Introduction
MicroRNAs (miRNAs) are small non-coding RNAs
implicated in post-transcriptional suppression of target genes in a
sequence-specific manner by suppressing mRNA translation and, less
frequently, causing degradation of the target mRNA (1). They have been implicated in the
regulation of as many as 30% of the human cell mRNAs, with
fundamental roles in cell proliferation, differentiation and
apoptosis. A single miRNA molecule has the capacity to target a
wide range of different mRNAs, some of which may have opposing
oncogenic or tumor-suppressive functions, depending on the context
(2). Furthermore, the growth and
spread of a cancer is both the combinations of a function of the
cancer cells themselves and various extrinsic factors, which
interact with the cancer cells, affecting their behavior. miRNAs
influence largely on each of the extrinsic factors such as the
immune system, tumor stromal cells, therapy and oncoviruses
(2).
Several studies implicate miR-146a as pleiotropic
regulator of carcinogenesis, as polymorphism and altered expression
have been linked with cancer risk and invasive and metastatic
capacity in diverse cancers (3);
however, its roles in carcinogenesis are not fully understood.
miR-146a-5p has been reported to be overexpressed in squamous cell
carcinoma (SCC) of the head and neck, cervix and lung (4–6), and
is implicated in oncogenicity of oral squamous cell carcinoma
(OSCC) (7). In vitro
studies indicate that transfection of miR-l46a precursor results in
cell proliferation (5);
additionally, transient transfection of a miRNA-146a mimic into
human bronchial epithelial cells can protect the cells from
apoptosis in response to the combination of inflammatory cytokines
and transforming growth factor-β1 (TGF-β1). However, it is
suppressive in many other malignancies, including breast, gastric,
prostatic and pancreatic carcinomas (8–11).
In fact, miR-146a seems to play multiple roles in the regulation of
different phenotypes by targeting a wide range of different genes
in various cellular contexts, leading to the controversy to its
contribution in cancer. Emerging evidence suggests that there is a
direct link between miRNAs and cancer (12), as well as, inflammation (13,14).
Chronic inflammation and the activation of nuclear factor kappa B
(NF-κB) are known to be associated with development of multiple
cancers (15), while inflammatory
cytokines are potent stimulants of miR-146a through NF-κB pathway
(16,17). Specifically, several studies report
that various miRNAs, including hsa-miR-20a, hsa-miR-142-3p,
hsa-miR-19a, hsa-let-7f, hsa-miR-203, hsa-miR-17, hsa-miR-223,
hsa-miR-146b and hsa-miR-146a, are highly expressed in patients
with periodontitis (18–20). Furthermore, through regulation of
interleukin-6 (IL-6)/signal transducer and activator of
transcription 3 (STAT3) signaling, miR-146a may link the chronic
inflammation and cancer (21).
Notably, miR-146a-5p has been shown to target Smad4
(22) and tumor necrosis factor
receptor-associated factor 6 (TRAF6) (23), which both serve as significant
mediators in the TGF-β pathway. TGF-β regulates a wide array of
cellular processes such as cell growth, differentiation, apoptosis,
migration and extracellular matrix production (24). It is a potent antitumor cytokine
because of its property as a strong inhibitor for the growth of
epithelial cells. TGF-β utilizes multiple different signaling
pathways in addition to the canonical Smad pathway (25). These non-canonical pathways include
mitogen-activated protein kinase (MAPK) pathways, downstream of
TRAF6. TGF-β receptors interact with TRAF6 and induce the formation
of K63-linked poly-ubiquitin chains on TRAF6, recruiting
TGF-β-activated kinase 1 (TAK1) to activate Jun N-terminal kinase
(JNK) and p38 MAPK (p38) (26,27).
Signal integration between JNK and p38 determines cell
type-specific and context-dependent proliferation, survival and
differentiation of human tumors and cancer cell lines (28).
TRAF6 plays important roles in the transcription and
expression of numerous inflammation- and apoptosis-related genes
(29). TRAF6 is known to promote
apoptosis by activation of caspase-8 (30), similarly to the action of Fas
associated death domain (FADD) on stimulation by Fas (31). Compellingly, FADD is also a target
gene suppressed by miR-146a-5p and the suppression is related to
resistance against activation-induced cell death (32). The third domain of FADD contains
the phosphorylation site at Ser194 residue partly contributing to
the activities of FADD in humans (33), and phosphorylated FADD is
translocated into the nucleus to perform apoptosis unrelated
functions (34).
In the present study, we demonstrated that
miR-146a-5p, which is frequently upregulated in OSCC cell lines and
in blood and saliva of OSCC patients compared with normal
counterparts, showed pleiotropic effects on proliferation and
apoptosis of OSCC cell lines. This was partially based on the
contextual responses of JNK, downstream of TRAF6 that was
suppressed by miR-146a-5p in SCC cell lines. Our data suggest that
miR-146a-5p affects cellular processes in a cellular
context-dependent manner, and potentiates proliferation of OSCC
cell lines.
Materials and methods
Cultures of primary human cells and OSCC
cell lines
Primary normal human oral keratinocytes (NHOKs) were
prepared and maintained as previously reported (35). Briefly, NHOKs were isolated from
human gingival tissue specimens obtained from healthy volunteers
(age range, 20–30 years) who were undergoing oral surgery. NHOKs
were cultured in keratinocyte growth medium containing 0.15 mM
calcium and a supplementary growth factor bullet kit (KGM;
Clonetics, San Diego, CA, USA). Keratinocytes in their second
passage were used in the described experiments. Primary normal
human dermal fibroblasts (NHDFs) were prepared and maintained as
previously described (36). The
HOK-16B line, human oral keratinocytes immortalized by transfecting
cells with the cloned HPV-16 genome (37), was also cultured in KGM. The cancer
cell lines SCC-4, SCC-9, FaDu and SiHa were purchased from the
American Type Culture Collection (ATCC; Rockville, MD, USA). SCC-4
and SCC-9 cell lines were cultured in Dulbecco's modified minimum
essential medium (DMEM)/Ham's F12 (Gibco-BRL, Bethesda, MD, USA)
supplemented with 10% fetal bovine serum (FBS) and 0.4 µg/ml
hydrocortisone. Among three oral cancer cell lines, CTHOK-16B-BaP
and CTHOK-16B-DMBA, which were tumorigenically transformed by
chronic exposure of the HOK-16B cells to benzo(a)pyrene and
7,12-dimethylbenz(a) anthracene, respectively (38), and Spt-HOK80, which was
spontaneously transformed from primary human oral kera-tinocytes
(39) were cultured in DMEM
supplemented with 10% FBS and 0.4 µg/ml hydrocortisone. Cell
lines such as CTHOK-16B-DMBA and Spt-HOK80 were established by our
group in 1995 and 2010, respectively (38,39).
HOK-16B-BaP cell line, the original name of which is CTHOK-16B-BaP,
was first reported by our group in 1995 (38). Other oral cancer cell lines
KOSCC-11, KOSCC-25A, KOSCC-25B, KOSCC-25C, KOSCC-25D, KOSCC-25E,
KOSCC-33A and KOSCC-33B (40) were
also cultured in DMEM supplemented with 10% FBS and 0.4
µg/ml hydrocortisone. The SiHa line was grown in DMEM
supplemented with 10% FBS, while the FaDu line was grown in DMEM
supplemented with 10% FBS and 0.4 µg/ml hydrocortisone.
Blood, saliva and OSCC tissue
samples
The whole blood, whole saliva and OSCC tissue
samples used in this study were obtained from OSCC patients who
underwent surgical resection at the Department of Oral and
Maxillofacial Surgery, Seoul National University Dental Hospital,
and healthy controls who did not have signs or symptoms of
inflammation of the gingival tissues and systemic diseases. In the
present study, the oral hygiene of oral cancer patients was not
taken into consideration. This was because OSCC patients with poor
oral hygiene followed by chronic inflammation, including
periodontitis, have high incidence of oral cancer clinically,
making it difficult to exclude patients with inflammation. All
procedures for obtaining human gingival tissue specimens, whole
blood, whole saliva and cancer specimens were reviewed and approved
by the Institutional Review Board on Human Subjects Research and
the Ethics Committee at Seoul National University Dental
Hospital.
RNA isolation in the whole blood, saliva
and cells
Four hundred microliters of the whole saliva were
used for RNA isolation. Saliva samples were extracted using the
mirVana™ miRNA Isolation kit (Ambion, Austin, TX, USA) according to
the manufacturer's instructions. For the initial lysis step, we
used 1 ml of lysis/binding solution per 400 µl saliva
sample. Total RNAs including small RNAs were isolated in whole
blood and cells by miRNeasy Mini kit (Qiagen, Valencia, CA, USA)
according to the manufacturer's instructions.
miRNA microarray
Cyanine 3-labeled complementary RNA was prepared
from total RNA including miRNA (500 ng) using Agilent's Low RNA
Input Linear Amplification kit (Agilent Technologies, Santa Clara,
CA, USA). Labeled cRNA was applied to microarray (Human miRNA
Microarray Release 19.0, 8×60K; Agilent Technologies) using
Agilent's Gene Expression Hybridization kit. Hybridized microarray
was washed using Agilent's Gene Expression Wash Buffer kit. The
microarray chip was scanned using Agilent's DNA micro-array
scanner, and the raw signal density was acquired from Feature
Extraction software. The differential gene expression was
classified by Benjamini-Hochberg's false discovery ratio method
using GeneSpring GX12 software.
TaqMan miRNA assay
Complementary DNAs (cDNAs) were synthesized from
total RNA including miRNA using TaqMan® MicroRNA reverse
transcription kit with gene-specific primers (Applied Biosystems,
Foster City, CA, USA). Reverse transcription reactions (for final
quantity or concentrations) contained 10 ng RNA samples, 1 mM of
dNTPs, 1X RT primer, 1X RT buffer, 3.8 U of RNase inhibitor and 50
U of reverse transcriptase. The 15 µl reactions were
incubated for 30 min at 16°C, 30 min at 42°C, 5 min at 85°C and
then held at 4°C. Quantitative real-time PCR (RT-qPCR)
quantification of miRNA expression was carried out using
TaqMan® MicroRNA assays kit (Applied Biosystems)
according to the manufacturer's instructions. The primers used
were: miR-146a-5p (ID, 000468) and RNU44 (ID, 001094) (Applied
Biosystems). The 20 µl PCR included 1.33 µl RT
product, 1X TaqMan® Universal Master Mix II without UNG,
and 1X TaqMan-primers mix (Applied Biosystems). Reactions were
incubated in a 96-well plate at 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec and 60°C for 1 min. The threshold cycle
(Ct) was determined using default threshold settings. All
experiments were done in quadruples. The RNU44 was used as a
control to normalize miRNA input in the RT-qPCR assay.
Determination of HPV infection
The presence of 'high-risk' HPV (type 16, 18 and 33)
DNA was determined by reverse transcription polymerase chain
reaction (RT-PCR). Total RNA was reverse transcribed to cDNA, which
was used for PCR amplification of HPV E5 and E6 (38
cycles) and GAPDH (28 cycles; denaturation at 95°C for 20
sec, annealing at 60°C for 10 sec and extension at 70°C for 10
sec). The primers (Table I)
amplified the E5 and E6 genes of the three types of
HPV. The PCR products were resolved on a 1.5% agarose gel, stained
with ethidium bromide and visualized using a UV illuminator.
 | Table IPrimers for E5 and E6 genes of the
three types of HPV used to assess HPV infection in RT-PCR
amplification. |
Table I
Primers for E5 and E6 genes of the
three types of HPV used to assess HPV infection in RT-PCR
amplification.
| Gene name (NCBI
ID) | Forward primer | Reverse primer | Product size
(bp) |
|---|
| HPV-16 E5
(AF120713.1) |
5′-TTGATACTGCATCCACAACATTACTG-3′ |
5′-TGCGTATGTAGACACAGACAAAAGC-3′ | 110 |
| HPV-18 E5
(NC_001357.1) |
5′-TTGTGTATGCATGTATGTGTGC-3′ |
5′-GCAGGGGACGTTATTACCAC-3′ | 111 |
| HPV-33 E5
(M12732.1) |
5′-CGTCCTTTAATACTTTCCATTTCTACCT-3′ |
5′-CACCCAAAGCAGCAATACCA-3′ | 66 |
| HPV-16 E6
(AF003018.1) |
5′-TGCACAGAGCTGCAAACAACT-3′ |
5′-CCCGAAAAGCAAAGTCATATACCT-3′ | 100 |
| HPV-18 E6
(M20324.1) |
5′-TACCTGATCTGTGCACGGAACT-3′ |
5′-ACCGCAGGCACCTCTGTAAG-3′ | 100 |
| HPV-33 E6
(X64085.1) |
5′-TGCCAAGCATTGGAGACAAC-3′ |
5′-CGTCGGGACCTCCAACAC-3′ | 109 |
| GAPDH
(NM_002046.3) |
5′-CCATCTTCCAGGAGCGAGATC-3′ |
5′-GCCTTCTCCATGGTGGTGAA-3′ | 100 |
Transfection
A non-specific control siRNA (Invitrogen, Carlsbad,
CA, USA) and siRNAs against human TRAF6 (40 nM; Sigma-Aldrich, St.
Louis, MO, USA) and FADD (40 nM; Santa Cruz Biotechnology, Santa
Cruz, CA, USA) were used for gene silencing. To regulate
miR-146a-5p expression in cells, specific miRVana mimic, inhibitor
(Ambion) and each respective negative control (Thermo Fisher
Scientific, Waltham, MA, USA; Applied Biosystems) were transfected
using Lipofectamine RNAiMAX transfection reagent (Invitrogen).
Cells were plated into 6-well plates at a density of
3×105 cells/well, cultured overnight, and then
transfected with 20 nM hsa-miR-146a-5p mimic or 20 nM
hsa-miR-146a-5p inhibitor using 8 µl of Lipofectamine
RNAiMAX transfection reagent. After 2 days, the transfected cells
were harvested and analyzed by cell viability assay, TUNEL assay
and immunoblotting.
Cell viability assay
The viabilities of cells were investigated using the
EZ-Cytox Cell Viability Assay kit (water-soluble tetrazolium salt
method; Daeil Lab Service Co., Ltd., Seoul, Korea). Cells
(3×103 cells/100 µl) were seeded onto a 96-well
microplate, adapted overnight and then transfected with
hsa-miR-146a-5p mimic or anti-miR hsa-miR-146a-5p inhibitor for 48
h at 37°C. The water-soluble tetrazolium salt reagent solution (10
µl) was added to each well, and the plate was incubated for
2 h at 37°C. The absorbance at 450 nm was then measured using a
microplate reader.
TUNEL assay
Apoptosis was analyzed in situ by the TUNEL
assay using the In Situ Cell Death Detection kit,
Fluorescein (Roche Applied Science, Mannheim, German) according to
the manufacturer's instructions. To ensure a representative count,
each cell culture slide was divided into quarters, and at least two
fields were photographed with Olympus BX50 fluorescence microscope.
The number of apoptotic cells was measured by counting the
TUNEL-positive cells. Average number and standard deviation were
calculated from four independent experiments.
Immunoblotting
Cells (1×106) were plated onto 60-mm
dishes and then washed with ice-cold phosphate-buffered saline
(PBS) and lysed with 150 µl RIPA buffer (50 mM Tris-HCl, pH
7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, 2 mM
Na3VO4 and 1 mM glycerol phosphate)
containing a protease inhibitor cocktail. The proteins present in
the RIPA lysates were denatured using SDS sample buffer, resolved
by SDS-PAGE and electroblotted onto nitrocellulose membranes. The
membranes were immunoblotted with primary antibodies against Smad4;
FADD; p-FADD (Ser194); NF-κB p65; JNK; p-JNK (Thr183/Tyr185); p38;
phospho-p38 (Thr180/Tyr182); Bcl-2 (Cell Signaling Technology,
Danvers, MA, USA); Mad (Abcam, Cambridge, MA, USA); p53 (Millipore,
Bedford, MA, USA); TRAF6; TGFβ1; p15; c-Jun; p-c-Jun (Santa Cruz
Biotechnology); or actin (Sigma-Aldrich). All blots were then
incubated with anti-rabbit horseradish peroxidase-conjugated
secondary antibodies (Cell Signaling Technology). The signals were
detected by electrochemiluminescence (iNtRON Biotechnology,
Seongnam-si, Korea).
Statistical analyses
Comparison of the distribution of −ΔCT values
between groups were analyzed by the language R (41), and ROC curves were obtained with
the package ROCR (42). All other
data are presented as the mean ± SD. The statistical analyses of
data were performed using the Statistica 6.0 software package
(StatSoft, Tulsa, OK, USA). Results were compared using analysis of
variance tests. When significant differences were found, pairwise
comparisons were performed using Scheffe's adjustment. Statistical
significance was also calculated using a two-tailed Student's
t-test. Differences with a P<0.05 were considered statistically
significant.
Results
miR-146a-5p is highly expressed in
OSCC
To explore possible miRNAs involved in the
transformation of normal oral keratinocytes, we analyzed the
difference in miRNA expression between NHOKs and
HPV-16-immortalized human oral keratinocytes, HOK-16B, by miRNA
microarray with cut-off values of 2-fold increase or decrease.
Twenty-three miRNAs were upregulated while 46 miRNAs were
downregulated in immortalized human oral keratinocytes compared to
NHOKs (Table II). Microarray also
revealed miR-3934-5p, miR-365b-5p, miR-146a-5p and miR-3138 being
most abun dant in immortalized human oral keratinocytes (Table II). On the basis of this, we
hypothesized that miR-146a-5p expression is overexpressed in OSCCs
and may underlie OSCC initiation and progression. Furthermore, the
miR-146a-5p and its target genes were of interest, because
miR-146a-5p regulates signal transduction of TGF-β, a potent
antitumor cytokine, by repressing Smad4 in human cancer (43) and its biological function in
carcinogenesis is not fully understood. To test our hypothesis, we
first examined miR-146a-5p expression in 13 OSCC cell lines and
other 2 SCC cell lines, and in matched 20 pairs of OSCC and normal
tissues, 30 whole blood and 21 saliva samples from pre-operative
OSCC patients and 10 healthy controls by a TaqMan miRNA assay.
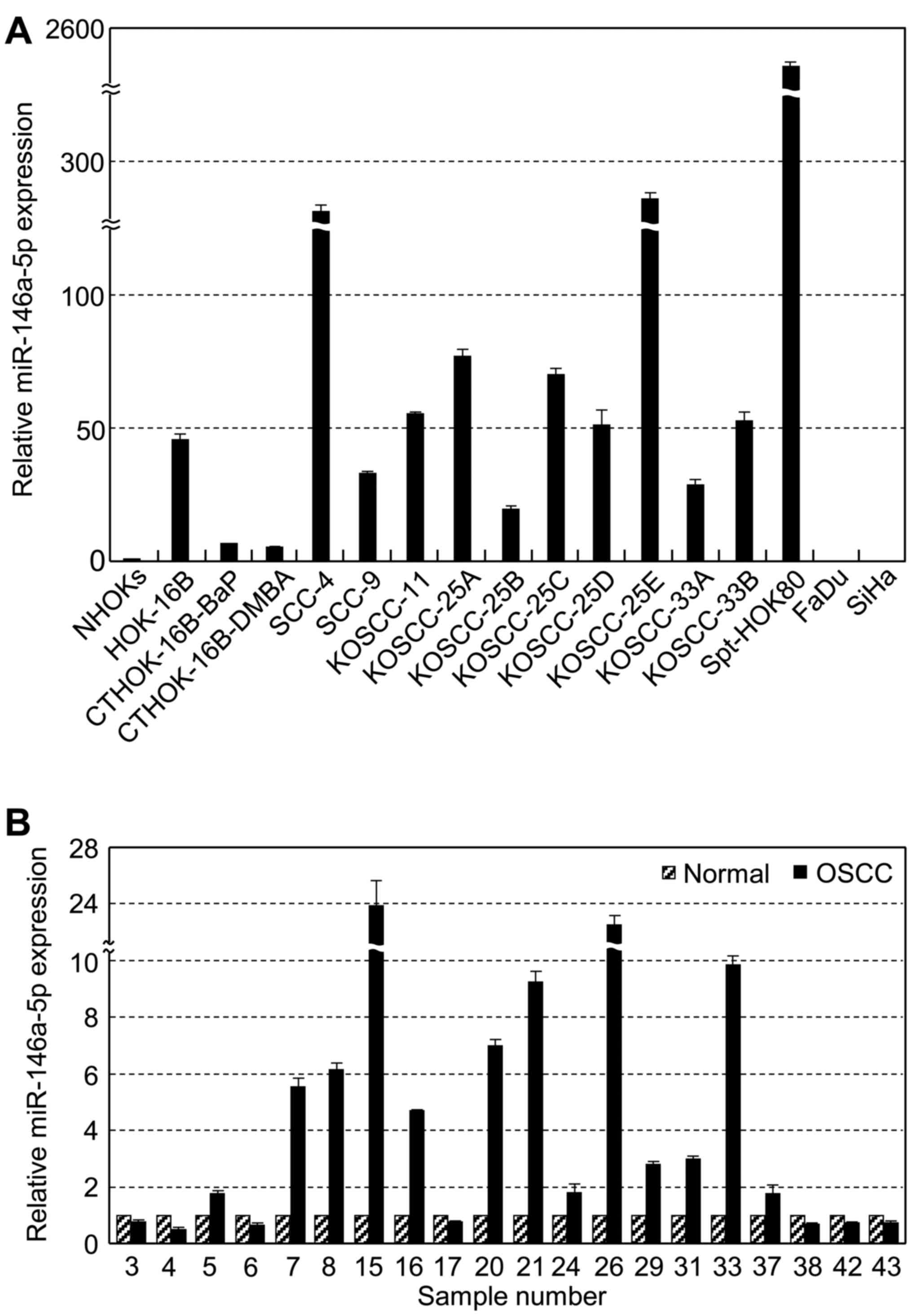
miR-146a-5p expression was significantly upregulated in all 13 OSCC
cell lines examined compared to the normal oral keratinocytes;
however, its expression in other 2 SCC cell lines established from
pharynx and cervix carcinomas was extremely lower (~10%) than that
observed in NHOKs (Fig. 1A).
Analysis of matched OSCC-normal tissue pairs demonstrated an
increase in miR-146a-5p expression in 65% (13/20) of the OSCCs
compared to the matched normal tissues (Fig. 1B). Blood miR-146a-5p levels were
significantly higher in OSCC patients compared to the healthy
controls with mean −ΔCT values of 2.2300363 and 0.1399096,
respectively (P=0.0007967) (Fig.
1C). Similarly, miR-146a-5p levels in saliva were significantly
abundant in OSCC patients compared to the healthy controls with
mean −ΔCT values of 0.7711158 and −2.7807178, respectively
(P=0.00007308) (Fig. 1D). However,
among the 18 OSCC patients whose whole blood and saliva were
collected, each miR-146a-5p level in blood and saliva showed
minimal evidence of a true correlation when tested by the Pearson
method (correlation coefficient =−0.08920913, P=0.7248). To
evaluate the diagnostic significance of miR-146a-5p in determining
OSCC status, receiver operating characteristic (ROC) curves were
obtained for each blood and saliva sample sets, and the area under
the curves (AUC) was 0.8033333 and 0.9004329, respectively
(Fig. 1C and D). Collectively,
these data demonstrate that miR-146a-5p is highly upregulated in
immortalized human oral keratinocytes and OSCC cell lines, and that
miR-146a-5p levels in OSCC tissues, whole blood and whole saliva
significantly increase in OSCC patients compared with healthy
controls.
| Table IImiRNA expression profile in
HPV-16-immortalized human oral keratinocytes relative to the NHOKs
by miRNA microarray. |
Table II
miRNA expression profile in
HPV-16-immortalized human oral keratinocytes relative to the NHOKs
by miRNA microarray.
Upregulated miRNAs
| Downregulated
miRNAs
|
|---|
| Systematic
name | Fold change | Systematic
name | Fold change | Systematic
name | Fold change |
|---|
|
hsa-miR-3934-5p | 15214.5 | hsa-let-7d-5p | 0.50 | hsa-let-7e-5p | 0.35 |
|
hsa-miR-365b-5p | 415.0 |
hsa-miR-365a-3p | 0.49 | hsa-let-7g-5p | 0.34 |
|
hsa-miR-146a-5p | 391.2 |
hsa-miR-130a-3p | 0.49 | hsa-miR-5100 | 0.33 |
| hsa-miR-3138 | 282.7 | hsa-miR-4286 | 0.48 | hsa-let-7a-5p | 0.31 |
| hsa-miR-4430 | 6.4 | hsa-miR-18a-5p | 0.48 | hsa-let-7i-5p | 0.31 |
| hsa-miR-150-3p | 5.8 | hsa-miR-4306 | 0.48 | hsa-miR-1260b | 0.31 |
| hsa-miR-378d | 4.1 | hsa-miR-20a-5p | 0.46 | hsa-let-7f-5p | 0.29 |
| hsa-miR-30b-3p | 3.7 | hsa-miR-99a-5p | 0.45 | hsa-miR-4284 | 0.28 |
| hsa-miR-6068 | 3.2 | hsa-miR-30c-5p | 0.45 | hsa-let-7c | 0.28 |
| hsa-miR-1275 | 3.0 | hsa-miR-455-3p | 0.44 | hsa-miR-21-5p | 0.28 |
| hsa-miR-378i | 3.0 |
hsa-miR-374b-5p | 0.43 | hsa-miR-455-5p | 0.27 |
|
hsa-miR-378a-3p | 2.9 | hsa-miR-31-3p | 0.42 | hsa-miR-29b-3p | 0.26 |
| hsa-miR-30d-5p | 2.8 | hsa-miR-185-5p | 0.41 | hsa-miR-1260a | 0.25 |
|
hsa-miR-1229-5p | 2.7 | hsa-miR-28-5p | 0.41 | hsa-miR-26b-5p | 0.25 |
| hsa-miR-5787 | 2.7 | hsa-miR-222-3p | 0.40 |
hsa-miR-125b-5p | 0.24 |
|
hsa-miR-3679-5p | 2.7 | hsa-miR-4465 | 0.39 | hsa-miR-96-5p | 0.24 |
|
hsa-miR-4787-5p | 2.6 | hsa-miR-210 | 0.37 |
hsa-miR-374a-5p | 0.23 |
| hsa-miR-205-3p | 2.5 | hsa-miR-29a-3p | 0.37 | hsa-miR-98-5p | 0.21 |
|
hsa-miR-6724-5p | 2.4 | hsa-miR-221-3p | 0.37 | hsa-miR-30a-5p | 0.20 |
|
hsa-miR-181a-5p | 2.2 | hsa-miR-3659 | 0.37 |
hsa-miR-1273g-3p | 0.16 |
| hsa-miR-638 | 2.2 | hsa-miR-203a | 0.37 | hsa-miR-100-5p | 0.10 |
| hsa-miR-4739 | 2.1 | hsa-miR-424-5p | 0.37 |
hsa-miR-135b-5p | 0.08 |
| hsa-miR-2861 | 2.0 | hsa-let-7b-5p | 0.36 | hsa-miR-30a-3p | 0.00 |
miR-146a-5p expression is not associated
with HPV E5 in SCC cell lines
Because HPV-16 E5 oncogene is reported to modulate
the expression of miR-146a-5p (44), we examined whether miR-146a-5p
expression is correlated with HPV E5 expression in OSCC and SCC
cell lines. We first examined whether cancer cell lines expressed
'high-risk' HPV E5 and E6 transcripts (type 16, 18 and 33) and
compared to their cellular miR-146a-5p levels. HPV-16 E5 and E6
transcripts were detected in HOK-16, CTHOK-16B-BaP, CTHOK-16B-DMBA
and SiHa cell lines (Fig. 1E).
Other 12 OSCC or SCC cell lines, however, proved to be free of HPV
DNA (data not shown). The miR-146a-5p expression was significantly
upregulated in HOK-16B, CTHOK-16B-BaP and CTHOK-16B-DMBA, which
expressed HPV-16 E5; however, its expression in SiHa, which
expressed HPV-16 E5, was extremely lower (~10%) than that observed
in NHOKs (Fig. 1A). Unexpectedly,
our data support that miR-146a-5p expression is not necessarily
correlated with HPV-16 E5 expression in human cancer cell
lines.
miR-146a-5p variously affects cell
proliferation and apoptosis in normal and SCC cells
In view of the observed widespread upregulation of
miR-146a-5p expression in OSCC cell lines and OSCC clinical
specimens, we assessed whether miR-146a-5p upregulation has
associated proliferation potential in NHOKs, SCC-9, HEK293 and
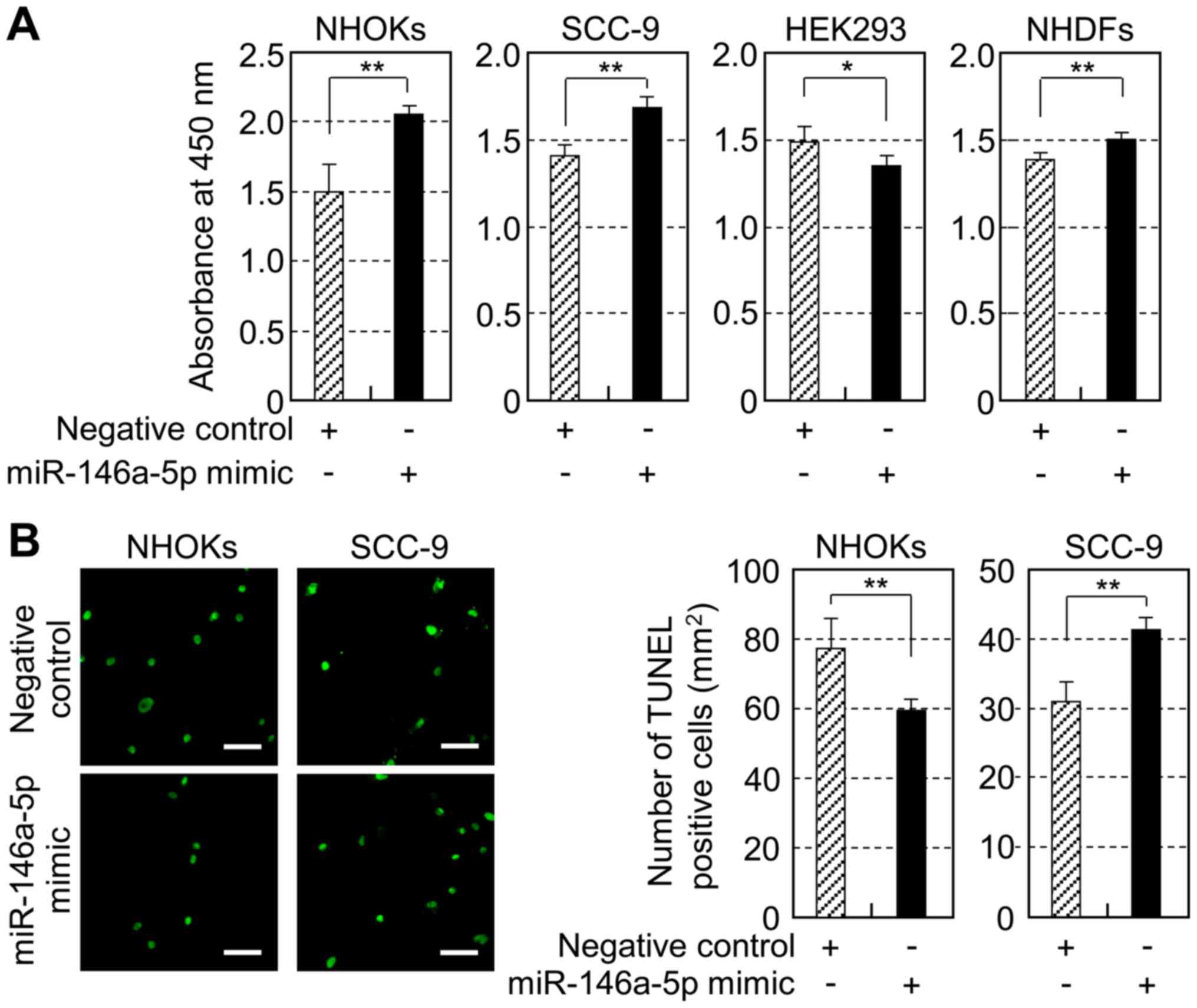
NHDFs. Compared to that in negative control-transfected cells, the
proliferation of NHOKs and SCC-9 was significantly enhanced by
introduction of exogenous miR-146a-5p (Fig. 2A), which was verified by a TaqMan
miRNA assay (data not shown). Additionally, NHDFs responded with
small but significant increase in cell proliferation.
Distinctively, exogenous miR-146a-5p expression inhibited the
proliferation of HEK293 cell line (Fig. 2A). Furthermore, we determined
whether high miR-146a-5p expression is associated with apoptosis in
NHOKs and SCC-9 using TUNEL assay. Exogenous miR-146a-5p expression
caused inhibition of apoptosis in NHOKs, while it increased
apoptosis in SCC-9 (Fig. 2B).
Collectively, overexpression of miR-146a-5p by transfection of
cells with miR-146a-5p mimic resulted in diverse effects on
proliferation and apoptosis depending on the cell types.
miR-146a-5p targets TRAF6 but not Smad4
or FADD in normal and SCC cells
To elucidate the underlying mechanisms associated
with the different outcomes of miR-146a-5p on proliferation and
apoptosis in different cell types, we examined the protein levels
of several known miR-146a-5p target genes, such as TRAF6, SMAD4 and
FADD and their downstream signaling molecules by immunoblotting.
After transfection of cells with miR-146a-5p mimic, TRAF6
expression was invariably downregulated in all cell types tested;
however, Smad4 expression remained constant in NHOKs and SCC-9,
while it decreased in HEK293 and NHDFs (Fig. 2C). FADD expression was slightly
downregulated only in HEK293 cells (Fig. 2C). Notably, exogenous miR-146a-5p
expression downregulated phosphorylated FADD at Ser194
(p-FADD-Ser194) in NHOKs and HEK293 cells but not in SCC-9 and
NHDFs (Fig. 2C). JNK and p38 are
MAPKs at the downstream of TRAF6 in TGF-β signaling that are both
activated by phosphorylation (27). Exogenous miR-146a-5p expression
deactivated JNK in SCC-9, but had no discernable effects in NHOKs,
HEK293 and NHDFs (Fig. 2C).
Similarly, exogenous miR-146a-5p expression also deactivated p38 in
SCC-9. These data suggest that miR-146a-5p hampers translation of
its target genes in a cell type-specific fashion.
miR-146a-5p confers proliferation
advantage to SCC-9
In the opposite approach, we suppressed endogenous
miR-146a-5p expression in SCC-9 followed by functional assays.
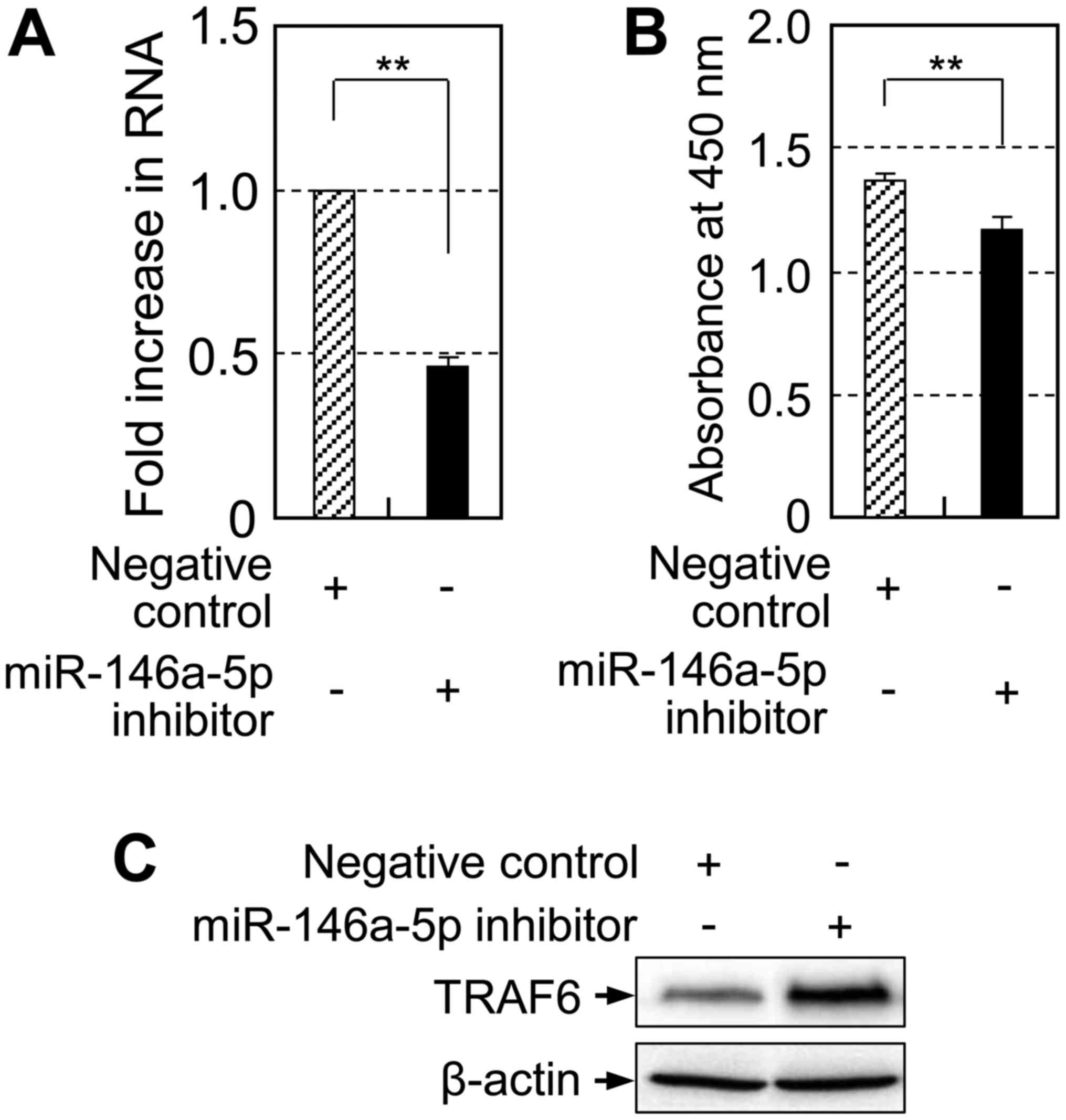
Suppression (~46%) of endogenous miR-146a-5p expression (Fig. 3A) by transfection of cells with
miR-146a-5p inhibitor significantly inhibited cell proliferation
compared to the negative control-transfected cells (Fig. 3B). SCC-9 rather than SCC-4 was
selected for the further studies to clearly elucidate the effects
of miR-146a-5p on cellular processes of OSCC cell lines in this
study. It is important to finely control the expression levels of
miR-146a-5p in the cell lines by its mimic or inhibitor. Since the
basal miR-146a-5p expression level in SCC-4 was too high (Fig. 1A), 54% reduction of its expression
by the miR-146a-5p inhibitor, as shown in Fig. 3A, seemed still high, making it
ambiguous to clarify its reduced function. On the other hand, SCC-9
had relatively lower, but significantly higher expression level of
miR-146a-5p compared to normal oral keratinocytes (Fig. 1A), making it more effective to
examine the clear effect of the miR-146a-5p mimic or inhibitor
transfection. Next, to assess the underlying mechanisms of
miR-146a-5p inhibitor-induced inhibition of cell proliferation, we
examined the protein level of TRAF6, one of the miR-146a-5p target
genes. miR-146a-5p inhibitor induced upregulation of TRAF6
(Fig. 3C). Our data suggest that
miR-146a-5p inhibitor inhibits proliferation of SCC-9 by modulating
the expression level of TRAF6.
TRAF6 silencing induces deactivation of
JNK and p38 in SCC-9 but not in NHOKs
To assess the functional significance of the
universal effect of exogenous miR-146a-5p expression, which led to
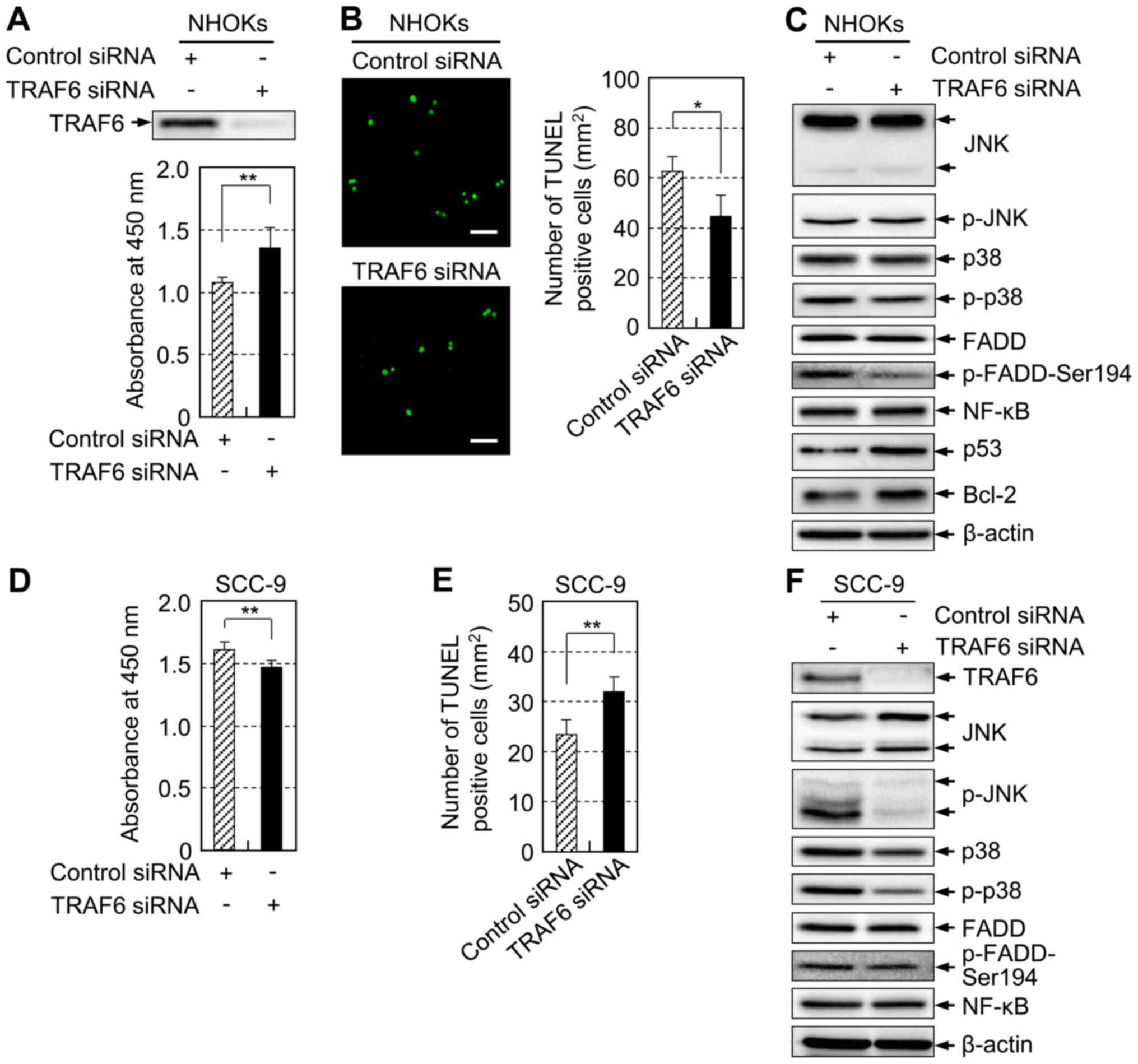
TRAF6 downregulation, we silenced endogenous TRAF6 expression in
exponentially proliferating NHOKs and SCC-9 by transfecting the
cells with a specific small interfering RNA (siRNA). TRAF6
knockdown (Fig. 4A, upper panel)
led to significantly increased cell proliferation (Fig. 4A, lower panel) and inhibited
apoptosis of NHOKs (Fig. 4B),
mimicking the effects of exogenous miR-146a-5p expression (Fig. 2A and B). By contrast, TRAF6
knockdown significantly decreased cell proliferation (Fig. 4D) and increased apoptosis (Fig. 4E) of SCC-9. Immunoblotting revealed
that TRAF6 knockdown resulted in marked deactivation of JNK and p38
in SCC-9 (Fig. 4F), giving very
similar results to those of the exogenous miR-146a-5p expression
(Fig. 2C). However, TRAF6
knockdown did not affect the phosphorylation of JNK and p38 but
decreased the amount of p-FADD-Ser194 in NHOKs (Fig. 4C). Overall, our data suggest two
points; first, TRAF6 knockdown can be either proliferative or
inhibitory in terms of cell proliferation depending on the cellular
context, and second, the TRAF6-JNK and TRAF6-p38 channels can be
constitutively active in SCC-9.
JNK plays a crucial role in the
proliferation of NHOKs and SCC-9
The phenotypically contradictory results of TRAF6
silencing imply antagonistic effectors downstream of TRAF6
signaling. Since JNK and p38 are activated by TRAF6 in certain
circumstances, we defined their roles using specific kinase
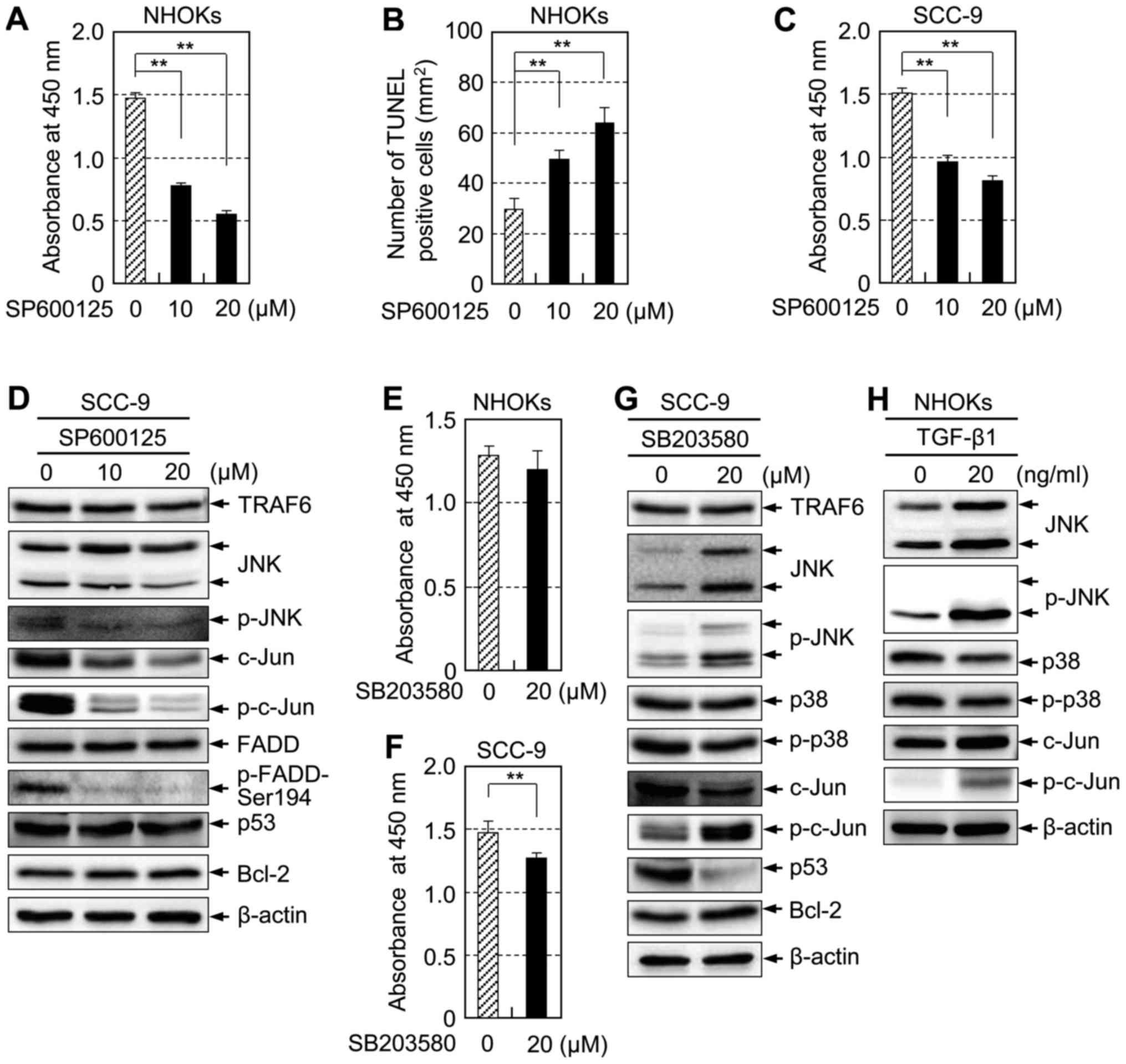
inhibitors targeting each of them. Treatment of NHOKs with
SP600125, a JNK specific inhibitor, significantly reduced cell
proliferation (Fig. 5A) and
increased apoptosis (Fig. 5B) in a
dose-dependent manner, as well as reduced proliferation of SCC-9,
similarly (Fig. 5C).
Immunoblotting revealed that SP600125 inhibited phosphorylation of
JNK and its substrate c-Jun in a dose-dependent manner;
compellingly, p-FADD-Ser194 was also reduced in SCC-9 (Fig. 5D). By contrast, SB203580, a p38
inhibitor, resulted in an insignificant reduction of proliferation
of NHOKs (Fig. 5E) but reduced
proliferation of SCC-9 (Fig. 5F).
Unexpectedly, reduction of phosphorylated p38 was marginal, and an
unanticipated upregulation of JNK and phosphorylated JNK was
observed in SCC-9 by immunoblotting. JNK activation by SB203580
also increased phosphorylated c-Jun (Fig. 5G). Similarly, TGF-β1 activated JNK
and c-Jun, but not p38 in exponentially proliferating NHOKs
(Fig. 5H). These likely secondary
effects could have dampened the effects of p38 suppression on cell
proliferation. Nevertheless, these data suggest that JNK/c-Jun
signaling pathway plays a key role in regulation of proliferation
of SCC-9.
FADD and p-FADD are not involved in the
proliferation of NHOKs and SCC-9
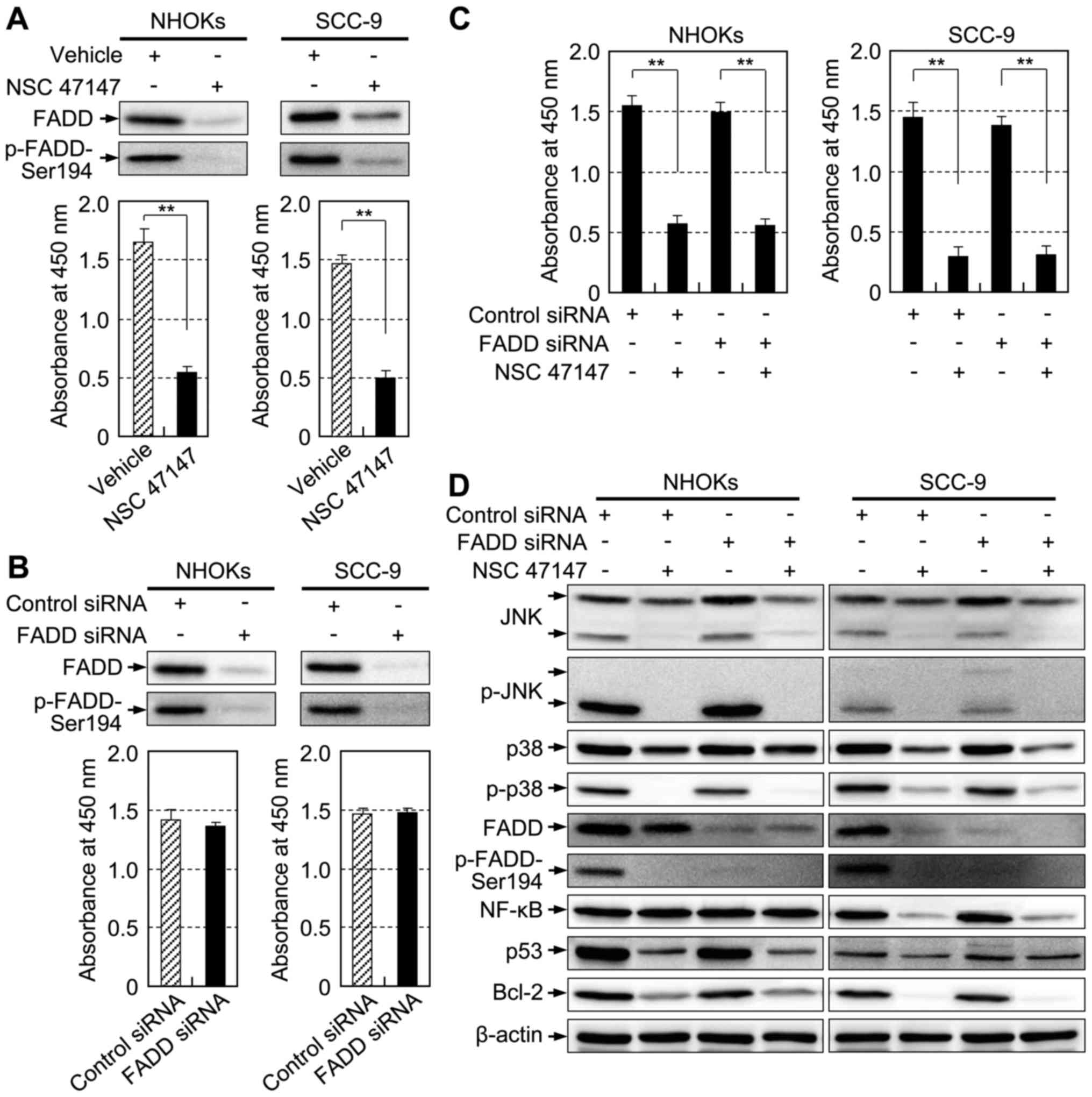
Since exogenous miR-146a-5p expression reduced the
expression of p-FADD-Ser194 in NHOKs and HEK293 (Fig. 2C), we opted to investigate the
functional relevance of p-FADD-Ser194 in cell proliferation.
Proliferation was drastically inhibited by treatment of NHOKs and
SCC-9 with NSC 47147 (Fig. 6A,
lower panel), a kinase inhibitor that is known to inhibit FADD
phosphorylation (45). Notably,
expression of both FADD and p-FADD was suppressed (Fig. 6A, upper panel). Thus, to confirm
the effects of simultaneous downregulation of both FADD and
p-FADD-Ser194 expression, we suppressed their expression by
transfection of cells with a FADD-specific siRNA. Unexpectedly,
simultaneous silencing of both FADD and p-FADD-Ser194 (Fig. 6B, upper panel) did not result in
cell proliferation blockage of NHOKs and SCC-9 (Fig. 6B, lower panel). Therefore, to
verify these conflicting results, cells transfected with a
FADD-specific siRNA were subsequently treated with NSC 47147. NSC
47147 was able to reduce cell proliferation in the absence of FADD
and p-FADD-Ser194 expression, and the inhibition pattern of cell
proliferation was strikingly similar to that of control
siRNA-transfected cells (Fig. 6C).
Immunoblotting revealed that NSC 47147 not only inhibited
phosphorylation of FADD, but also inhibited phosphorylation of JNK
and p38, suggesting a broad inhibition of MAPK pathway (Fig. 6D). Overall, our data suggest that
FADD and p-FADD-Ser194 expression has a minimal effect on the
proliferation of NHOKs and SCC-9.
Discussion
Overexpression of miR-146a-5p has been previously
identified in OSCC tissues (7,46),
blood of OSCC patients (7), and
oral precancerous lesions (46).
In the present study, we widened the view with salivary abundance
of the miR-146a-5p. The miR-146a-5p in saliva seemed not to
originate from blood, which was suggested by the poor correlation
of −∆CT values in matched samples. The tumor could contribute to
miR-146a-5p levels in blood and saliva, respectively, by direct
shedding of the miRNA. Both levels of miR-146a-5p in blood and
saliva of OSCC patients proved themselves to be a possible
candidate as a diagnostic marker of OSCC (47,48).
However, clinical application of miR-146a-5p as a diagnostic marker
requires further validation, because disease status other than
OSCC, such as oral lichen planus and psoriasis, may also have high
levels of miR-146a-5p in their blood or saliva (49).
miR-146a-5p was highly upregulated in immortalized
human oral keratinocytes and all the OSCC cell lines examined, but
not in other SCC cell lines. Although upregulation of miR-146 in
cervial carcinomas is previously reported (5), its expression was almost negligible
in SiHa cell line established from cervical carcinoma. This is
supported by the fact that all seven cervical cancer cell lines
examined expressed no detectable miR-146a (5). In addition, differently from a
previous study (44), we found
that miR-146a-5p expression is not necessarily correlated with
'high-risk' HPV E5 expression in human cancer cell lines. These
findings partially explain the discrepancy between low incidence of
HPV and overexpression of miR-146a-5p in OSCC. Otherwise,
contributing factors of miR-146a-5p upregulation possibly includes
cytokines that activate NF-κB. TGF-β upregulated miR-146a-5p
expression in NHOKs (50), which
may also contribute to the overexpression of the miRNA in OSCC.
Other possible candidates include tumor necrosis factor and IL-1.
Thus, overexpression of miR-146a-5p in OSCC cell lines may reflect
the constitutive activation of NF-κB in these cells.
Despite of its wide involvement in cell biology and
increasing research, miRNAs are getting more and more thrown into
disarray owing to their functionally complicated nature. miR-146a/b
has been previously reported to be linked with cancer risk and
invasive and metastatic capacity in diverse cancers (3); however, its roles in carcinogenesis
are not fully understood yet. In the present study, we demonstrated
two additional layers of complexity that require attention to
apprehend the significance of the miRNA. The first layer of miRNA
complexity comprised of cell type- and/or context-dependent miRNA
target selection, which resulted in sparing of a certain target
while suppressing another universally. For instance, the range of
target mRNAs of a single miRNA such as miR-146a-5p differed by cell
type. The gold standard for miRNA functionality is verification of
the protein expression pattern of target mRNAs. Indeed, our data
demonstrated that a validated miRNA target in a specific cell type
can dodge from the miRNA-mediated post-transcriptional suppression
in another cell type. This evasion from post-transcriptional
suppression by miRNA could impart variable contexts that
phenomenalize the effects of a miRNA to the cells expressing
it.
Specifically, for human oral keratinocytes,
miR-146a-5p spared Smad4 from post-transcriptional suppression.
Smad4 plays a crucial role in the canonical TGF-β signaling pathway
and is implicated in epithelial-mesenchymal transition by TGF-β
(51). A considerable body of
evidence indicates that Smad4 is a valid target gene of miR-146a-5p
(22,43); however, exogenous miR-146a-5p
expression did not affect the expression levels of Smad4 in NHOKs
and SCC cell lines. This observation suggests that miR-146a-5p
selectively disarms the non-canonical, TRAF6-mediated branch of the
TGF-β signaling in human oral keratinocytes. One theoretical
consequence of sparing Smad4 involvement in human oral
keratinocytes is that the discriminative downregulation by
miR-146a-5p may selectively confer resistance against cytostatic
and pro-apoptotic effect of TGF-β signaling, while preserving other
arms of the pathway, possibly the epithelial-mesenchymal transition
inducing arm, to the cells. Future studies need to address this
possibility to clarify the significance of miR-146a-5p upregulation
on metastasis.
The second layer of miRNA complexity arose from the
functional pleiotropism of a miRNA target gene, which in this study
was exemplified by TRAF6. TRAF6 was a universal target of
miR-146a-5p in all cells and cell lines tested; however, TRAF6
suppression by a TRAF6-specific siRNA resulted in contradictory
functional consequences in NHOKs and SCC-9. There seemed to be
conflicting downstream signals under the common signal transducer
TRAF6, which comprised of a proliferative signal exemplified by JNK
in SCC-9 and an anti-proliferative signal that suppress the
expression of Bcl-2 in NHOKs. Our results from TRAF6-specific siRNA
experiments showed that the JNK arm is dependent on TRAF6 in SCC-9
but not in NHOKs. Namely, TRAF6 downregulation by both miR-146a-5p
and TRAF6-specific siRNA deactivated JNK in SCC-9 but not in NHOKs.
Nevertheless, the causative signaling that constitutively activates
TRAF6-mediated JNK activity is currently undefined. One of the
possible candidates seems to be TGF-β signaling, because TRAF6
suppression is implicated in the abrogation of TGF-β signaling by
miR-146a-5p (50). TGF-β signaling
encompasses the MAPK pathway, which starts from the interaction
between TRAF6 and TAK1 and results in JNK activation (26,27).
As expected, we observed that TGF-β1 activated JNK and its
substrate c-Jun in exponentially proliferating NHOKs.
FADD expression was not affected by miR-146a-5p in
all cells and cell lines tested. This represented additional
support to the functional diversity of the miR-146a-5p, because
FADD expression was repressed by the miRNA in HEK293. Since
miR-146a-5p reduced the proportion of p-FADD in several cells, we
tested the function of FADD and p-FADD in proliferation of NHOKs
and SCC-9. We showed that suppression of both FADD and p-FADD by
combined treatment of FADD-specific siRNA and NSC 47147 was not
implicated in proliferation control, at least in steady state of
NHOKs and SCC-9. Rather, the anti-proliferative effect of NSC 47147
was associated with inactivation of JNK, which further support the
crucial role of the kinase in proliferation of NHOKs and SCC-9.
Further studies are required to verify the role of periodontitis in
miR-146a-5p expression in oral cancer patients, and to investigate
the role of miR-146a-5p in oral cancer using in vivo animal
models.
In summary, our findings showed that miR-146a-5p was
upregulated in OSCC and granted proliferative advantage to normal
oral keratinocytes and OSCC cell lines in a context-dependent
manner. Our data also suggested that miR-146a-5p selectively
disarmed the TRAF6-mediated branch of the TGF-β signaling in OSCC
cell lines by sparing Smad4 involvement.
Acknowledgments
The present study was supported by the Midcareer
Researcher Program through a grant from the National Research
Foundation of Korea, funded by the Ministry of Science, ICT and
Future Planning, Korean government (2016R1A2B2007246), and the
Korea Healthcare Technology R&D Project, funded by the Ministry
for Health, Welfare & Family Affairs, Republic of Korea
(HI15C2455) (to B.-M.M.).
References
|
1
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Svoronos AA, Engelman DM and Slack FJ:
OncomiR or tumor suppressor? The duplicity of microRNAs in cancer.
Cancer Res. 76:3666–3670. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elsarraj HS, Stecklein SR, Valdez K and
Behbod F: Emerging functions of microRNA-146a/b in development and
breast cancer: microRNA-146a/b in development and breast cancer. J
Mammary Gland Biol Neoplasia. 17:79–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chang SS, Jiang WW, Smith I, Poeta LM,
Begum S, Glazer C, Shan S, Westra W, Sidransky D and Califano JA:
MicroRNA alterations in head and neck squamous cell carcinoma. Int
J Cancer. 123:2791–2797. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang X, Tang S, Le SY, Lu R, Rader JS,
Meyers C and Zheng ZM: Aberrant expression of oncogenic and
tumor-suppressive microRNAs in cervical cancer is required for
cancer cell growth. PLoS One. 3:e25572008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Raponi M, Dossey L, Jatkoe T, Wu X, Chen
G, Fan H and Beer DG: MicroRNA classifiers for predicting prognosis
of squamous cell lung cancer. Cancer Res. 69:5776–5783. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hung PS, Liu CJ, Chou CS, Kao SY, Yang CC,
Chang KW, Chiu TH and Lin SC: miR-146a enhances the oncogenicity of
oral carcinoma by concomitant targeting of the IRAK1, TRAF6 and
NUMB genes. PLoS One. 8:e799262013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hurst DR, Edmonds MD, Scott GK, Benz CC,
Vaidya KS and Welch DR: Breast cancer metastasis suppressor 1
up-regulates miR-146, which suppresses breast cancer metastasis.
Cancer Res. 69:1279–1283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kogo R, Mimori K, Tanaka F, Komune S and
Mori M: Clinical significance of miR-146a in gastric cancer cases.
Clin Cancer Res. 17:4277–4284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin SL, Chiang A, Chang D and Ying SY:
Loss of mir-146a function in hormone-refractory prostate cancer.
RNA. 14:417–424. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Y, Vandenboom TG II, Wang Z, Kong D,
Ali S, Philip PA and Sarkar FH: miR-146a suppresses invasion of
pancreatic cancer cells. Cancer Res. 70:1486–1495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kloosterman WP and Plasterk RHA: The
diverse functions of microRNAs in animal development and disease.
Dev Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marques-Rocha JL, Samblas M, Milagro FI,
Bressan J, Martínez JA and Marti A: Noncoding RNAs, cytokines, and
inflammation-related diseases. FASEB J. 29:3595–3611. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Connell RM, Rao DS and Baltimore D:
microRNA regulation of inflammatory responses. Annu Rev Immunol.
30:295–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kundu JK and Surh YJ: Inflammation:
Gearing the journey to cancer. Mutat Res. 659:15–30. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perry MM, Moschos SA, Williams AE,
Shepherd NJ, Larner-Svensson HM and Lindsay MA: Rapid changes in
microRNA-146a expression negatively regulate the IL-1β-induced
inflammatory response in human lung alveolar epithelial cells. J
Immunol. 180:5689–5698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu X, Nelson A, Wang X, Kanaji N, Kim M,
Sato T, Nakanishi M, Li Y, Sun J, Michalski J, et al: MicroRNA-146a
modulates human bronchial epithelial cell survival in response to
the cytokine-induced apoptosis. Biochem Biophys Res Commun.
380:177–182. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie YF, Shu R, Jiang SY, Liu DL and Zhang
XL: Comparison of microRNA profiles of human periodontal diseased
and healthy gingival tissues. Int J Oral Sci. 3:125–134. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Irwandi RA and Vacharaksa A: The role of
microRNA in periodontal tissue: A review of the literature. Arch
Oral Biol. 72:66–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olsen I, Singhrao SK and Osmundsen H:
Periodontitis, pathogenesis and progression: miRNA-mediated
cellular responses to Porphyromonas gingivalis. J Oral Microbiol.
9:13333962017. View Article : Google Scholar :
|
|
21
|
Preedy VR and Hunter RJ: Cytokine.
MicroRNAs and Cytokines. Liu XD: CRC Press; New York: pp. 10–16.
2011
|
|
22
|
Xiao B, Zhu ED, Li N, Lu DS, Li W, Li BS,
Zhao YL, Mao XH, Guo G, Yu PW, et al: Increased miR-146a in gastric
cancer directly targets SMAD4 and is involved in modulating cell
proliferation and apoptosis. Oncol Rep. 27:559–566. 2012.
|
|
23
|
Paik JH, Jang JY, Jeon YK, Kim WY, Kim TM,
Heo DS and Kim CW: MicroRNA-146a downregulates NF-κB activity via
targeting TRAF6 and functions as a tumor suppressor having strong
prognostic implications in NK/T cell lymphoma. Clin Cancer Res.
17:4761–4771. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang YE: Non-Smad pathways in TGF-β
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar :
|
|
26
|
Sorrentino A, Thakur N, Grimsby S,
Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH and
Landström M: The type I TGF-β receptor engages TRAF6 to activate
TAK1 in a receptor kinase-independent manner. Nat Cell Biol.
10:1199–1207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamashita M, Fatyol K, Jin C, Wang X, Liu
Z and Zhang YE: TRAF6 mediates Smad-independent activation of JNK
and p38 by TGF-β. Mol Cell. 31:918–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vaiopoulos AG, Papachroni KK and
Papavassiliou AG: Colon carcinogenesis: Learning from NF-κB and
AP-1. Int J Biochem Cell Biol. 42:1061–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He L, Wu X, Siegel R and Lipsky PE: TRAF6
regulates cell fate decisions by inducing caspase 8-dependent
apoptosis and the activation of NF-κB. J Biol Chem.
281:11235–11249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peter ME and Krammer PH: Mechanisms of
CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 10:545–551.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Curtale G, Citarella F, Carissimi C,
Goldoni M, Carucci N, Fulci V, Franceschini D, Meloni F, Barnaba V
and Macino G: An emerging player in the adaptive immune response:
microRNA-146a is a modulator of IL-2 expression and
activation-induced cell death in T lymphocytes. Blood. 115:265–273.
2010. View Article : Google Scholar
|
|
33
|
Chen G, Bhojani MS, Heaford AC, Chang DC,
Laxman B, Thomas DG, Griffin LB, Yu J, Coppola JM, Giordano TJ, et
al: Phosphorylated FADD induces NF-κB, perturbs cell cycle, and is
associated with poor outcome in lung adenocarcinomas. Proc Natl
Acad Sci USA. 102:12507–12512. 2005. View Article : Google Scholar
|
|
34
|
Hua ZC, Sohn SJ, Kang C, Cado D and Winoto
A: A function of Fas-associated death domain protein in cell cycle
progression localized to a single amino acid at its C-terminal
region. Immunity. 18:513–521. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oh JE, Kook JK and Min BM: βig-h3 induces
keratinocyte differentiation via modulation of involucrin and
transglutaminase expression through the integrin α3β1 and the
phosphatidylinositol 3-kinase/Akt signaling pathway. J Biol Chem.
280:21629–21637. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yeo IS, Oh JE, Jeong L, Lee TS, Lee SJ,
Park WH and Min BM: Collagen-based biomimetic nanofibrous
scaffolds: Preparation and characterization of collagen/silk
fibroin bicomponent nanofibrous structures. Biomacromolecules.
9:1106–1116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park NH, Min BM, Li SL, Huang MZ, Cherick
HM and Doniger J: Immortalization of normal human oral
keratinocytes with type 16 human papillomavirus. Carcinogenesis.
12:1627–1631. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Min B, Woo K, Baek J, Lee G and Park N:
Malignant transformation of hpv-immortalized human oral
keratinocytes by chemical carcinogens. Int J Oncol. 7:249–256.
1995.PubMed/NCBI
|
|
39
|
Jang DH, Oh JE, Kang HK, Kim OB, Min SK,
Jung SY, Hong SD, Lee JI and Min BM: Spontaneous tumorigenicity of
primary human oral keratinocytes with human papillomavirus
negativity and impaired apoptosis. Int J Oncol. 36:1491–1501.
2010.PubMed/NCBI
|
|
40
|
Lee G, Kim YB, Kim JH, Kim MS, Shin KH,
Won YS, Lee JI, Choung PH, Hyun BH and Min BM: Characterization of
novel cell lines established from three human oral squamous cell
carcinomas. Int J Oncol. 20:1151–1159. 2002.PubMed/NCBI
|
|
41
|
R Development Core Team: 2011, A Language
and Environment for Statistical Computing. Vienna, Austria: the R
Foundation for Statistical Computing; ISBN: 3-900051-07-0Available
online at http://www.R-project.org/.
|
|
42
|
Sing T, Sander O, Beerenwinkel N and
Lengauer T: ROCR: Visualizing classifier performance in R.
Bioinformatics. 21:3940–3941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Geraldo MV, Yamashita AS and Kimura ET:
MicroRNA miR-146b-5p regulates signal transduction of TGF-β by
repressing SMAD4 in thyroid cancer. Oncogene. 31:1910–1922. 2012.
View Article : Google Scholar
|
|
44
|
Greco D, Kivi N, Qian K, Leivonen SK,
Auvinen P and Auvinen E: Human papillomavirus 16 E5 modulates the
expression of host microRNAs. PLoS One. 6:e216462011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schinske KA, Nyati S, Khan AP, Williams
TM, Johnson TD, Ross BD, Tomás RP and Rehemtulla A: A novel kinase
inhibitor of FADD phosphorylation chemosensitizes through the
inhibition of NF-κB. Mol Cancer Ther. 10:1807–1817. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cervigne NK, Reis PP, Machado J, Sadikovic
B, Bradley G, Galloni NN, Pintilie M, Jurisica I, Perez-Ordonez B,
Gilbert R, et al: Identification of a microRNA signature associated
with progression of leukoplakia to oral carcinoma. Hum Mol Genet.
18:4818–4829. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mitchell PS, Parkin RK, Kroh EM, Fritz BR,
Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant
KC, Allen A, et al: Circulating microRNAs as stable blood-based
markers for cancer detection. Proc Natl Acad Sci USA.
105:10513–10518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Park NJ, Zhou H, Elashoff D, Henson BS,
Kastratovic DA, Abemayor E and Wong DT: Salivary microRNA:
Discovery, characterization, and clinical utility for oral cancer
detection. Clin Cancer Res. 15:5473–5477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Arão TC, Guimarães ALS, de Paula AMB,
Gomes CC and Gomez RS: Increased miRNA-146a and miRNA-155
expressions in oral lichen planus. Arch Dermatol Res. 304:371–375.
2012. View Article : Google Scholar
|
|
50
|
Min SK, Jung SY, Kang HK, Jo SB, Kim MJ
and Min BM: MicroRNA-146a-5p limits elevated TGF-β signal during
cell senescence. Mol Ther Nucleic Acids. 7:335–338. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|