Introduction
Gastric carcinoma (GC), the fifth most common
malignancy and second leading cause of cancer-related death
worldwide, remains an important public health challenge especially
in East Asian countries including China (1,2).
Though therapeutic strategies have been improved in recent years,
GC still has low 5-year survival rate owing to late diagnosis, high
relapse and metastatic rates, multidrug resistance, and severe
toxicities (3,4). The process of GC growth and
metastasis is complex and involves a large number of oncogenes and
tumor suppressor genes. As a result, development of more effective
chemotherapeutic agents for GC therapy is urgent.
Wnt/β-catenin signaling pathway, or the canonical
Wnt pathway, plays a fundamental role in many human physiological
processes as well as tumor progression (5). The abnormality of Wnt/β-catenin
pathway is a common feature in many human cancers and promotes cell
proliferation and metastasis (6–11).
Particularly, aberrant activation of β-catenin signaling is found
in ~30–50% of GC tissues and various gastric cancer cell lines and
indicates poor prognosis (12–14).
Moreover, via β-catenin pathway, H. pylori induced gastric
stem cell generation and expansion, promoting gastric cancer
initiation and progression (15).
β-catenin, the crucial molecule in β-catenin pathway and frequently
detected in GC, promotes the transcription of several oncogenic
target genes related to cancer evolution (16,17).
Epithelial-mesenchymal transition (EMT), by which epithelial cells
lose their polarity and cell-cell adhesion and acquire the
properties of mesenchymal cells, plays a pivotal role in cancer
migration, invasion and metastasis (18,19).
β-catenin pathway is also one of the major signalings involved in
EMT and then plays a critical role in metastasis (20). Consequently, β-catenin pathway has
emerged as a promising target for GC therapy.
MicroRNAs (miRNAs) are a family of small (~22
nucleotides in length) and endogenous noncoding RNAs processed from
double-stranded hairpin precursors (21). Several studies showed that miRNAs
have a capacity to act as tumor suppressors and their deregulation
is associated with initiation and progression of many human cancers
(22,23). The miR-200 family, comprising 5
members (miR-200a, -200b, -200c, -141 and -429), is found
significantly downregulated in prostate cancer, breast cancer, lung
adenocarcinoma and GC and may become a potential prognostic
predictor of GC (24–29). Downregulation of miR-200 family
also leads to reduced E-cadherin expression, which is a crucial
step in the carcinogenesis of EBV-associated GC (30). miR-200a shows anti-oncogenic
features in many cancers including GC (31–33).
Elevated miR-200a in SGC-7901 cells inhibited cell growth and
invasion and induced G0/G1 phase arrest (33). More importantly, recent studies
demonstrated that miR-200a inhibited GC growth and EMT through
suppressing β-catenin signaling while miR-200b and miR-200c, two
additional members in miR-200 family, showed no impact on β-catenin
expression (33–35). Accordingly, targeting miR-200a and
β-catenin is a promising therapeutic option for GC.
Toosendanin (TSN), as shown in Fig. 1A, is a triterpenoid extracted from
the bark or fruits of Melia toosendan Sieb et Zucc, which
mainly grows in China and India. Mounting evidence has indicated
that TSN plays a variety of biological activities including
analgesic, insecticidal and anti-inflammatory functions (36). Antitumor effect is another
important property of TSN which can kill multiple human cancer
cells in vitro with half maximal inhibitory concentration
(IC50) ranging from 5.4 to 900 nM (37–39).
In addition, TSN can induce tumor apoptosis by regulating
mitochondrial pathway in vitro and suppress hepatocellular
carcinoma growth in BALB/C mice in vivo (36,37).
Our previous study documented that TSN inhibited growth and induces
apoptosis in colorectal cancer (CRC) cells through suppression of
AKT/GSK-3β/β-catenin pathway (40).
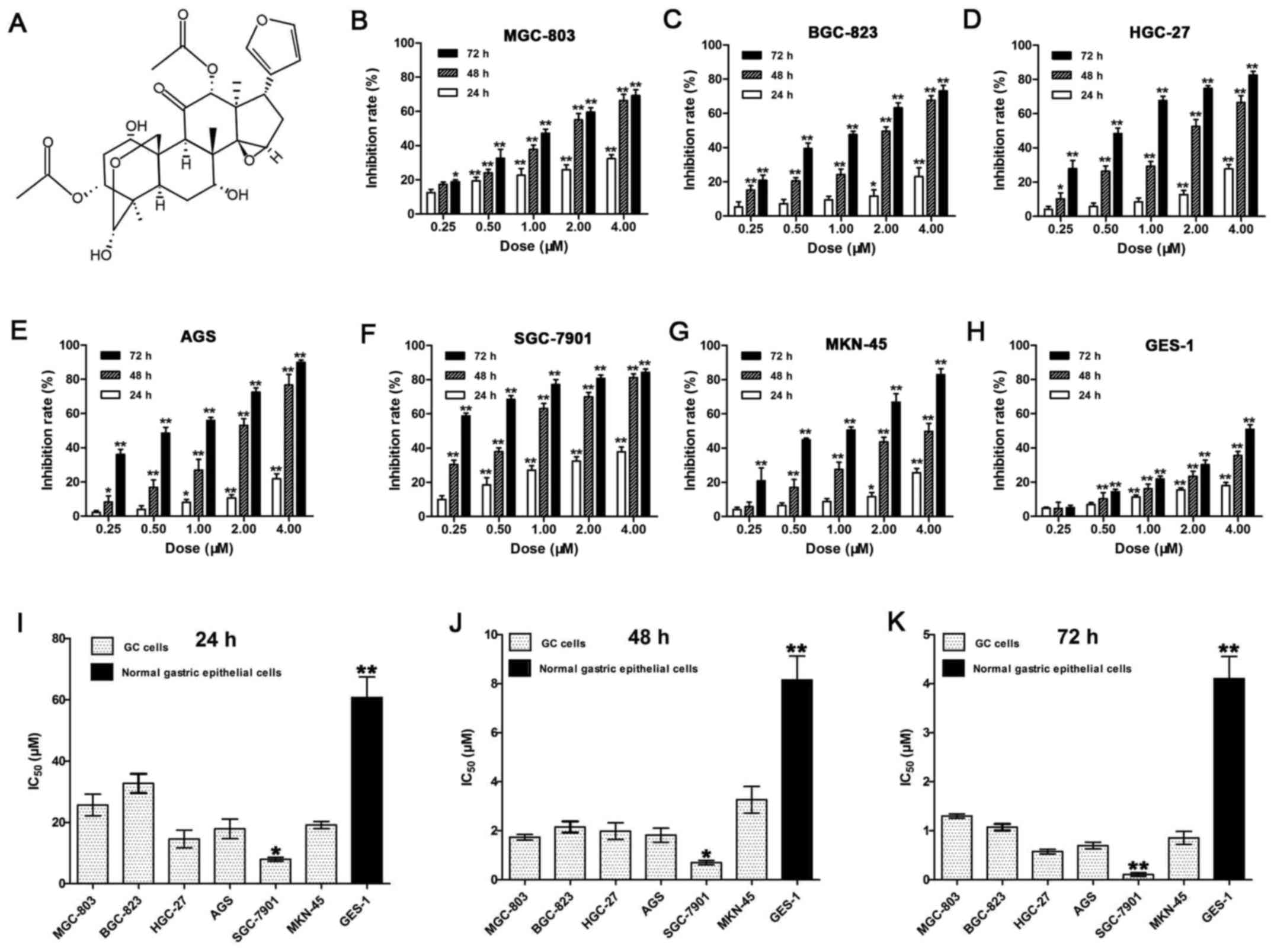 | Figure 1The chemical structure of toosendanin
and inhibitory effects of TSN on the proliferation of human GC cell
lines and GES-1 cells. (A) The molecular structural formula of
toosendanin (C30H38O11, molecular
weight = 574.63). (B-H) Inhibition rates of proliferation were
determined by MTT assay and calculated using formula: 1 − OD
(experiment) / OD (control). The cells treated by TSN (0, 0.25,
0.5, 1, 2 and 4 µM) for 24, 48 and 72 h contained GC cell
lines MGC-803 (B), BGC-823 (C), HGC-27 (D), AGS (E), SGC-7901 (F)
and MKN-45 (G) as well as normal human gastric epithelial cell
lines GES-1 (H). (I-K) IC50 (µM) of TSN on
various human GC cell lines and GES-1 cells for 24 h (I), 48 h (J)
and 72 h (K) were calculated on the basis of inhibition rates. Data
shown are statistical analysis of three independent experiments
(*p<0.05, **p<0.01). |
Nevertheless, effects of TSN on GC and their
sophisticated molecular mechanisms have not been reported before.
In the present study, we revealed antitumor effects of TSN on GC
and their novel mechanism. The results demonstrated that TSN
inhibits GC growth, invasion, migration, EMT and liver metastasis
in vitro and in vivo by stimulating expression of
miR-200a which attenuates activation of β-catenin pathway.
Knockdown of miR-200a or raising β-catenin level in GC cells
weakened the above function of TSN. Our data suggested that
suppression of GC growth and metastasis by TSN may be associated
with miR-200a-mediated downregulation of β-catenin pathway, thereby
showing a promising preclinical activity of TSN for GC therapy.
Materials and methods
Drugs and reagents
TSN (purity ≥99%) was purchased from Shanghai Yuanye
Biotechnology Co., Ltd. (Shanghai, China). 3-(4,5)-dimethylthiazol(-2-yl)-2,5-diphenyltetrazolium
bromide (MTT), Annexin V/propidium iodide (PI) apoptosis detection
kit, cell cycle analysis kit and enhanced chemiluminescence
(ECL)-Plus/kit were all from KeyGen Biotech Co., Ltd. (Nanjing,
China). TRIzol reagent were from Invitrogen (Carlsbad, CA, USA).
SYBR Green Master Mixture was obtained from Takara (Otsu, Japan).
miScript Reverse Transcription kit and miScript SYBP Green PCR kit
were all from Qiagen (Japan). β-catenin, c-Myc, cyclin D1, Bcl-2,
MMP-9, AKT, p-AKT(Ser473), GSK3β, p-GSK3β
(Ser9) and β-actin polyclonal antibodies were all from
ABclonal Biotech Co. (Wuhan, China). Histone H3, E-cadherin,
N-cadherin and vimentin monoclonal antibodies were all from Cell
Signaling Technology (Beverly, MA, USA). ERK1/2 and p-ERK1/2
(Thr202/Tyr204) polyclonal antibodies were
both from Hangzhou HuaAn Biotechnology Co., Ltd. (Hangzhou,
China).
Cell culture
Human GC cell lines MGC-803, BGC-823, HGC-27, AGS,
SGC-7901 and MKN-45 and normal human gastric epithelial cells GES-1
were obtained from the Type Culture Collection, Chinese Academy of
Sciences (Shanghai, China), cultured in RPMI-1640 medium
supplemented with 10% heat-inactivated fetal bovine serum (FBS),
100 U/ml penicillin and 100 µg/ml streptomycin and in a
humidified atmosphere containing 5% CO2 at 37°C.
Vector construction and cell
infection
Lentivirus-mediated β-catenin overexpression vector
(LV-CTNNB1), negative control vector (CON238), lentivirus-mediated
hsa-miR-200a-3p knockdown vector (LV-hsa-miR-200a-3p-inhibition)
and negative control vector (CON137) were all from GeneChem Co.,
Ltd. (Shanghai, China). The correct sequences and insertions were
confirmed by DNA sequencing. Transfection was performed according
to the manufacturer's instructions. On the day of vector
transduction, SGC-7901 cells were cultured at 5×104
cells/well in 24-well plates containing serum-free medium with
polybrene (5 mg/ml). At 50% confluence, cells were transfected with
recombinant experimental virus or control virus at the optimal MOI
(multiplicity of infection) of 50, and cultured at 37°C and 5%
CO2 for 4 h. Then supernatant was discarded and medium
containing serum was added. Positive and stable transfectants were
selected and expanded for further study.
Cell proliferation assay
Cell proliferation was analyzed using the MTT assay.
Briefly, cells were cultured in 96-well plates at a density of
5×103 cells/well. After adhering to the plate surface,
cells were treated with TSN of different doses for various times,
followed by 20 µl MTT (5 mg/ml) incubation for further 4 h
and subsequent 150 µl DMSO dissolution for 5 min. The
optical densities (ODs) were measured at 570 nm using an enzyme
immunoassay analyzer (Bio-Rad, Hercules, CA, USA). The cell
proliferation inhibition rates were calculated using the following
formula: 1 − OD (experiment) / OD (control). The experiment was
performed in triplicate.
Flow cytometric analysis
Annexin V/PI apoptosis detection kit was used to
detect cell apoptosis. Briefly, cells (1×106) treated
with or without TSN for 48 h were harvested and suspended in
binding buffer according to the instruction of the apoptosis kit.
Approximately 5 µl Annexin V and 5 µl PI were then
added to the fixed cells for 20 min in darkness at room
temperature. Then, Annexin V binding buffer was added to the
mixture before analysis by fluorescent activated cell sorting
(fACS) on a flow cytometer. Three separate experiments were
performed for each clone.
For cell cycle distribution analysis, cells were
incubated in serum-free medium for 24 h followed by treatment with
TSN for another 48 h. Then cells were trypsinized, washed with PBS,
fixed with 75% cold ethanol overnight at 4°C and washed with PBS
again. Afterwards, the fixed cells were stained with PI in the
presence of RNase A for 30 min in the dark. The samples were then
analyzed on FACsort flow cytometer (Becton-Dickinson, Mountain
View, CA, USA). The DNA content in cells could be read according to
PI fluorescence. ModFit3.0 software (Verity Software House,
Topsham, ME, USA) was used for cell cycle analysis. The experiment
was performed in triplicate.
Transwell invasion and wound-healing
assay
SGC-7901 cells were suspended in serum-free medium
with or without TSN after starvation in serum-free medium for 24 h.
The suspension were then added to the upper chamber coated with
Matrigel (3.9 µg/µl, 80 µl), while the lower
chamber was filled with RPMI-1640 medium containing 10% bovine
serum. After 24-h incubation, the non-invaded cells on the upper
side of the chamber were washed away and cells attached to the
bottom were fixed with 75% alcohol and stained with crystal violet.
The number of cells invading through the Matrigel were counted
using microscope in three random fields of each group.
Cell migration was assessed by wound-healing assay.
Briefly, the adherent cells in a 6-well plate
(1×106/well) were wounded with a standard 200-µl
pipette tip to form a 1-mm wide strip across the well. Then the
wounded monolayers were washed twice with PBS before incubation in
medium with or without TSN. After 48-h incubation, the wound
closure was observed and photographed with a microscope.
RNA extraction and qRT-PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen) according to the manufacturer's protocol. Total RNA (1
µg) was used to synthesize cDNA by reverse transcription
using M-MLV reverse transcriptase and cDNA amplification was
performed using the Power SYBR Green Master Mix kit (for AKT, ERK,
GSK3β, β-catenin, c-Myc, cyclin D1, Bcl-2 and MMP-9). MiScript
Reverse Transcription kit and miScript SYBP Green PCR kit were for
miR-200a according to the manufacturer's instructions. The primer
sequences are shown in Table I.
The expression of human β-actin or U6 was used as internal control.
Cycle threshold (Ct) values were obtained graphically for the
target genes, miR-200a, U6 and β-actin. ΔCt = Ct (target genes or
miR-200a) − Ct (β-actin or U6). ΔΔCt = ΔCt (experimental group) −
ΔCt (control group). Data were analyzed using the comparative Ct
method (2−ΔΔCt). Each sample was tested in
triplicate.
 | Table ISequences of the primers used in the
real-time PCR amplifications. |
Table I
Sequences of the primers used in the
real-time PCR amplifications.
| Gene primer | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| miR-200a |
AACACTGTCTGGTAACGATGTCGT | miScript SYBP Green
PCR Kit Universal primer |
| U6 |
GGATTAACGATACAGAGAAGATT | miScript SYBP Green
PCR Kit Universal primer |
| AKT |
GGGACAGAGGAGCAAGGT |
CGACAGCGGAAAGGTTAA |
| ERK |
TACACCAACCTCTCGTACATCG |
CATGTCTGAAGCGCAGTAAGATT |
| GSK-3β |
GACTAAGGTCTTCCGACCCC |
AAGAGTGCAGGTGTGTCTCG |
| β-catenin |
GGCCATATCCACCAGAGTGAA |
GCCAATGGCTTGGAATGAGA |
| c-myc |
CACCAGCAGCGACTCTGA |
GATCCAGACTCTGACCTTTTGC |
| Cyclin D1 |
GAAGTTGCAAAGTCCTGGAGC |
ATGGTTTCCACTTCGCAGCA |
| Bcl-2 |
TCGCCCTGTGGATGACTGAG |
CAGAGTCTTCAGAGACAGCCAGGA |
| MMP-9 |
CGCGCTGGGCTTAGATCAT |
GGTGCCGGATGCCATTC |
| ACTB |
GGCCAACCGCGAGAAGAT |
CGTCACCGGAGTCCATCA |
Western blot analysis
Cytoplasmic proteins were extracted with RIPA buffer
and nuclear protein extraction was conducted using Nuclear
Extraction kit (Life Technologies, Carlsbad, CA, USA). Protein
concentrations were determined using Bradford assay and cell
extracts were boiled for 5 min in loading buffer. Equal amount of
proteins were separated on 12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis before translocation onto
polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA,
USA). Then the membranes were blocked with 5% skim milk for 1 h.
The primary antibodies against AKT, p-AKT, ERK, p-ERK, GSK3β,
p-GSK3β, β-catenin, histone H3, c-Myc, cyclin D1, Bcl-2 and MMP-9
were diluted according to the manufacturer's instructions and the
membranes were incubated overnight at 4°C. Afterwards, the
membranes were washed three times followed by incubation in
HRP-labeled goat anti-rabbit IgG secondary antibodies at a dilution
ratio of 1:4,000 at room temperature for 1 h. Immunodetection was
performed with an ECL-Plus/kit.
In vivo orthotopic xenograft tumor
model
All animals were purchased from Shanghai Laboratory
Animal Center of Chinese Academy Sciences (Shanghai, China) and
were adapted to cages with 12-h light/dark cycle in a
temperature-controlled environment for study. SGC-7901 cells with
or without transduction were subcutaneously injected and maintained
by passage in the hypodermis of female BALB/c/nu/nu mice (4–5 weeks
of age, 18–20 g). The female BALB/c/nu/nu mice (4–5 weeks of age,
18–20 g) were randomly divided into 4 groups (n=5/group). Mice in
groups 1 and 2 were orthotopically implanted in the stomach with
subcutaneous GC tissue pieces formed by SGC-7901 cells without
transduction while SGC-7901 cells transfected with LV-CTNNB1 or
LV-hsa-miR-200a-3p inhibition belonged to groups 3 and 4,
respectively. Before sugery, the female mice were administered with
rhabarber and glauber to further reduce the immunity and improve
the rate of tumor formation. The surgical procedure of gastric
orthotopic transplantation was performed as previously reported
(41). Briefly, subcutaneous GC
tissue pieces (~2.0 cm × 2.0 cm × 1.0 cm) were harvested and minced
into small pieces (~1 mm3). The female nude mouse
stomach was gently exteriorized via a left-side upper abdominal
incision and one small tissue pocket in the middle wall of the
greater curvature was cut. One tumor piece was placed into the
pocket and fixed with a drop of medical tissue glue (gifts from
Department of General Surgery, Shanghai Jiao Tong University
Affiliated Shanghai Sixth People's Hospital, Shanghai, China). The
stomach was then relocated into the abdominal cavity followed by
the abdominal closure with 4-0 absorbable sutures. Seven days after
surgery, TSN was dissolved in RPMI-1640 medium and
intraperitoneally given to all mice in groups 2, 3 and 4 at a dose
of 0.20 mg/kg/day and 0.2 ml once every other day for 35 days.
Meanwhile, the equal volume of RPMI-1640 medium was administered to
mice in group 1 in the same way. All mice were sacrificed by
cervical dislocation and dissected 24 h after the final medicine.
The orthotopically implanted tumor tissues were removed from
stomach and was measured with a caliper and calculated with the
formula volume = (length × width2)/2. The metastatic
nodules in each liver were observed and counted. All in vivo
animal studies were approved by the Animal Ethics and Research
Committee of Shanghai Jiao Tong University and in accordance with
the Internal Biosafety and Bioethics Guidelines of School of
Medicine, Shanghai Jiao Tong University.
Immunohistochemistry (IHC)
Immunohistochemistry was performed to assay the
expression of tissue proteins in β-catenin pathway. Briefly, the
gastric tumor tissue slides were deparaffinized, rehydrated,
antigen-retrieved (performed with 10 mM sodium citrate buffer, pH
6.0, at 90°C for 30 min), blocked and antibody-incubated in turn.
The slides were preincubated with 0.04% bovine serum albumin to
block non-specific binding. Subsequently, the slides were incubated
with primary polyclonal antibodies (ABclonal Biotech Co., Ltd.) at
a dilution of 1:200 overnight at 4°C and a secondary antibody
(KeyGen Biotech Co.) at room temperature for 1 h. Finally, the
sections were stained with DAB (3,3-diaminoben-zidine) and
counterstained with hematoxylin. Images were visualized under a
microscope (Olympus, Tokyo, Japan).
Statistical analysis
SPSS 20.0 was used for the statistical analysis. All
of the values were recorded as the mean ± standard deviation (SD).
Independent-sample t-test and one-way analysis of variance (ANOVA)
were used to analyze statistical differences of experimental data
between two groups and more than two groups, respectively.
Chi-square test (χ2) test was used to compare the
differences of proportions between two groups. Significance was
defined as p<0.05 or p<0.01.
Results
Effects of TSN on proliferation of human
GC cells and human normal gastric epithelial cells
We examined the effects of TSN on the viability of
various GC cells (MGC-803, BGC-823, HGC-27, AGS, SGC-7901 and
MKN-45) and normal human gastric epithelial cells (GES-1) using MTT
assay. As shown in Fig. 1B–H, TSN
produced marked proliferation inhibition of cells in a dose- and
time-dependent manner. The calculated IC50 values on the
basis of inhibition rates were 7.95±0.65 to 60.74±6.73 µM
(24 h), 0.70±0.09 to 8.15±0.97 µM (48 h) and 0.11±0.04 to
4.10±0.45 µM (72 h) (Fig.
1I–K). Among these cell lines, SGC-7901 cells were most
sensitive to the killing effect of TSN, whereas GES-1 was the least
sensitive. The results indicate that at a certain dosage range, TSN
might selectively kill human GC cells rather than normal human
gastric epithelial cells.
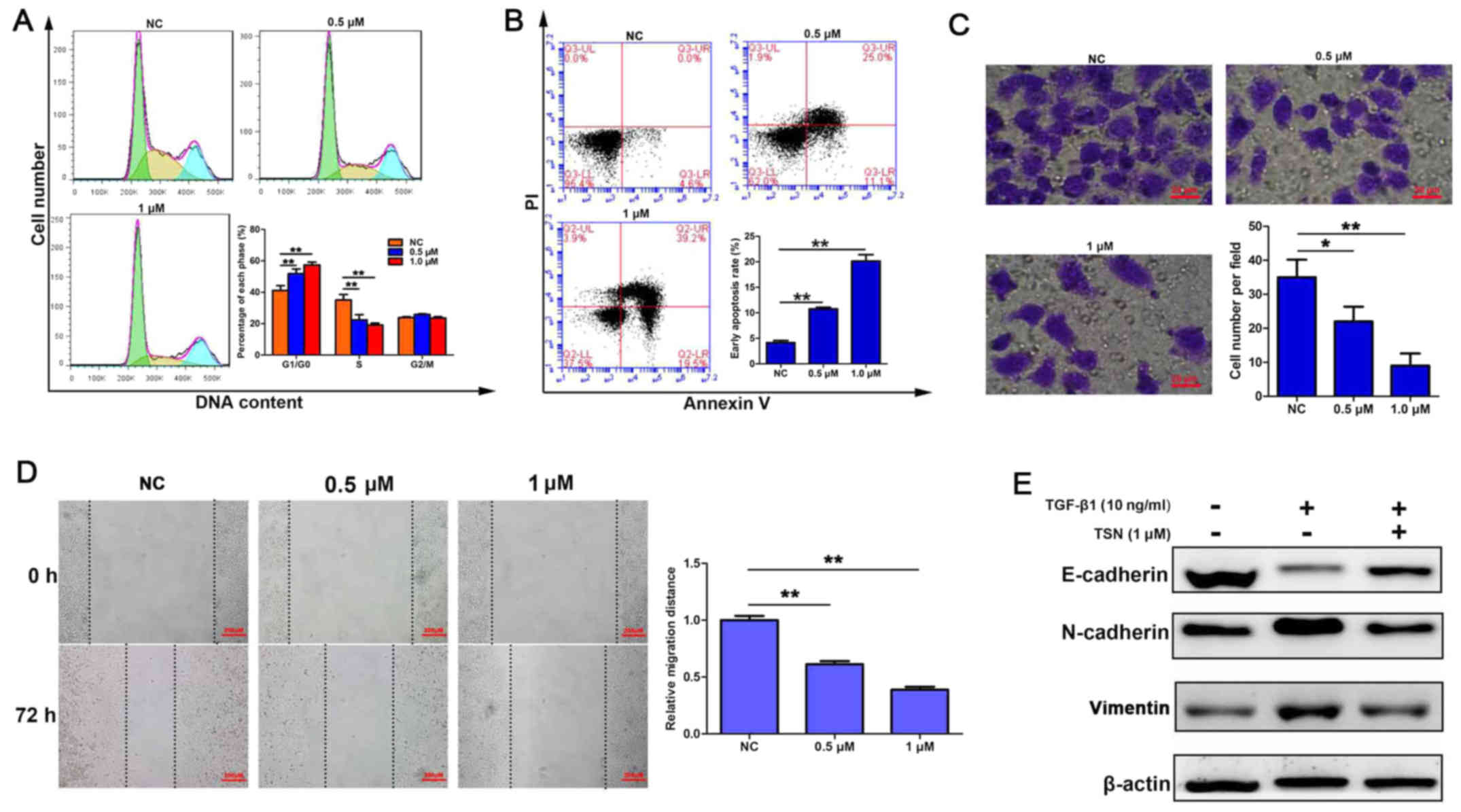
TSN induces cell cycle arrest and
apoptosis and inhibits invasion, migration and TGF-β1-induced EMT
in SGC-7901 cells
Since TSN exerted the most powerful cytotoxicity on
SGC-7901 cells, we next used PI staining and flow cytometric
analysis to determine if TSN influenced distribution of the cell
cycle. As shown in Fig. 2A, the
proportion of cells at G0/G1 phase increased while that at S phase
decreased after treatment with TSN (0.5 and 1 µM) for 48 h
in a dose-dependent manner. Then Annexin V/PI double staining and
flow cytometric analysis were performed to assess the rate of
apoptosis. As indicated in Fig.
2B, after treating for 48 h, TSN induced a dose-dependent
increase of cells undergoing early apoptosis. Furthermore, invasion
potential was determined by Transwell assay and the results showed
that TSN suppressed cell invasive capacity also in a dose-dependent
manner (Fig. 2C). The
wound-healing assay confirmed the slower migration of SGC-7901
cells by TSN treatment (Fig. 2D).
In addition, western blot analysis showed that expression of
E-cadherin, which was an epithelial marker and reduced by TGF-β1
(10 ng/ml), was increased by TSN. TSN also led to significant
downregulation of mesenchymal markers (vimentin and N-cadherin)
expression elevated by TGF-β1 (Fig.
2E).
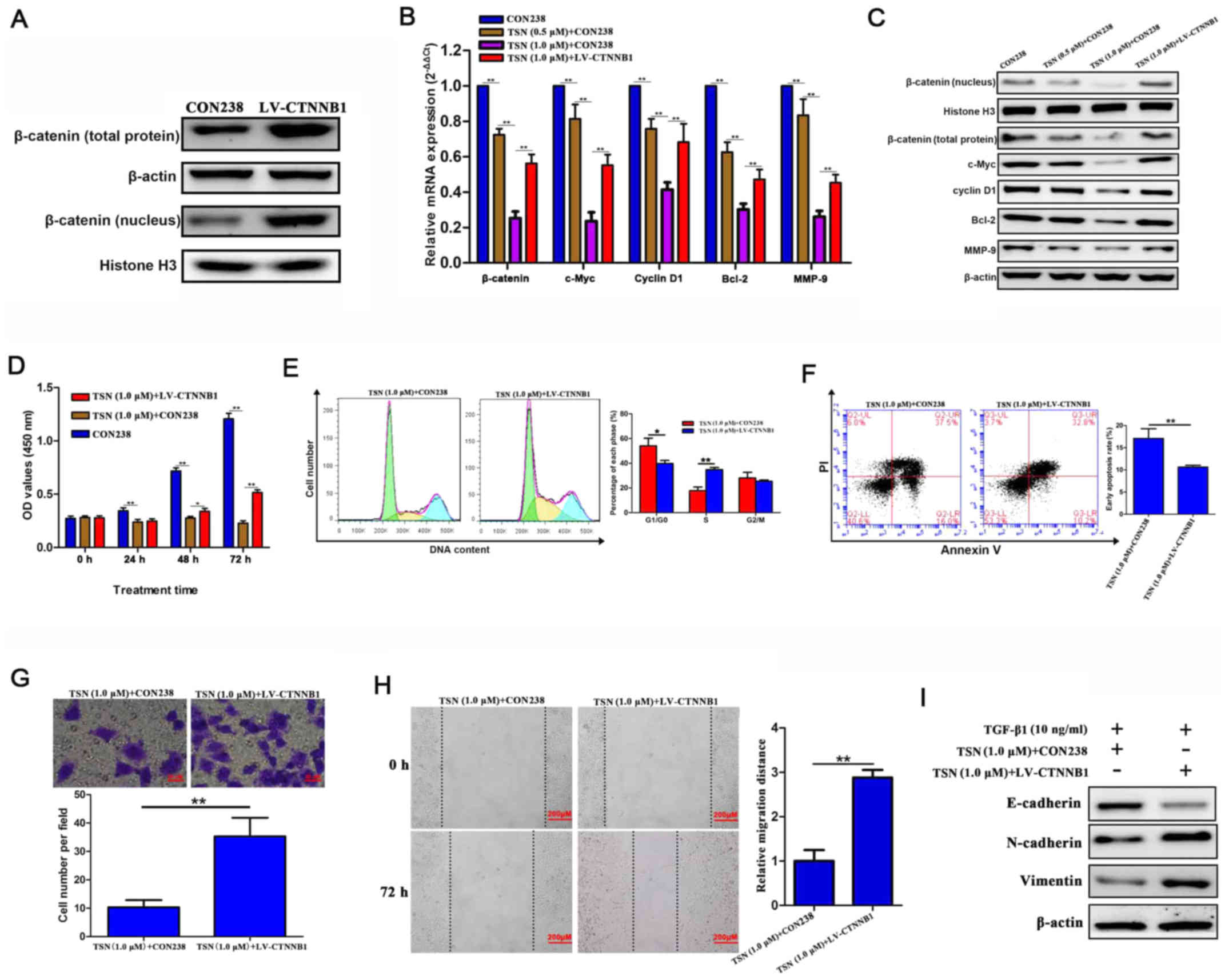
TSN has antitumor effects on SGC-7901
cells via β-catenin signaling
We hypothesized effects of TSN on SGC-7901 cells
could be mediated by β-catenin signaling. β-catenin and its
downstream target genes were assessed by qRT-PCR and western
blotting. The β-catenin protein expression (including that in the
nucleus) in SGC-7901 cells transfected by LV-CTNNB1 significantly
increased comparing with those without transfection (Fig. 3A). TSN could significantly decrease
the mRNA and protein levels of genes in β-catenin pathway
(β-catenin, c-Myc, cyclin D1, Bcl-2 and MMP-9) in SGC-7901 cells in
a dose-dependent manner and exogenous β-catenin overexpression
abolished the effects (Fig. 3B and
C). functionally, increasing the levels of β-catenin reversed
the effects of TSN on biological behavior of SGC-7901 cells,
including proliferation, cell cycle progression, apoptosis,
invasion, migration and TGF-β1-induced EMT (Fig. 3D–I). These findings suggest that
TSN might possess antitumor effects on SGC-7901 cells through
repressing β-catenin signaling in vitro.
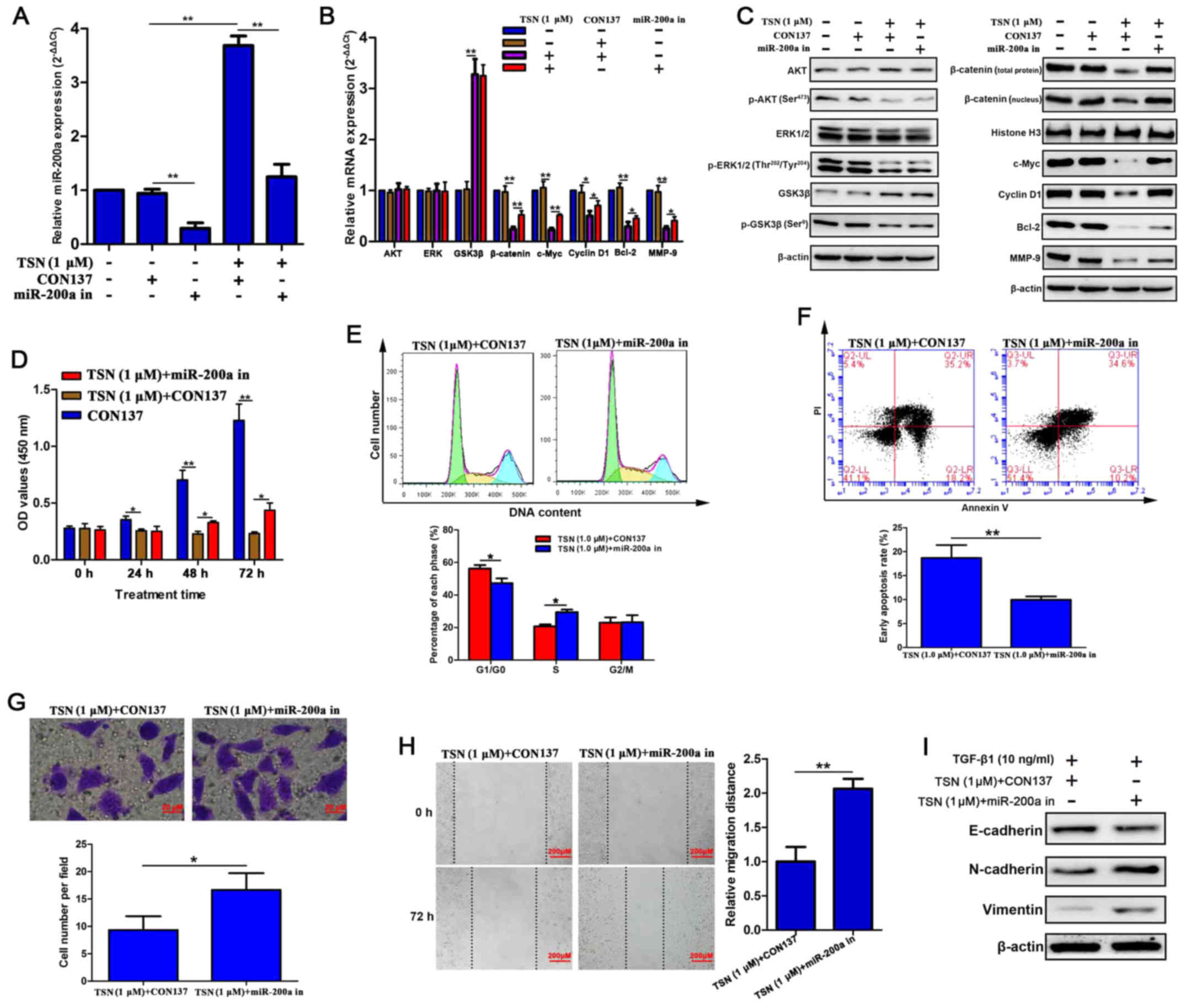
TSN suppresses β-catenin pathway partly
through upregulating miR-200a
To further verify whether miR-200a mediates the
inhibitory effects of TSN on β-catenin pathway, we established
stable SGC-7901 cell lines transfected with
LV-hsa-miR-200a-3p-inhibition in which the level of miR-200a was
lower compared with that in SGC-7901 cells without transfection
(Fig. 4A). After treating for 48
h, TSN dramatically elevated miR-200a expression which was
abrogated by miR-200a silencing (Fig.
4A). More importantly, knockdown of miR-200a attenuated
suppressive effects of TSN on mRNA and protein expression of
β-catenin, c-Myc, cyclin D1, Bcl-2 and MMP-9 (Fig. 4B and C). Nevertheless, it is worth
noting that miR-200a silencing did not influence protein level of
upstream regulators of β-catenin (p-AKT, p-ERK and p-GSK3β) which
were downregulated by TSN (Fig.
4C). Results also showed that the mRNA and protein level of AKT
and ERK were not influenced by any of the above treatments.
Knockdown of miR-200a did not affect protein level of GSK3β which
raised after TSN treatment due to the reduction of p-GSK3β protein
level (Fig. 4C). Altogether, these
data strongly indicated that TSN suppresses β-catenin pathway by
activating AKT and ERK signaling as well as facilitating miR-200a
which might only target β-catenin and then influence its downstream
genes.
TSN-induced impaired biological behaviors
of SGC-7901 cells were relieved by miR-200a silencing
To investigate the role of miR-200a in inhibitory
effects of TSN on SGC-7901 cells, MTT, flow cytometry, Transwell,
wound healing, and western blot analysis were used. Our results
displayed that when cells transfected with CON137 (negative control
for LV-hsa-miR-200a-3p-inhibition) were exposed to TSN (1
µM) for 48 h, significant cell cycle arrest at G0/G1 phase
and apoptosis were found as well as decline of proliferation,
invasion, migration and TGF-β1-induced EMT. Moreover, miR-200a
silencing enabled SGC-7901 cells to overcome the above effects of
TSN (Fig. 4D–I). Taken together,
these findings supported that miR-200a mediated suppressive effect
of TSN on β-catenin pathway and biological behaviors in SGC-7901
cells.
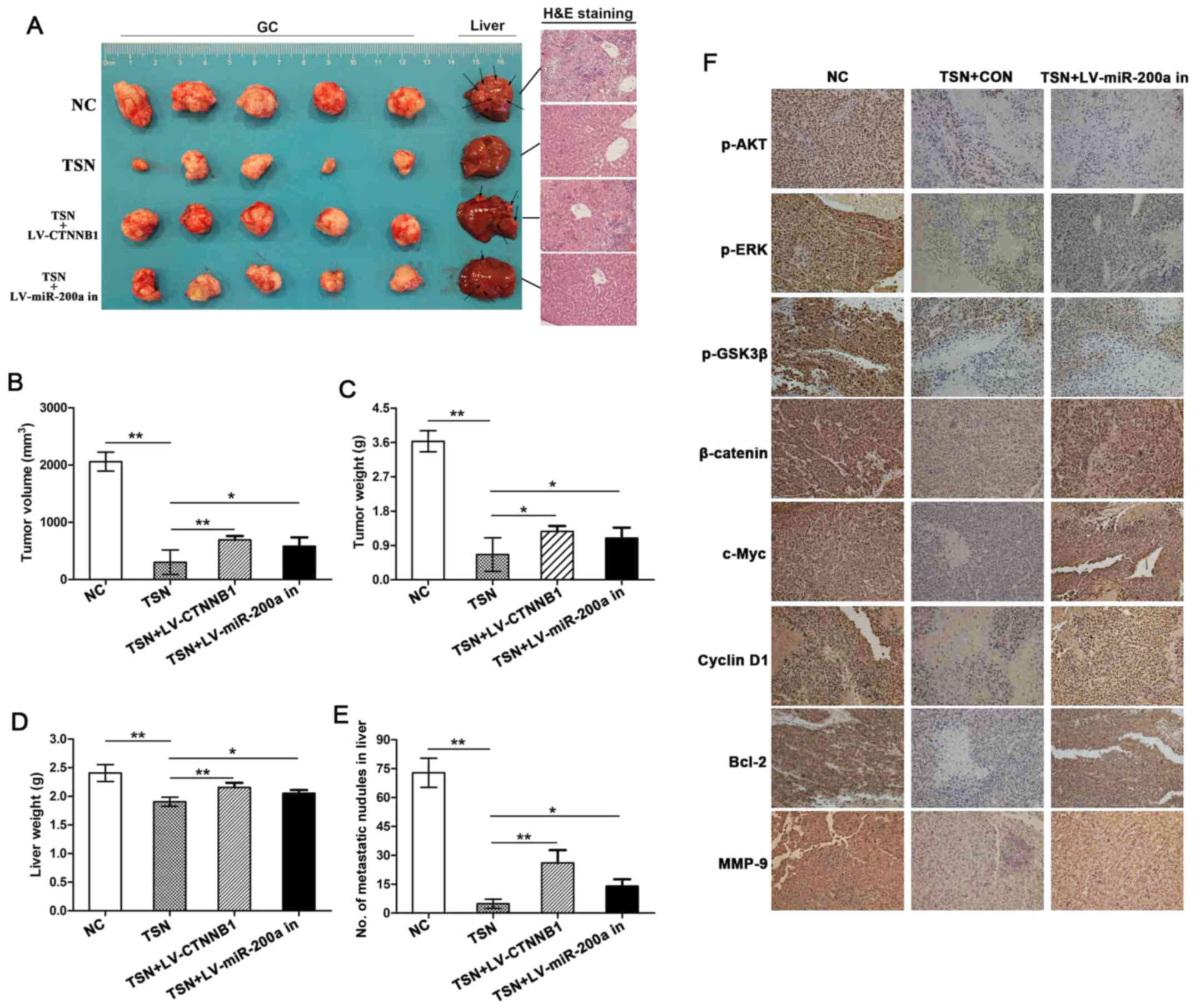
TSN inhibits GC growth and liver
metastasis via miR-200a/β-catenin axis in vivo
Given that TSN has antitumor effects on GC in
vitro, potential effects of TSN on GC growth and liver
metastasis in vivo were further investigated by establishing
orthotopic transplantation GC model in nude mice. The results
showed that mice administered by TSN had lower average volume and
weight of GC tissues, less number of metastatic nodules in liver
and lower liver weight than those of animals in control group. More
importantly, in groups of models established with SGC-7901 cells
transfected with LV-CTNNB1 or LV-hsa-miR-200a-3p inhibition, the
above curative effect of TSN was significantly weakened (Fig. 5A–E). In addition, as indicated in
Fig. 5f, IHC assay showed that TSN
reduced the expression of p-AKT, p-ERK, p-GSK3β, β-catenin, c-Myc,
cyclin D1, Bcl-2 and MMP-9 in GC tissues. Consistent with
observations in vitro, miR-200a depletion abolished
suppressive effects of TSN on the above proteins excluding p-AKT,
p-ERK and p-GSK3β in tissues. These data provided evidence that TSN
inhibits GC growth and liver metastasis via miR-200a/β-catenin axis
in vivo.
Discussion
Although progress has been made for GC therapeutic
strategy, clinical improvement is still marginal for cancer
metastasis and severe drug toxicities and resistance (3). As a result, increasing natural
products and their derivatives which have rich structural diversity
and promising therapeutic applications have appealed to
pharmacologists (42,43). TSN is such a derivative possessing
many biological functions including anticancer activity. We have
previously reported toxicity of TSN on CRC cells (40) and in this study, TSN inhibited
proliferation of a series of GC cells but exerted less cytotoxicity
on normal gastric epithelial cell GSE-1. SGC-7901 cells showed the
most sensitivity to TSN which suppressed proliferation, arrested
cell cycle at G0/G1 phase, induced apoptosis and inhibited invasion
and migration of SGC-7901 cells in a dose-dependent manner.
Moreover, administration of TSN also significantly inhibited GC
growth and liver metastasis in nude mice in vivo. EMT, by
which cells lose their epithelial phenotype, and acquire a
migratory mesenchymal phenotype, plays a pivotal role in GC
migration and invasion (18,44).
Transforming growth factor β1 (TGF-β1), a molecular member of TGF-β
signaling pathway whose dysregulation can result in tumor
development, cooperates with signaling pathways, such as Ras and
Wnt, to induce EMT in multiple human cancers (45–47).
When gastric cancer cells receive signals from their
microenvironment, such as TGF-β1, EMT occur and provide cells a
distinct advantage in tumor progression and metastasis (48). The present study manifested that
TGF-β1 could induce EMT in SGC-7901 cells by downregulating
epithelial marker E-cadherin expression and upregulating
mesenchymal marker N-cadherin and vimentin expression, consistent
with previous reports (49–51).
TSN treatment reversed the above effects of TGF-β1. All these
results suggested that TSN inhibited bioactivity of SGC-7901 cells
from different angles.
Aberrant activation of Wnt/β-catenin pathway can
cause uncontrolled cell growth and cell malignant transformation
(8,9). The central event is the accumulation
of β-catenin in the cytoplasm followed by translocating into the
nucleus, binding with T cell factor/lymphoid enhancer factor
(TCF/LEF)-1 proteins and causing transactivation of downstream
target oncogenes such as c-Myc, cyclin D1, Bcl-2 and MMP-9
(52–54). β-catenin was also found to regulate
EMT as an E-cadherin-binding protein involved in the regulation of
cell to cell adhesion (55). We
have previously found AKT/GSK-3β/β-catenin pathway mediated
inhibitory effects of TSN on CRC cells (40). Herein, we reported that TSN
significantly decreased the mRNA and protein levels of β-catenin,
c-Myc, cyclin D1, Bcl-2 and MMP-9 in a dose-dependent manner in
SGC-7901 cells. Since c-Myc, cyclin D1, Bcl-2 and MMP-9 were
closely associated with proliferation, cell cycle progression,
apoptosis and metastasis of GC (56–59),
we speculated that inactivation of β-catenin pathway contributed to
impaired activities of SGC-7901 cells treated by TSN. Our
hypothesis was further verified by gain-of-function analysis which
revealed that β-catenin overexpression reversed the effects of TSN
on GC in vitro and in vivo. This evidence supported
that TSN exerts antitumor effects on SGC-7901 cells via β-catenin
pathway.
In recent studies, miR-200a was confirmed to be a
tumor suppressor and a negative regulator of β-catenin signaling
and β-catenin mRNA can be downregulated by miR-200a (24,33,34,60).
In view of the fact that miRNA can also widely promote target mRNA
degradation in animals including human (61,62),
the mechanism in it may be related to β-catenin mRNA degradation by
miR-200a which interacted with 3′-UTR of β-catenin mRNA. Consistent
with this, the present study revealed that TSN significantly
upregulated level of miR-200a and then decreased β-catenin mRNA in
SGC-7901 cells. In vitro and in vivo findings also
showed that knockdown of miR-200a attenuated the inhibitory effects
of TSN on mRNA and protein of β-catenin and its downstream
molecules. The reason for this may be that knockdown of miR-200a
can weaken the degradation of β-catenin mRNA which results in
increasing level of β-catenin mRNA and protein. However, silencing
of miR-200a only partly reversed suppressive effects of TSN on mRNA
expression of β-catenin. As a result, we speculate that besides
miR-200a, other TSN-regulated molecules may also participate in
decreased β-catenin mRNA by TSN treatment. More complicated
molecular mechanism involved in it will be explored in the
future.
Interestingly, p-AKT, p-ERK and p-GSK3β, the
upstream regulators of protein of β-catenin, were also repressed by
TSN but not abolished by miR-200a silencing subsequently. The
reason for this may be that targets of miR-200a include β-catenin
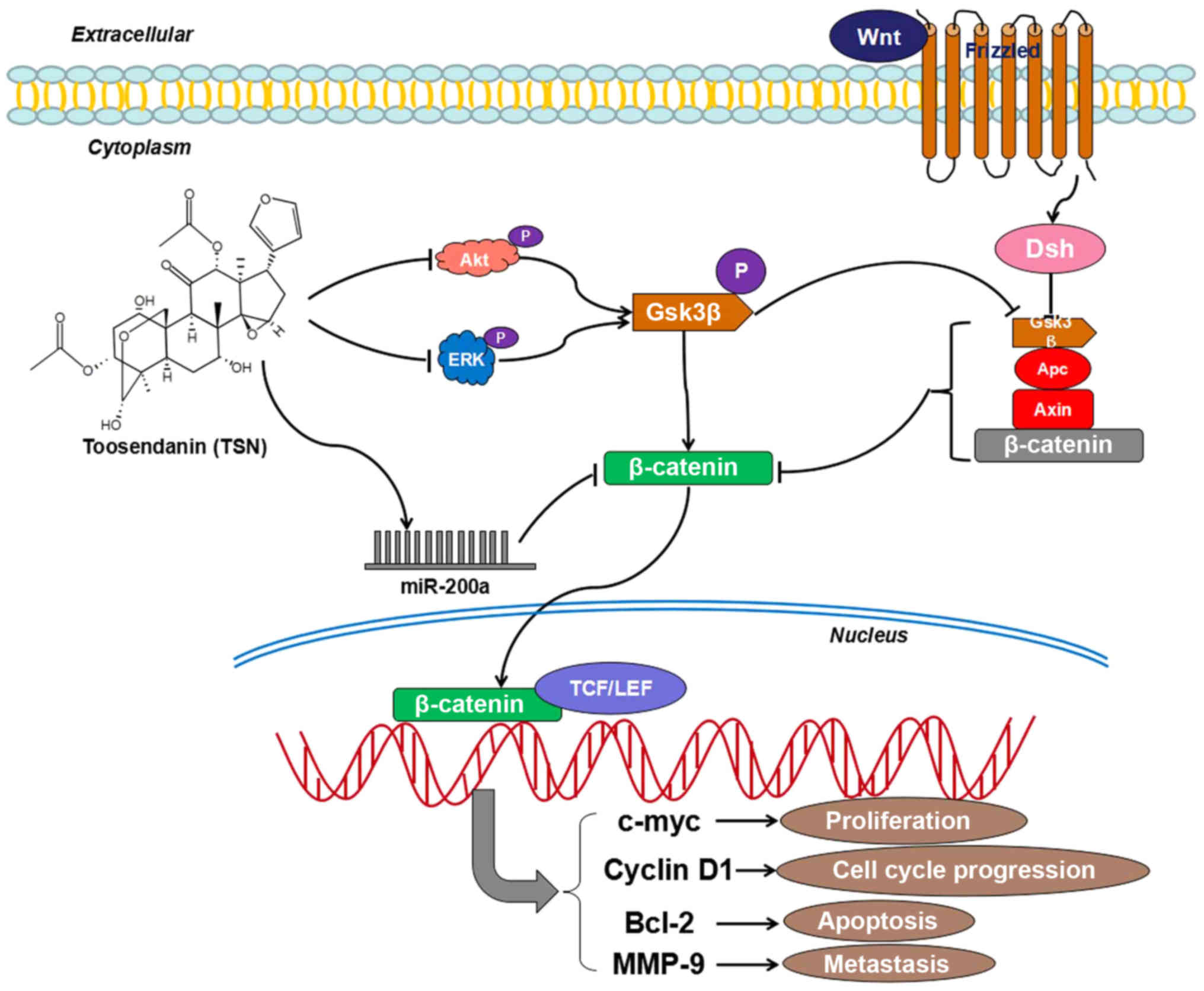
but not p-AKT, p-ERK and p-GSK3β. Taken together, TSN blocked
β-catenin signaling by means, on one hand, of raising expression of
miR-200a which in turn targets β-catenin, on the other hand, by
suppressing ERK/GSK3β/β-catenin and AKT/GSK3β/β-catenin signaling
pathways (Fig. 6). Furthermore, it
was observed that knockdown of miR-200a attenuated TSN-induced
suppression of biological behavior of SGC-7901 cells in
vitro and in vivo. This was consistent with the role of
miR-200a/β-catenin axis in anticancer effects of TSN on SGC-7901
cells.
In conclusion, our data demonstrated that TSN,
derived from Melia toosendan Sieb et Zucc, suppressed GC by
inhibiting proliferation, invasion, migration and TGF-β1-induced
EMT and inducing cell apoptosis and cell cycle arrest. The
molecular mechanisms involved may include miR-200a-mediated
downregulation of β-catenin pathway. All these findings highlight
the potential of TSN as a chemotherapeutic agent for GC therapy.
Notably, IC50 value of GC cells (0.11–32.71 µM)
was not much lower than that of normal gastric epithelial cells
(GES-1, 4.10–60.74 µM). It indicated the potential side
effects of TSN administration such as gastrointestinal reaction in
the body. Although obvious adverse reactions in nude mice
administrated by TSN (0.20 mg/kg/day) were not found in our in
vivo study, increased vigilance might be necessary when higher
dose of TSN is used. Therefore, pharmacokinetics and toxicology of
TSN in vivo are the future research objectives.
Acknowledgments
This study was supported by National Nature Science
Foundation of China (no. 81573747) and Hong Kong Scholars Program
(no. XJ2015033).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fodale V, Pierobon M, Liotta L and
Petricoin E: Mechanism of cell adaptation: When and how do cancer
cells develop chemoresistance? Cancer J. 17:89–95. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Croce JC and McClay DR: Evolution of the
Wnt pathways. Methods Mol Biol. 469:3–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu M, Ting DT, Stott SL, Wittner BS,
Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, et
al: RNA sequencing of pancreatic circulating tumour cells
implicates WNT signalling in metastasis. Nature. 487:510–513. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fu L, Zhang C, Zhang LY, Dong SS, Lu LH,
Chen J, Dai Y, Li Y, Kong KL, Kwong DL, et al: Wnt2 secreted by
tumour fibroblasts promotes tumour progression in oesophageal
cancer by activation of the Wnt/β-catenin signalling pathway. Gut.
60:1635–1643. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pinto D, Gregorieff A, Begthel H and
Clevers H: Canonical Wnt signals are essential for homeostasis of
the intestinal epithelium. Genes Dev. 17:1709–1713. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sansom OJ, Reed KR, Hayes AJ, Ireland H,
Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H,
Nathke IS, et al: Loss of Apc in vivo immediately perturbs Wnt
signaling, differentiation, and migration. Genes Dev. 18:1385–1390.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ooi CH, Ivanova T, Wu J, Lee M, Tan IB,
Tao J, Ward L, Koo JH, Gopalakrishnan V, Zhu Y, et al: Oncogenic
pathway combinations predict clinical prognosis in gastric cancer.
PLoS Genet. 5:e10006762009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clements WM, Wang J, Sarnaik A, Kim OJ,
MacDonald J, Fenoglio-Preiser C, Groden J and Lowy AM: beta-catenin
mutation is a frequent cause of Wnt pathway activation in gastric
cancer. Cancer Res. 62:3503–3506. 2002.PubMed/NCBI
|
|
14
|
Ikenoue T, Ijichi H, Kato N, Kanai F,
Masaki T, Rengifo W, Okamoto M, Matsumura M, Kawabe T, Shiratori Y,
et al: Analysis of the beta-catenin/T cell factor signaling pathway
in 36 gastrointestinal and liver cancer cells. Jpn J Cancer Res.
93:1213–1220. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song X, Xin N, Wang W and Zhao C:
Wnt/β-catenin, an oncogenic pathway targeted by H. pylori in
gastric carcinogenesis. Oncotarget. 6:35579–35588. 2015.PubMed/NCBI
|
|
16
|
Kolligs FT, Bommer G and Göke B:
Wnt/beta-catenin/tcf signaling: A critical pathway in
gastrointestinal tumorigenesis. Digestion. 66:131–144. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
18
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
López-Novoa JM and Nieto MA: Inflammation
and EMT: An alliance towards organ fibrosis and cancer progression.
EMBO Mol Med. 1:303–314. 2009. View Article : Google Scholar
|
|
20
|
Talbot LJ, Bhattacharya SD and Kuo PC:
Epithelial-mesenchymal transition, the tumor microenvironment, and
metastatic behavior of epithelial malignancies. Int J Biochem Mol
Biol. 3:117–136. 2012.PubMed/NCBI
|
|
21
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar
|
|
24
|
Liu J, Ruan B, You N, Huang Q, Liu W, Dang
Z, Xu W, Zhou T, Ji R, Cao Y, et al: Downregulation of miR-200a
induces EMT phenotypes and CSC-like signatures through targeting
the β-catenin pathway in hepatic oval cells. PLoS One.
8:e794092013. View Article : Google Scholar
|
|
25
|
Chen Z, Saad R, Jia P, Peng D, Zhu S,
Washington MK, Zhao Z, Xu Z and El-Rifai W: Gastric adenocarcinoma
has a unique microRNA signature not present in esophageal
adenocarcinoma. Cancer. 119:1985–1993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang L, Guo F, Huo B, Lv Y, Wang Y and
Liu W: Expression and clinical significance of the microRNA-200
family in gastric cancer. Oncol Lett. 9:2317–2324. 2015.PubMed/NCBI
|
|
27
|
Barron N, Keenan J, Gammell P, Martinez
VG, Freeman A, Masters JR and Clynes M: Biochemical relapse
following radical prostatectomy and miR-200a levels in prostate
cancer. Prostate. 72:1193–1199. 2012. View Article : Google Scholar
|
|
28
|
Guttilla IK, Adams BD and White BA: ERα,
microRNAs, and the epithelial-mesenchymal transition in breast
cancer. Trends Endocrinol Metab. 23:73–82. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roybal JD, Zang Y, Ahn YH, Yang Y, Gibbons
DL, Baird BN, Alvarez C, Thilaganathan N, Liu DD, Saintigny P, et
al: miR-200 Inhibits lung adenocarcinoma cell invasion and
metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 9:25–35. 2011.
View Article : Google Scholar :
|
|
30
|
Shinozaki A, Sakatani T, Ushiku T, Hino R,
Isogai M, Ishikawa S, Uozaki H, Takada K and Fukayama M:
Downregulation of microRNA-200 in EBV-associated gastric carcinoma.
Cancer Res. 70:4719–4727. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cao Q, Mani RS, Ateeq B, Dhanasekaran SM,
Asangani IA, Prensner JR, Kim JH, Brenner JC, Jing X, Cao X, et al:
Coordinated regulation of polycomb group complexes through
microRNAs in cancer. Cancer Cell. 20:187–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mateescu B, Batista L, Cardon M, Gruosso
T, de Feraudy Y, Mariani O, Nicolas A, Meyniel JP, Cottu P,
Sastre-Garau X, et al: miR-141 and miR-200a act on ovarian
tumorigenesis by controlling oxidative stress response. Nat Med.
17:1627–1635. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Su J, Zhang A, Shi Z, Ma F, Pu P, Wang T,
Zhang J, Kang C and Zhang Q: MicroRNA-200a suppresses the
Wnt/β-catenin signaling pathway by interacting with β-catenin. Int
J Oncol. 40:1162–1170. 2012.PubMed/NCBI
|
|
34
|
Cong N, Du P, Zhang A, Shen F, Su J, Pu P,
Wang T, Zjang J, Kang C and Zhang Q: Downregulated microRNA-200a
promotes EMT and tumor growth through the wnt/β-catenin pathway by
targeting the E-cadherin repressors ZEB1/ZEB2 in gastric
adenocarcinoma. Oncol Rep. 29:1579–1587. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang K, Zhang JX, Han L, You YP, Jiang T,
Pu PY and Kang CS: MicroRNA roles in beta-catenin pathway. Mol
Cancer. 9:2522010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He Y, Wang J, Liu X, Zhang L, Yi G, Li C,
He X, Wang P and Jiang H: Toosendanin inhibits hepatocellular
carcinoma cells by inducing mitochondria-dependent apoptosis.
Planta Med. 76:1447–1453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang MZ, Wang ZF and Shi YL: Involvement
of cytochrome c release and caspase activation in
toosendanin-induced PC12 cell apoptosis. Toxicology. 201:31–38.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu JC, Min ZD and Ip NY: Melia toosendan
regulates PC12 Cell differentiation via the activation of protein
kinase A and extracellular signal-regulated kinases. Neurosignals.
13:248–257. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang B, Wang ZF, Tang MZ and Shi YL:
Growth inhibition and apoptosis-induced effect on human cancer
cells of toosendanin, a triterpenoid derivative from Chinese
traditional medicine. Invest New Drugs. 23:547–553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang G, Feng CC, Chu SJ, Zhang R, Lu YM,
Zhu JS and Zhang J: Toosendanin inhibits growth and induces
apoptosis in colorectal cancer cells through suppression of
AKT/GSK-3β/β-catenin pathway. Int J Oncol. 47:1767–1774. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Li B, Zhang Y, Xiang CP, Li YY and
Wu XL: Serial observations on an orthotopic gastric cancer model
constructed using improved implantation technique. World J
Gastroenterol. 17:1442–1447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Clardy J and Walsh C: Lessons from natural
molecules. Nature. 432:829–837. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mishra BB and Tiwari VK: Natural products:
An evolving role in future drug discovery. Eur J Med Chem.
46:4769–4807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang ZY and Ge HY: Micrometastasis in
gastric cancer. Cancer Lett. 336:34–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Janda E, Lehmann K, Killisch I, Jechlinger
M, Herzig M, Downward J, Beug H and Grünert S: Ras and TGF[beta]
cooperatively regulate epithelial cell plasticity and metastasis:
Dissection of Ras signaling pathways. J Cell Biol. 156:299–313.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nawshad A, Lagamba D, Polad A and Hay ED:
Transforming growth factor-beta signaling during
epithelial-mesenchymal transformation: Implications for
embryogenesis and tumor metastasis. Cells Tissues Organs.
179:11–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shinto O, Yashiro M, Kawajiri H, Shimizu
K, Shimizu T, Miwa A and Hirakawa K: Inhibitory effect of a TGFbeta
receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous
gastric cancer cells. Br J Cancer. 102:844–851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhu Y, Liu Y, Qian Y, Dai X, Yang L, Chen
J, Guo S and Hisamitsu T: Research on the efficacy of Celastrus
Orbiculatus in suppressing TGF-β1-induced epithelial-mesenchymal
transition by inhibiting HSP27 and TNF-α-induced NF-κB/Snail
signaling pathway in human gastric adenocarcinoma. BMC Complement
Altern Med. 14:4332014. View Article : Google Scholar
|
|
50
|
Ye Z, Li J, Han X, Hou H, Chen H, Zheng X,
Lu J, Wang L, Chen W, Li X, et al: TET3 inhibits TGF-β1-induced
epithelial-mesenchymal transition by demethylating miR-30d
precursor gene in ovarian cancer cells. J Exp Clin Cancer Res.
35:722016. View Article : Google Scholar
|
|
51
|
Ma M, He M, Jiang Q, Yan Y, Guan S, Zhang
J, Yu Z, Chen Q, Sun M, Yao W, et al: miR-487a promotes
TGF-β1-induced EMT, the migration and invasion of breast cancer
cells by directly targeting MAGI2. Int J Biol Sci. 12:397–408.
2016. View Article : Google Scholar :
|
|
52
|
Cai C and Zhu X: The Wnt/β-catenin pathway
regulates self-renewal of cancer stem-like cells in human gastric
cancer. Mol Med Rep. 5:1191–1196. 2012.PubMed/NCBI
|
|
53
|
Park EJ, Chung HJ, Park HJ, Kim GD, Ahn YH
and Lee SK: Suppression of Src/ERK and GSK-3/β-catenin signaling by
pinosylvin inhibits the growth of human colorectal cancer cells.
Food Chem Toxicol. 55:424–433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cai J, Guan H, Fang L, Yang Y, Zhu X, Yuan
J, Wu J and Li M: MicroRNA-374a activates Wnt/β-catenin signaling
to promote breast cancer metastasis. J Clin Invest. 123:566–579.
2013.PubMed/NCBI
|
|
55
|
Kikuchi A, Yamamoto H, Sato A and
Matsumoto S: New insights into the mechanism of Wnt signaling
pathway activation. Int Rev Cell Mol Biol. 291:21–71. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Seo JH, Jeong ES and Choi YK: Therapeutic
effects of lentivirus-mediated shRNA targeting of cyclin D1 in
human gastric cancer. BMC Cancer. 14:1752014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Burlacu A: Regulation of apoptosis by
Bcl-2 family proteins. J Cell Mol Med. 7:249–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang F, Xue X, Zheng L, Bi J, Zhou Y, Zhi
K, Gu Y and Fang G: Long non-coding RNA GHET1 promotes gastric
carcinoma cell proliferation by increasing c-Myc mRNA stability.
FEBS J. 281:802–813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang BG, Du T, Zang MD, Chang Q, Fan ZY,
Li JF, Yu BQ, Su LP, Li C, Yan C, et al: Androgen receptor promotes
gastric cancer cell migration and invasion via AKT-phosphorylation
dependent upregulation of matrix metalloproteinase 9. Oncotarget.
5:10584–10595. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Saydam O, Shen Y, Würdinger T, Senol O,
Boke E, James MF, Tannous BA, Stemmer-Rachamimov AO, Yi M, Stephens
RM, et al: Downregulated microRNA-200a in meningiomas promotes
tumor growth by reducing E-cadherin and activating the
Wnt/beta-catenin signaling pathway. Mol Cell Biol. 29:5923–5940.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Giraldez AJ, Mishima Y, Rihel J, Grocock
RJ, Van Dongen S, Inoue K, Enright AJ and Schier AF: Zebrafish
MiR-430 promotes deadenylation and clearance of maternal mRNAs.
Science. 312:75–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lim LP, Lau NC, Garrett-Engele P, Grimson
A, Schelter JM, Castle J, Bartel DP, Linsley PS and Johnson JM:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|