Introduction
Gallbladder cancer (GBC) is the most common and
aggressive cancer of the biliary tract, it constitutes 80–85% of
total biliary tract cancer. GBC shows a striking geographical
variation in its incidence, with high incidence has been reported
from Asia, South America and some part of central Europe (1). GBC incidence is relatively low
(2.2/100,000) (2). However, GBC is
a notoriously lethal disease and has a poor prognosis, the mean
overall survival time is 6 months and 5-year survival rate is 5%
(3). Thus, it is critical to
investigate the molecular mechanisms of GBC progression, which may
be helpful to identify novel diagnostic and therapeutic target.
The bromodomain and extra terminal (BET) proteins,
which include BRD2, BRD3, BRD4 and BRDT, regulate gene expression
as the acetylated lysine binding domain in chromatin (4,5). As
the best-studied member of the BET family, Bromodomain containing
protein 4 (BRD4) has been demonstrated to play an essential role in
cell cycle and cell proliferation (6,7).
BRD4 functions as a transcription regulator through interactions
with P-TEFb, MYC and NF-κB (8,9).
Deregulation of BRD4 protein is increasingly found in several
different types of cancers such as colorectal cancer, breast
cancer, pancreatic cancer and lung cancer (5,10–12).
BRD4 is considered to be a compelling therapeutic target. However,
the role of BRD4 has not been well studied for gallbladder
cancer.
In this study, we investigated the expression level
and potential roles of BRD4 in GBC, aiming to provide novel insight
into GBC diagnosis and therapy.
Materials and methods
Patient samples
All specimens were achieved from 51 GBC patients
with pathologically confirmed GBC after surgery between 2013 and
2014 in Xinhua Hospital Affiliated to Shanghai Jiaotong University.
None of the patients received radiotherapy or chemotherapy before
surgery. The tissue samples were staged on the basis of the
AGCC-TNM Classification of Malignant Tumors (7th edition). The
pathological classification was reviewed separately by two
experienced pathologists. Tissue samples were embedded in paraffin
prior to immunohistochemistry analysis. For quantitative real-time
PCR, part of specimens were immediately frozen in liquid nitrogen
after resection. Informed consent was obtained in all cases.
Ethical approval was obtained from the ethics committee of the
Xinhua Hospital Affiliated to Shanghai Jiao Tong University School
of Medicine.
Immunohistochemistry
Immunohistochemistry (IHC) staining was performed
according to the standard immunoperoxidase staining procedure
(13). Immunoreactivity was scored
as follows: 0, <5% immunoreactive cells; 1, 5–25% immunoreactive
cells; 2, 25–50% immunoreactive cells; 3, 50–75% immunoreactive
cells and 4 for >50% immunoreactive cells. The staining
intensity of BRD4 expression was visually scored as follows: 0,
negative staining; 1, weak staining; 2, moderate staining and 3 for
strong staining. The final score was the sum of extent and
intensity. The specimens were classified as negative (0–1), weak
(2–3), moderate (4–5), and
strong (6–7) staining.
Quantitative real-time PCR
Total RNA was extracted from GBC tissues and cells
using TRIzol reagent. cDNA was synthesized by using PrimeScript
Reverse Transcriptase (Takara, Osaka, Japan) following the
manufacturer's instructions. The expression level of BRD4 was
measured using the SYBR-Green method, and products were detected by
StepOnePlus™ Real-time PCR system (Applied Biosystems, Foster City,
CA, USA). The primers used for amplification of BRD4: BRD4 (forward
primer) 5′-ACAACAAGCCTGGAGATGACA-3′, BRD4 (reverse primer)
5′-GTTTGGTACCGTGGAAACGC-3′; GAPDH (forward primer)
5′-CAACAGCCTCAAGATCATCAGC-3′, GAPDH (reverse primer)
5′-TTCTAGACGGCAGGTCAGGTC-3′. The relative gene expression levels
were calculated using the 2−ΔΔCT analysis method
(14).
Cell culture
The human GBC cell lines NOZ, EH-GB1, GBC-SD,
SGC-996 and OCUG were obtained from the Shanghai Cell Institute
National Cell Bank. NOZ and SGC-996 were cultured in Williams'
medium E and RPMI-1640 medium (HyClone, Logan, UT, USA),
respectively. The EH-GB1, GBC-SD and OCUG cells were cultured in
DMEM (Gibco, Grand Island, NY, USA). Cells were supplemented with
15% fetal bovine serum (FBS; Gibco) at 37°C in a 5% CO2
incubator.
Immunofluorescence analysis
Cells were fixed in 4% paraformaldehyde for 15 min
at room temperature and then permeabilized in 0.5% Triton for 5
min. 3% BSA was used for blocking. The cells were incubated with
anti-BRD4 antibody at 4°C for 12 h. Then, the cells were incubated
with goat anti-rabbit IgG (Beyotime, Shanghai, China) for 30 min,
and counterstained with Hoechst 33342 (Beyotime). A fluorescence
microscope (Leica DMI3000B; Leica, Wetzlar, Germany) was used for
observation.
siRNA, plasmid and Lv-shRNA
transfection
BRD4 siRNAs were designed and synthesized by Biotend
(Shanghai, China). The siRNAs targeting human BRD4 were:
BRD4-siRNA1: 5′-CCUGAUUACUAUAAGAUCAdTdT-3′, BRD4-siRNA2:
5′-CAGUGACAGUUCGACUGAUdTdT-3′. Cells were plated onto 6-well plates
at 30% density and transfected with BRD4-siRNA or NC-siRNA using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's protocol.
The pcDNA4c hBrd4 was obtained from Addgene (Plasmid
14441), and the amino acid change T249P was modified by Longqian
Biotech. Viafect Transfection Reagent was used for transfection.
BRD4 expression level was detected by qRT-PCR and western
blotting.
pGMLV-SC5 RNAi vectors encoding BRD4
(5′-CCTGATTACTATAAGATCA-3′) were constructed by Genomeditech
(Shanghai, China). NOZ cells were infected at a multiplicity of
infection (MOI) of 80 in complete culture medium for 24 h. qRT-PCR
and western blotting were used to examine the knockdown
efficiency.
In vitro cell proliferation assays
The CCK-8 (Cell Counting Kit-8; Dojindo, Kumamoto,
Japan) assay was performed to evaluate cell proliferation. NOZ and
EH-GB1 cells were plated at a density of 1×103 cells per
well in 96-well plates. The absorbance value (OD) was measured at
450 nm using a Spectra Max 190 (Molecular Devices, Sunnyvale, CA,
USA).
In the colony formation assay, cells were planted in
6-well plates with 600 cells per well and were cultured for 9 and
14 days, respectively. Then, the colonies were fixed in 4%
paraformaldehyde and stained with 0.5% crystal violet (Beyotime).
The number of colonies was photographed and counted under a
microscope.
Migration and invasion assays
Cell migration and invasion were performed using
24-well Transwell chambers (Corning Inc., Corning, NY, USA).
Briefly, the upper chamber was seeded with 2×104 cells
in 200 μl serum-free medium, while the lower chamber was
filled with 500 μl basal medium containing 15% FBS. After 20
h of incubation, cells were fixed with 4% paraformaldehyde for 20
min and stained with crystal violet for 20 min. Then cells on the
upper surface of the upper chambers were scraped. Five fields were
chosen randomly for analysis.
Apoptosis assay
Apoptosis was inspected using an Annexin V-FITC
Apoptosis Detection kit (BD Pharmingen, San Diego, CA, USA). Cells
were harvested and washed three times with PBS. Next, 100 μl
of Annexin V binding buffer (1X), 5 μl of PI (50
μg/ml) and 5 μl of FITC (BD Pharmingen) were added to
the cell suspensions and incubated for 15 min at room temperature
in the dark. Apoptosis array was analyzed using FACS Canto II (BD
Pharmingen).
Western blotting
Total protein was extracted using RIPA buffer (Cell
Signaling, Danvers, MA, USA). Cell proteins were separated by
SDS-PAGE and then transferred onto PVDF membranes (Millipore Corp.,
Billerica, MA, USA). Blots were blocked with 5% skim milk at room
temperature for 2 h. The PVDF membranes were reacted with a series
of primary antibodies at 4°C overnight. The anti-BRD4 antibody was
acquired from Abcam (Cambridge, MA, USA). Primary antibodies
against E-cadherin, vimentin, N-cadherin, Bax, Bad, Bcl-2, PI3K,
p-PI3K, AKT, p-AKT, JNK, p-JNK, GAPDH were obtained from Cell
Signaling Technology. Then, the blots were reacted with the
appropriate HRP-conjugated secondary antibody and quantified using
a Gel Doc 2000 (Bio-Rad, Hercules, CA, USA).
Inhibitor reagent
PI3K/AKT pathway inhibitor GDC-0941 was purchased
from MCE. Cells were treated with different GDC-0941 concentrations
of 0, 1, 3, 10 μM. Total proteins were isolated for western
blotting after 72 h incubation.
Nude mouse subcutaneous xenograft
model
Male nude mice (four-week-old) were purchased from
the Shanghai Laboratory Animal Center of the Chinese Academy of
Sciences (Shanghai, China). All procedures were approved by the
Ethics Committee of Xinhua Hospital Affiliated to Shanghai Jiaotong
University School of Medicine. NOZ cells (Lv-shNC/Lv-shBRD4 group,
2×106) were subcutaneously injected into the left axilla
of nude mice (n=5 mice/group). The tumour volumes were measured
(1/2 × width2 × length) weekly. The nude mice were
sacrificed after 4 weeks and tumors were weighed. Tumor tissues
were stored for western blotting and qRT-PCR analysis.
Statistical analysis
Statistical analyses were performed with SPSS
software (version 22.0). The Student's t-test and nonparametric
tests were used when necessary. The Kaplan-Meier test was used for
the overall survival analysis. Subsequently, the Cox proportional
hazards model was used for independent prognostic factors analysis.
All assays were independently performed 3 times. P<0.05 was
considered to indicate a statistically significant difference.
Results
High expression of BRD4 correlates to
poor prognosis of GBC patients
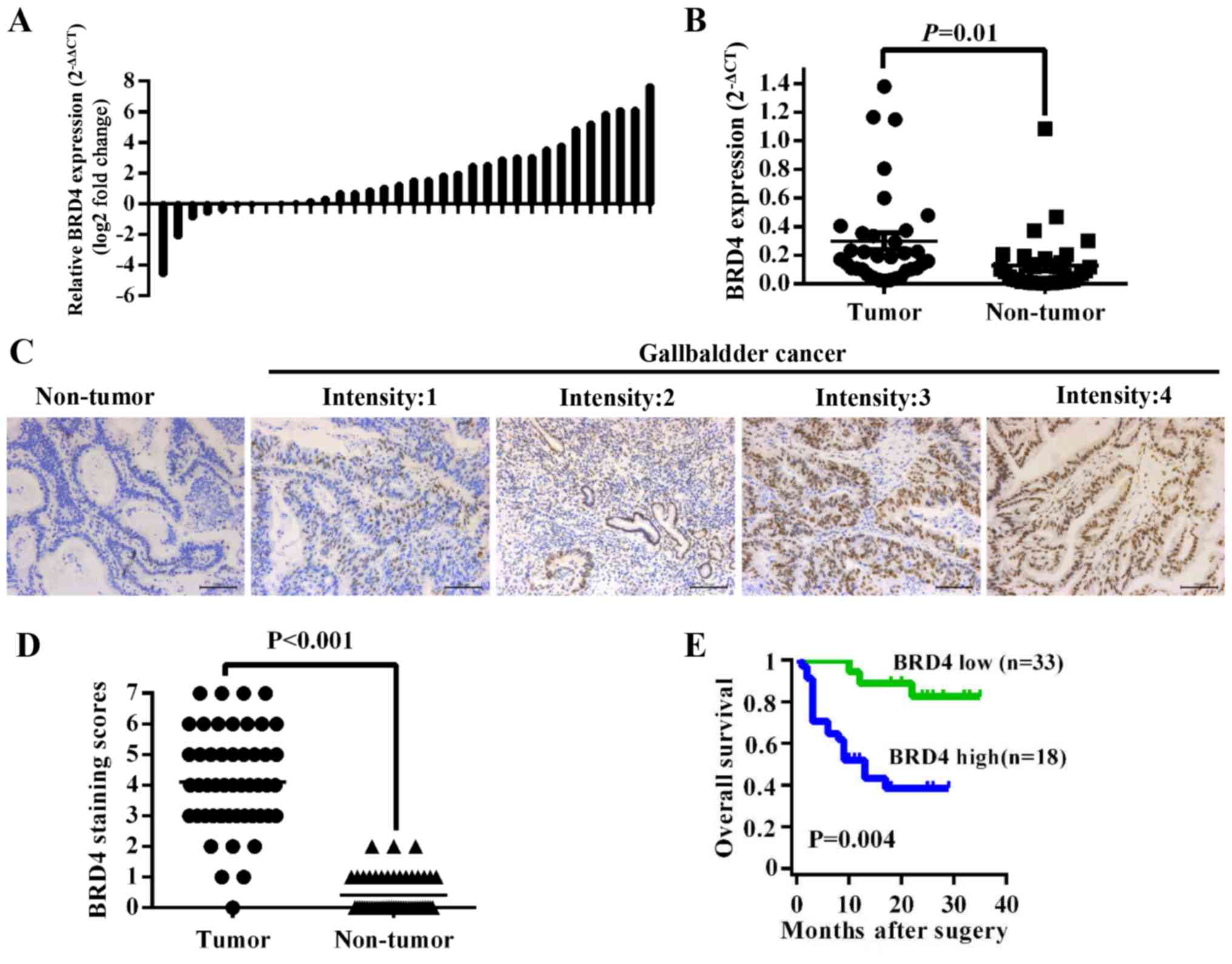
To investigate the expression level of BRD4 in GBC
tissues, we first compared BRD4 mRNA expression in 34 pairs of GBC
tissues. The results showed that BRD4 expression was higher in
tumor tissues than in adjacent non-tumor tissues (P=0.01) (Fig. 1A and B). Then we evaluated BRD4
protein levels in 51 archived paraffin-embedded specimens of GBC
tissues using immunohistochemistry (Fig. 1C). As shown in Fig. 1D, the BRD4 expression level was
higher in tumor tissues than in their corresponding adjacent normal
tissues (P<0.001). We further analyzed the correlation between
BRD4 levels and clinicopathological data of GBC patients. The BRD4
levels were significantly related to pathologic T stage
(P<0.001), lymph node metastasis (P=0.025) and TNM stage
(P=0.002) (Table I). The mean
overall survival time of GBC patients with medium or high [4–7]
BRD4 expression was 10.00±6.91 months, significantly lower than
patients with negative or low [0–3] BRD4 expression (24.67±6.72
months, P<0.001) (Fig. 1E).
Multivariate Cox regression analysis indicated that BRD4 expression
level was an independent prognostic factor in GBC patients
(P=0.028, Table II).
 | Table IRelationship between BRD4 expression
and relevant factors in GBC patients. |
Table I
Relationship between BRD4 expression
and relevant factors in GBC patients.
| Factors | Cases | BRD4 expression
| P-value |
|---|
| Low | High |
|---|
| Sex | | | | 0.727 |
| Male | 10 | 4 | 6 | |
| Female | 41 | 14 | 27 | |
| Age (years) | | | | 0.028 |
| <60 | 10 | 7 | 3 | |
| ≥60 | 41 | 11 | 30 | |
| Histology | | | | 0.574 |
| Well/moderate | 45 | 17 | 28 | |
| Poor | 6 | 1 | 5 | |
| T | | | | <0.001 |
| Tis+T1+T2 | 20 | 14 | 6 | |
| T3+T4 | 31 | 4 | 27 | |
| N | | | | 0.025 |
| Absent | 26 | 13 | 13 | |
| Present | 25 | 5 | 20 | |
| M | | | | 0.141 |
| Negative | 45 | 18 | 27 | |
| Positive | 6 | 0 | 6 | |
| Tumor stage | | | | 0.002 |
| 0–I | 6 | 6 | 0 | |
| II–IV | 45 | 12 | 33 | |
| Table IIUnivariate and multivariate analysis
for overall survival in GBC patients. |
Table II
Univariate and multivariate analysis
for overall survival in GBC patients.
| Variables | Univariate analysis
| Multivariate
analysis
|
|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Sex
(male/female) | 1.006
(0.341–2.967) | 0.991 | | |
| Age
(<60/≥60) | 1.062
(0.392–2.879) | 0.906 | | |
| Histology (well +
moderate/poor) | 0.801
(0.187–3.427) | 0.765 | | |
| T (Tis + T1 + T2/T3
+ T4) | 2.833
(1.088–7.379) | 0.033 | 1.217
(0.396–3.743) | 0.732 |
| N
(absent/present) | 2.286
(0.968–5.402) | 0.059 | | |
| M
(negative/positive) | 4.012
(1.410–11.411) | 0.009 | 2.413
(0.823–7.078) | 0.109 |
| Tumor stage
(0–I/II–IV) | 4.353
(0.584–32.453) | 0.151 | | |
| BRD4 expression
(low/high) | 5.474
(1.740–17.215) | 0.004 | 4.336
(1.173–16.027) | 0.028 |
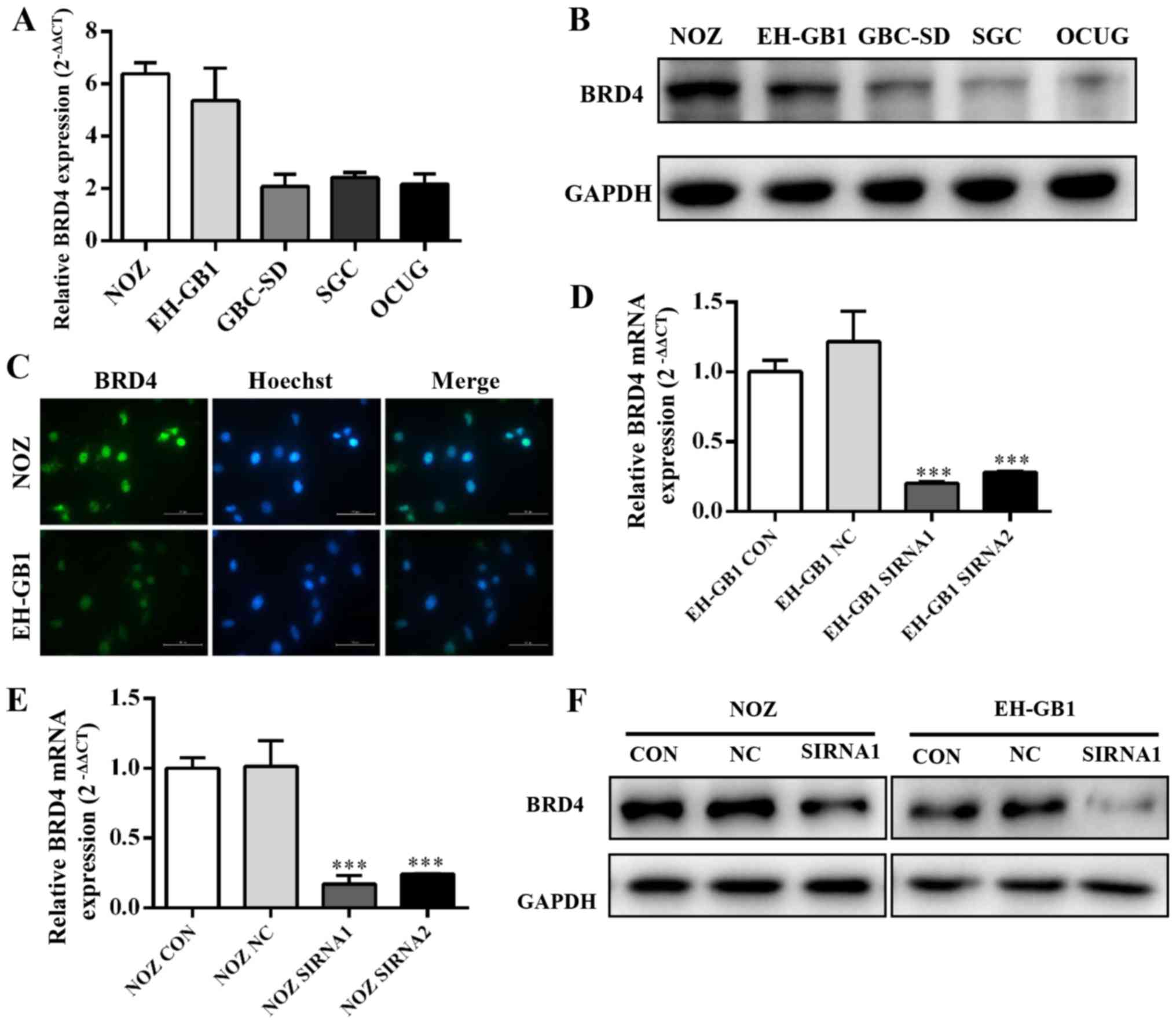
BRD4 expression in GBC cell lines
Five established gallbladder cancer cell lines (NOZ,
EH-GB1, GBC-SD, SGC-996, OCUG) were analyzed for expression of BRD4
by qRT-PCR and western blotting. Fig.
2A and B showed that BRD4 expression was significantly
increased in GBC cell lines, particularly in the NOZ and EH-GB1
lines. Immunocytochemical staining confirmed that BRD4 protein
mainly expressed on the cell nucleus of GBC cells (Fig. 2C).
Then we transfected gallbladder cancer cell lines
with siRNAs against BRD4. We evaluated the efficiency of siRNA
transfection by qRT-PCR and western blotting. The results showed
that BRD4 mRNA and protein expression were clearly suppressed after
transfection with BRD4-siRNA (Fig. 2D
and E).
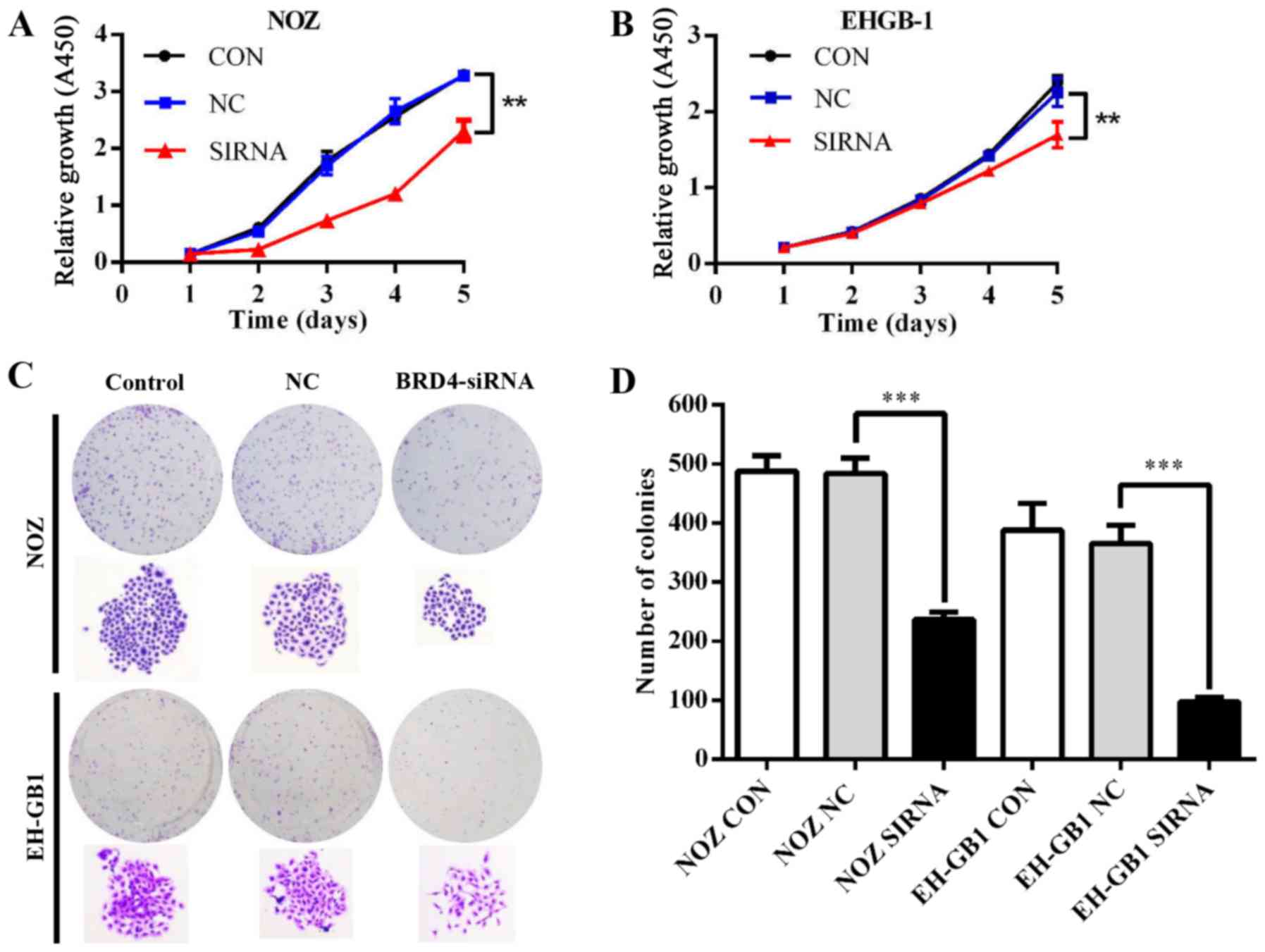
Effect of BRD4 on proliferation in GBC
cells
To explore the role of BRD4 in GBC cell
proliferation, we conducted CCK-8 and colony formation assays.
Fig. 3A and B showed that the
proliferative ability was inhibited in NOZ and EH-GB1 cells when
silencing BRD4 (P<0.01). Meanwhile, downregulation of BRD4
reduced colony formation capacity of GBC cells (P<0.001)
(Fig. 3C and D).
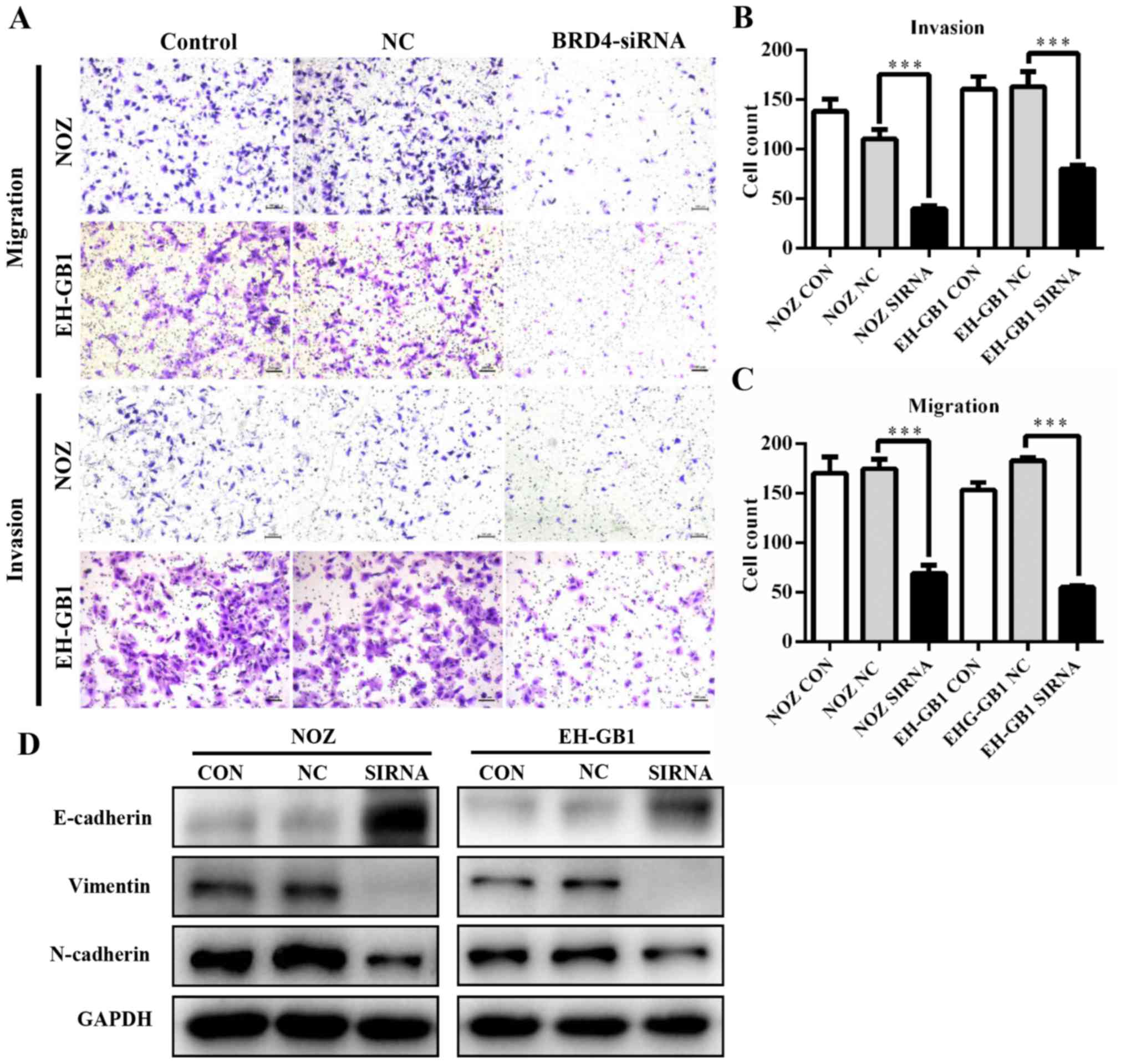
Knockdown of BRD4 inhibits GBC cell
metastasis
To determine whether BRD4 deregulation affects GBC
metastasis, we performed transwell assays. As shown in Fig. 4A–C, knockdown of BRD4 significantly
decreased the migration and invasion abilities of GBC cells. This
suggested that BRD4 could enhance the metastatic capacity of GBC.
Then we examined proteins relative to epithelial-mesenchymal
transition (EMT). Results revealed that when BRD4 was depleted, the
expression of epithelial marker E-cadherin increased, while the
expression of Vimentin and N-cadherin (mesenchymal markers)
decreased (Fig. 4D).
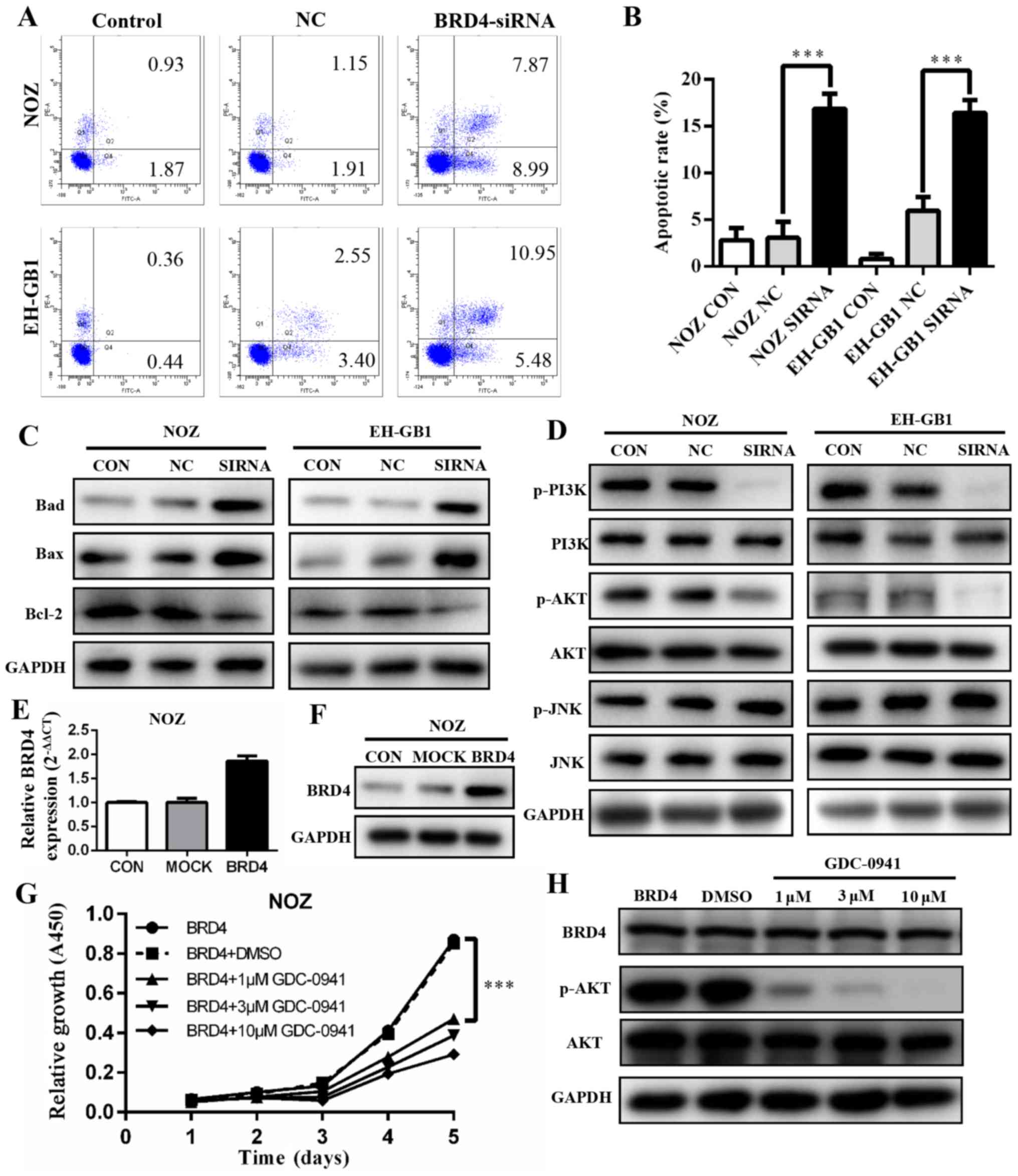
BRD4 silencing induces apoptosis in GBC
cell lines via PI3K/AKT pathway
To explore if and how BRD4 affects apoptosis, we
conducted flow cytometry analysis. The results showed that
apoptotic and dead cells increased significantly in the cells with
BRD4 siRNA transfection (P<0.001). The apoptosis rates of BRD4
siRNA groups were 16.86 and 16.43% in NOZ cells and EH-GB1 cells,
respectively (Fig. 5A and B). This
indicated that the inhibition of BRD4 affected GBC cell apoptosis.
Next, we detected the protein levels of several Bcl-2 family
proteins. As shown in Fig. 5C,
upregulation of Bax and Bad protein and downregulation of Bcl-2
were induced when transfected with BRD4 siRNA in GBC cells.
The PI3K/AKT and JNK/SAPK signaling pathways have
been demonstrated to play essential roles in Bad regulation
(15,16). We found that the expression levels
of p-PI3K and p-AKT decreased significantly when BRD4 was silenced.
However, JNK and p-JNK protein expression levels were not changed
compared with controls (Fig.
5D).
To verify our findings, we transfected BRD4
full-length plasmid into NOZ cell lines, the expression level of
BRD4 was markedly upregulated (Fig. 5E
and F). Then cells were treated with a PI3K/AKT inhibitor
(GDC-0941). The cell proliferation was inhibited (Fig. 5G) while BRD4 expression remained
unchanged (Fig. 5H).
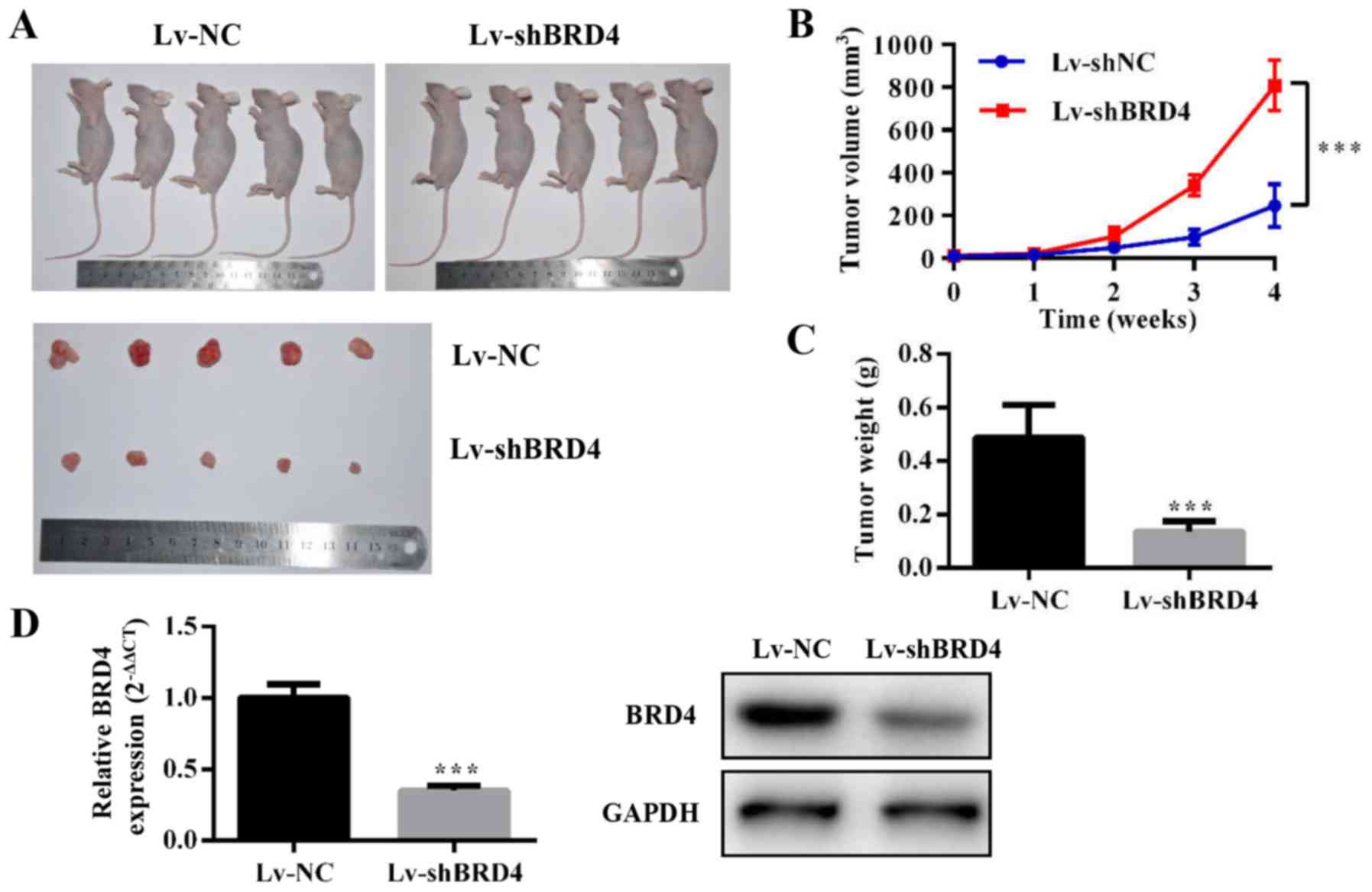
Knockdown of BRD4 inhibits tumor growth
in vivo
To evaluate the effect of BRD4 on GBC growth in
vivo, we constructed NOZ cell lines stably expressing
shRNA-BRD4 or the negative control. Then, we injected
subcutaneously Lv-shBRD4 and negative control NOZ cells into nude
mice (Fig. 6A). The tumor volume
and weight were significantly inhibited in the Lv-shBRD4 group
compared to the negative control group (Fig. 6B and C). The mRNA and protein level
of BRD4 decreased in the BRD4-silenced group compared with negative
control group (Fig. 6D). These
results confirmed that BRD4 is essential for GBC growth
regulation.
Discussion
BRD4 has been proved to possess a significant effect
on cell biological activity as an important BET family member. In
recent years, the effect of BRD4 has been observed in various types
of cancer tissues and cell lines (6,17–20).
In this study, for the first time, we evaluated BRD4 expression in
GBC tissues. We found that BRD4 was significantly up regulated in
GBC tissues compared to their adjacent non-cancerous tissues.
Moreover, high BRD4 expression level was correlated with poor
prognosis in GBC patients. This indicates that high BRD4 protein
expression may be a crucial prognostic indicator for GBC. We also
demonstrated the significant suppression of GBC by downregulating
the expression of BRD4 both in vitro and in vivo.
EMT has been implicated as an integral part of
metastasis, and clarifying the underlying molecular mechanism of
the latter is crucial for clinical therapies (21). Twist is a key member of
EMT-activating transcriptional factors. Twist expression represses
E-cadherin-mediated cell-cell adhesive processes and activates
mesenchymal markers (22). The
available evidence shows that di-acetylated Twist binds the second
bromodomain of BRD4 in basal-like breast cancer (23). Another study shows that the
administration of BRD4 inhibitor readily abolished TGF-induced EMT
in small cell lung cancer (21).
In this study, knockdown of BRD4 in GBC cells enhanced protein
expression of the epithelial marker E-cadherin and reduced the
protein expression of mesenchymal markers N-cadherin and vimentin.
Our data suggest that BRD4 downregulation inhibits migration and
invasion of GBC cells by inducing EMT.
The initiation of apoptosis results in cell death.
Bcl-2 family which includes pro- and anti-apoptotic molecules act
as regulatory protein (24,25).
All the BH3-only proteins, which include Bcl-2-associated death
promoter (Bad), can activate Bcl-2-associated X protein (Bax)
indirectly by binding to and inhibiting the BCL-2 (26). Our data further supported that
cancer cell death occurred due to decreased expression of Bcl-2 and
increased expression of Bad and Bax in BRD4 depleted GBC cells. The
previous study indicates that PI3K/AKT is involved in Bad
inactivation, whereas the JNK pathway is associated with Bad
activation (27). In our study,
depletion of BRD4 inhibited the expression of phosphorylation AKT,
but we did not find expression change for JNK. Moreover, BRD4
expression level remained unchanged after using PI3K/AKT inhibitor,
which confirmed that downregulation of BRD4 in GBC cells induced
apoptosis via PI3K/AKT pathway.
In summary, we demonstrated that BRD4 deregulation
is involved in GBC progression. Knockdown of BRD4 could effectively
affect cell proliferation, metastasis and apoptosis in GBC cell
lines and inhibit tumor growth in vivo. We hypothesize that
BRD4 plays an essential role in GBC apoptosis by downregulating
phosphorylation levels of AKT via the PI3K/AKT pathway. Inhibition
of BRD4 expression may be a novel therapeutic strategy for GBC.
Therefore, BRD4 may be a novel promising prognostic marker and an
antitumor target in the treatment of GBC.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 31501127), Shanghai Sailing
Program (no. 16YF1407100), Project Funded by China Postdoctoral
Science Foundation (no. 2015M571577), and Nanjing Medical
University Science and Technology Development Fund (no.
2016NJMU132).
References
|
1
|
Garg PK, Pandey D and Sharma J: The
surgical management of gallbladder cancer. Expert Rev Gastroenterol
Hepatol. 9:155–166. 2015. View Article : Google Scholar
|
|
2
|
Müller BG, De Aretxabala X and González
Domingo M: A review of recent data in the treatment of gallbladder
cancer: what we know, what we do, and what should be done. Am Soc
Clin Oncol Educ Book. e165–e170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu W, Hu Y, Ma Q, Zhou L, Jiang L, Li Z,
Zhao S, Xu Y, Shi W, Li S, et al: miR-223 increases gallbladder
cancer cell sensitivity to docetaxel by downregulating STMN1.
Oncotarget. 7:62364–62376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dhalluin C, Carlson JE, Zeng L, He C,
Aggarwal AK and Zhou MM: Structure and ligand of a histone
acetyltransferase bromodomain. Nature. 399:491–496. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu Y, Zhou J, Ye F, Xiong H, Peng L, Zheng
Z, Xu F, Cui M, Wei C, Wang X, et al: BRD4 inhibitor inhibits
colorectal cancer growth and metastasis. Int J Mol Sci.
16:1928–1948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andrieu G, Tran AH, Strissel KJ and Denis
GV: BRD4 regulates breast cancer dissemination through
Jagged1/Notch1 signaling. Cancer Res. 76:6555–6567. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dey A, Chitsaz F, Abbasi A, Misteli T and
Ozato K: The double bromodomain protein Brd4 binds to acetylated
chromatin during interphase and mitosis. Proc Natl Acad Sci USA.
100:8758–8763. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shahbazi J, Liu PY, Atmadibrata B, Bradner
JE, Marshall GM, Lock RB and Liu T: The bromodomain inhibitor JQ1
and the histone deacetylase inhibitor panobinostat synergistically
reduce N-Myc expression and induce anticancer effects. Clin Cancer
Res. 22:2534–2544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu SY, Lee AY, Lai HT, Zhang H and Chiang
CM: Phospho switch triggers Brd4 chromatin binding and activator
recruitment for gene-specific targeting. Mol Cell. 49:843–857.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marcotte R, Sayad A, Brown KR,
Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE,
Bader GD, Mills GB, et al: Functional genomic landscape of human
breast cancer drivers, vulnerabilities, and resistance. Cell.
164:293–309. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sahai V, Kumar K, Knab LM, Chow CR, Raza
SS, Bentrem DJ, Ebine K and Munshi HG: BET bromodomain inhibitors
block growth of pancreatic cancer cells in three-dimensional
collagen. Mol Cancer Ther. 13:1907–1917. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao YF, Wu YB, Long X, Zhu SQ, Jin C, Xu
JJ and Ding JY: High level of BRD4 promotes non-small cell lung
cancer progression. Oncotarget. 7:9491–9500. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meseure D, Vacher S, Alsibai KD, Nicolas
A, Chemlali W, Caly M, Lidereau R, Pasmant E, Callens C and Bieche
I: Expression of ANRIL-polycomb complexes-CDKN2A/B/ARF genes in
breast tumors: Identification of a two-gene (EZH2/CBX7) signature
with independent prognostic value. Mol Cancer Res. 14:623–633.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Z, Chen Y, Wang X, Zhang H, Zhang Y,
Gao Y, Weng M, Wang L, Liang H, Li M, et al: LASP-1 induces
proliferation, metastasis and cell cycle arrest at the G2/M phase
in gallbladder cancer by down-regulating S100P via the PI3K/AKT
pathway. Cancer Lett. 372:239–250. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bhakar AL, Howell JL, Paul CE, Salehi AH,
Becker EB, Said F, Bonni A and Barker PA: Apoptosis induced by
p75NTR over-expression requires Jun kinase-dependent
phosphorylation of Bad. J Neurosci. 23:11373–11381. 2003.PubMed/NCBI
|
|
16
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Segura MF, Fontanals-Cirera B,
Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, González-Gomez
P, Morante M, Jubierre L, Zhang W, et al: BRD4 sustains melanoma
proliferation and represents a new target for epigenetic therapy.
Cancer Res. 73:6264–6276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shimamura T, Chen Z, Soucheray M,
Carretero J, Kikuchi E, Tchaicha JH, Gao Y, Cheng KA, Cohoon TJ, Qi
J, et al: Efficacy of BET bromodomain inhibition in Kras-mutant
non-small cell lung cancer. Clin Cancer Res. 19:6183–6192. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tögel L, Nightingale R, Chueh AC,
Jayachandran A, Tran H, Phesse T, Wu R, Sieber OM, Arango D,
Dhillon AS, et al: Dual targeting of bromodomain and extraterminal
domain proteins, and WNT or MAPK signaling, inhibits c-MYC
expression and proliferation of colorectal cancer cells. Mol Cancer
Ther. 15:1217–1226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang
M, Wang L, Luo G, Li Y, Zeng F, et al: BRD4 regulates EZH2
transcription through upregulation of C-MYC and represents a novel
therapeutic target in bladder cancer. Mol Cancer Ther.
15:1029–1042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang H, Liu Y, Xue M, Liu H, Du S, Zhang
L and Wang P: Synergistic action of master transcription factors
controls epithelial-to-mesenchymal transition. Nucleic Acids Res.
44:2514–2527. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang
Q, Lin Y, Li J, Kang T, Tao M, et al: Disrupting the interaction of
BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like
breast cancer. Cancer Cell. 25:210–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang H, Li Z, Chu B, Zhang F, Zhang Y, Ke
F, Chen Y, Xu Y, Liu S, Zhao S, et al: Upregulated LASP-1
correlates with a malignant phenotype and its potential therapeutic
role in human cholangiocarcinoma. Tumour Biol. 37:8305–8315. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Delbridge AR and Strasser A: The BCL-2
protein family, BH3-mimetics and cancer therapy. Cell Death Differ.
22:1071–1080. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kamada H, Nito C, Endo H and Chan PH: Bad
as a converging signaling molecule between survival PI3-K/Akt and
death JNK in neurons after transient focal cerebral ischemia in
rats. J Cereb Blood Flow Metab. 27:521–533. 2007. View Article : Google Scholar
|