Introduction
Colorectal cancer (CRC) is the third most common
type of cancer and the leading cause of cancer mortality worldwide,
accounting for an estimated 10 million new cases and 5 million
deaths (1,2). Therefore, a better understanding of
the molecular mechanisms underlying the onset and progression of
CRC is essential in order to improve the diagnosis, develop new
therapies, and achieve an accurate prognosis. Hence, in order to
decrease the incidence of late prognosis and prevent subsequent
mortality, the identification of additional biomarkers for CRC is
paramount.
AKAP12/Gravin (A-kinase anchor protein 12), which
was first isolated from the serum of patients with myasthenia
gravis (3), is a member of the
cyclic AMP-dependent kinase-anchoring protein (AKAP) family and
acts as a scaffold protein that assembles multiple signaling
molecules at their functional subcellular locations, including
protein kinase A, protein kinase C, cyclin D and calmodulin
(4,5). Moreover, AKAP12 suppresses
Src-induced oncogenic proliferation, invasiveness and cell death by
directly scaffolding Src from growth factor receptor and FAK
complexes to lipid rafts, without altering the intrinsic kinase
activity of Src. In addition, the rodent homologue of AKAP12
impacts the blood-brain barrier via the regulation of vascular
endothelial growth factor of astrocytes (6). AKAP12 is located at 6q24-25.2, a
depletion hotspot that is commonly expressed in tumors of the
prostate, breast and ovary. However, accumulating evidence
indicates that AKAP12 mRNA is underexpressed or lost in the
majority of CRC tissues (7).
Moreover, DNA hypermethylation of the AKAP12 promoter region
causing the subsequent downregulation of mRNA expression occurs in
the majority of human cancers (8–11).
Hence, the underexpression or loss of AKAP12 suggests that it may
be a potential indicator associated with oncogenesis.
CRC progression is characterized by epigenetic
alterations, including promoter DNA hypermethylation and
post-translational modifications of histones. Histone deacetylases
(HDACs) are transcriptional co-repressors that remove acetyl groups
from histones (12). Previous
studies have suggested that the deregulation of HDACs suppresses
transcription by tightening the chromatin structure, which further
contributes to tumorigenesis. There are three classes of HDACs:
Class I, which includes HDAC1, 2, 3 and 8; class II, composed of
HDAC4-6, 9 and 10; class III, which includes members of the SIR2
family; and class IV, which has only one member, HDAC11 (12). HDACs catalyze the removal of an
acetyl group from the ε-amino group of lysine side chains of the
histone molecules H2A, H2B, H3 and H4, thereby reconstituting a
positive charge to the lysine moiety (13). Emerging data suggest that the
increased expression of HDACs occurs in several types of cancer
other than CRC (14), including
cancers of the stomach (15),
liver (16), breast (17) and prostate (18), as well as in hematological
malignancies (19).
HDAC3, in addition to HDAC1, 2 and 8, is a class I
HDAC that is expressed extensively in the majority of cancers
(20–22). Functionally, HDAC3 has the ability
to induce proliferation by activating vitamin D signaling, reduce
apoptotic death, and promote invasion and metastasis by interacting
with WD Repeat Domain 5 (WDR5) in a broad spectrum of cancer cells.
Furthermore, HDAC3 is considered to play a critical role in the
suppression of gene transcription by unliganded or antagonist-bound
nuclear hormone receptors, and reportedly forms multi-protein
complexes with SMRT and N-CoR (23). Biologically, HDAC3 has been noted
for its ability to induce growth, promote metastasis and reduce the
apoptotic death of CRC cells by suppressing p21 expression.
Mechanically, HDAC3 also influences the phosphorylation of signal
transducer and activator of transcription 3 (STAT3) at serine 727
by interacting with protein phosphatase 2 (PP2, also known as PP2A)
(24) and regulating the
deacetylation of histone and non-histone target proteins (25). Despite there being significant
evidence that HDAC3 plays an important role in cancer
initialization and progression, there is limited information on the
association between HDAC3 and the tumor suppressor factor, AKAP12,
in CRC.
The aim of this study was to determine whether HDAC3
and AKAP12 expression play a role in the regulation of CRC
progression and metastasis. The results of this study demonstrated
that HDAC3 expression was significantly upregulated in CRC cells
and tissues, whereas that of AKAP12 was decreased. Importantly, the
co-silencing of HDAC3 and AKAP12 inhibited the growth, enhanced the
apoptosis and suppressed the migration of CRC cells to a greater
extent than the silencing of each factor alone. These findings may
prove to be helpful for the further understanding of the unique
roles of HDAC3 and AKAP12 interactions in the onset and progression
of CRC.
Materials and methods
Patients and samples
All specimens were collected from patients with CRC
at Huashan Hospital and Shanghai Tenth People's Hospital (Shanghai,
China). Written consent was obtained from all subjects and the
study protocol was approved by the Ethics Committees of Huashan
Hospital and Shanghai Tenth People's Hospital. In total, 96 paired
non-tumor adjacent normal tissues and tumor tissues were collected
from 96 patients with CRC following surgical resection. All samples
were stored in an ultra-low temperature refrigerator.
Cell culture and primary CRC cell
isolation
Three CRC cell lines (LoVo, SW480 and LS174T) were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA). The LoVo cells were cultured in F-12K medium
(Gibco/Invitrogen Corp., Grand Island, NY, USA) supplemented with
10% fetal bovine serum (FBS; Gemini Bio Products, Broderick, CA,
USA) and incubated under an atmosphere of 5% CO2 at
37°C. The SW480 cells were cultured in L-15 medium containing 10%
FBS without 5% CO2 and the LS174T cells were maintained
in Dulbecco's modified Eagle's medium (DMEM) (both from Gibco)
containing 10% FBS at 37°C in a humidified incubator containing 5%
CO2. Approximately 1×106 cells were seeded
per square in 6-well plates. Following 24 h of incubation, fresh
culture medium with or without Trichostatin A (TSA; 5
µmol/l, V900931; Sigma-Aldrich Corp., St. Louis, MO, USA)
and RGFP966 (10 µM, S7229; Selleckchem, Houston, TX, USA)
were added and the cells were further incubated for an additional
48 h. Two inhibitors specifically targeting AKT, MK2206 (2
µM, S1078) and AZD5363 (10 µM, S8019) (both from
Selleckchem), were applied in the presence of si-HDAC3, si-AKAP12
or si-HDAC3 plus si-AKAP12 in the SW480 cells for 6 h. The pmyr-AKT
vector was kindly provided by Professor Lei Chen from Eastern
Hepatobiliary Surgery Hospital. The exogenous expression of AKT was
performed by transient transfection with pmyr-AKT plasmid (1
µg for each well of 6-well plate). Cisplatin was obtained
from Selleckchem (S1166). After 48 h of transfection, the cells
were treated with cisplatin (10 µg/ml) for an additional 16
h.
Two human CRC tissues were collected immediately
following resection and placed into a sterile tube contain DMEM
supplemented with 10% FBS and 1% penicillin/streptomycin. The
tissues were cut in 1 ml of DMEM plus FBS and antibiotic
(penicillin/streptomycin; Invitrogen Corp.). The tissues were then
transferred into a 15-ml falcon tube containing 2 ml of medium
supplemented with 1,000 U/ml collagenase II incubated for 10 min
(37 °C), and mixed on a vortex for 10 sec. Subsequently, 2 ml of
medium/well was removed and added to an equal volume of FBS. This
procedure was repeated 3 times. The dissociated cells were
harvested by centrifugation (250 × g) for 10 min, resuspended in
DMEM supplement with 20% FBS and 1% penicillin/streptomycin, and
plated in a 6-well plate. After 24 h, primary CRC cells were
transiently transfected with siRNAs, and western blot analysis was
performed 48 h after siRNA transfection.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA (1 µg) was extracted using TRIzol
reagent (Invitrogen Corp.) and reverse transcribed into cDNA using
the PrimeScript™ reagent kit (Takara Bio, Inc., Shiga, Japan). The
synthesized cDNA was amplified using the KAPA™ SYBR®
Fast qPCR kit (Kapa Biosystems, Inc., Wilmington, MA, USA) and the
Applied Biosystems 7500 Fast Real-Time PCR system (Applied
Biosystems, Carlsbad, CA, USA) according to the following protocol:
Initial denaturation at 95°C for 3 min, then 45 cycles at 95°C for
10 sec, 60°C for 34 sec, and a final extension at 72°C for 5 min.
With the reference gene, glyceraldehyde 3-phosphate dehydrogenase
(GAPDH), as an internal control, the following forward and reverse
primer sequences, respectively, were used for RT-qPCR: HDAC3,
5′-GGA GCT GGA CAC CCT ATG AA-3′ and 5′-TAT TGG TGG GGC TGA CTC
TC-3′; AKAP12, 5′-GTC TCC TTC ATT CGC AGG CT-3′ and 5′-CAT GGC TCC
TCC GCA CTT CTC-3′; GAPDH, 5′-GAA GGT GAA GGT CGG AGT CA-3′ and
5′-GAA GAT GGT GAT GGG ATT TC-3′; matrix metallopeptidase (MMP)2,
5′-TAC AGG ATC ATT GGC TAC ACA CC-3′ and 5′-GGT CAC ATC GCT CCA GAC
T-3′; MMP7, 5′-GAG TGA GCT ACA GTG GGA ACA-3′ and 5′-CTA TGA CGC
GGG AGT TTA ACA T-3′; TIMP2, 5′-AAG CGG TCA GTG AGA AGG AAG-3′ and
5′-GGG GCC GTG TAG ATA AAC TCT AT-3′. All primers were synthesized
by Sangon Biotech Co. Ltd. (Shanghai, China). Each reaction was
performed in triplicate and the 2-ΔΔCq method was used
to calculate relative mRNA abundance (26,27).
A positive or negative change in expression of 2-fold or greater
was considered significant.
Chromatin immunoprecipitation (ChIP)
assay
Chromatin immunoprecipitation was carried out as
previously described (28).
Briefly, the SW480 cells were fixed in 1% formaldehyde and protease
inhibitor cocktail 1 (PIC1, comprised of 1 µg/ml leupeptin,
1.4 µg/ml pepstatin, 0.2 mg/ml PMSF, 1 mM EGTA and 1 mM
EDTA) for 10 min with gentle rotation at room temperature for 10
min at room temperature. The cross-linking reaction was quenched
with Glycine Stop-Fix solution (Shandong New Beiyang Information
Technology Co., Ltd., Beijing, China). The pellet was resuspended
in ice-cold lysis buffer and dounced on ice with ~10 to 15 strokes
to aid nuclei release. Nuclei were released after 30 strokes using
a dounce homogenizer and collected following centrifugation (2,1382
× g, 4°C, 15 min). The pellets were resuspended in 6 ml
homogenization buffer (10 mM HEPES, pH 7.6, 25 mM KCl, 1 mM EDTA, 1
mM EGTA, 1 M sucrose, 10% glycerol, 0.15 mM spermine, supplemented
with PIC1) and layered onto 3 ml of the same buffer. The nuclei
were then pelleted at 5,031 × g for 1 h (SW41 rotor; Beckman
Coulter, Irving, TX, USA) and stored at −80°C. The nuclear pellets
were re-suspended in 0.3 ml nuclear lysis buffer (50 mM Tris pH
7.6, 10 mM EDTA and 1% SDS), and diluted with 0.6 ml
immunoprecipitation (IP) dilution buffer (0.01% SDS, 1.1% Triton
X-100, 167 mM NaCl, 16.7 mM Tris pH 7.6, 1.2 mM EDTA). For
sonication, 0.3 ml (1/3) of nuclear lysate was sonicated for 25–30
cycles 30 sec on 30 sec at 4°C with a BioRuptor twin sonicator
(Diagenode). Sonicated chromatin was then further diluted to 1 ml
with IP dilution buffer, which is sufficient for 3 ChIP reactions.
The average size of the fragments was ~120–150 bp. The SW480 cell
chromatin fragments were immunoprecipitated with anti-HDAC3
antibody (dilution, 1:2,000; ab96005; Abcam, Cambridge, UK). Primer
pairs for HDAC3 binding sites in AKAP12 intron-1 regions (pair-2)
and for the right (pair-3) or left (pair-1) adjacent to HDAC3
binding sites were used to investigate the binding of HDAC3. The
primer pairs are shown as follows: Pair-1 forward,
5′-GTCGCCCTGTTAGAAACGGG-3′ and reverse, 5′-GCACAGCCCCAGACACGCCC-3′
(PCR product, 134 bp); Pair-2 forward, 5′-AGATGCTGCTGCAGGGCGTG-3′
and reverse, 5′-ACGCGCTCGCGGCAAACTCC-3′ (PCR product, 137 bp);
Pair-3 forward, 5′-CGGAGGCTAAGAGGTGGCC-3′ and reverse,
5′-CCCGCGAGGTCCCGAGAGCGCC-3′ (PCR product, 138 bp).
Luciferase assay
Generally, the SW480 cells were transfected with a
firefly luciferase reporter gene construct (pGL3-CMV; E1751;
Promega, Madison, WI, USA). Cell extracts were prepared 48 h
following transfection with the luciferase vector, and the relative
luciferase activity was measured using the Dual Luciferase Reporter
Assay System (E1910; Promega), and was normalized to Renilla
luciferase activity. The intron-1 region of AKAP12 was firstly
amplified with the following primers: Forward,
5′-GTCGGCTGCAGCAGAAGCTC-3′ and reverse,
5′-CCCGCGAGGTCCCGAGAGCGCC-3′ and then inserted into the upstream of
CMV sequence of pGL3-CMV (E1751; Promega) construct (named as
pGL3-CMV-AKAP-Intron-1). In addition, the E and GC Box deleted
intron-1 construct were named as pGL3-CMV-AKAP-Intron-Mutant. The
relative luciferase activity was measured as described above. In
addition, the above-mentioned experiments were performed at least 3
times before drawing any conclusions.
Transient transfection with siRNA
All siRNAs (S1, S2, S3 and a negative control) used
to silence HDACs and AKAP12 were synthesized by Shanghai GenePharma
Co., Ltd. (Shanghai, China). siRNA targeting the human Bcl-2 gene
(S1915) was purchased from Thermo Fisher Scientific Inc., Waltham,
MA, USA). The cells were grown to confluency, trypsinized and
resuspended in Opti-MEM™ I Reduced Serum Media (Gibco).
Non-targeting control, HDACs, or AKAP12 siRNA were transfected into
the cells using Lipofectamine 2000 reagent (Invitrogen-Life
Technologies) with Opti-MEM at a concentration of 100 nM. Following
transfection for 24 h, RNA was isolated and quantified by
RT-qPCR.
Western blot analysis
Total protein was extracted from the CRC cell lines
using Cell Lysis Buffer for Western and Immunoprecipitation
(Beyotime Institute of Biotechnology, Haimen, China) and the
protein concentration was measured using a BCA Protein Reagent kit
(Beyotime Institute of Biotechnology). Subsequently, 30–50
µg of protein from each sample were separated by sodium
dodecyl sulfate polyacrylamide gel electrophoresis on 10 and 6%
gels, respectively, and transferred onto a polyvinylidene fluoride
membrane (EMD Millipore, Temecula, CA, USA). The membrane was then
incubated with the following antibodies: HDAC3 (dilution, 1:2,000;
ab96005), AKAP12 (dilution, 1:5,000; ab49849) (both from Abcam),
total AKT (#2920), phospho-AKT (Ser473) (#4051), phospho-AKT
(Thr308) (#9275), PI3K-110α (#4249), PI3K-110β (#3011) and p85
(#4257) (dilution for all, 1:1,000; all from Cell Signaling
Technology, Inc., Danvers, MA, USA) and β-actin rabbit monoclonal
antibody (dilution, 1:2,000; #8457; Cell Signaling Technology,
Inc.) overnight at 4°C. After washing, the membrane was incubated
with the secondary antibody (dilution, 1:5,000, ab191866 and
ab222759; Abcam) at room temperature for 2 h. Proteins were
visualized using ECL Plus Western Blotting detection reagents (EMD
Millipore) and measured using the LAS-3000 Imaging System with Fuji
Image Quant software (Fuji Film, Tokyo, Japan) according to the
manufacturer's instructions.
Cell proliferation assay
All cell lines were seeded and transfected in 6-well
plates, and then transferred to 96-well plates at 1,000 cells per
well 24 h following transfection. After 24, 48, 72, 96 and 120 h of
incubation in the 96-well plates, the numbers of viable cells were
detected using Cell Counting kit-8 reagents (Dojindo Laboratories,
Kyushu Island, Japan), following the manufacturer's instructions.
The relative viable cell numbers were measured using a microplate
reader (Multiscan GO; Thermo Fisher Scientific, Waltham, MA, USA)
at an absorbance optical density at 450 nm.
Flow cytometric assay
All cell lines transfected with siRNA were seeded in
6-well plates at 1×106 cells per well and then incubated
at 37°C overnight. The cells were stained with propidium iodide
(PI) conjugated with Annexin V-FITC (BD Biosciences, Franklin
Lakes, NJ, USA) for flow cytometric analysis. First, all the cells
were washed twice with cold phosphate-buffered saline and
resuspended in 1X binding buffer. The cells were then transferred
to a 5-ml tube containing 5 µl each of Annexin V-FITC and
PI, and then incubated in the dark for 30 min at room temperature.
Finally, 300 ml of 1X binding buffer were added and ~10,000
apoptotic cells were collected. For cell cycle analysis, the cells
treated with siRNA were incubated at 37°C for 48 and 72 h, stained
with PI solution in the dark for 30 min, and then subjected to flow
cytometry. Data were analyzed using FlowJo software (FlowJo, LLC,
Ashland, OR, USA).
Transwell assay
Approximately 5×104 SW480 cells were
plated into the upper chambers (PET, 8-µm pore size) of the
apparatus (Corning Inc., Corning, NY, USA) with serum-free medium.
The lower chambers were filled with complete L-15 medium,
supplemented with 10% FBS. Following 48 h of incubation, the cells
in the upper chambers were washed, fixed with 95% ethanol, and
stained with 0.1% crystal violet (Sigma-Aldrich Corp.). Finally,
images of the the cells that had migrated through the membrane to
the lower surface of the upper chamber were captured under an
inverted microscope (Nikon Corp., Tokyo, Japan).
Immunohistochemical (IHC) analysis
The tissues were fixed in 4% formalin, embedded in
paraffin, and sectioned at a thickness of 4 µm. The sections
were deparaffinized by washing several times with xylene and then
rehydrated in graded alcohol solutions. The sections were
permeabilized in citrate buffer (pH 6.0; Maixin Biotech Co., Ltd.,
Fuzhou, China) for 10 min and then incubated with normal goat serum
for 1 h. Subsequently, the sections were incubated with anti-HDAC3
polyclonal antibody (dilution, 1:100, ab7030) and anti-AKAP12
monoclonal antibody (dilution, 1:100, ab49849) (both from Abcam)
for 1 h at 37°C. The following day, the slides were washed and
incubated with the corresponding secondary antibody (dilution,
1:500, ab191866; Abcam). Finally, the expression levels of HDAC3
and AKAP12 in the non-tumor and tumor tissues were
calculated: The whole immunohistochemical evaluation and scoring
were performed by two independent pathologists. The staining
percentages were determined by randomly selecting 4–5 fields for
each tissue section, and after counting the total number of
nucleus, the tissues with the numbers of positive nuclei over the
5% total nuclei were considered as positive, or otherwise negative.
In the positive group, the staining status was designated as '+',
'++' and '+++' according to the staining density, of which '+' and
'++' were regarded as positive-medium, whereas '+++' as
positive-high. Images were acquired under an inverted microscope
(Nikon Corp.). All specimens were collected from patients with CRC
at Huashan Hospital and Shanghai Tenth People's Hospital. Written
consent was obtained from all subjects and the study protocol was
approved by the Ethics Committees of Huashan Hospital and Shanghai
Tenth People's Hospital.
Statistical analysis
One-way ANOVA and the χ2 test were used
to compare differences between various groups to assess statistical
significance. The gray values of the western blot bands were
analyzed using ImageJ software (https:-imagej-nih-gov/ij/). Following one-way ANOVA,
the LSD method was applied to further analyze two individual groups
as needed. All statistical analyses were performed using SPSS 20.0
software (IBM-SPSS, Inc., Chicago, IL, USA). Probability (P)-values
<0.05 and <0.01 were considered to indicate statistically
significant differences.
Results
Negative correlation between HDAC3 and
AKAP12 expression in clinical CRC tissues
To investigate the expression of HDAC3 in CRC, 96
paired CRC tissues and adjacent non-cancerous tissues were
subjected to IHC analysis. As shown in Fig. 1A, positive immunostaining of the
HDAC3 protein was observed in the nuclei of 84 cancerous tissues.
Among these positive cases, 53 (63.1%) exhibited high expression
levels and 31 (36.9%) exhibited moderate expression levels in tumor
tissues. Representative examples displaying positive HDAC3 staining
indicated that the level of HDAC3 in the CRC tissues was much
higher than that in non-tumor tissues. As shown in Fig. 1C, HDAC3 protein expression was
markedly upregulated in the tumor tissues (χ2=5.658,
P=0.0174).
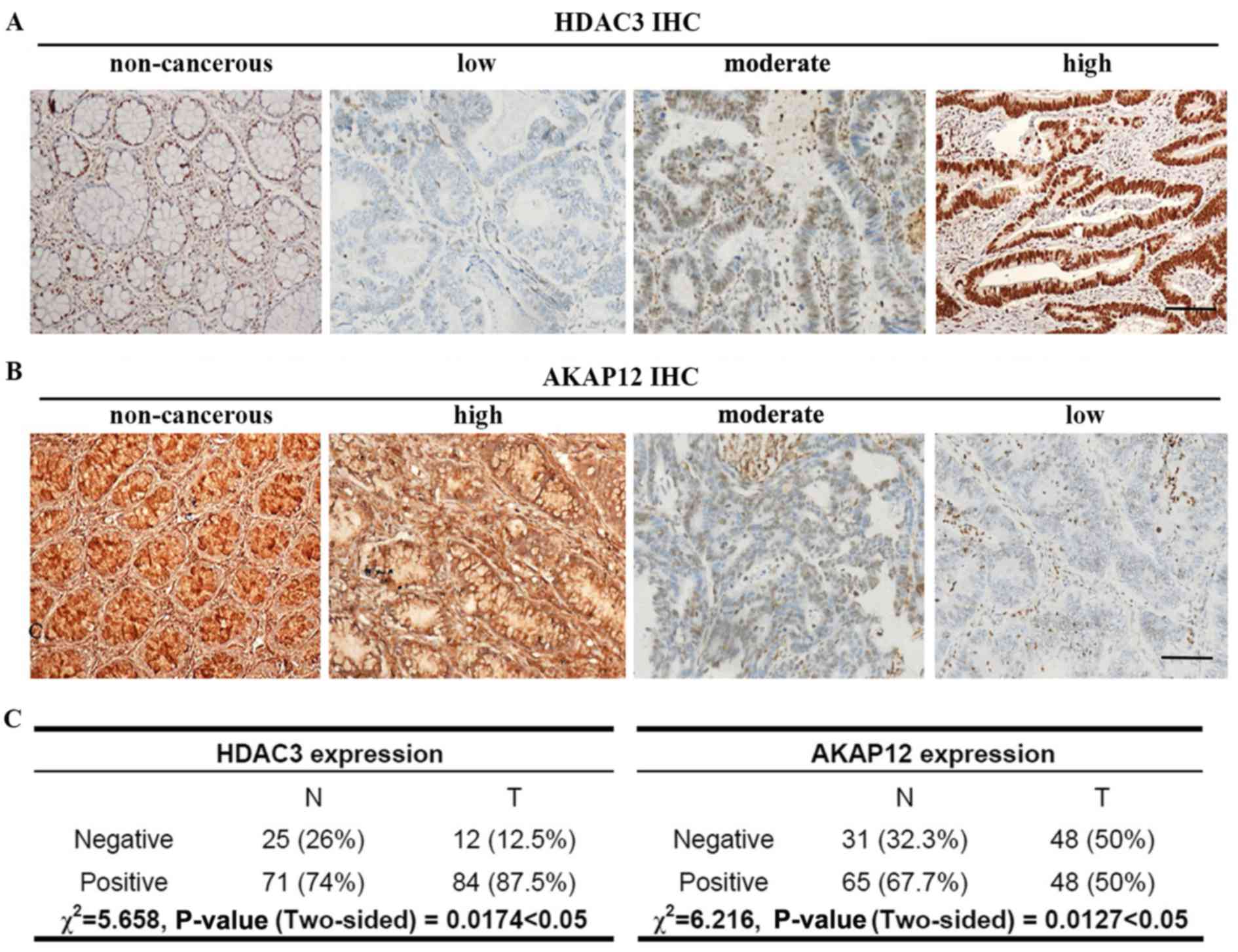 | Figure 1Representative immunostaining images
of colorectal adenocarcinoma tissues. (A) HDAC3 expression in
colorectal tumor tissues and adjacent noncancerous tissues was
classified as low, moderate, or high. (B) The expression of AKAP12
in colorectal tumor tissues and adjacent non-cancerous tissues was
also classified as low, moderate, or high. (C) The χ2
test was used to analyze the protein expression levels of HDAC3
(n=96, χ2=5.658, P<0.05) and AKAP12 (n=96,
χ2=6.216, P<0.01) in tumor and non-tumorous
colorectal tissues: −, negative (low); +, positive (moderate and
hight). Scale bar, 100 µm. |
AKAP12 functions as a tumor suppressor gene in
various types of cancer. Thus, we then examined the expression of
AKAP12 in CRC tissues. Representative images of IHC-stained
colorectal adenocarcinoma tissues are shown in Fig. 1B. The expression level of AKAP12
was lost in 48 (50%) tumor tissues, 28 (29.2%) displayed moderate
expression, and only 20 (20.8%) exhibited a high expression. In
total, AKAP12 exhibited a lower expression in 69 (71.8%) of the 96
tumor tissues when compared with the levels in their matched
non-tumor adjacent tissues, whereas all the non-cancerous tissues
exhibited a relatively high AKAP12 expression (data not shown). The
data obtained from IHC analysis were analyzed statistically using
the χ2 test. As shown in Fig. 1C, AKAP12 protein expression was
markedly decreased in the tumor tissues (χ2=6.216,
P=0.0127). As shown in Tables I
and II, the expression of AKAP12
or HDAC3 in the tumor tissues exhibited significant differences
with a number of the clinical characteristics.
 | Table ICorrelation between AKAP12 expression
and clinicopathological characteristics. |
Table I
Correlation between AKAP12 expression
and clinicopathological characteristics.
| Characteristic | Total (n=96) | Negative
(n=48) | Positive
(n=48) | P-value |
|---|
| Age (years) | | | | 0.8379 |
| <65 | 45 | 23 | 22 | |
| >65 | 51 | 25 | 26 | |
| Sex | | | | 0.1530 |
| Male | 49 | 21 | 28 | |
| Female | 47 | 27 | 20 | |
| Location | | | | 0.4332 |
| Left | 45 | 25 | 20 | |
| Transverse | 5 | 1 | 4 | |
| Right | 27 | 12 | 15 | |
| Rectum | 19 | 10 | 9 | |
| T stage | | | | 0.1396 |
| T1/2 | 8 | 2 | 6 | |
| T3/4 | 88 | 46 | 42 | |
| N stage | | | | <0.0001 |
| N0 | 47 | 7 | 40 | |
| N1/2/3 | 49 | 41 | 8 | |
| AJCC stage
(TNM) | | | | <0.0001 |
| I + II | 44 | 4 | 40 | |
| III + IV | 52 | 44 | 8 | |
| Histological
grade | | | | 0.0007 |
| Well | 10 | 1 | 9 | |
| Medium | 71 | 34 | 37 | |
| Poor | 15 | 13 | 2 | |
| Venous and
lymphatic metastasis | | | | <0.0001 |
| No | 47 | 7 | 40 | |
| Yes | 49 | 41 | 8 | |
| Liver
metastasis | | | | 0.0154 |
| No | 86 | 39 | 47 | |
| Yes | 10 | 9 | 1 | |
| Table IICorrelation between HDAC3 and
clinicopathological characteristics. |
Table II
Correlation between HDAC3 and
clinicopathological characteristics.
| Characteristic | Total (n=96) | Negative
(n=12) | Positive
(n=84) | P-value |
|---|
| Age (years) | | | | 0.3149 |
| <65 | 45 | 4 | 41 | |
| >65 | 51 | 8 | 43 | |
| Sex | | | | 0.9385 |
| Male | 49 | 6 | 43 | |
| Female | 47 | 6 | 41 | |
| Location | | | | 0.1337 |
| Left | 45 | 6 | 39 | |
| Transverse | 5 | 2 | 3 | |
| Right | 27 | 1 | 26 | |
| Rectum | 19 | 3 | 16 | |
| T stage | | | | 0.0008 |
| T1/2 | 8 | 4 | 4 | |
| T3/4 | 88 | 8 | 80 | |
| N stage | | | | 0.0016 |
| N0 | 47 | 11 | 36 | |
| N1/2/3 | 49 | 1 | 48 | |
| AJCC stage
(TNM) | | | | 0.0053 |
| I + II | 44 | 10 | 34 | |
| III + IV | 52 | 2 | 50 | |
| Histological
grade | | | | 0.0004 |
| Well | 10 | 5 | 5 | |
| Moderately | 71 | 7 | 64 | |
| Poor | 15 | 0 | 15 | |
| Venous and
lymphatic metastasis | | | | 0.0016 |
| No | 47 | 11 | 36 | |
| Yes | 49 | 1 | 48 | |
| Liver
metastasis | | | | 0.2067 |
| No | 86 | 12 | 74 | |
| Yes | 10 | 0 | 10 | |
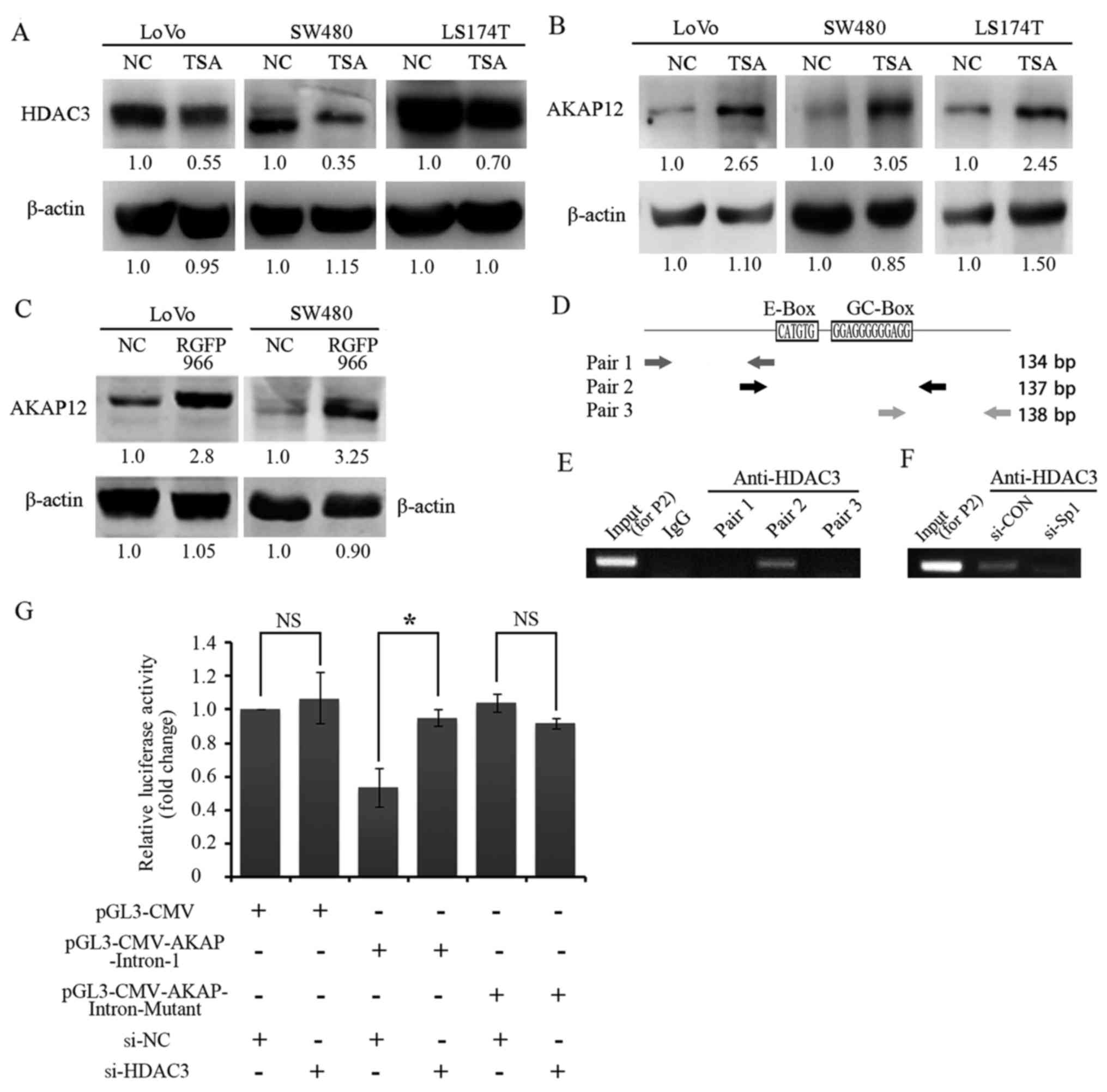
Treatment with an HDAC inhibitor induces
AKAP12 expression in SW480 cells
To validate the effects of HDAC3 on AKAP12
expression in CRC, the expression levels in 3 human CRC cell lines
(LoVo, SW480 and LS174T) were examined by western blot analysis.
TSA is a classical HDAC inhibitor, which inhibits HDACs in a
non-competitive and reversible manner. TSA inhibits the
proliferation and promotes the apoptosis of a variety of cancer
cells, such as colorectal, prostate and breast cancer cells
(29–31). Thus, TSA was applied to clarify the
potential effect of HDAC3 on AKAP12. As expected, a negative
correlation between HDAC3 and AKAP12 expression was observed in all
3 CRC cell lines (Fig. 2A and B)
in the presence of TSA. The use of the HDAC3 specific inhibitor,
RGFP966, markedly increased the protein level of AKAP12 in both the
LoVo and SW480 cell lines (Fig.
2C). Importantly, a regulatory element containing the E/GC-box
within intron-1 of the AKAP12 gene loci was first identified, and
chromatin immunoprecipitation assay revealed that the presence of
the E and GC-box (Pair-2) is indispensable for the binding of HDAC3
at intron-1 loci (Fig. 2D and E).
It has been documented that Sp1 is also necessary for the DNA
binding activity of HDAC3 (32);
we thus examined the effects of Sp1 on the HDAC3/DNA complex and
found that the binding of HDAC3 with DNA was attenuated in the
absence of Sp1 (Fig. 2F). More,
the intron-1 of AKAp12 (wild-type or E/GC-box deleted mutant form)
was inserted into the pGL3-CMV construct to verify the effects of
HDAC3 on gene expression. As shown in Fig. 2G, the luciferase activity of the
pGL3-CMV-AKAP-Intron-1 construct was decreased by <50% as
compared with that of the pGL3-CMV construct. However, transfection
with siRNA targeting HDAC3 restored the luciferase activity. As
expected, no significant changes were observed in the
pGL3-CMV-AKAP-Intron-mutant-transfected cells in comparison with
the pGL3-CMV cells following transfection with si-NC or si-HDAC3
(Fig. 2G). Taken together, these
data indicate that HDAC3 may suppress the expression of AKAP12 by
directly binding with the E/GC-box region of AKAP12 gene loci in
CRC cells.
HDAC3-mediated AKAP12 downregulation
promoted cell proliferation and G2/M transition, and prevented cell
apoptosis
In order to examine the biological roles of HDAC3
and AKAP12 in colorectal tumorigenesis, the expression of HDAC3,
AKAP12, or HDAC3 and that of AKAP12 was suppressed via RNA
interference (RNAi). The efficacy of RNAi on the SW480 cells was
confirmed by RT-qPCR and western blot analysis. The most efficient
siRNA was applied for the following experiments (Fig. 3A and B). As shown in Fig. 3C, transfection with si-HDAC3
resulted in the inhibition of cell proliferation, whereas
transfection with si-AKAP12 treatment led to an increase in cell
proliferation. Of note, the co-silencing of HDAC3 and AKAP12
yielded a similar result as that observed with transfection with
si-AKAP12, suggesting that HDAC3 may promote cell proliferation by
negatively regulating the expression of AKAP12. Similarly, the
results of a colony formation assay revealed that transfection with
si-HDAC3 inhibited the growth of the colorectal cells, whereas
transfection with si-AKAP12 or si-HDAC3 together with si-AKAP12
markedly increased the number and size of colonies in vitro
(Fig. 3D).
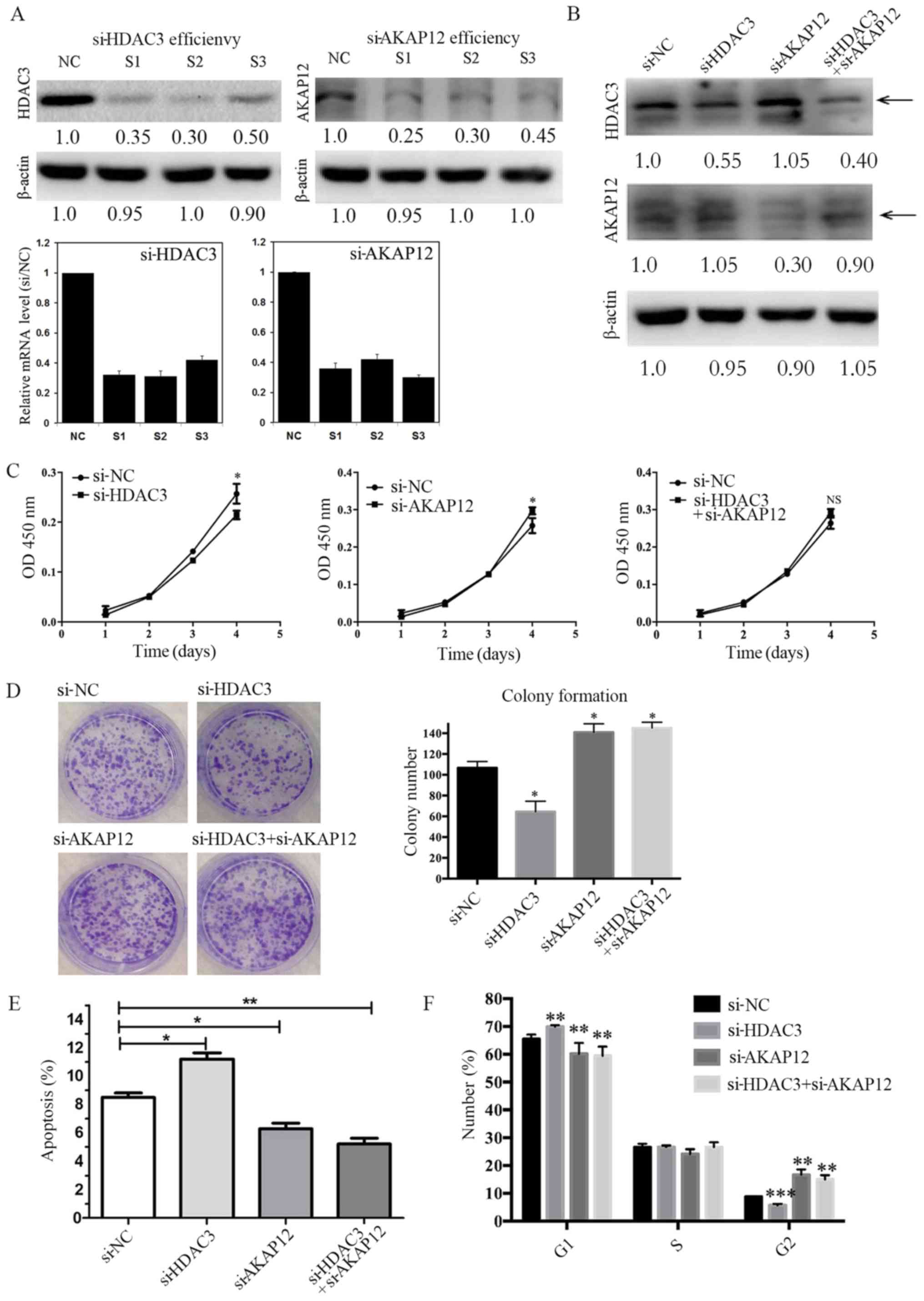 | Figure 3HDAC3 and AKAP12 co-silencing inhibit
the proliferation, induce apoptosis, and promote G2/M checkpoint
arrest in SW480 cells. (A and B) The expression of HDAC3 and
AKAP12 in SW480 cells transfected with siRNA was detected by
RT-qPCR (24 h, lower panel) and western blot analysis (48 h, upper
panel). (C) The growth of SW480 cells after the silencing of HDAC3
and AKAP12 was examined by CCK-8 assay on the indicated days.
Significant differences were found after the co-silencing of HDAC3
and AKAP12. (D) The SW480 cells, following silencing of HDAC3 and
AKAP12, were subjected to colony formation assays following
transfection and culture for 14 days. (E) The SW480 cells, after
the silencing of HDAC3 and AKAP12, were subjected to flow cytometry
to examine cell apoptosis. All experiments were performed in
triplicate. (F) The SW480 cells, after the silencing of HDAC3 and
AKAP12, were cultured in an incubator for 48 h and subjected to
flow cytometric analysis. Data are presented as the means ± SD.
One-way ANOVA test: *P<0.05; **P<0.01;
***P<0.001; NS, no significant difference. |
Based on the observation that the silencing of
AKAP12 or AKAP12 with that of HDAC3 induced cell proliferation and
colony formation, the anti-apoptotic activity of the CRC cells was
investigated. As shown in Fig. 3E,
the percentage of apoptotic cells was much lower following HDAC3
and AKAP12 co-silencing or AKAP12 silencing alone, as compared with
that observed with si-HDAC3 silencing alone by applying PI and
Annexin V staining. Moreover, flow cytometric analysis revealed an
increased proportion of cells in the G2/M phase and a slight
reduction in the number of cells in the G1 phase following
transfection with si-AKAP12 or si-HDAC3 together with si-AKAP12
(Fig. 3F). Taken together, these
results indicate that HDAC3 may function as a pro-tumor gene by
inhibiting AKAP12 expression in CRC.
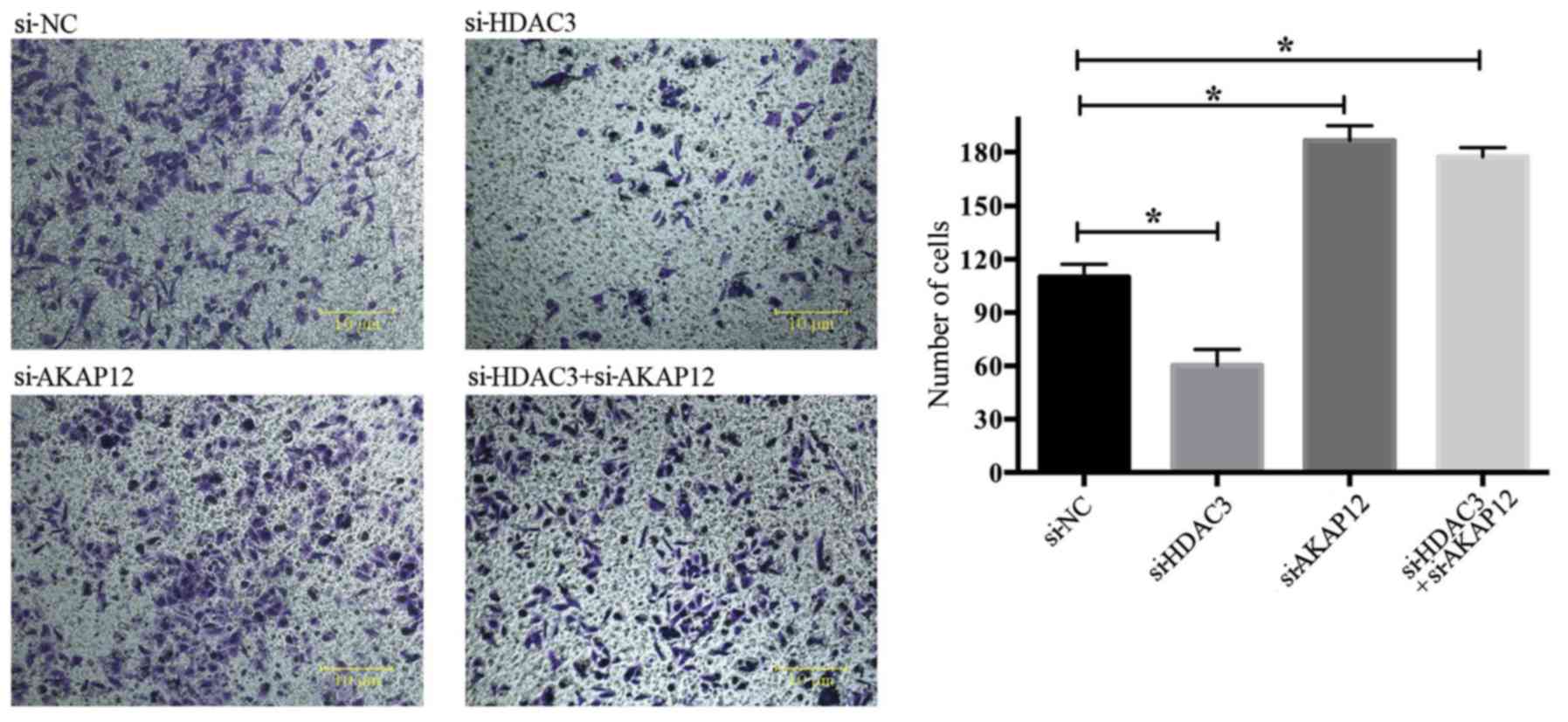
Negative regulation of cell migration by
AKAP12
To further assess the role of AKAP12 in cell
migration, Transwell assays were performed to examine cell
migration following HDAC3 and AKAP12 knockdown. As shown in
Fig. 4, the number of metastatic
cells was markedly decreased following transfection with si-HDAC3,
whereas transfection with si-AKAP12 or with both si-AKAP12 and
si-HADC3 enhanced cell metastasis in vitro.
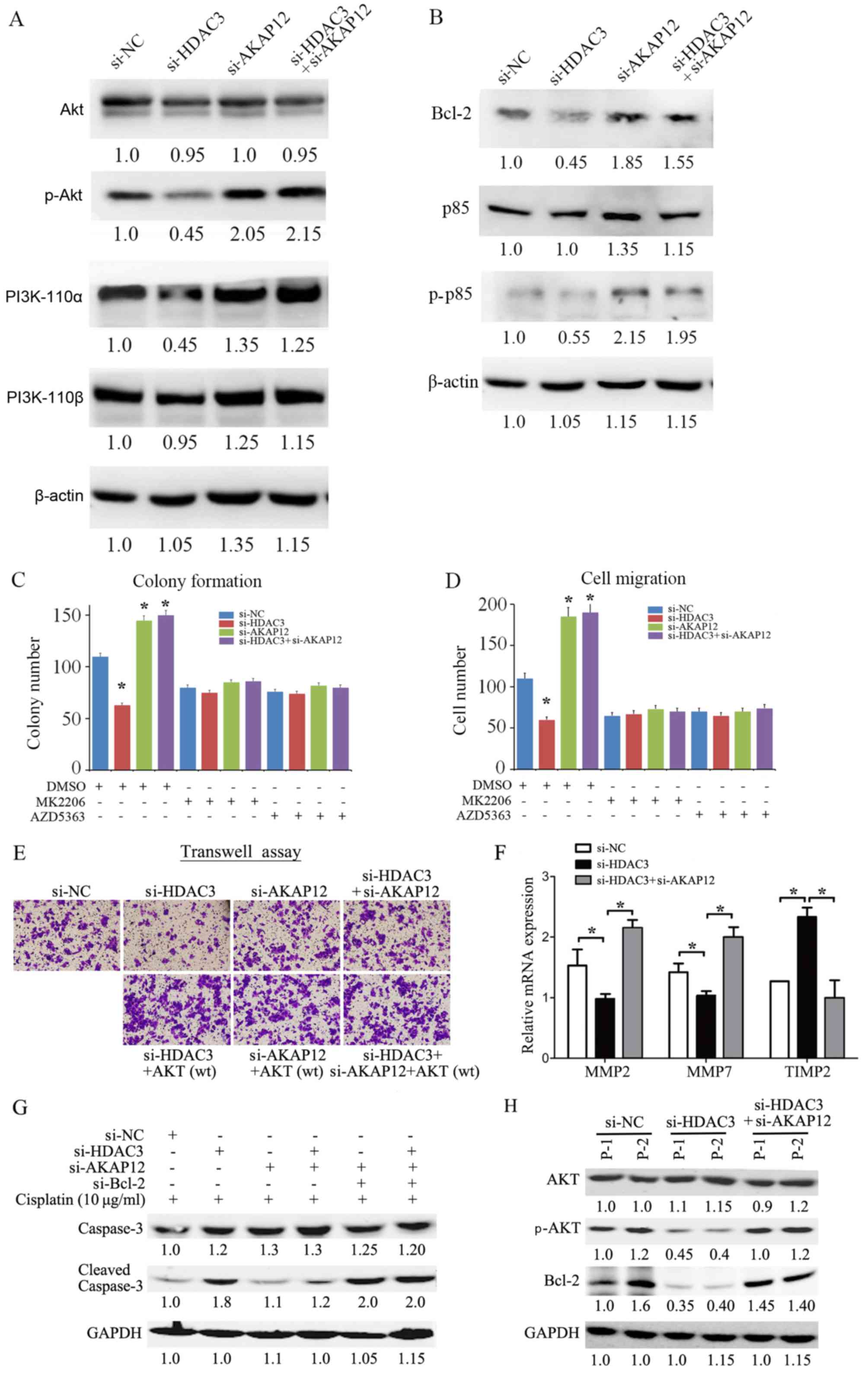
Increased protein levels of p-AKT in
response to transfection iwth si-AKAP12 are necessary for colony
formation and migration
Previous findings on endothelial cells have
suggested that the tumor suppressor gene, AKAP12, is epigenetically
regulated by HDAC7, which may regulate STAT3 (33). Other data have also shown that
HDAC3 modulates the JAK/STAT3 pathway in multiple myeloma cells
(34). Thus, the ability of HDAC3
to enhance cell malignant phenotypes via the modulation of the
AKAP12-JAK-STAT3 axis was investigated. We found no significant
changes in STAT3 signaling upon HDAC3 or AKAP12 knockdown in the
CRC cells (data not shown). As the PI3K/AKT pathway has been widely
documented as the major pathway involved in tumor progression by
controlling cell proliferation, apoptosis, aggressiveness and
metastasis (35), the level of
p-AKT in the CRC cells was examined. The results revealed that
transfection with si-HDAC3 decreased the level of p-AKT. Moreover,
the expression level of p-AKT was significantly increased in
response to si-AKAP12 transfection. Importantly, transfection with
si-AKAP12 reversed the si-HDAC-3-mediated inactivation of AKT
(Fig. 5A).
Mechanistically, no significant changes in the
levels of PI3K subunits, including PI3K-110α, PI3K-110β and p85,
were observed following transfection with si-AKAP12 or si-AKAP12
plus si-HDAC3 in comparison to transfection with si-NC (Fig. 5A). Of note, the level of
phosphorylated p85, a regulatory subunit of PI3K, was increased in
cells transfected with si-AKAP12 or si-AKAP12 together with
si-HDAC3, indicating that the underlying mechanisms may involve
AKAP12-regulated AKT activity. In addition, the expression of the
anti-apoptotic protein, Bcl-2, was increased in response to
si-AKAP12 transfection, suggesting that increased Bcl-2 expression
may be necessary for AKAP12-mediated cell apoptosis (Fig. 5B).
To further verify the potential effects of AKT
signaling on the regulation of the cell phenotype by AKAP12, two
inhibitors specifically targeting AKT, MK2206 and AZD5363, were
applied in the presence of si-HDAC3, si-AKAP12 or si-HDAC3 plus
si-AKAP12 in the SW480 cells. Both colony formation and migration
assay revealed that the use of the AKT inhibitors significantly
reduced the number of colonies and migrating cells in the si-AKAP12
group and in the si-HDAC3 plus si-AKAP12 group in comparison with
the si-NC group (Fig. 5C and D).
In addition, we also found the exogenous expression of AKT restored
si-HDAC3-modulated cell migration in an AKAP12-dependent manner
(Fig. 5E). Mechanistically, the
decreased expression of MMP2 and MMP7 and the increased expression
of TIMP2 was observed following transfection with si-HDAC3, and
this effect was by transfection with si-AKAP12 simultaneously
(Fig. 5F). Further, transfection
with siRNA against Bcl-2 sensitized the cells to cisplatin-induced
caspase-3 cleavage and cell apoptosis in both the si-AKAP12 and
si-HDAC3- plus si-AKAP12-transfected cells (Fig. 5G). Taken together with the data
from two primary CRC cells (Fig.
5H), it was thus suggested that HDAC3-AKAP12 manipulate cell
malignant phenotypes via the PI3K/AKT/MMP pathway and the Bcl-2
gene in CRC cells.
Discussion
Numerous studies have demonstrated that AKAP12 is
epigenetically reduced or lost in many types of cancer, including
prostate cancer (36), ovarian
cancer (37), hepatocellular
cancer (38), CRC (39) and acute myeloid leukemia (9). However, the biological roles of
AKAP12 in CRC progression and the intrinsic mechanisms of AKAP12
depletion associated with the progression of CRC are not yet
completely understood. The results of the present study provide
evidence that AKAP12 functions as a tumor suppressor in CRC. First,
the expression profiles of AKAP12 were examined at both the mRNA
and protein levels, and the results revealed a significant
upregulation following TSA treatment. Second, AKAP12 was found to
significantly inhibit CRC cell growth and migration, while
promoting apoptotic cell death.
Emerging studies have demonstrated a deficiency in
AKAP12, which plays an important role in tumor progression and
metastasis, in many types of cancer (40–44).
In human gastric cancer cells, AKAP12 can lead to reduced colony
formation and the induction of cell apoptosis (40). Consistent with the findings of
previous studies, in this study, AKAP12 expression was
downregulated in 45 (46.9%) of 96 CRC tissues as compared with
their matched non-tumor tissues in this study. The proliferation
and apoptosis of the SW480 cells was then investigated to evaluate
the potential regulatory effects of AKAP12 on CRC cells by CCK-8
assay and flow cytometry. The results revealed a potentially
suppressive role of AKAP12 in the development of CRC.
The HDAC family of transcriptional co-repressors,
which regulates a large variety of genes and functional regulatory
proteins, has emerged as an important regulator of maturation and
transformation. The overexpression of HDACs is a well-documented
phenomenon in a number of malignancies, particularly in CRC
(9–15). A recent meta-analysis of a variety
of human cancers indicated that HDAC3 may be one of the most
frequently upregulated genes in cancer cells (45). The overexpression of HDAC3
reportedly has the capability to inhibit basal and butyrate-induced
p21 transcription in a Sp1/Sp3-dependent manner, whereas the
silencing of HDAC3 stimulates p21 promoter activity and expression
(32). Godman et al found
that HDAC3 knockdown significantly suppressed β-catenin
translocation from the plasma membrane to the nucleus in the Wnt
and vitamin D signaling pathway (20). In addition, HDAC3 also specifically
inhibited NF-κB-mediated cell metastasis via interacting with CREB3
(22). Similarly, the findings of
the present study suggested that HDAC3 knockdown induced apoptotic
cell death, inhibited cell growth, and decreased the ability of CRC
cells to metastasize. Therefore, HDAC3 may be regarded as a
biological marker for the prediction of metastasis in CRC.
In the present study, the association between HDAC3
and AKAP12 in CRC cells was examined. First, a significant negative
correlation between AKAP12 and HDAC3 in CRC was found. AKAP12
expression was increased prominently following treatment with the
HDAC inhibitor, TSA, suggesting that deacetylation may play a key
role in the downregulation of AKAP12 expression in CRC. Generally,
HDAC manipulates transcriptional activity through binding with the
DNA sequences beside or upstream the transcription start site. It
is noteworthy that we found that the binding of the HDAC over
AKAP12 intron led to the transcriptional suppression by ChIP and
luciferase assay. We hence speculated that some co-factors or
unknown chromosome architecture may be involved. Further studies
are warranted to explore the potential mechanisms involved.
The PI3K/AKT pathway is a major contributor to CRC
progression (35,46). PI3K/AKT can be recruited and
stimulated by the serine/threonine kinases PKD1 and AKT, whereby
PKD1 phosphorylates AKT on threonine 308 and a second
phosphorylation, catalyzed by mTORC2, on serine 473 activates AKT.
AKT provides signals that lead to cell growth and differentiation,
and angiogenesis. It prevents apoptosis in CRC as phosphorylated
AKT stimulates a multitude of downstream targets, such as BAD,
mTOR, FOXO proteins, MDM2 and VEGF, with the exception of the tumor
suppressors, PTEN and GSK-3β. A previous study illustrated that
HDAC inhibition exerted concentration-dependent anti-proliferative
effects in CRC cells, when combined with an EGFR/HER2 kinase
inhibitor, via the RAS/RAF/MEK/MAPK and PI3K/AKT pathways (47). Ye et al (48) found that the tumor suppressor,
PIB5PA, blocked AKT activation via the downregulation of
phosphorylation of the serine 473 residue, and mediated HDAC2 and
HDAC3 hypoacetylation levels by binding to the Sp1 transcription
promoter. In line with previous studies, the findings of the
present study suggested that the co-silencing of HDAC3 and AKAP12
increased the level of AKT phosphorylation, similar to the results
achieved by AKAP12 knockdown alone. These findings indicate a
negative regulatory effect of AKAP12 on the PI3K/AKT signaling
pathway.
In summary, these findings demonstrate that AKAP12,
which is epigenetically regulated by HDAC3, is a suppressive
regulator with the capability to inhibit cell growth and migration,
and promote the apoptosis of CRC cells. Moreover, the
downregulation of AKAP12 by HDAC3 is indispensable for
HDAC3-induced PI3K/AKT activation and consequent cell metastasis.
Further studies are warranted to clarify the underlying mechanisms
of AKAP12-induced PI3K/AKT activation in CRC.
Acknowledgments
The authors would like to thank the Department of
Pathology, Huashan Hosiptal affiliated to Fudan University and the
Shanghai Tenth People's Hospital affiliated to Tongji University
for providing the CRC specimens.
Notes
[1]
Funding
This study was supported by grants from the
Outstanding Young Talent Plan of Shanghai (grant no. XYQ2013095),
the National Science Foundation (grant nos. 81370067 and 81572061)
and a grant from Shanghai Shenkang Hospital Development Center
(grant no. SHDC22014006).
[2] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
WWL and FYS mainly designed the research. PH and KL
mainly performed the research. SBL and MG analyzed the data and TTH
helped to collect the data. All authors have read and approved the
final manuscript.
[4] Ethics
approval and consent to participate
Written consent was obtained from all subjects and
the study protocol was approved by the Ethics Committees of Huashan
Hospital and Shanghai Tenth People's Hospital.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Ellina MI, Bouris P, Aletras AJ,
Theocharis AD, Kletsas D and Karamanos NK: EGFR and HER2 exert
distinct roles on colon cancer cell functional properties and
expression of matrix macromolecules. Biochim Biophys Acta.
1840:2651–2661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim HJ, Yu MH, Kim H, Byun J and Lee C:
Noninvasive molecular biomarkers for the detection of colorectal
cancer. BMB Rep. 41:685–692. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gordon T, Grove B, Loftus JC, O'Toole T,
McMillan R, Lindstrom J and Ginsberg MH: Molecular cloning and
preliminary characterization of a novel cytoplasmic antigen
recognized by myasthenia gravis sera. J Clin Invest. 90:992–999.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nauert JB, Klauck TM, Langeberg LK and
Scott JD: Gravin, an autoantigen recognized by serum from
myasthenia gravis patients, is a kinase scaffold protein. Curr
Biol. 7:52–62. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Finger EC, Castellini L, Rankin EB,
Vilalta M, Krieg AJ, Jiang D, Banh A, Zundel W, Powell MB and
Giaccia AJ: Hypoxic induction of AKAP12 variant 2 shifts
PKA-mediated protein phosphorylation to enhance migration and
metastasis of melanoma cells. Proc Natl Acad Sci USA.
112:4441–4446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee SW, Kim WJ, Choi YK, Song HS, Son MJ,
Gelman IH, Kim YJ and Kim KW: SSeCKS regulates angiogenesis and
tight junction formation in blood-brain barrier. Nat Med.
9:900–906. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu W, Guan M, Su B, Ye C, Li J, Zhang X,
Liu C, Li M, Lin Y and Lu Y: Quantitative assessment of AKAP12
promoter methylation in colorectal cancer using
methylation-sensitive high resolution melting: Correlation with
Duke's stage. Cancer Biol Ther. 9:862–871. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Agrawal A, Murphy RF and Agrawal DK: DNA
methylation in breast and colorectal cancers. Mod Pathol.
20:711–721. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mostafa MR, Yahia RS, Abd El Messih HM,
El-Sisy E and El Ghannam DM: Gravin gene expression in acute
myeloid leukemia. Med Oncol. 30:5482013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guan M, Zhou X, Soulitzis N, Spandidos DA
and Popescu NC: Aberrant methylation and deacetylation of deleted
in liver cancer-1 gene in prostate cancer: Potential clinical
applications. Clin Cancer Res. 12:1412–1419. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin Z, Hamilton JP, Yang J, Mori Y, Olaru
A, Sato F, Ito T, Kan T, Cheng Y, Paun B, et al: Hypermethylation
of the AKAP12 promoter is a biomarker of Barrett's-associated
esophageal neoplastic progression. Cancer Epidemiol Biomarkers
Prev. 17:111–117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sikandar S, Dizon D, Shen X, Li Z,
Besterman J and Lipkin SM: The class I HDAC inhibitor MGCD0103
induces cell cycle arrest and apoptosis in colon cancer initiating
cells by upregulating Dickkopf-1 and non-canonical Wnt signaling.
Oncotarget. 1:596–605. 2010.
|
|
13
|
Richon VM, Sandhoff TW, Rifkind RA and
Marks PA: Histone deacetylase inhibitor selectively induces p21WAF1
expression and gene-associated histone acetylation. Proc Natl Acad
Sci USA. 97:10014–10019. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spurling CC, Godman CA, Noonan EJ,
Rasmussen TP, Rosenberg DW and Giardina C: HDAC3 overexpression and
colon cancer cell proliferation and differentiation. Mol Carcinog.
47:137–147. 2008. View
Article : Google Scholar
|
|
15
|
Weichert W, Röske A, Gekeler V, Beckers T,
Ebert MP, Pross M, Dietel M, Denkert C and Röcken C: Association of
patterns of class I histone deacetylase expression with patient
prognosis in gastric cancer: A retrospective analysis. Lancet
Oncol. 9:139–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Du C, Lv Z, Ding C, Cheng J, Xie H,
Zhou L and Zheng S: The up-regulation of histone deacetylase 8
promotes proliferation and inhibits apoptosis in hepatocellular
carcinoma. Dig Dis Sci. 58:3545–3553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soung YH, Pruitt K and Chung J: Epigenetic
silencing of ARRDC3 expression in basal-like breast cancer cells.
Sci Rep. 4:38462014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lovaas JD, Zhu L, Chiao CY, Byles V,
Faller DV and Dai Y: SIRT1 enhances matrix metalloproteinase-2
expression and tumor cell invasion in prostate cancer cells.
Prostate. 73:522–530. 2013. View Article : Google Scholar
|
|
19
|
Heller G, Schmidt WM, Ziegler B, Holzer S,
Müllauer L, Bilban M, Zielinski CC, Drach J and Zöchbauer-Müller S:
Genome-wide transcriptional response to 5-aza-2′-deoxycytidine and
trichostatin a in multiple myeloma cells. Cancer Res. 68:44–54.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Godman CA, Joshi R, Tierney BR, Greenspan
E, Rasmussen TP, Wang HW, Shin DG, Rosenberg DW and Giardina C:
HDAC3 impacts multiple oncogenic pathways in colon cancer cells
with effects on Wnt and vitamin D signaling. Cancer Biol Ther.
7:1570–1580. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu MZ, Tsai YP, Yang MH, Huang CH, Chang
SY, Chang CC, Teng SC and Wu KJ: Interplay between HDAC3 and WDR5
is essential for hypoxia-induced epithelial-mesenchymal transition.
Mol Cell. 43:811–822. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim HC, Choi KC, Choi HK, Kang HB, Kim MJ,
Lee YH, Lee OH, Lee J, Kim YJ, Jun W, et al: HDAC3 selectively
represses CREB3-mediated transcription and migration of metastatic
breast cancer cells. Cell Mol Life Sci. 67:3499–3510. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guenther MG, Barak O and Lazar MA: The
SMRT and N-CoR corepressors are activating cofactors for histone
deacetylase 3. Mol Cell Biol. 21:6091–6101. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Togi S, Kamitani S, Kawakami S, Ikeda O,
Muromoto R, Nanbo A and Matsuda T: HDAC3 influences phosphorylation
of STAT3 at serine 727 by interacting with PP2A. Biochem Biophys
Res Commun. 379:616–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Das C and Kundu TK: Transcriptional
regulation by the acetylation of nonhistone proteins in humans - a
new target for therapeutics. IUBMB Life. 57:137–149. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arocho A, Chen B, Ladanyi M and Pan Q:
Validation of the 2-DeltaDeltaCt calculation as an alternate method
of data analysis for quantitative PCR of BCR-ABL P210 transcripts.
Diagn Mol Pathol. 15:56–61. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Yang Y, Lin X, Lu X, Luo G, Zeng T, Tang
J, Jiang F, Li L, Cui X, Huang W, et al: Interferon-microRNA
signalling drives liver precancerous lesion formation and
hepatocarcinogenesis. Gut. 65:1186–1201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Xu J, Wang H, Wu L, Yuan W, Du J
and Cai S: Trichostatin A, a histone deacetylase inhibitor,
reverses epithelial-mesenchymal transition in colorectal cancer
SW480 and prostate cancer PC3 cells. Biochem Biophys Res Commun.
456:320–326. 2015. View Article : Google Scholar
|
|
30
|
Fortson WS, Kayarthodi S, Fujimura Y, Xu
H, Matthews R, Grizzle WE, Rao VN, Bhat GK and Reddy ES: Histone
deacetylase inhibitors, valproic acid and trichostatin-A induce
apoptosis and affect acetylation status of p53 in ERG-positive
prostate cancer cells. Int J Oncol. 39:111–119. 2011.PubMed/NCBI
|
|
31
|
Chatterjee N, Wang WL, Conklin T, Chittur
S and Tenniswood M: Histone deacetylase inhibitors modulate miRNA
and mRNA expression, block metaphase, and induce apoptosis in
inflammatory breast cancer cells. Cancer Biol Ther. 14:658–671.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilson AJ, Byun DS, Popova N, Murray LB,
L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH and
Mariadason JM: Histone deacetylase 3 (HDAC3) and other class I
HDACs regulate colon cell maturation and p21 expression and are
deregulated in human colon cancer. J Biol Chem. 281:13548–13558.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Turtoi A, Mottet D, Matheus N, Dumont B,
Peixoto P, Hennequière V, Deroanne C, Colige A, De Pauw E,
Bellahcène A, et al: The angiogenesis suppressor gene AKAP12 is
under the epigenetic control of HDAC7 in endothelial cells.
Angiogenesis. 15:543–554. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Minami J, Suzuki R, Mazitschek R, Gorgun
G, Ghosh B, Cirstea D, Hu Y, Mimura N, Ohguchi H, Cottini F, et al:
Histone deacetylase 3 as a novel therapeutic target in multiple
myeloma. Leukemia. 28:680–689. 2014. View Article : Google Scholar
|
|
35
|
Jiang QG, Li TY, Liu DN and Zhang HT:
PI3K/Akt pathway involving into apoptosis and invasion in human
colon cancer cells LoVo. Mol Biol Rep. 41:3359–3367. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Akakura S, Huang C, Nelson PJ, Foster B
and Gelman IH: Loss of the SSeCKS/Gravin/AKAP12 gene results in
prostatic hyperplasia. Cancer Res. 68:5096–5103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bateman NW, Jaworski E, Ao W, Wang G,
Litzi T, Dubil E, Marcus C, Conrads KA, Teng PN, Hood BL, et al:
Elevated AKAP12 in paclitaxel-resistant serous ovarian cancer cells
is prognostic and predictive of poor survival in patients. J
Proteome Res. 14:1900–1910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hayashi M, Nomoto S, Kanda M, Okamura Y,
Nishikawa Y, Yamada S, Fujii T, Sugimoto H, Takeda S and Kodera Y:
Identification of the A kinase anchor protein 12 (AKAP12) gene as a
candidate tumor suppressor of hepatocellular carcinoma. J Surg
Oncol. 105:381–386. 2012. View Article : Google Scholar
|
|
39
|
Suren D, Yildirim M, Alikanoglu AS, Kaya
V, Yildiz M, Dilli UD and Sezer C: Lack of relation of AKAP12 with
p53 and Bcl-2 in colorectal carcinoma. Asian Pac J Cancer Prev.
15:3415–3418. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Choi MC, Jong HS, Kim TY, Song SH, Lee DS,
Lee JW, Kim TY, Kim NK and Bang YJ: AKAP12/Gravin is inactivated by
epigenetic mechanism in human gastric carcinoma and shows growth
suppressor activity. Oncogene. 23:7095–7103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Burnworth B, Pippin J, Karna P, Akakura S,
Krofft R, Zhang G, Hudkins K, Alpers CE, Smith K, Shankland SJ, et
al: SSeCKS sequesters cyclin D1 in glomerular parietal epithelial
cells and influences proliferative injury in the glomerulus. Lab
Invest. 92:499–510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gelman IH: Suppression of tumor and
metastasis progression through the scaffolding functions of
SSeCKS/Gravin/AKAP12. Cancer Metastasis Rev. 31:493–500. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Radeva MY, Kugelmann D, Spindler V and
Waschke J: PKA compartmentalization via AKAP220 and AKAP12
contributes to endothelial barrier regulation. PLoS One.
9:e1067332014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schott MB, Gonowolo F, Maliske B and Grove
B: FRET biosensors reveal AKAP-mediated shaping of subcellular PKA
activity and a novel mode of Ca(2+)/PKA crosstalk. Cell Signal.
28:294–306. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pilarsky C, Wenzig M, Specht T, Saeger HD
and Grützmann R: Identification and validation of commonly
overexpressed genes in solid tumors by comparison of microarray
data. Neoplasia. 6:744–750. 2004. View Article : Google Scholar
|
|
46
|
Setia S, Nehru B and Sanyal SN: The
PI3K/Akt pathway in colitis associated colon cancer and its
chemoprevention with celecoxib, a Cox-2 selective inhibitor. Biomed
Pharmacother. 68:721–727. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
LaBonte MJ, Wilson PM, Fazzone W, Russell
J, Louie SG, El-Khoueiry A, Lenz HJ and Ladner RD: The dual
EGFR/HER2 inhibitor lapatinib synergistically enhances the
antitumor activity of the histone deacetylase inhibitor
panobinostat in colorectal cancer models. Cancer Res. 71:3635–3648.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ye Y, Jin L, Wilmott JS, Hu WL, Yosufi B,
Thorne RF, Liu T, Rizos H, Yan XG, Dong L, et al: PI(4,5)P2
5-phosphatase A regulates PI3K/Akt signalling and has a tumour
suppressive role in human melanoma. Nat Commun. 4:15082013.
View Article : Google Scholar : PubMed/NCBI
|