Introduction
Breast cancer is associated with the highest
incidence and mortality rates of malignancies affecting women
worldwide and the World Health Organization (WHO) estimates that
the incidence of breast cancer will increase in the next 10 years
(1). The triple-negative phenotype
(TNP) pertaining to the absence or very low expression of estrogen,
progesterone and epidermal growth factor receptors, is one of the
most aggressive breast cancer subtypes that correlates with a poor
prognosis (2). These
characteristics combined with limited therapeutic choices
frequently lead to chemoresistance and metastasis (3,4).
Therefore, the development of novel therapeutic strategies is
urgently required.
Simvastatin (SIM), a lipophilic statin widely used
as an anti-cholesterolemic drug, has shown potential for use in the
suppression of the proliferation and metastasis of colon, prostate,
melanoma, lung, lymphoma, liver and breast cancer cells (5–10).
In addition, the protective effects of SIM have been reported in a
prospective Danish study where women prescribed with SIM exhibited
reduced breast cancer risk for several years (11). This agent acts by inhibiting the
3-hydroxy-3-methylglutaryl-coenzyme a (HMG-CoA) reductase in the
mevalonate pathway, blocking the formation of intermediary products
in the biosynthesis of cholesterol, such as geranylgeranyl and
farnesyl pyrophosphate, and affecting the prenylation of small
GTPases, such as Rho, Ras and others (12). SIM deregulates isoprenylation
compounds, resulting in a direct effect against the activation of
cell proliferation, adhesion and survival signaling cascades
(13). Some data suggest that SIM
can reverse acquired resistance to cetuximab, bortezomib or
doxorubicin, and is capable of suppressing cancer stem-like
populations in human breast cancer (6,14–16).
Pentoxifylline (PTX), a methylxanthine derivate, is
well known for its effects as a sensitizer to chemo-radiotherapy
(17). It has been used in
clinical practice as a hemorheologic agent for more than 35 years,
due to its abilities of improving oxygenation, lower platelet
aggregation and thrombus formation (18). PTX has also been reported to have
biological activities, such as those of a phosphodiesterase
inhibitor, inflammatory cytokine regulator, immunomodulator,
antioxidant and antifibrotic agent (19). Oncological studies have indicated
that PTX in combination with thiotepa, cisplatin, melphalan,
doxorubicin, vincristine or the proteasome inhibitor, MG132, exerts
anti-proliferative effects probably by stimulating pro-apoptotic
signals (20–22). Recent studies have also
demonstrated its capacity to modify the expression profiles of
matrix metalloproteinase (MMP)-2 and MMP-9, adhesion molecules, and
chemokine receptors in melanoma cells and MDA-MB-231 breast cancer
cells (23,24).
Recently, an increasing number of novel target
molecules and novel drugs have made readily available in clinical
practice or are currently undergoing clinical trials or are under
development. However, their therapeutic effects seem less than
satisfactory. On the other hand, rational drug use and the best
combination regimen would also lead to better anticancer therapy.
Thus, the aims of this study were to determine an effective
combination treatment, in an aim to help the clinician to establish
an ideal protocol for improving the management of breast cancer
characterized by TNP. The drugs SIM and PTX have been proven to
exert antitumor effects individually; however, to date, at least to
the best of our knowledge, their combined effects on breast cancer
cells with the TNP have not yet been examined. Therefore, this
study was carried out to evaluate the hypothesis that the
concomitant administration of both these compounds may strengthen
their anticancer effects.
Materials and methods
Cell culture and treatment
The MDA-MB-231 human breast cancer cells (HTB-26,
ATCC®) were cultured at 37°C with 5% of CO2
in RPMI-1640 medium (Eurobio, Les Ulis, France) containing 10% FBS,
1% glutamine and 0.1% antibiotics (penicillin/streptomycin). The
cells were treated alone or in combination with SIM (Sigma)
dissolved in DMSO and PTX (Sigma) in PBS at the indicated
concentrations. Cells treated with DMSO (<0.1%) were used as
controls.
Cell viability and clonogenic assay
The sensitivity to cytotoxic drugs was evaluated by
colorimetric MTT (Alfa Aesar, Ward Hill, MA, USA) assay. Following
overnight seeding into 96-well culture plates, the cells were
treated with 0–50 µM of SIM or 0–50 mM of PTX for 24–48 h.
Subsequently, 10 µl of MTT reagent were added to each well
followed by incubation of 3 h. Absorbance was measured on an ELISA
reader (BioTek Instruments, Inc., Winooski, VT, USA) at a 550 nm
wavelength. Cell viability percentages were calculated taking into
account the control as 100%. The half maximal inhibitory
concentration (IC50) value was estimated in each case
employing GraphPad Prism v6 statistical software. Subtoxic
concentrations (0.5 mM for PTX and 0.5 µM for SIM) were
selected for combination studies and their cytotoxicity was
evaluated by MTT assay. The combination index values were
calculated based on the Chou-Talalay median-effect equation
(25,26) permitting the mathematical
prediction of the response nature as synergy <1, additive =1 and
antagonism >1 using fractional effects of IC25,
IC50, IC75 and IC90.
SIM at 0.5 µM and PTX at 0.5 mM were also
used to treat the cells in the presence or absence of the autophagy
blocker, 3-methyladenine 1 mM (Sigma-Aldrich, St. Louis, MO, USA)
for 24 h. The survival rates of these cells were then analyzed by
MTT assay.
Colony formation assay was performed to evaluate the
long-term effects on cell recovery and the ability of the cells to
proliferate and form new colonies. The colony formation assay was
performed with washed drug-treated cells (after 24 and 48 h of
treatment with SIM at 0.5 µM and PTX at 0.5 mM) in drug-free
medium. The cells were then re-seeded in 6 well-plates (200
cells/well) and incubated in drug-free medium for a further 14
days. Subsequently, the cells were fixed with 4% paraformaldehyde
and stained with crystal violet 0.2% (Sigma-Aldrich); colonies
consisting of at least 50 cells were counted under an inverse
microscope (ZEISS-Axiovert 135, Zeiss, Oberkochen, Germany). Data
are expressed in terms of survival fraction (SF) which corresponds
to the quotient between sample plating efficiency (PE) and control
PE (27). In the figures, images
are representative of colonies formed by surviving cells after 14
days and bar charts (%) represent the colony numbers versus the
initial seeded cell number.
Cell cycle analysis
The effects of the treatments on cell cycle
distribution were examined by propidium iodide (PI) staining
detected by flow cytometry. Following treatment for 24–48 h, the
cells were trypsinized and collected by centrifugation, fixed in
70% of cold ethanol, washed with PBS and were then diluted in 100
µl of mixture solution containing PI (1 mg/ml) and RNase (50
µg/ml), and incubated in the dark for 30 min. Acquisition
samples were made using a FACSCalibur™ cytometer (BD Biosciences,
San Jose, CA, USA) in FL-2 channel and data were analyzed using
FlowJo V.x.0.7 software (Tree Star Inc., Ashland, OR, USA). Pre-G0
was defined as the signals at the left of the G0/G1 pick,
essentially known as the cell debris in the flow cytometer.
Annexin V-FITC labeling
To accomplish cytotoxic knowledge, quantitative and
descriptive tests were carried out to reveal the nature and degree
of cell death caused by the treatment agents. Apoptosis was
quantified following the specifications of the Annexin V-FITC PI
detection kit (4A Biotech Co., Beijing, China). Briefly, the cells
were seeded in 6 well-plates following by treatments for 12, 24 and
36 h. The cells were then collected by trypsinization, washed and
incubated for 15 min with 100 µl of staining buffer.
Acquisition and analysis were carried out on an FACSCalibur™
cytometer (BD Biosciences) using FL-1 and FL-3 channels.
Caspase 3 activity assay
The activity of the apoptotic caspase 3 enzyme was
measured using Ac-DEVD-AMC substrate (Enzo Life Sciences,
Farmingdale, NY, USA), a fluorogenic molecule that contains the
same amino acid sequence site of the PARP cleavage, and it is
weakly fluorescent, but following proteolytic cleavage by caspase 3
it becomes highly fluorescent. After 24–48 h of incubation with the
drugs, the cells were lysed in 200 µl of ice-cold lysis
buffer (without protease inhibitors) and 25 µl of
homogenized cell lysate was transferred to a black multi-well plate
format containing 175 µl of reaction buffer per well and 10
µl of substrate were added to each well. The fluorescence
intensity was measured within 2 h using a plate reader with an
excitation and emission maximum of 360 and 440 nm, respectively.
Percentage of activity was normalized vs. the control without
treatment.
Cell death detection and DNA
fragmentation examined by ELISA
The cell death detection ELISAPLUS
(Roche, Indianapolis, IN, USA) kit was used to quantify the DNA
fragments induced by apoptosis processes and present inside
cytoplasm or apoptotic bodies, as the kit quantifies the
histone-complexed DNA fragments (mono- and oligonucleosomes) out of
the cytoplasm of cells after the induction of apoptosis. Briefly,
the cells were treated for 24–48 h and were then lysed for 30 min
at room temperature and centrifuged at 200 × g for 10 min. ELISA
was carried out using 20 µl of supernatants and 80 µl
of immunoreagent placed into a well of a strepvidin-coated
microplate. The samples were incubated for 2 h at room temperature,
washed and re-incubated for 15 min with ABTS substrate. Stop
reaction solution was added and the density optical values were
obtained using 405 nm of wavelength on ELISA reader (BioTek
Instruments, Inc.). The enrichment of mono- and oligonucleosomes in
the cytoplasm were calculated and normalized vs. the control.
Autophagy analysis
The percentage of programmed cell death type II or
autophagy was determined using the Cyto-ID® autophagy
detection kit (Enzo Life Sciences) which labels specifically
autophagosomes vacuoles or autophagolysosomes. The cells were
processed identically as for apoptosis detection, but stained for
30 min with Cyto-ID reagent diluted in 500 µl of medium
without phenol red enriched with 5% FBS. Data were obtained using
the same resources as those for cell cycle assay using the FL-1
channel. Other autophagy markers (LC3A/B) were also determined by
western blot analysis using anti LC3A/B antibody
(Abcam®) as described below.
Detection of intracellular ROS
production
The general redox-stage in the treated cells was
detected using dichlorodihydrofluorescein diacetate (DCFH-DA) dye.
This probe is based on the formation and intracellular accumulation
of fluorescent products (green) resulting from H2DCF-DA
(non-fluorescent) oxidation by reactive oxygen species (ROS). After
24 h of cell seeding, the medium was replaced with phenol red-free
RPMI containing 10 µM of H2DCF-DA and the respective drug
concentrations followed by incubation for 1 h in standard culture
conditions at 37°C. Subsequently, following trypsinization, the
cellular suspension was immediately analyzed by flow cytometry
using the FL-1 channel.
Cytokine array
Cytokine expression profiles were evaluated
following the instructions of the manufacturer of the
RayBio® Human Cytokine Antibody Array (RayBiotech,
Norcross, GA, USA). Briefly, supernatants of the cells exposed to
treatments for 24 h were recovered by centrifugation (3,500 × g).
Each experimental membrane (provided with the kit) was then blocked
prior to incubation with the samples overnight at 4°C. After
washes, the membranes were incubated for 1 h with biotin-coupled
primary antibodies (provided with the kit by the manufacturer),
washed and covered with HRP-conjugated streptavidin for 2 h. The
detection was made by exposing the membranes to chemiluminescence
in the Gene Snap Syngene v7.09.17 (Ingenius Bioimaging) instrument
and signal intensities were quantified by densitometry.
Western blot analysis
After 24 h of treatment, the cells were lysed using
RIPA solution (containing protease inhibitor) under 10 pulses of
sonication with 60% amplitude. The lysate was allowed to stand for
30 min and proteins were collected by centrifugation at 3,500 × g.
All preparations were processed on ice or 4°C. Protein
concentrations were determined by Bradford method and western blot
analysis was carried out according to conventional methods. The
primary antibodies against p38k (9212), phosphor-p38k (9211), mTOR
(2983), phosphor-mTOR (5536), p65 (8242), phosphor-p65 (3033), IKBα
(4814), phosphor-IKBα (2859), IKKα (11930), IKKβ (8943),
phosphor-IKKαβ (2697), ERK1/2 (9102), phosphor-ERK1/2 (9106) were
purchased from Cell Signaling Technology (Danvers, MA, USA).
Antibodies against AKT1/2 (sc-1619), phosphor-AKT1/2 (sc-7985-R),
PI3K (sc-1637) were from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). β-actin antibody was from Sigma-Aldrich Co. (A2228) and
LC3A/B (ab128025) antibody was from Abcam (Cambridge, MA, USA).
Statistical analysis
Data are expressed as the means ± standard deviation
of 2 or 3 independent experiments carried out in triplicate.
Differences between groups were determined by one or two
statistical methods: ANOVA followed by post hoc correction using
GraphPad Prism 6 software. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
Synergistic inhibitory effects of PTX and
SIM on cell growth
Both apoptosis and autophagy are dynamic processes
which occur prior to cell death. Therefore, it was necessary to
examine the effects at different time points for the different
tests. We usually selected the time points of 12–24 h for
intracellular signaling analysis and 36 or 48 h for the observation
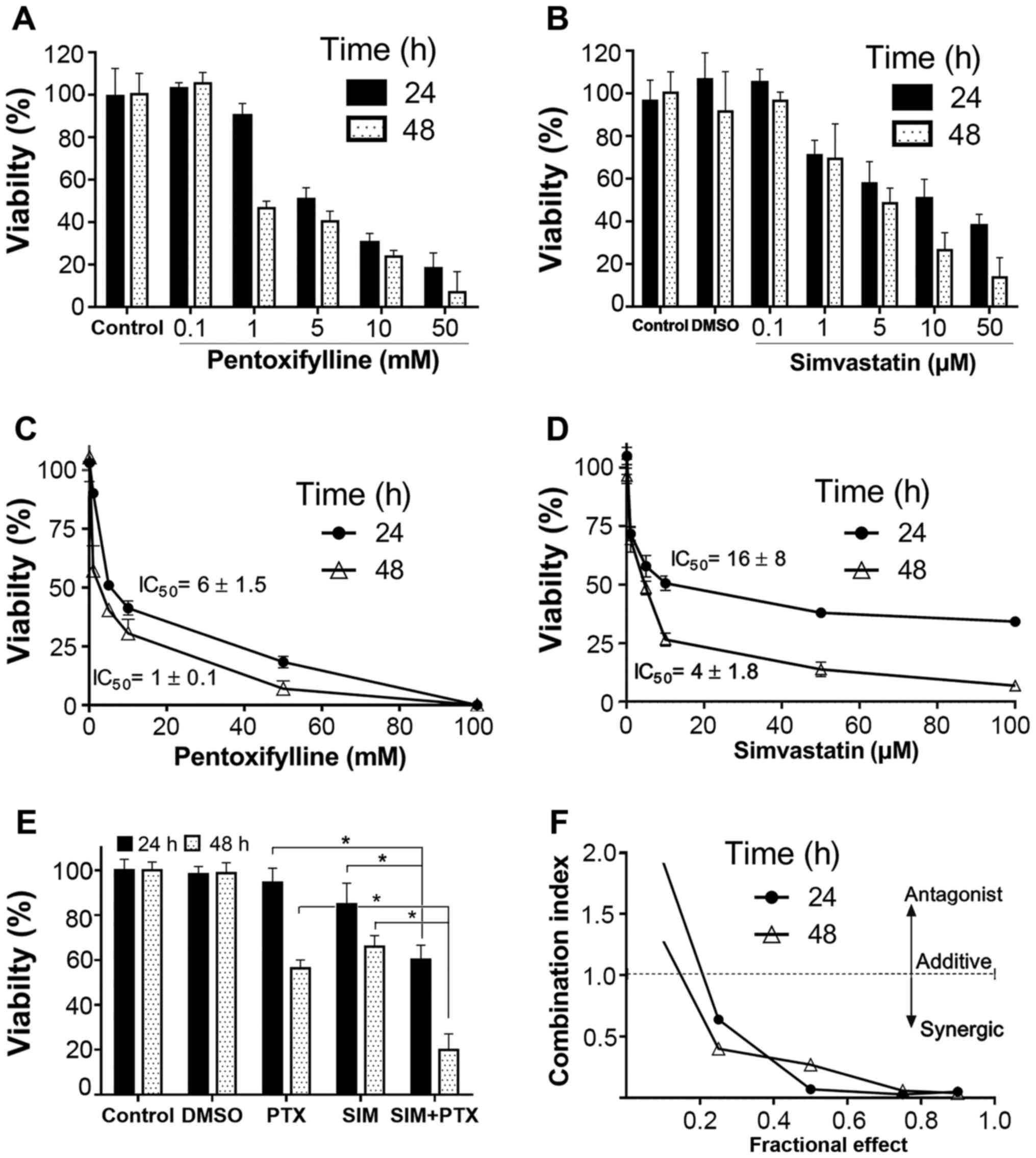
of the effects on cell viability or cell death. When used alone,
both SIM and PTX inhibited cell proliferation in a dose-dependent
manner (Fig. 1A and B). The
IC50 values were 16±8 and 4±1.8 µM for SIM, and
were 6±1.5 and 1±0.1 mM for PTX after 24 and 48 h of treatment,
respectively (Fig. 1C and D). When
the cells were treated with 0.5 mM of PTX in combination with 0.5
µM of SIM, as shown in Fig.
1E, cell proliferation was inhibited by >38% and 80% at 24
and 48 h, as compared to the single drug-treated controls (15–42%).
This indicated that the inhibitory effect was approximately doubled
as compared to treatment with SIM and PTX alone. Mathematical data
validation suggested a strong synergistic inhibitory effect on cell
growth by the combined use of the two drugs for 24 and 48 h
(Fig. 1F). In order to make data
analysis easier, the cells were treated with fixed doses of 0.5 mM
PTX and 0.5 µM SIM in the following experiments.
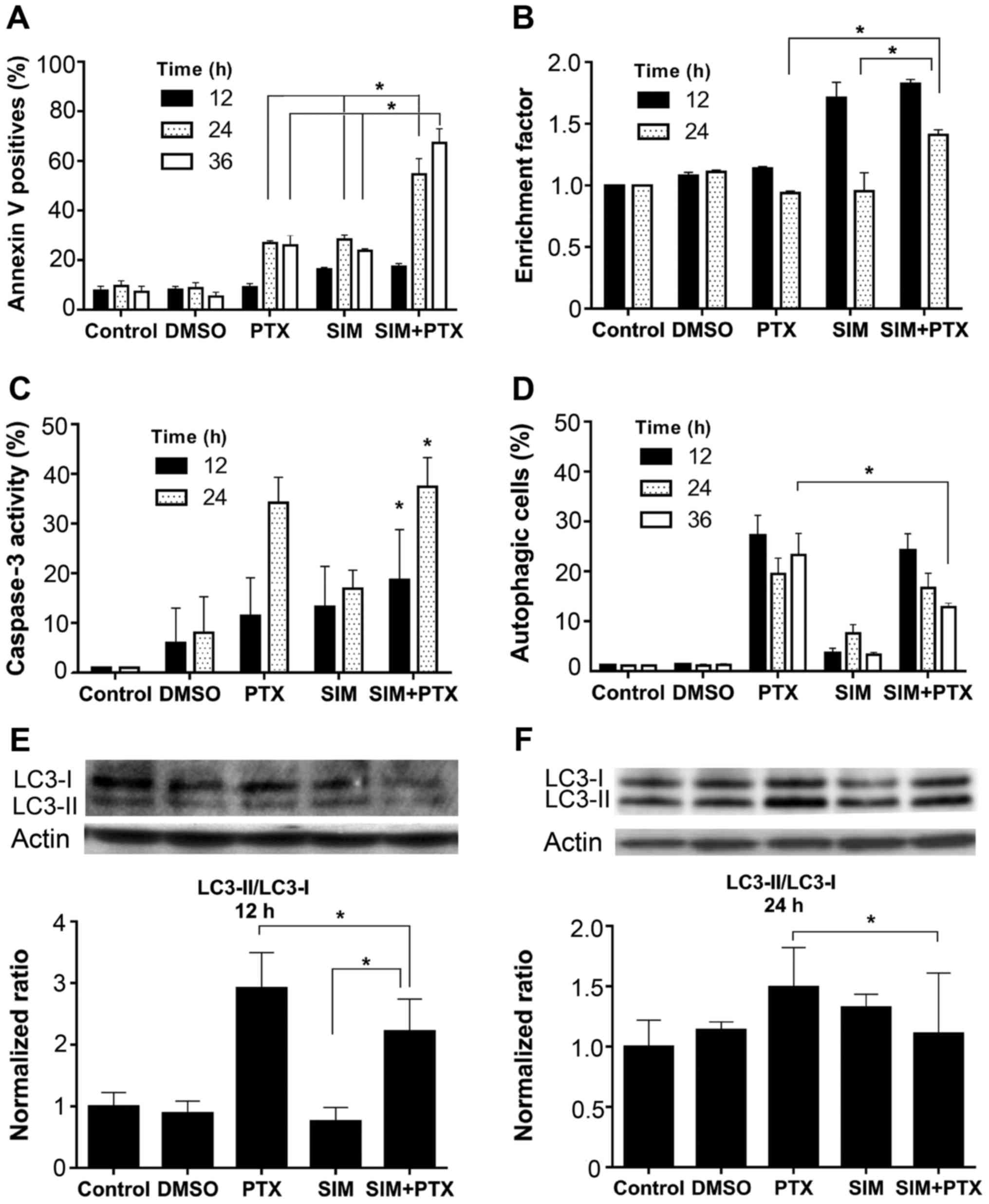
Cell apoptosis was analyzed in both early and late
stages using different assays. In Annexin V labeling detection, the
positive cell number in the mono-treated groups increased to
approximately 25% at 24 and 36 h, whereas combined treatment
increased the apoptosis to approximately 65%, and approximately
>2-fold as compared to treatment with SIM or PTX alone (Fig. 2A). Consistently, increased levels
of DNA fragmentation were observed in the SIM + PTX-treated cells
at 24 h (Fig. 2B). In accordance
with the Annexin V and DNA fragmentation results, caspase 3
activity was elevated at 24 h in all groups and the highest levels
were observed in the combination group (Fig. 2C). Thus PTX and SIM induced
apoptosis and the combined use of both agents further amplified
cell apoptosis.
PTX and SIM combination treatment reduces
cell autophagy
Autophagy was analyzed with the cells after
treatment. As shown in Fig. 2D,
0.5 mM PTX induced approximately 20–28% of cell autophagy, whereas
0.5 µM SIM treatment led to a much lower induction (only
approximately 3%). However, when PTX was used in combination with
SIM, the autophagic cell level was significantly diminished to 13%
after 36 h of treatment. This was confirmed by western blot
analysis following treatment with anti-LC3A/B antibody, a marker of
autophagy (Fig. 2E and F), showing
a decrease in the LC3-II/LC3-i ratio. When the cells were treated
with SIM for 24 h, a very low percentage of autophagic cells was
observed (Fig. 2D); however, a
relatively high LC3-II/LC3-ratio was observed (Fig. 2F). It was suggested that the
process of autophagy was also terminated after the formation of
LC3-associated autophagosomes. The possible mechanisms may be the
inhibition of autophagosome fusion with lysosomes or other
unidentified causes.
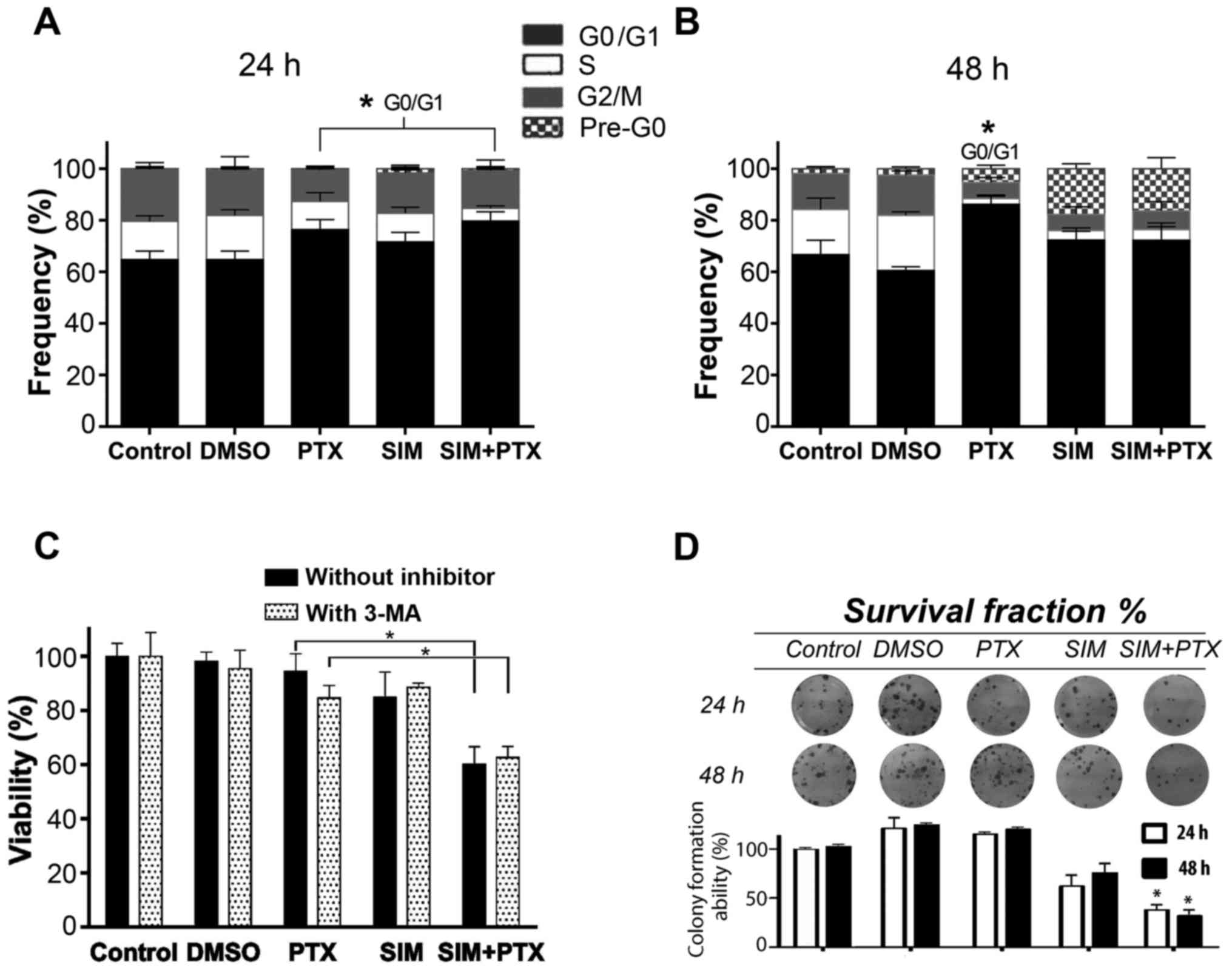
Cell cycle blockage at the G0/G1 phase is
enhanced by combination treatment
Following centrifugation, the supernatants
containing very small dead cell fragments were washed out and the
cell pellets were used for cell cycle analysis. The 3 treatment
groups showed visible blockage at the G0/G1 phase after 24 h of
treatment, especially the combination group (Fig. 3A). After 48 h of treatment, cell
death occurred, indicating an important appearance of the pre-G0
phase which was composed of cell debris in the SIM and SIM+PTX
groups (P<0.01 vs. PTX group), with a significant decrease in
the number of cells in the S and G2M phases. The cells in the PTX
group had accumulated in the G0/G1 phase instead of the pre-G0
phase after death (Fig. 3B).
Autophagy blockage enhances cell
mortality following combination treatment
To examine whether autophagy is suppressed by SIM,
the autophagy inhibitor, 3-MA, was used to treat the PTX- and
SIM-treated cells. As shown in Fig.
3C, cell viability was reduced by approximately 10% in the
PTX-treated cells following the addition of 3-MA; however, the
cells in the SIM-treated group were not affected. When 3-MA was
added to the PTX + SIM-treated cells, no further inhibition was
observed. The absence of the additional inhibition by 3-MA in the
PTX + SIM group indicated that PTX-induced autophagy was
efficiently suppressed by SIM. Therefore, autophagy seemed to
promote cancer resistance and acted as a pro-survival factor.
Combination treatment suppresses
long-term cell survival
Cell clonogenic assay was performed to further
establish the association between cell growth inhibition and cell
survival. The cells were cultured with the drugs for either 24 or
48 h, washed and then seeded into plates. After 15 days following
these treatments, as shown in Fig.
3D, the colony formation ability of the cells was examined and
the colony numbers were approximately 115–120% for the PTX group,
62–75% for the SIM group and 38–32% for the combination group at 24
and 48 h, respectively. These results indicated that the marked PTX
pro-autophagic effect was associated with an unusual cell survival
and resumed growth ability. This observation was in favor of the
hypothesis that the addition of SIM to PTX counterbalanced the PTX
pro-autophagic effect.
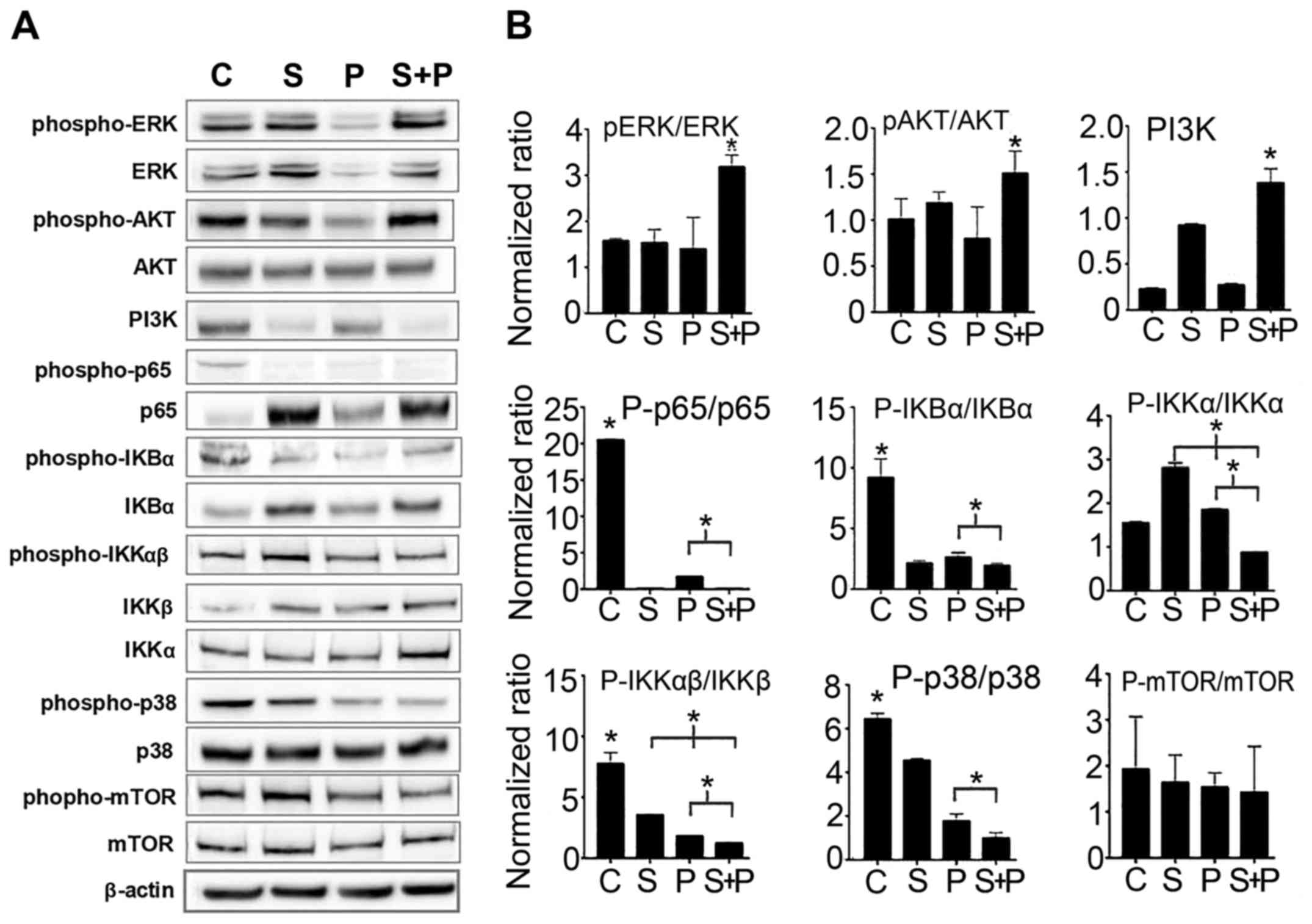
Cell signaling is deregulated by combined
treatment with PTX and SIM
In order to investigate the conceivable molecular
mechanisms implicated in the combined effects of PTX and SIM,
different cell signaling molecules were probed. As shown in
Fig. 4, the results of western
blot analysis revealed that ERK and AKT were significantly
activated following combination treatment as compared to the
untreated control cells and to the cells treated with PTX or SIM
alone (increase of >50% by examining the p-ERK/ERK ratio),
although neither PI3K nor mTOR expression was elevated.
Furthermore, the NF-κB signaling pathway in the cells treated with
both agents was downregulated, as evidenced by the decreased p65
(p-p65/p-65) and IKK (p-IKKα/IKKα and p-IKKβ/IKKβ) activation
levels compared to the cells treated with PTX alone. The NF-κB
signaling pathway is known to rescue cells from apoptosis; its
downregulation in the cells treated with both agents may decrease
chances of survival. Another signaling molecule p38 also showed a
significant downregulation in the cells treated with both agents
compared to the cells treated with PTX or SIM alone. However, the
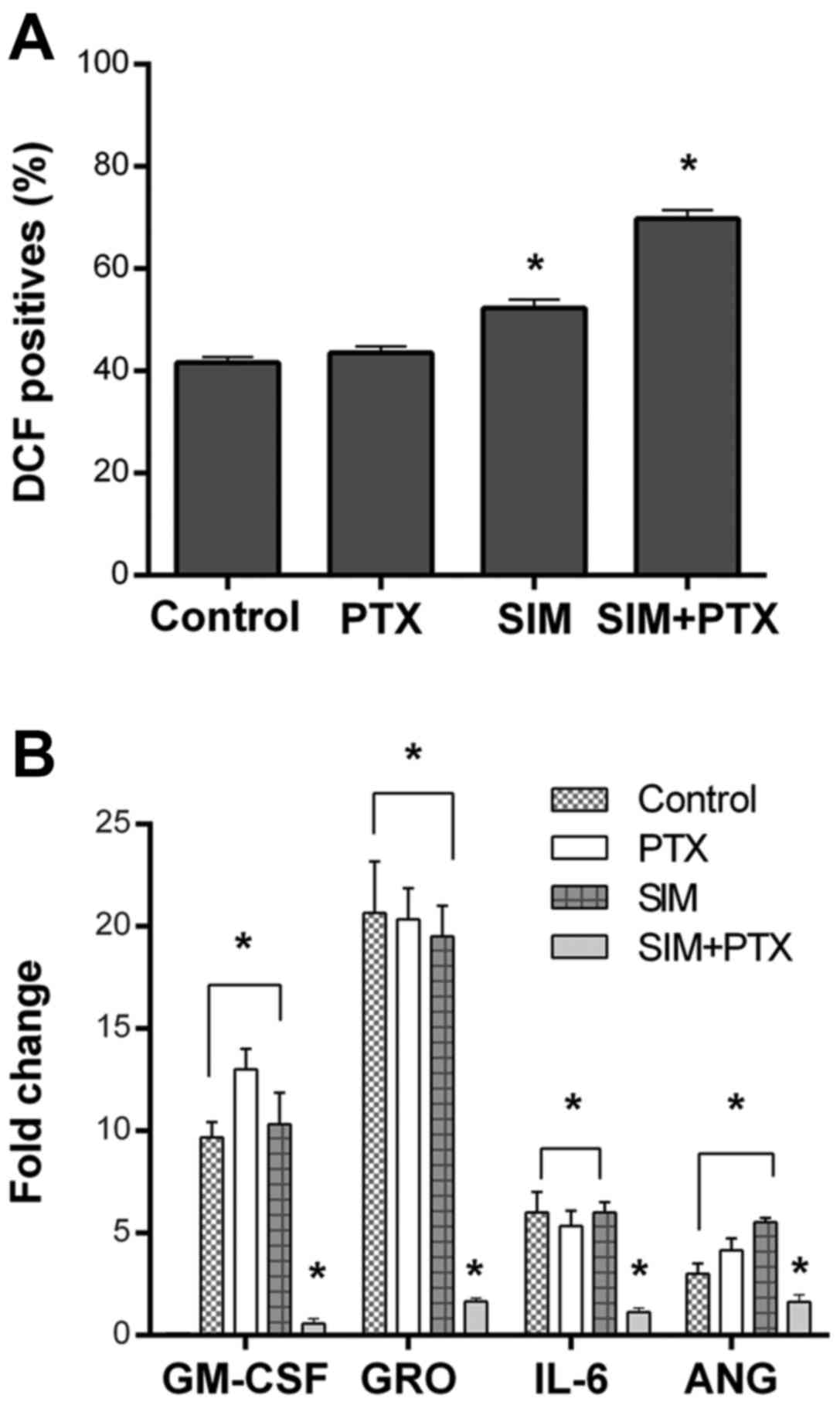
levels of reactive oxygen species (ROS) were upregulated in the SIM
+ PTX group when compared with the mono treatments (Fig. 5A).
Cytokine array assays revealed a significant
downregulation of GM-CSF, GRO, IL-6 and angiotensin when the cells
were treated with SIM + PTX. These cytokines are known to play key
roles in local inflammation (Fig.
5B) (28).
Discussion
In this study, to the best of our knowledge, we
report for the first time an alternative approach with which to
enhance the sensitivity of the triple-negative breast cancer cell
line, MDA-MB-231, involving the co-administration of SIM and PTX.
The results revealed that the cell growth and cell cycle were
inhibited by either SIM or PTX mono-treatments; however, the
combination of both drugs synergistically augmented these effects.
Our results revealed that PTX induced mostly G0/G1 cell cycle
arrest as shown by flow cytometry, as also previously reported
(22). We further observed
(Fig. 3B) that in combination with
SIM, PTX decreased the number of cells in the G0/G1 phase and the
cells began to shrink into the pre-G0 phase with cell fragmentation
after 48 h of treatment. This explained the accelerated cell death
at 24 h to 48 h, as observed in Fig.
1E. We also observed a shift in the balance between apoptosis
and autophagy at the 24 h time point. SIM induced mainly cell
apoptosis, while PTX induced both cell autophagy and apoptosis.
When the two drugs were used in combination, the autophagic rates
were reduced, while the apoptotic levels were inversely increased.
These results strongly support the existence of a well-regulated
balance between apoptosis and autophagy. Therefore, our study
revealed most likely an interesting pharmaceutical property of SIM,
indicating that it is capable of breaking the balance, altering
cell destination and inducing cell death.
The experiments with the autophagy inhibitor, 3-MA,
revealed that the addition of 3-MA affected the survival of
PTX-treated cells, but not that of SIM or PTX + SIM-treated cells.
This indicated an efficient blockage of autophagy by SIM. Autophagy
is considered a cell self-adaptation mechanism against
environment-associated stress and starvation (29). It is known to be closely connected
to apoptosis within cells since both the stimulation and inhibition
of autophagy have been reported to promote tumor cell apoptosis
(30–32). In this study, we performed
clonogenic experiments to determine whether PTX-induced autophagy
can attenuate or counterbalance apoptosis as a pro-survival factor
in MDA-MB-231 cells. The results revealed that the PTX-treated
cells re-grew very well and rapidly formed new colonies after
withdrawing PTX, demonstrating that autophagic tumor cells were
able to escape from the death following treatment by PTX.
Therefore, these results are consistent with the notion that
autophagy activation plays a role in tumor cell resistance to
drugs. This phenomenon is believed to be relevant to clinical
cancer resistance, particularly due to the growth of residue
dormant cancer cells following chemotherapy (32,33).
An attempt was made to elucidate the plausible
molecular mechanisms of cell signaling pathways of combination
treatment. In the combination-treated cells, we observed the
activation of the ERK1/2 and AKT pathways, but not the
pro-autophagic mTOR pathway as compared to the mono treatments, as
the p-ERK1/2 and p-AKT ratio was increased, but not mTOR expression
was not. To date, the role of ERK in the induction of apoptosis is
still under debate (34). ERK
inhibition has been reported to be necessary for apoptosis in
MDA-MB-231 and other tumor cells (17,32,35–38).
However, others have shown that ERK activation is required for the
induction of apoptosis (39–43).
The reason for the discrepancy of these opposing observations
remains unclear, and both could be true due to their complex
connections to different downstream signal molecules in different
cells. Our study suggested the involvement of ERK/AKT activation
using combined treatment. We further demonstrated that the
ERK-downstream targets, ROS and p38, also reacted in this manner
(ROS was upregulated but p-p38 was downregulated).
We further investigated ERK-downstream targets by
examining the NF-κB signaling pathway, since NF-κB is known to
prevent cell apoptosis. Indeed, in the combined treatment group,
NF-κB signaling was found to be inhibited, as shown by the low or
suppressed levels of the activation of p65 and IKK proteins. Since
the NF-κB signaling pathway rescues cells from apoptosis, its
absent response to ROS stress and further downregulation in the
combination-treated cells may be a mechanism with which to decrease
chances of cell survival.
In fact, the two drugs may affect tumor growth by
other mechanisms, such as cell-cell dialogues, local inflammatory
reaction and tumor angiogenesis (44,45).
Our results of cytokine array supported such an assumption, as the
combination treatment downregulated the secretion of several
inflammatory cytokines, such as IL-6, GRO, GM-SCF and angiotensin
by the tumor cells. The reduced secretion of these cytokines can be
expected to lessen tumor-induced local inflammation. As local
inflammation is known to promote tumor angiogenesis and metastasis,
therefore, an additional benefit of PTX + SIM treatment could be
expected from the diminution of inflammation and angiogenesis
within tumors.
Taken together, our results demonstrated a potential
benefit of combined therapy of PTX and SIM, which transforms
autophagic cancer cells into apoptotic cells, more efficiently
prevents tumor cell growth, and more efficiently suppresses
triple-negative breast cancer cells. At present, although many
mechanisms involved in this effect still remain to be investigated,
we hope that our results may be useful for further studies and
beneficial to clinical cancer therapy.
Acknowledgments
Y.C. Castellanos-Esparza was financially supported
by a grant from the National Polytechnic Institute (IPN) and the
National Council for Science and Technology (CONACyT) from Mexico.
S. Wu was financially supported by the Chinese Scholarship Council.
We are grateful to Mrs. Elizabeth Le Grand, Dr T. Simon, Dr
Alexander Petit and Dr Isabel Dubus for their kind assistance.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chacón RD and Costanzo MV: Triple-negative
breast cancer. Breast Cancer Res. 12((Suppl) 2): S32010. View Article : Google Scholar :
|
|
3
|
Reddy KB: Triple-negative breast cancers:
An updated review on treatment options. Curr Oncol. 18:e173–e179.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dawson SJ, Rueda OM, Aparicio S and Caldas
C: A new genome-driven integrated classification of breast cancer
and its implications. EMBO J. 32:617–628. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee J, Lee I, Han B, Park JO, Jang J, Park
C and Kang WK: Effect of simvastatin on cetuximab resistance in
human colorectal cancer with KRAS mutations. J Natl Cancer Inst.
103:674–688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoque A, Chen H and Xu XC: Statin induces
apoptosis and cell growth arrest in prostate cancer cells. Cancer
Epidemiol Biomarkers Prev. 17:88–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Favero GM, F Otuki M, Oliveira KA, Bohatch
MS Jr, Borelli P, Barros FE, Maria DA, Fernandes D and Bydlowski
SP: Simvastatin impairs murine melanoma growth. Lipids Health Dis.
9:1422010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chan KK, Oza AM and Siu LL: The statins as
anticancer agents. Clin Cancer Res. 9:10–19. 2003.PubMed/NCBI
|
|
10
|
Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS,
Cai DQ, Wu Z, Qin JW, Yu YH and Kim SK: HMG-CoA reductase
inhibitors induce apoptosis of lymphoma cells by promoting ROS
generation and regulating Akt, Erk and p38 signals via suppression
of mevalonate pathway. Cell Death Dis. 4:e5182013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ahern TP, Pedersen L, Tarp M,
Cronin-Fenton DP, Garne JP, Silliman RA, Sørensen HT and Lash TL:
Statin prescriptions and breast cancer recurrence risk: A Danish
nationwide prospective cohort study. J Natl Cancer Inst.
103:1461–1468. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Graaf MR, Richel DJ, van Noorden CJ and
Guchelaar HJ: Effects of statins and farnesyltransferase inhibitors
on the development and progression of cancer. Cancer Treat Rev.
30:609–641. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rohilla A, Rohilla S, Kumar A, Khan MU and
Deep A: Pleiotropic effects of statins: A boulevard to
cardioprotection. Arab J Chem. 9:S21–S27. 2016. View Article : Google Scholar
|
|
14
|
Gopalan A, Yu W, Sanders BG and Kline K:
Eliminating drug resistant breast cancer stem-like cells with
combination of simvastatin and gamma-tocotrienol. Cancer Lett.
328:285–296. 2013. View Article : Google Scholar
|
|
15
|
Fuchs D, Berges C, Opelz G, Daniel V and
Naujokat C: HMG-CoA reductase inhibitor simvastatin overcomes
bortezomib-induced apoptosis resistance by disrupting a
geranylgeranyl pyro-phosphate-dependent survival pathway. Biochem
Biophys Res Commun. 374:309–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sadeghi-Aliabadi H, Minaiyan M and
Dabestan A: Cytotoxic evaluation of doxorubicin in combination with
simvastatin against human cancer cells. Res Pharm Sci. 5:127–133.
2010.PubMed/NCBI
|
|
17
|
Barancik M, Bohacova V, Gibalova L, Sedlak
J, Sulova Z and Breier A: Potentiation of anticancer drugs: Effects
of pentoxifylline on neoplastic cells. Int J Mol Sci. 13:369–382.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Słoczyńska K, Kózka M, Pękala E, Marchewka
A and Marona H: In vitro effect of pentoxifylline and lisofylline
on deformability and aggregation of red blood cells from healthy
subjects and patients with chronic venous disease. Acta Biochim
Pol. 60:129–135. 2013.
|
|
19
|
Goel PN and Gude RP: Delineating the
anti-metastatic potential of pentoxifylline in combination with
liposomal doxorubicin against breast cancer cells. Biomed
Pharmacother. 68:191–200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bravo-Cuellar A, Hernández-Flores G,
Lerma-Díaz JM, Domínguez-Rodríguez JR, Jave-Suárez LF, De
Célis-Carrillo R, Aguilar-Lemarroy A, Gómez-Lomeli P and
Ortiz-Lazareno PC: Pentoxifylline and the proteasome inhibitor
MG132 induce apoptosis in human leukemia U937 cells through a
decrease in the expression of Bcl-2 and Bcl-XL and phosphorylation
of p65. J Biomed Sci. 20:132013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fan S, Smith ML, Rivet DJ II, Duba D, Zhan
Q, Kohn KW, Fornace AJ Jr and O'Connor PM: Disruption of p53
function sensitizes breast cancer MCF-7 cells to cisplatin and
pentoxifylline. Cancer Res. 55:1649–1654. 1995.PubMed/NCBI
|
|
22
|
Goel PN and Gude RP: Unravelling the
antimetastatic potential of pentoxifylline, a methylxanthine
derivative in human MDA-MB-231 breast cancer cells. Mol Cell
Biochem. 358:141–151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kamran MZ and Gude RP: Preclinical
evaluation of the anti-metastatic efficacy of Pentoxifylline on
a375 human melanoma cell line. Biomed Pharmacother. 66:617–626.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goel PN and Gude RP: Pentoxifylline
regulates the cellular adhesion and its allied receptors to
extracellular matrix components in breast cancer cells. Biomed
Pharmacother. 68:93–99. 2014. View Article : Google Scholar
|
|
25
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bhat UG, Pandit B and Gartel AL: ARC
synergizes with ABT-737 to induce apoptosis in human cancer cells.
Mol Cancer Ther. 9:1688–1696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
28
|
Brasier AR: The nuclear
factor-kappaB-interleukin-6 signalling pathway mediating vascular
inflammation. Cardiovasc Res. 86:211–218. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Viry E, Paggetti J, Baginska J,
Mgrditchian T, Berchem G, Moussay E and Janji B: Autophagy: An
adaptive metabolic response to stress shaping the antitumor
immunity. Biochem Pharmacol. 92:31–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y, Liu J, Qiu Y, Jin M, Chen X, Fan
G, Wang R and Kong D: ZSTK474, a specific class I
phosphatidylinositol 3-kinase inhibitor, induces G1 arrest and
autophagy in human breast cancer MCF-7 cells. Oncotarget.
7:19897–19909. 2016.PubMed/NCBI
|
|
31
|
Giuliano S, Cormerais Y, Dufies M, Grépin
R, Colosetti P, Belaid A, Parola J, Martin A, Lacas-Gervais S,
Mazure NM, et al: Resistance to sunitinib in renal clear cell
carcinoma results from sequestration in lysosomes and inhibition of
the autophagic flux. Autophagy. 11:1891–1904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chu PM, Chen LH, Chen MT, Ma HI, Su TL,
Hsieh PC, Chien CS, Jiang BH, Chen YC, Lin YH, et al: Targeting
autophagy enhances BO-1051-induced apoptosis in human malignant
glioma cells. Cancer Chemother Pharmacol. 69:621–633. 2012.
View Article : Google Scholar
|
|
33
|
Tucci M, Stucci S, Savonarola A, Resta L,
Cives M, Rossi R and Silvestris F: An imbalance between Beclin-1
and p62 expression promotes the proliferation of myeloma cells
through autophagy regulation. Exp Hematol. 42:897–908. e8912014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar
|
|
35
|
Sharma K, Ishaq M, Sharma G, Khan MA,
Dutta RK and Majumdar S: Pentoxifylline triggers autophagy via ER
stress response that interferes with Pentoxifylline induced
apoptosis in human melanoma cells. Biochem Pharmacol. 103:17–28.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang T, Seah S, Loh X, Chan CW, Hartman M,
Goh BC and Lee SC: Simvastatin-induced breast cancer cell death and
deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by
metabolic products of the mevalonate pathway. Oncotarget.
7:2532–2544. 2016.
|
|
37
|
Palanivel K, Kanimozhi V, Kadalmani B and
Akbarsha MA: Verrucarin A induces apoptosis through ROS-mediated
EGFR/MAPK/Akt signaling pathways in MDA-MB-231 breast cancer cells.
J Cell Biochem. 115:2022–2032. 2014.PubMed/NCBI
|
|
38
|
Zhang S, He Y, Tong Q, Chen Q, Wu X and
Huang W: Deltonin induces apoptosis in MDA-MB-231 human breast
cancer cells via reactive oxygen species-mediated mitochondrial
dysfunction and ERK/AKT signaling pathways. Mol Med Rep.
7:1038–1044. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pathania AS, Kumar S, Guru SK, Bhushan S,
Sharma PR, Aithagani SK, Singh PP, Vishwakarma RA, Kumar A and
Malik F: The synthetic tryptanthrin analogue suppresses STAT3
signaling and induces caspase dependent apoptosis via ERK up
regulation in human leukemia HL-60 cells. PLoS One. 9:e1104112014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Singh S, Upadhyay AK, Ajay AK and Bhat MK:
p53 regulates ERK activation in carboplatin induced apoptosis in
cervical carcinoma: A novel target of p53 in apoptosis. FEBS Lett.
581:289–295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pettersson F, Couture MC, Hanna N and
Miller WH: Enhanced retinoid-induced apoptosis of MDA-MB-231 breast
cancer cells by PKC inhibitors involves activation of ERK.
Oncogene. 23:7053–7066. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung
Y, Komarov AP, Keyomarsi K, Yarden Y and Seger R: Taxol-induced
apoptosis depends on MAP kinase pathways (ERK and p38) and is
independent of p53. Oncogene. 20:147–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Landskron G, De La Fuente M, Thuwajit P,
Thuwajit C and Hermoso MA: Chronic inflammation and cytokines in
the tumor microenvironment. J Immunol Res. 2014:1491852014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu
Y, Gong Z, Zhang S, Zhou J, Cao K, et al: Role of tumor
microenvironment in tumorigenesis. J Cancer. 8:761–773. 2017.
View Article : Google Scholar : PubMed/NCBI
|