPC-3 cell line was mainly used because this PC cell
line is intermediately sensitive to erlotinib with no Akt2
amplification or high expression (Figs. 2 and 3A). The stably EGFP-expressed cell
line was used as a control. Akt2-overexpression was
confirmed using a western blot analysis (Fig. 4A). Then, we performed a growth
inhibition assay using this cell line and found that the
BxPC-3/Akt2 cell line was resistant to erlotinib (Fig. 4B). In contrast, Akt2-overexpression
was not associated with gemcitabine resistance. In western blot
analyses, erlotinib inhibited the phosphorylation of EGFR and Akt
in a dose-dependent manner in the control cells. In contrast, the
phosphorylation of Akt strongly persisted in the BxPC-3/Akt2 cell
line, although erlotinib inhibited the phosphorylation of EGFR
(Fig. 4C). The erlotinib-induced
elevation in the expression level of cleaved caspase-3 in the
BxPC-3/Akt2 cell line was lower than that in the control cells
(Fig. 4D). Furthermore, similar
experiments were also performed in other cell lines (Ma-1 and
CCK81), demonstrating the resistance to anti-EGFR therapies in
Akt2-overexpressed cell lines (Fig. 4E). These results indicate that Akt2
might be related to resistance to anti-EGFR therapies.
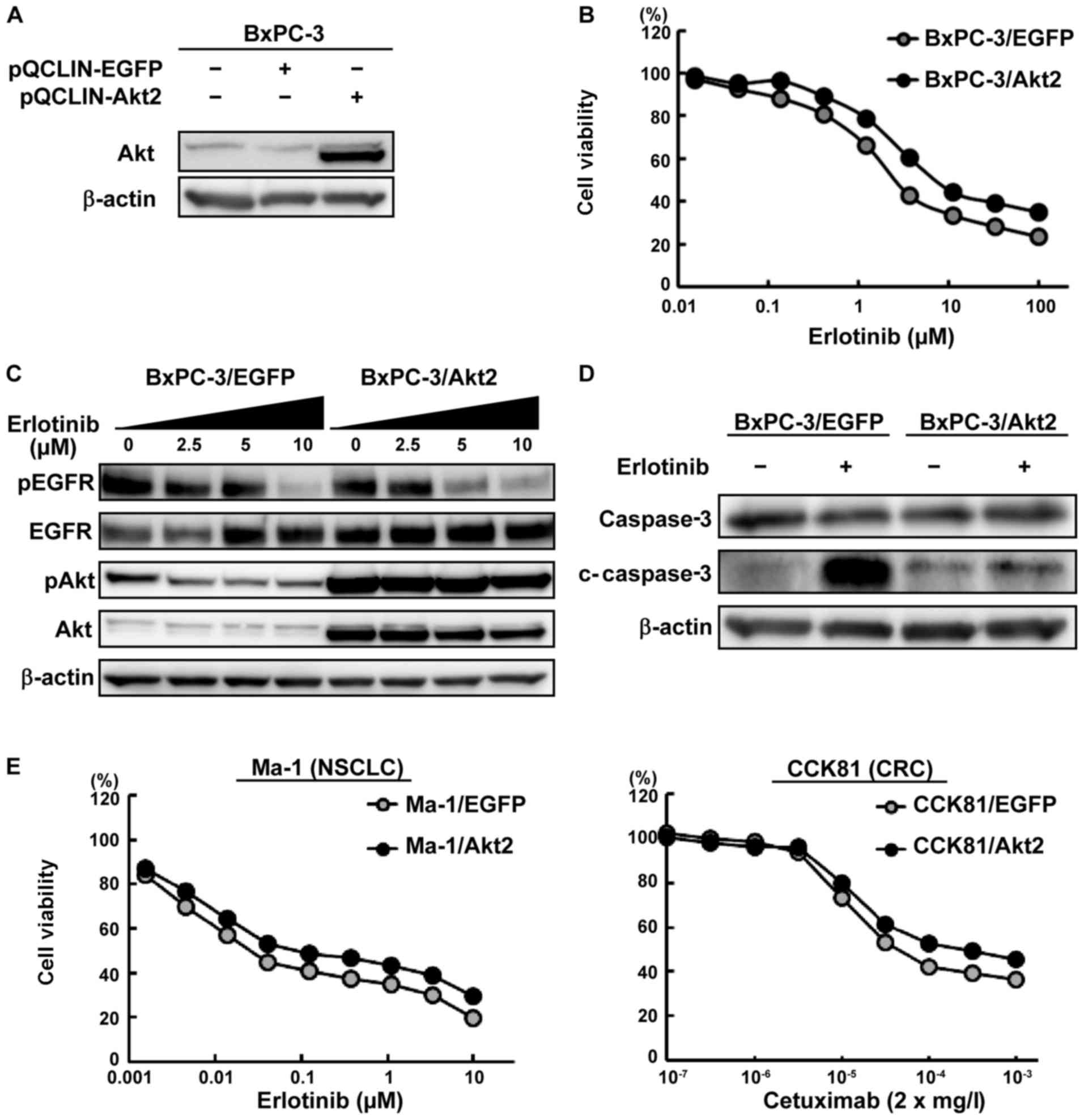 | Figure 4Influence of
Akt2-overexpression on the efficacy of anti-EGFR therapies
against PC, colorectal cancer (CRC) or non-small cell lung cancer
(NSCLC) cell lines. (A) Akt2 protein expression in the BxPC-3 cell
lines. To investigate the influence of Akt2, an
Akt2-overexpressed cell line was retrovirally created. An
EGFP-overexpressed cell line was used as a control. The
BxPC-3/Akt2 cell line exhibited the overexpression of Akt2 protein.
β-actin was used as an internal control. (B) Growth inhibitory
effects of erlotinib in the BxPC-3 cell lines. The cells were
exposed to each concentration of erlotinib for 72 h, and the growth
inhibitory effects were evaluated using an MTT assay. The
sensitivity to erlotinib was weakened in the BxPC-3/Akt2 cell line,
compared with the control. Lines, mean of independent triplicate
experiments. (C) Phosphorylation of EGFR and Akt in the Bx-PC3 cell
lines. Three hours after the cells were treated with the indicated
concentration of drugs, the samples were collected. Erlotinib
inhibited the phosphorylation of EGFR and Akt in a dose-dependent
manner in the BxPC-3/EGFP cell line. In contrast, the
phosphorylation of Akt strongly persisted in the BxPC-3/Akt2 cell
line regardless of the inhibition of the phosphorylation of EGFR.
β-actin was used as an internal control. pEGFR, phospho-EGFR; pAkt,
phopho-Akt. (D) Expression of an apoptosis-related molecule in the
BxPC-3 cell lines. Twenty-four hours after the cells were treated
with the drug (erlotinib, 2.5 μM), the samples were
collected. Erlotinib increased the expression of cleaved caspase-3
to a greater extent in the BxPC-3/EGFP cell line but did not
increase the expression in the BxPC-3/Akt2 cell line. β-actin was
used as an internal control. c-caspase-3, cleaved caspase-3. (E)
Growth inhibitory effects in NSCLC and CRC cell lines. The
Ma-1/Akt2 cell line (human NSCLC cell line) and CCK81/Akt2 cell
line (human CRC cell line) were resistant to erlotinib and
cetuximab, respectively. The cells were exposed to each
concentration of erlotinib/cetuximab for 72 h, and the growth
inhibitory effects were evaluated using an MTT assay. Lines, mean
of independent triplicate experiments. |
Combined effect of anti-EGFR therapies
and a PI3K inhibitor in the Akt2-overexpressed cell lines
To investigate the effect of a PI3K inhibitor on the
drug resistance induced by Akt2 overexpression, we performed
growth inhibition assays using erlotinib/cetuximab and BYL719 in
the Akt2-overexpressed lines. The combined treatment with
erlotinib/cetuximab and BYL719 considerably inhibited the cellular
growth, compared with the monotherapy (Fig. 5A). The IC50 values of
erlotinib/cetuximab were significantly lower in the combination
therapy than in the monotherapy (Fig.
5B). In the BxPC-3/Akt2 cell line, the phosphorylation of Akt
was also markedly inhibited by the combined treatment with BYL719
compared with erlotinib alone (Fig.
5C). Compared with monotherapy, the combined treatment with
BYL719 increased the expression of cleaved caspase-3 to a greater
extent (Fig. 5D). These results
indicate that Akt2 can be associated with resistance to anti-EGFR
therapies via the PI3K-Akt pathway and that such resistance can be
overcome by a PI3K inhibitor.
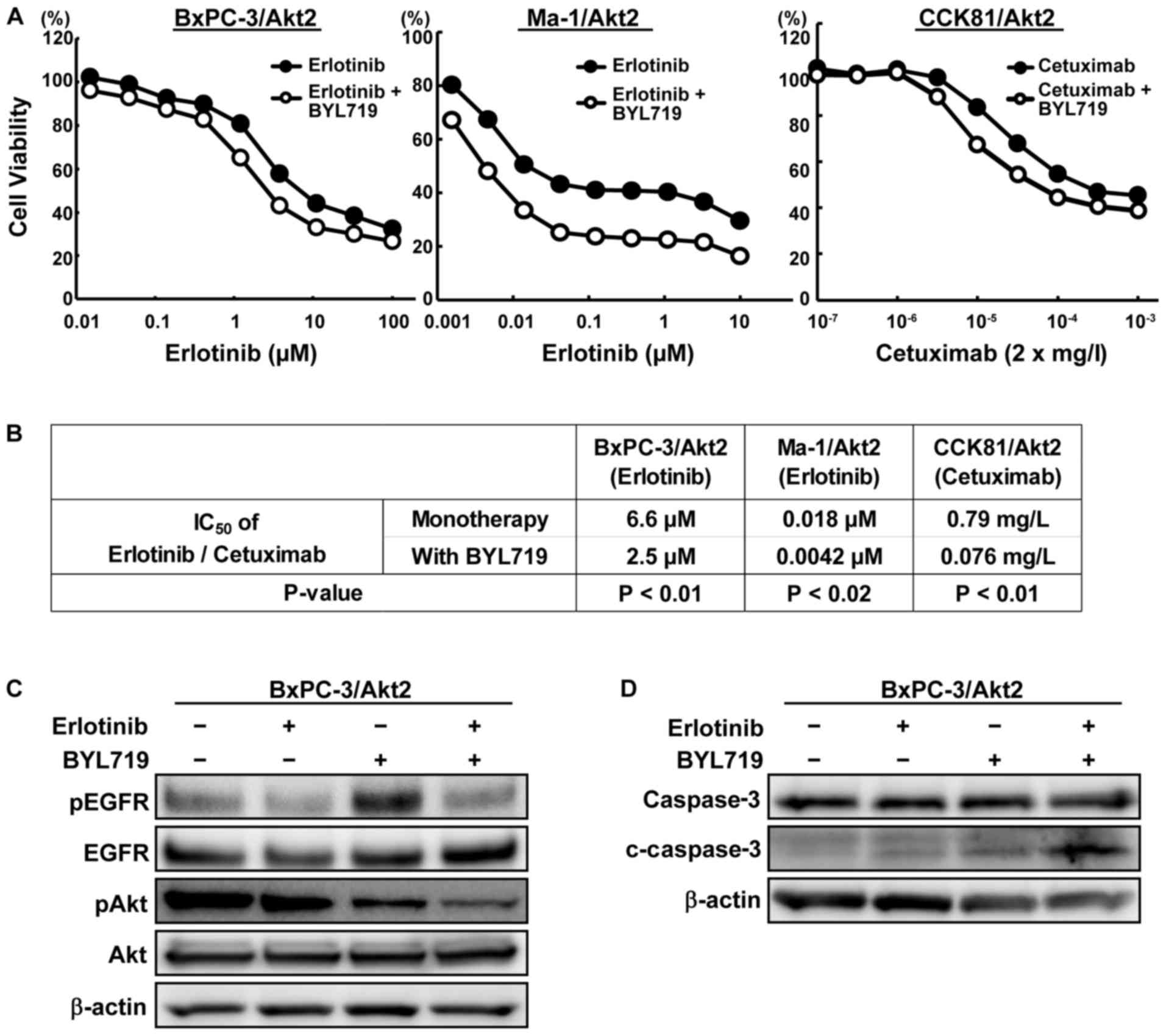 | Figure 5Combined effect of anti-EGFR therapy
and BYL719 in the Akt2-overexpressed cell lines. (A) Growth
inhibitory effects of the combined treatment of erlotinib/cetuximab
and BYL719 in the Akt2-overexpressed cell lines. Cells were
exposed to each concentration of erlotinib/cetuximab with or
without BYL719 (BxPC-3, 4 μM; CCK81 and Ma-1, 2 μM)
for 72 h, and the growth inhibitory effects were evaluated using an
MTT assay. The combined treatment with erlotinib/cetuximab and
BYL719 strongly inhibited the cellular growth compared with
erlotinib/cetuximab monotherapy. Lines, mean of independent
triplicate experiments. (B) IC50 values of
erlotinib/cetuximab with or without BYL719 in the
Akt2-overexpressed cell lines. The IC50 values of
erlotinib/cetuximab were significantly lower in the combination
therapy than in the monotherapy. (C) Phosphorylation of EGFR and
Akt in the BxPC-3/Akt2 cell line. Three hours aftter the cells were
exposed to the drugs (erlotinib, 5 μM; BYL719, 4 μM),
the samples were collected. The phosphorylation of Akt was
inhibited to a greater degree by the combined treatment than the
erlotinib monotherapy. β-actin was used as an internal control.
pEGFR, phospho-EGFR; pAkt, phopho-Akt. (D) Expression of an
apoptosis-related molecule in the BxPC-3/Akt2 cell line.
Twenty-four hours after the cells were exposed to the drugs
(erlotinib, 5 μM; BYL719, 4 μM), the samples were
collected. In contrast to erlotinib or BYL719 monotherapy, the
combined treatment increased the expression of cleaved caspase-3 to
a greater extent. β-actin was used as an internal control.
c-caspase-3, cleaved caspase-3. |
Akt2 amplification and high Akt2
expression levels in several cancers from TCGA datasets
Next, we analyzed the TCGA database to investigate
the frequencies of Akt2 amplification and high expression
levels in several cancers. Data for PC, lung adenocarcinoma, lung
squamous cell carcinoma, CRC, and head-and-neck squamous cell
carcinoma (HNSCC) were analyzed since EGFR-TKIs or anti-EGFR
monoclonal antibodies are clinically used for the treatment of
these cancers. A high Akt2 expression level was defined as a
Z-score >2.0 using RNA sequencing. Samples with a high
Akt2 expression level were found at a relative high
frequency of 7–14%. In contrast, the frequency of Akt2
amplification was not so high, except for PC (8%), and Akt2
amplification was correlated with the Akt2 expression level.
Among all the cancers that were examined, the frequencies of both
Akt2 amplification and a high expression level were the
highest for PC (Table II).
| Table IIHigh RNA expression and gene
amplification of Akt2 in TCGA database. |
Table II
High RNA expression and gene
amplification of Akt2 in TCGA database.
| Cancer type | Amplification
| RNA expression
|
|---|
| No. of samples | No. of samples with
amplification (%) | No. of samples | No. of samples with
a high expression level (%) |
|---|
| PC | 145 | 11 (8) | 178 | 25 (14) |
| LAd | 230 | 3 (1.3) | 230 | 21 (9.1) |
| LSq | 178 | 8 (4.5) | 178 | 24 (13.5) |
| CRC | 212 | 3 (1.4) | 244 | 17 (7) |
| HNSCC | 279 | 4 (1.4) | 279 | 27 (9.7) |
Discussion
In this study, we revealed the possible association
between high Akt2 expression levels and erlotinib resistance in
clinical specimens of advanced PC. In addition, in vitro
experiments also demonstrated this association and the efficacy of
combined treatment with a PI3K inhibitor for overcoming resistance.
Although a previous in vitro study demonstrated an
association between Akt2 and erlotinib resistance (21), to the best of our knowledge, this
is the first study to show this association in clinical specimens
of advanced PC.
EGFR is a cell membrane growth factor receptor
characterized by tyrosine kinase activity that plays a crucial role
in the control of key cellular transduction pathways (27). At present, targeting EGFR is one of
the most effective anticancer strategies available, and anti-EGFR
therapies are now widely used for the treatment of PC, NSCLC, CRC,
and HNSCC (27). For advanced PC,
erlotinib is the first drug for which a superiority of combination
therapy with gemcitabine in terms of OS and PFS, has been
documented in a large randomized trial, but the achievable
improvement has remained limited (3). This unsatisfactory outcome has
encouraged a number of studies which have attempted to identify
molecular markers capable of predicting the efficacy of erlotinib
in patients with PC (1,27). EGFR mutation or
amplification has been a potential predictor of the response to
EGFR-TKI treatment or patient prognosis in several cancers
(27), and EGFR-TKIs are known to
be effective against NSCLC harboring EGFR mutations
(28). Several studies, however,
have demonstrated that neither EGFR amplification nor
EGFR mutation is a predictive biomarker for the response to
erlotinib in PC (29,30). KRAS mutations, which act
downstream of the EGFR pathway, have been characterized as a
predictive biomarker for resistance to anti-EGFR antibodies in CRC
(31). In PC, however,
K-ras mutations, which occur in 90% of PC, have not been
recog-nized as a predictive biomarker for resistance to erlotinib
in combination with gemcitabine (29). These previous studies highlight the
need to explore alternative explanations for responses to erlotinib
and to identify markers that can predict the efficacy of erlotinib
in patients with PC. Recently, alterations of the PI3K-Akt pathway,
which is active downstream of the EGFR pathway, have been
implicated as potential mechanisms of resistance to anti-EGFR
therapies in CRC and lung adenocarcinoma (32–35).
Amplification and high expression levels of Akt2 are also
reportedly associated with resistance to erlotinib in PC in
vitro (21). The present study
showed a tendency toward a poorer response and a shorter PFS and OS
for erlotinib plus gemcitabine in patients with advanced PC
harboring high Akt2 expression. Futhermore, in vitro
experiments showed that Akt2 is associated with the resistance to
anti-EGFR therapies, and a TCGA dataset showed that the frequencies
of both Akt2 amplification and high expression levels are
relatively high in PC. These findings suggest that Akt2 expression
could be a predictive biomarker for resistance to erlotinib in PC.
In addition, in our in vitro experiments, the combined
treatment of erlotinib and a PI3K inhibitor was able to overcome
this resistance, indicating that this combined treatment might be a
promising starategy for the treatment of patients who are resistant
to anti-EGFR therapies because of high Akt2 expression levels.
A previous report showed that a high Akt2 expression
detected using immunohistochemistry was observed in 40% of cases,
consistent with the results of the present study, but a correlation
with the survival of patients with PC was not seen (14). In the previous study, all the
patients had received curative surgical resections and did not
receive erlotinib treatment (14).
In contrast, we focused on patients with advanced PC treated with
erlotinib plus gemcitabine and showed a tendency toward a poorer
outcome among patients with high Akt2 expression levels. The
PI3K/Akt/mTOR pathway regulates cell survival and apoptosis
(9), and Akt2 expression is
reportedly associated with prognosis and aggressiveness in other
cancers (16–19). In addition, Akt2, which is active
downstream of the EGFR pathway, seems to be associated with
erlotinib resistance. Therefore, the tendency toward a poorer
outcome among patients with advanced PC with high Akt2 expression
levels who had been treated with erlotinib plus gemcitabine, as
observed in the present study, seems reasonable.
The present study had some limitations. First, the
study was relatively small and was performed retrospectively, and
we could not show a significant correlation between Akt2 expression
and the response to erlotinib. Only a few patients with advanced PC
are eligible for surgical resection (1), and cytological examinations such as
EUS or CT-guided FNA, which is an invasive method, are not
rountinely performed when cancer is almost certainly diagnosed
using dynamic CT imaging (36).
Consequently, predictive biomarkers in advanced PC are difficult to
investigate and studies like ours are very rare. At our institute,
however, EUS-FNA is commonly conducted (37). Therefore, the present study might
be valuable from the aspect of its use of rare biopsy specimens
despite the small number of available samples. In addition, the
present study, showing Akt2 expression as a biomarker candiate,
encourages the use of aggresive biopsies in patients with advanced
PC. Recently, evaluation of gene amplification has become feasible
using a liquid biopsy, which detects cell-free circulating tumor
DNA in the blood (38,39). Liquid biopsies are less invasive
than EUS-FNA and might be useful for the detection of biomarkers
such as gene amplification.
As a second limitation, we could not evaluate Akt2
gene amplification and expression because of the poor sample
conditions. Due to the small amount of biopsied tissue obtained
using EUS-FNA, we could not extract a sufficient amount of DNA/RNA
for such analyses. Instead, we analyzed the TCGA database to
investigate the frequencies of Akt2 amplification and high
expression levels. The frequencies of Akt2 amplification and
high expression in PC are not as high as those of other gene
alterations, such as those of the K-ras mutation. Recently,
however, molucular targeted therapy for low-frequency alterations
has been developed. For example, the frequencies of ALK
rearrangements and uncommon EGFR mutations in NSCLC are as
low as 5 and 10%, respectively (40,41).
Patients with ALK rearrangements are well known to respond
dramatically to crizotinib (40),
and in a recent post hoc analysis and our in vitro studies,
afatinib was reported to be effective for patients harboring
uncommon EGFR mutations, including exon 18 mutations, the
S768I mutation, and the L861Q mutation (42–44).
Thus, the low frequencies of gene alterations shoud not be
overlooked, and effective biomarkers for the detection and
treatment of PC are eagerly anticipated, but not yet defined.
Therefore, Akt2 expression in PC might be valuable in treatment
with erlotinib. To eliminate these limitations and to confirm our
findings, further analyses including multi-center studies are
desirable.
In conclusion, we found an association between high
Akt2 expression levels and resisitance to erlotinib in both
clinical specimens of advanced PC and several cell lines. A high
Akt2 expression level might be a predictive biomarker for
resistance to anti-EGFR therapies, especially erlotinib, in
patients with PC. Our present study was, however, very small and
was performed retrospectively. Therefore, further large-scale
studies are needed to confirm these findings.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
CT
|
computed tomography
|
|
EGFP
|
enhanced green fluorescent protein
|
|
EGFR
|
epidermal growth factor receptor
|
|
EUS-FNA
|
endoscopic ultrasonography-guided fine
needle aspiration
|
|
FBS
|
fetal bovine serum
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
LAd
|
lung adenocarcinoma
|
|
LSq
|
lung squamous cell carcinoma
|
|
NSCLC
|
non-small cell lung cancer
|
|
OS
|
overall survival
|
|
PC
|
pancreatic cancer
|
|
PFS
|
progression-free survival
|
|
TCGA
|
the Cancer Genome Atlas
|
|
TKI
|
tyrosine kinase inhibitor
|
|
SD
|
standard deviations
|
Acknowledgments
We thank Mr. Shinji Kurashimo, Mr. Yoshihiro Mine,
Ms. Eiko Honda, Ms. Tomoko Kitayama, and Ms. Ayaka Kurumatani for
their technical assistance, and we thank Dr Yoshimi Hosono for the
pathological review. This study was supported in part by
Grant-in-Aid for Research Activity start-up (15H06754).
References
|
1
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al
National Cancer Institute of Canada Clinical Trials Group:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al Groupe Tumeurs Digestives of Unicancer;
PRODIGE Intergroup: FOLFIRINOX versus gemcitabine for metastatic
pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang-Gillam A, Li CP, Bodoky G, Dean A,
Shan YS, Jameson G, Macarulla T, Lee KH, Cunningham D, Blanc JF, et
al NAPOLI-1 Study Group: Nanoliposomal irinotecan with fluorouracil
and folinic acid in metastatic pancreatic cancer after previous
gemcitabine-based therapy (NAPOLI-1): A global, randomised,
open-label, phase 3 trial. Lancet. 387:545–557. 2016. View Article : Google Scholar
|
|
7
|
Kozak G, Blanco FF and Brody JR: Novel
targets in pancreatic cancer research. Semin Oncol. 42:177–187.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Macgregor-Das AM and Iacobuzio-Donahue CA:
Molecular pathways in pancreatic carcinogenesis. J Surg Oncol.
107:8–14. 2013. View Article : Google Scholar :
|
|
9
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar :
|
|
10
|
Ocana A, Vera-Badillo F, Al-Mubarak M,
Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD,
Seruga B, Pandiella A and Amir E: Activation of the PI3K/mTOR/AKT
pathway and survival in solid tumors: Systematic review and
meta-analysis. PLoS One. 9:e952192014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nitulescu GM, Margina D, Juzenas P, Peng
Q, Olaru OT, Saloustros E, Fenga C, Spandidos DA, Libra M and
Tsatsakis AM: Akt inhibitors in cancer treatment: The long journey
from drug discovery to clinical use (Review). Int J Oncol.
48:869–885. 2016.
|
|
12
|
Martini M, De Santis MC, Braccini L,
Gulluni F and Hirsch E: PI3K/AKT signaling pathway and cancer: An
updated review. Ann Med. 46:372–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruggeri BA, Huang L, Wood M, Cheng JQ and
Testa JR: Amplification and overexpression of the AKT2 oncogene in
a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog.
21:81–86. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamamoto S, Tomita Y, Hoshida Y, Morooka
T, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H,
et al: Prognostic significance of activated Akt expression in
pancreatic ductal adenocarcinoma. Clin Cancer Res. 10:2846–2850.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dobashi Y, Kimura M, Matsubara H, Endo S,
Inazawa J and Ooi A: Molecular alterations in AKT and its protein
activation in human lung carcinomas. Hum Pathol. 43:2229–2240.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rychahou PG, Kang J, Gulhati P, Doan HQ,
Chen LA, Xiao SY, Chung DH and Evers BM: Akt2 overexpression plays
a critical role in the establishment of colorectal cancer
metastasis. Proc Natl Acad Sci USA. 105:20315–20320. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakayama K, Nakayama N, Kurman RJ, Cope L,
Pohl G, Samuels Y, Velculescu VE, Wang TL and Shih IeM: Sequence
mutations and amplification of PIK3CA and AKT2 genes in purified
ovarian serous neoplasms. Cancer Biol Ther. 5:779–785. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bacus SS, Altomare DA, Lyass L, Chin DM,
Farrell MP, Gurova K, Gudkov A and Testa JR: AKT2 is frequently
upregulated in HER-2/neu-positive breast cancers and may contribute
to tumor aggressiveness by enhancing cell survival. Oncogene.
21:3532–3540. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bellacosa A, de Feo D, Godwin AK, Bell DW,
Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, et
al: Molecular alterations of the AKT2 oncogene in ovarian and
breast carcinomas. Int J Cancer. 64:280–285. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Edling CE, Selvaggi F, Buus R, Maffucci T,
Di Sebastiano P, Friess H, Innocenti P, Kocher HM and Falasca M:
Key role of phosphoinositide 3-kinase class IB in pancreatic
cancer. Clin Cancer Res. 16:4928–4937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wong MH, Xue A, Julovi SM, Pavlakis N,
Samra JS, Hugh TJ, Gill AJ, Peters L, Baxter RC and Smith RC:
Cotargeting of epidermal growth factor receptor and PI3K overcomes
PI3K-Akt oncogenic dependence in pancreatic ductal adenocarcinoma.
Clin Cancer Res. 20:4047–4058. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Velasco MA, Tanaka M, Yamamoto Y,
Hatanaka Y, Koike H, Nishio K, Yoshikawa K and Uemura H: Androgen
deprivation induces phenotypic plasticity and promotes resistance
to molecular targeted therapy in a PTEN-deficient mouse model of
prostate cancer. Carcinogenesis. 35:2142–2153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arao T, Ueshima K, Matsumoto K, Nagai T,
Kimura H, Hagiwara S, Sakurai T, Haji S, Kanazawa A, Hidaka H, et
al: FGF3/FGF4 amplification and multiple lung metastases in
responders to sorafenib in hepatocellular carcinoma. Hepatology.
57:1407–1415. 2013. View Article : Google Scholar
|
|
24
|
Togashi Y, Kogita A, Sakamoto H, Hayashi
H, Terashima M, de Velasco MA, Sakai K, Fujita Y, Tomida S, Kitano
M, et al: Activin signal promotes cancer progression and is
involved in cachexia in a subset of pancreatic cancer. Cancer Lett.
356(2 Pt B): 819–827. 2015. View Article : Google Scholar
|
|
25
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mitsudomi T and Yatabe Y: Mutations of the
epidermal growth factor receptor gene and related genes as
determinants of epidermal growth factor receptor tyrosine kinase
inhibitors sensitivity in lung cancer. Cancer Sci. 98:1817–1824.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
da Cunha Santos G, Dhani N, Tu D, Chin K,
Ludkovski O, Kamel-Reid S, Squire J, Parulekar W, Moore MJ and Tsao
MS: Molecular predictors of outcome in a phase 3 study of
gemcitabine and erlotinib therapy in patients with advanced
pancreatic cancer: National Cancer Institute of Canada Clinical
Trials Group Study PA.3. Cancer. 116:5599–5607. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tzeng CW, Frolov A, Frolova N, Jhala NC,
Howard JH, Buchsbaum DJ, Vickers SM, Heslin MJ and Arnoletti JP:
Epidermal growth factor receptor (EGFR) is highly conserved in
pancreatic cancer. Surgery. 141:464–469. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD,
Robitaille S, et al: K-ras mutations and benefit from cetuximab in
advanced colorectal cancer. N Engl J Med. 359:1757–1765. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sartore-Bianchi A, Martini M, Molinari F,
Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P,
De Dosso S, Mazzucchelli L, et al: PIK3CA mutations in colorectal
cancer are associated with clinical resistance to EGFR-targeted
monoclonal antibodies. Cancer Res. 69:1851–1857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sos ML, Koker M, Weir BA, Heynck S,
Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P,
et al: PTEN loss contributes to erlotinib resistance in EGFR-mutant
lung cancer by activation of Akt and EGFR. Cancer Res.
69:3256–3261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jeannot V, Busser B, Brambilla E, Wislez
M, Robin B, Cadranel J, Coll JL and Hurbin A: The PI3K/AKT pathway
promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by
a deacetylase-dependent mechanism. Int J Cancer. 134:2560–2571.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tamm EP, Bhosale PR and Lee JH: Pancreatic
ductal adenocarcinoma: Ultrasound, computed tomography, and
magnetic resonance imaging features. Semin Ultrasound CT MR.
28:330–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kitano M, Kudo M, Yamao K, Takagi T,
Sakamoto H, Komaki T, Kamata K, Imai H, Chiba Y, Okada M, et al:
Characterization of small solid tumors in the pancreas: The value
of contrast-enhanced harmonic endoscopic ultrasonography. Am J
Gastroenterol. 107:303–310. 2012. View Article : Google Scholar
|
|
38
|
Diaz LA Jr and Bardelli A: Liquid
biopsies: Genotyping circulating tumor DNA. J Clin Oncol.
32:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heitzer E, Ulz P and Geigl JB: Circulating
tumor DNA as a liquid biopsy for cancer. Clin Chem. 61:112–123.
2015. View Article : Google Scholar
|
|
40
|
Gridelli C, Peters S, Sgambato A, Casaluce
F, Adjei AA and Ciardiello F: ALK inhibitors in the treatment of
advanced NSCLC. Cancer Treat Rev. 40:300–306. 2014. View Article : Google Scholar
|
|
41
|
Roengvoraphoj M, Tsongalis GJ, Dragnev KH
and Rigas JR: Epidermal growth factor receptor tyrosine kinase
inhibitors as initial therapy for non-small cell lung cancer: Focus
on epidermal growth factor receptor mutation testing and
mutation-positive patients. Cancer Treat Rev. 39:839–850. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang JC, Sequist LV, Geater SL, Tsai CM,
Mok TS, Schuler M, Yamamoto N, Yu CJ, Ou SH, Zhou C, et al:
Clinical activity of afatinib in patients with advanced
non-small-cell lung cancer harbouring uncommon EGFR mutations: A
combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung
6. Lancet Oncol. 16:830–838. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kobayashi Y, Togashi Y, Yatabe Y, Mizuuchi
H, Jangchul P, Kondo C, Shimoji M, Sato K, Suda K, Tomizawa K, et
al: EGFR exon 18 mutations in lung cancer: Molecular predictors of
augmented sensitivity to afatinib or neratinib as compared with
first- or third-generation TKIs. Clin Cancer Res. 21:5305–5313.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Banno E, Togashi Y, Nakamura Y, Chiba M,
Kobayashi Y, Hayashi H, Terashima M, de Velasco MA, Sakai K, Fujita
Y, et al: Sensitivities to various epidermal growth factor
receptor-tyrosine kinase inhibitors of uncommon epidermal growth
factor receptor mutations L861Q and S768I: What is the optimal
epidermal growth factor receptor-tyrosine kinase inhibitor? Cancer
Sci. 107:1134–1140. 2016. View Article : Google Scholar : PubMed/NCBI
|