Introduction
Recurrent additional sex combs-like 1 (ASXL1)
mutations are frequent in myeloid malignancies, including
myelodysplastic syndrome (MDS), acute myeloid leukemia (AML),
myelofibrosis and chronic myelomonocytic leukemia, and they are
associated with disease progression and prognosis (1–6). In
particular, ASXL1 mutations are associated with high-risk
MDS and leukemic transformation in MDS (4). In AML, ASXL1 mutations are
more frequent in patients with aberrant karyotypes, particularly
trisomy 8 (7–9). Our previous study revealed that
ASXL1 mutations were frequent in patients with aplastic
anemia, a disease in which a minority of patients later develops
MDS and AML; ASXL1 mutations grouped with other unfavorable
mutations were associated with a poor prognosis (10). Cumulative evidence has suggested
that ASXL1 acts as a tumor suppressor, which leads to
leukemic transformation when mutated.
The ASXL1 gene is a polycomb family member,
which serves roles in activation and repression of homeobox genes
by regulating the polycomb and trithorax groups of proteins
(11–13). The most prominent isoform of
ASXL1 (isoform 1) encodes a protein containing 1,541 amino
acids, which comprises several domains, such as the putative
N-terminal DNA-binding domain, the ASX homology domains and the
C-terminal typical plant homeodomain (PHD) (14). ASXL1 associates with the
deubiquitinating enzyme BRCA1-associated protein 1 to promote gene
expression through the removal of H2A lysine 119 ubiquitination
induced by polycomb repressive complex (PRC)1 (15). In addition, ASXL1 may interact with
members of PRC2 to inhibit gene expression by promoting
trimethylation of histone H3 lysine 27 (H3K27). ASXL1
deletion impairs PRC2-mediated gene expression and causes severe
inhibition of H3K27 trimethylation in myeloid hematopoietic cells,
thus leading to malignant transformation (16). In addition, ASXL1 knockdown
triggers apoptosis of human hematopoietic stem and progenitor
cells, which leads to a reduction in stem cell frequency and
decreased cell expansion along the myeloid lineage (17). Loss of ASXL1 function in
mice causes embryonic lethality and introduces an MDS-like
phenotype after a long latency (18–20).
In addition, missense and frameshift mutations of ASXL1 in
cell lines and patient specimens promote myeloid transformation
(2,7); these findings are consistent with the
tumor suppressor role of ASXL1 (21,22).
The Clustered Regularly Interspaced Short
Palindromic Repeats (CRISPR)/CRISPR-associated protein-9 nuclease
(Cas9) system is a powerful genome editing technique. It utilizes a
single guide RNA (gRNA) molecule to specifically alter DNA
sequences, allowing study of gene functions by generating targeted
knockin and knockout mutations of any gene. While some clinical and
biological consequences of ASXL1 mutations have been
characterized, precise molecular mechanisms remain to be
elucidated. Our previous study conducted CRISPR/Cas9-engineered
gene editing of a cell line, which disclosed novel and important
biological consequences of DNA methyltransferase 3α mutations in
K562 cells (23). Therefore, in
the present study, ASXL1-mutated human hematopoietic cell
lines were generated using CRISPR/Cas9 gene editing as a model
system for functional studies. The ASXL1-mutated U937 cells
used in the present study should be useful to obtain insights into
myelomonocytic mechanisms of ASXL1 action, and to identify
therapeutic strategies for hematopoietic malignancies associated
with ASXL1 mutations.
Materials and methods
Generation of ASXL1-mutated U937 cell
lines
The human U937 leukemic cell line was purchased from
the American Type Culture Collection (Manassas, VA, USA). U937 and
its derivative cell lines were maintained in RPMI-1640 medium
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented
with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), 1% L-glutamine, 100 U/ml penicillin and
100 µg/ml streptomycin (Thermo Fisher Scientific, Inc.) at
37°C. Using the Amaxa® Cell Line
Nucleofector® kit C (Lonza, Inc., Allendale, NJ, USA),
1×106 U937 cells were transfected with a pU6-gRNA
plasmid (1 µg; target ID: HS0000388833, target site:
GCCACGCCGATGGCGAGAGCGG; Sigma-Aldrich; Merck KGaA) and a
pCMV-CG-Cas9-green fluorescence protein (GFP) plasmid (1 µg;
product number: CAS9GFPP-1EA; Sigma-Aldrich; Merck KGaA), according
to the manufacturer's protocols. GFP-positive cells were separated
by fluorescence-activated cell sorting 24 h post-transfection and
were seeded as single cells in 96-well plates. After ~2 weeks,
surviving single cell clones were selected and expanded. Aliquots
of these single cell clones were stored in liquid nitrogen using
freezing medium and the remaining cells were subjected to genomic
DNA extraction, in order to validate gene mutations by
sequencing.
Validation of ASXL1 mutations
Single cell clones were disrupted in lysis buffer
[10 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.5% Triton X-100 and 100
µg/ml Proteinase K] and were incubated at 56°C for 1 h in
order to liberate total genomic DNA. Subsequently, cell lysates
were treated at 96°C for 5 min to inactivate proteinase K and
subjected to polymerase chain reaction (PCR) amplification using
the Takara LA TaqDNA Polymerase with GC Buffer (Takara Bio USA,
Inc., Mountain View, CA, USA) as follows: 94°C for 3 min; 45 cycles
at 94°C for 30 sec, 58°C for 50 sec and 72°C for 1 min; and 72°C
for 5 min. Following purification with the QIAquick PCR
purification kit (Qiagen, Inc., Valencia, CA, USA), amplicons were
sequenced using the BigDye Terminator version 3.1 in the ABI Prism
3100 analyzer (both from Applied Biosystems; Thermo Fisher
Scientific, Inc.) in both directions to confirm ASXL1 gene
mutations in exon 8 (containing a targeted site of the gRNA). The
following primers were used for PCR and sequencing: Forward 1,
5′-gccagaccatgaagtggtggtttc-3′; forward 2,
5′-cagaccatgaagtggtggtttctc-3′; reverse 1, 5′-
ctggtaaaggaattggaatagaag-3′ a nd reverse 2,
5′-gacatcatcttctcactaggcctg-3′. All procedures were performed
according to the manufacturers' protocols. According to sequencing
results, transfected single cell clones were categorized into
transfected wild-type (WT) clones (including WT1 and WT2 used in
further experiments), and ASXL1-mutated (MT) clones
(including MT+/− and MT−/−).
Cell growth and cell cycle analyses
To generate growth curves, cells in the logarithmic
phase were seeded into 96-well plates at 2.5×104
cells/well (200 µl) in triplicate, and viable cells were
counted by trypan blue exclusion over 8 consecutive days. All
experiments were conducted alongside passage-matched parental U937
cells. For cell cycle analysis, 1×106 cells suspended in
1 ml NuCycl propidium iodide (PI) (Exalpha Biologicals, Inc.,
Shirley, MA, USA) were incubated in the dark for 20 min at 37°C,
after which they were placed on ice and cell cycle progression was
determined within 30 min by flow cytometry.
5-Fluorouracil (5-FU)-induced cell growth
inhibition and apoptosis
To examine 5-FU-induced cell growth inhibition,
cells were plated at 2×104 cells/well (200 µl) in
a 96-well plate in triplicate and were treated with various
concentrations of 5-FU (0, 2.5, 5, 10, 20, 50 and 100 µM). A
total of 48 h post-treatment, cell proliferation was evaluated
using the Cell Counting kit-8 (CCK-8; Sigma-Aldrich; Merck KGaA)
according to manufacturer's protocol. Briefly, 10 µl of
CCK-8 solution was added to each well, and the cells were incubated
at 37°C for 2 h. Subsequently, the absorbance was measured at 450
nm using the PerkinElmer Victor3 V Plate Counter (Conquer
Scientific, San Diego, CA, USA). The inhibitory rate was calculated
as follows: [1 - (absorbance of treated cells/absorbance of control
cells)] x 100%. To analyze 5-FU-induced apoptosis, cells were
seeded at 2×105 cells/well (1 ml) in 24-well plates and
were treated with various concentrations of 5-FU (0, 2.5, 5, 10, 20
and 40 µM). After 48 h, cells were harvested, washed in cold
PBS, and stained with 5 µl of fluorescein isothiocyanate
(FITC) Annexin V and 10 µl of propidium iodide (PI) solution
in 100 µl of Annexin V Binding Buffer (FITC Annexin V
Apoptosis Detection kit with PI; BioLegend, Inc., San Diego, CA,
USA) at room temperature in the dark for 15 min, according to the
manufacturer's protocol, followed by flow cytometry.
Phorbol 12-myristate 13-acetate
(PMA)-induced differentiation
Cells were cultured at 1×105 cells/well
(1 ml) in 6-well plates for flow cytometry, or in slide chambers
(Nunc Lab-Tek II Chamber Slide; Thermo Fisher Scientific, Inc.) for
Wright-Giemsa staining, and were subjected to treatment with
various concentrations of PMA (0, 1, 5, 10, 50, 100 and 200 ng/ml).
After 96 h, adhesive cells in 6-well plates were harvested using
cell scrapers, washed with cold PBS and stained with anti-cluster
of differentiation (CD)11b-phycoerythrin antibody (Ab) (clone
M1/70, 101208; BioLegend, Inc.) at 0.02 mg/ml on ice for 30 min,
followed by flow cytometry. For Wright-Giemsa staining, adherent
cells in slide chambers were washed with PBS and stained with
StainRITE Wright-Giemsa Stain Solution (Polysciences, Inc.,
Warrington, PA, USA) to examine their morphologies using a Zeiss
Axioskope 2 Plus microscope (Carl Zeiss AG, Oberkochen, Germany)
and to assess differentiation.
Gene expression analyses by RNA
sequencing (RNA-seq) and reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the RNeasy Plus Mini
kit (Qiagen, Inc.), and was subsequently used for RNA-seq and
RT-qPCR. RNA-seq was performed by Genewiz (South Plainfield, NJ,
USA). Briefly, samples underwent ribosomal RNA depletion, stranded
RNA library preparation, multiplexing and cluster generation,
followed by Illumina HiSeq2500 (Illumina, San Diego, CA, USA) at
2×100 base pair for paired-end sequencing in a high output mode.
Sequence reads were trimmed and then aligned to the human reference
genome (hg19) using Tophat2, and the featureCounts function within
the Rsubread package was applied to summarize data to gene-level
read counts with USCS refseq annotation (24). Fragments per kilobase of transcript
per million mapped reads (FPKM) values were calculated with
cufflinks (25).
EdgeR was used to identify differentially expressed
genes between groups (26) and
topGO was used for annotation (27). Differentially expressed genes,
which were identified using threshold P-value <0.05, fold change
>2 and false discovery rate (FDR)-corrected P-value <0.20,
were further subjected to Ingenuity Pathway Analysis (IPA).
Heatmaps containing representative top-regulated genes were
generated using R (https://www.r-project.org). Gene Set Enrichment
Analysis (GSEA) was performed using online free software http://software.broadinstitute.org/gsea/index.jsp
from the Broad Institute (Cambridge, MA, USA). Enriched gene sets
were defined according to the following criteria: P<0.05 and
FDR-corrected P-value <0.05. miso (28) was used to detect RNA splicing
differences, specifically differentially regulated exons or
isoforms.
For RT-qPCR, total RNA was reverse transcribed to
cDNA using the SuperScript III First-Strand Synthesis Super Mix
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. cDNA was subjected to qPCR using the TaqMan Gene
Expression Master Mix and the following TaqMan probes with FAM-MGB
(Thermo Fisher Scientific, Inc.): Calcium voltage-gated channel
auxiliary subunit α2δ3 (CACNA2D3; Hs01045030_m1), cytochrome
B-245 β chain (CYBB; Hs00166163_m1), cathepsin G
(CTSG; Hs01113415_g1), actin-like 8 (ACTL8; Hs00380546_m1),
NLR family apoptosis inhibitory protein (NAIP;
Hs03037952_m1), oxidation resistance 1 (OXR1;
Hs00757844_m1), chondroitin sulfate proteoglycan 4 (CSPG4;
Hs00361541_g1), C-type lectin domain family 5, member A
(CLEC5A; Hs00183780_m1), and ASXL1 (Hs00392415_m1 and
Hs00899495_g1). A housekeeping gene, β-actin (ACTB), was
measured using the ACTB probe (Hs99999903_m1) with VIC-MGB,
as an endogenous control to normalize differences in input cDNA
amounts. Triplicate samples were analyzed using the 7500 Fast
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) as follows: Stage 1, 95°C for 20 sec; stage 2, 40 cycles at
95°C for 3 sec and 60°C for 30 sec; data were collected at stage 2
step 2. Relative quantification was calculated using the ΔΔCq
method (29).
Protein extraction, immunoprecipitation
and immunoblotting
For immunoblotting of CYBB and ACTL8, cells were
lysed using M-PER Mammalian Protein Extraction Reagent (Thermo
Fisher Scientific, Inc.) containing cOmplete Mini EDTA-free
Protease Inhibitor Cocktail (Sigma-Aldrich; Merck KgaA). The
proteins were then resolved by SDS-PAGE (4-12% gel). For immunoblot
analysis of ASXL1, cells were lysed with the
radioimmunoprecipitation assay lysis buffer system (sc-24948; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), and proteins were
subjected to immunoprecipitation with anti-ASXL1 mouse Ab (6E2,
sc-293204; Santa Cruz Biotechnology, Inc.) using Dynabeads Protein
G Immunoprecipitation kit (Thermo Fisher Scientific, Inc.),
followed by separation with 8% SDS-PAGE. Protein concentrations
were quantified using the Micro Bicinchoninic Acid Protein Assay
kit (Thermo Fisher Scientific, Inc.), and 100 µg extracted
total proteins (for CYBB and ACTL8) and immunoprecipitated ASXL1
from 3.5 mg total proteins were loaded into individual wells of
gels. After protein transfer onto polyvinylidene fluoride
membranes, immunoblotting was performed with the following primary
Abs: Anti-CYBB rabbit Ab (NOX2/gp91phox, ab129068), anti-ACTL8
rabbit Ab (ab184562) (both from Abcam, Cambridge, MA, USA),
anti-ASXL1 Ab (6E2, sc-293204; Santa Cruz Biotechnology, Inc.) and
anti-GAPDH Rabbit Ab (#2118; Cell Signaling Technology, Inc.,
Danvers, MA, USA). After incubation with horseradish peroxidase
(HRP)-conjugated anti-rabbit immunoglobulin G (IgG) Ab (sc-2004;
Santa Cruz Biotechnology, Inc.) or anti-mouse IgG Ab (m-IgGκ
BP-HRP; sc-516102), signals were detected using the SuperSignal
West Dura Extended Duration Substrate and/or the SuperSignal West
Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific,
Inc.) and blots were visualized using the ImageQuant LAS 4000
system (GE Healthcare Life Sciences, Pittsburgh, PA, USA). GAPDH
was used as a loading control, and GAPDH expression of remaining
cell lysates was measured as a loading control for
immunoprecipitated ASXL1 protein fractions.
Flow cytometry and statistical
analyses
Intracellular staining of CYBB was performed
according to Novus Biologicals General Intracellular Cytoplasmic
Target Flow Protocol (https://www.novusbio.com/support/support-by-application/general-intracellular-cytoplasmic-target-flow-protocol).
Briefly, cells were fixed with 4% paraformaldehyde prior to
permeabilization with 1X PBS + 0.5% Tween-20. Anti-CYBB/NOX2-PE Ab
(NL7, NBP1-41012PE; Novus Biologicals, LLC, Littleton, CO, USA) and
anti-MDL-1/CLEC5A-Alexa Fluor 488 Ab (FAB2384G; R&D Systems,
Minneapolis, MN, USA) were used to detect CYBB and CLEC5A
expression. For all flow cytometry experiments, data acquisition
was performed using BD Fortessa (BD Biosciences, Franklin Lakes,
NJ, USA) and data were subjected to analysis using FlowJo software
version 7.6.4 (FlowJo, LLC, Ashland, OR, USA). Experiments were
conducted in triplicate unless otherwise indicated, data are
presented as the means ± standard error of the mean. Statistical
analysis was performed using one-way analysis of variance for
comparisons between three or more groups (GraphPad Prism, version
7.02; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Generation of ASXL1-mutated U937 cell
lines using CRISPR/Cas9
ASXL1 mutations were introduced into U937
cells using CRISPR/Cas9 gene editing. The ASXL1 gene was initially
sequenced in wild-type bulk parental (WTblk) U937 cells and
confirmed the absence of pathogenic mutations, with the exception
of a heterozygous substitution (c.604C>A, p.Pro602Thr) reported
as a polymorphism by comparison with its published nucleotide
sequence (isoform 1, NM_015338). The gRNA used in the present study
targeted a specific site (nt1010-1031) of exon 8 in ASXL1,
which contains 13 exons in total. Using Sanger sequencing, the
present study observed a c.594insA (Ser199Glufsx55) mutation, which
creates a short, truncated protein followed by 55 additional amino
acids, due to a premature termination codon (Fig. 1A–C), thus resulting in a protein
consisting of only the N-terminal ASXN domain. A total of 6 MT cell
lines, including MT+/− (MT1, MT2 and MT3) and
MT−/− (MT11, MT12 and MT13), and transfected WT cell
lines (WT1 and WT2 derived from transfecting single cell clones
with WT ASXL1) were used for further experiments. Introduced
ASXL1 mutations were observed in mRNA sequences of
individual cell lines using RNA-seq data (data not shown). ASXL1
protein expression was also decreased in MT+/− cell
lines and was not detectable in MT−/− cell lines
(Fig. 1D). The cell morphology of
ASXL1-mutated U937 cell lines appeared indistinguishable from that
of WT cells, as determined by conventional microscopy (Fig. 1E).
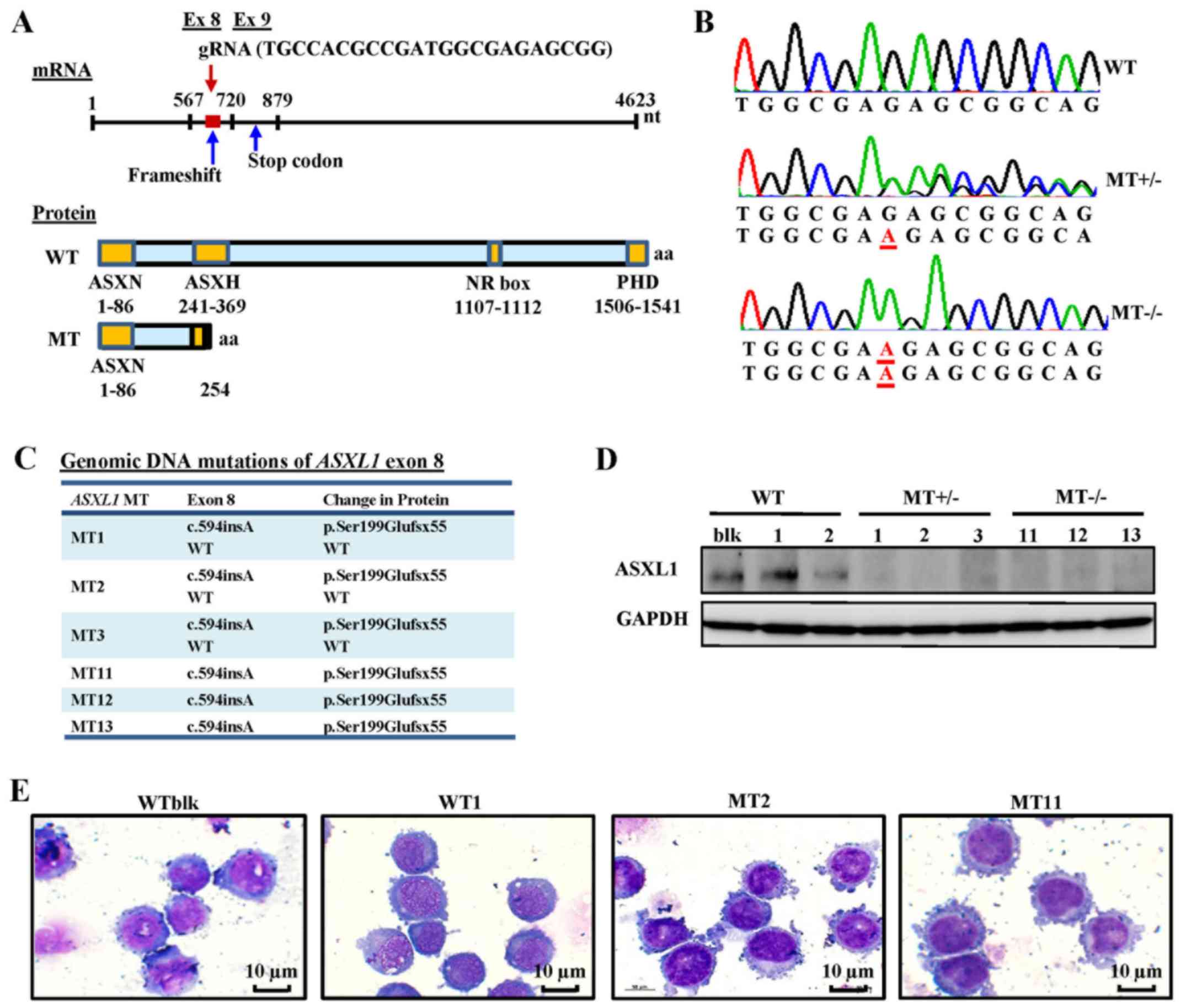 | Figure 1Generation of ASXL1-mutated
U937 cell lines using the CRISPR/Cas9 system. (A) Schematic
diagrams of ASXL1 mRNA, and ASXL1-WT and
ASXL1-MT proteins. The target site of gRNA, frameshift
mutation and truncated proteins are indicated. (B) Representative
electropherograms, as determined by Sanger sequencing, for the
evaluation of ASXL1 mutations in exon 8 introduced by
CRISPR/Cas9. Inserted nucleotides are underlined in red. (C)
ASXL1 mutations in exon 8 of all mutated cell lines with
associated changes in ASXL1 protein. All genomic and protein
sequence variants are represented using the Human Genome Variation
Society nomenclature. WT indicates a WT sequence without mutations.
(D) Immunoblot analysis of ASXL1 in WT and MT cell lines.
Immunoprecipitated protein samples were subjected to 8% SDS-PAGE
and immunoblot analysis. (E) Representative images of
Wright-Giemsa-stained WTblk, WT1, MT2 and MT11 U937 cells in
logarithmic growth phase. ASXL1, additional sex combs-like 1;
CRISPR/Cas9, Clustered Regularly Interspaced Short Palindromic
Repeats/CRISPR-associated protein-9 nuclease; Ex, exon; gRNA, guide
RNA; NR, nuclear receptor; PHD, plant homeodomain; MT, mutated; WT,
wild-type; WTblk, wild-type bulk parental. |
ASXL1 mutations have little impact on
cell proliferation and cell cycle progression of U937 cells
The present study hypothesized that
ASXL1-mutated U937 cells may possess abnormalities in
essential cellular functions, including proliferation, cell cycle
progression, apoptosis and resistance to cytotoxic drugs. Growth
curves were similar among the WT (WTblk and WT1), MT+/−
(MT1, MT2 and MT3) and MT−/− (MT11, MT12 and MT13) cell
lines (Fig. 2A). Distribution
among the G1, S and G2 phases of the cell
cycle exhibited modest variations among the individual cell lines
(Fig. 2B). 5-FU is an inhibitor of
thymidylate synthesis that interferes with DNA replication by
inducing double-strand breaks. The present study examined
susceptibility of ASXL1-mutated U937 cells to 5-FU. The
results indicated that their growth inhibition rates were elevated
with increasing 5-FU concentration, plateauing at ~20 µM,
and variations were detected among the ASXL1-mutated cell
lines and the WTblk and WT1 cells (Fig. 2C, left panel). However, average
inhibition rates were similar among the WT, MT+/− and
MT−/− groups (Fig. 2C,
right panel). Similarly, apoptosis following exposure to 5-FU was
not significantly different among almost all of the cell lines
(Fig. 2D, left panel), or among
the WT, MT+/− and MT−/− groups (Fig. 2D, right panel).
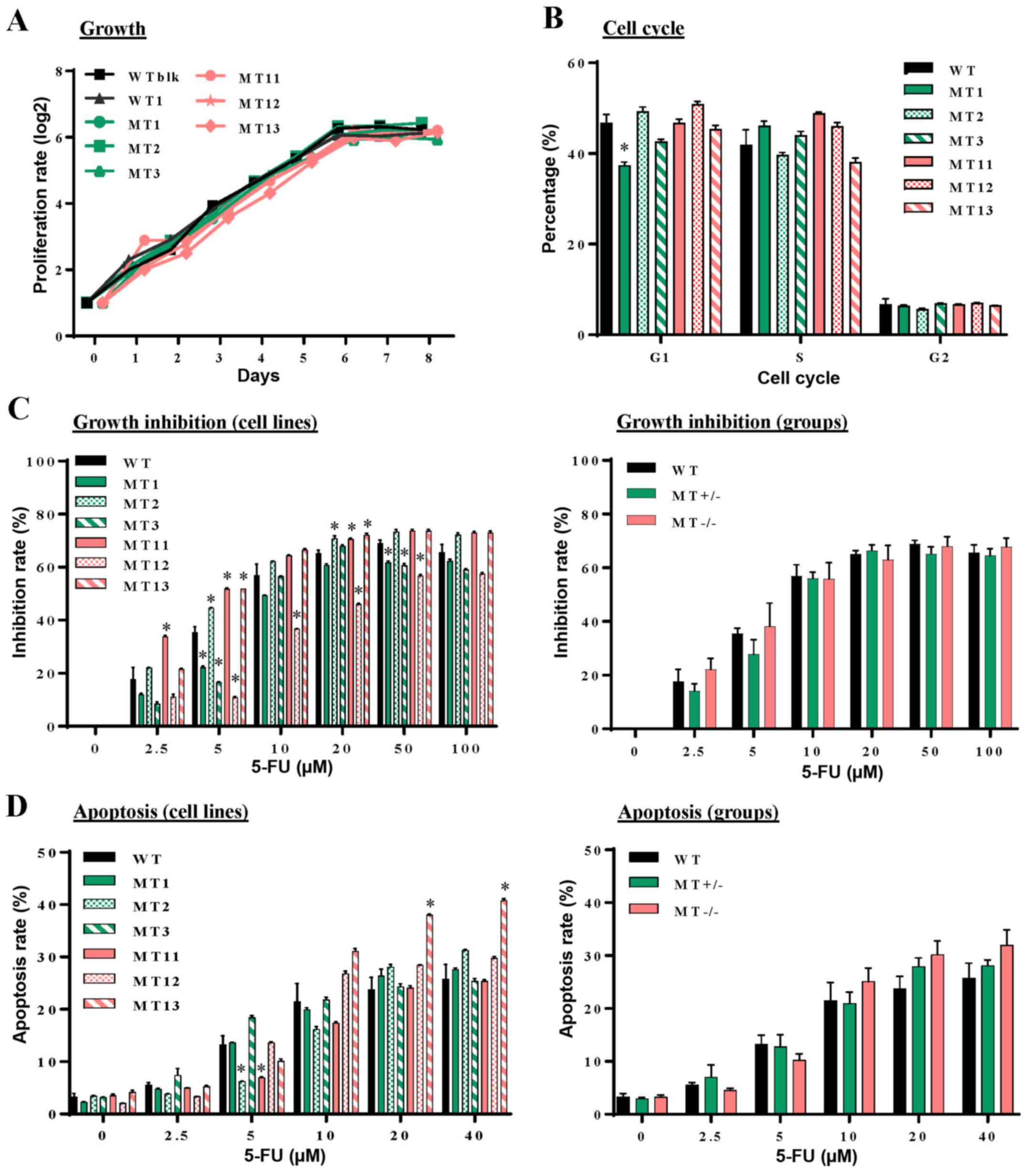 | Figure 2Effects of CRISPR/Cas9
nuclease-mediated additional sex combs-like 1 mutations on cell
proliferation, cell cycle progression, and 5-FU induced growth
inhibition and apoptosis. (A) Proliferation of individual WT,
MT+/− and MT−/− U937 cell lines was measured
at different time-points in culture by counting viable cells using
a trypan blue exclusion procedure. (B) Flow cytometric analysis of
cell cycle progression with PI DNA staining was performed at
logarithmic growth phase during the culture of WT, MT+/−
and MT−/− cells. (C) Following treatment of WT,
MT+/− and MT−/− cells with various
concentrations of 5-FU for 48 h, cell proliferation was measured
using cell counting kit-8 and inhibition rates were calculated
compared with untreated samples of the corresponding cells. Left
panel, inhibition rates of individual cell lines; right panel,
average inhibition rates of WT, MT+/− and
MT−/− U937 cell groups. (D) Following treatment with
various concentrations of 5-FU, cell apoptosis was assessed by flow
cytometry with Annexin V and PI staining. Left panel, percentages
of apoptotic cells in individual cell lines; right panel, average
percentages of individual groups. WT in (B-D) indicates average
values derived from WTblk, WT1 and WT2 cells. Data are presented as
the means ± standard error of the mean. *P<0.05 vs.
the WT group, one-way analysis of variance. 5-FU, 5-fluorouracil;
CRISPR/Cas9, clustered regulaly interspaced short palindromic
repeats/CRISPR-associated protein-9 nuclease; PI, propidium iodide;
MT, mutated; WT, wild-type; WTblk, wild-type bulk parental. |
ASXL1 mutations impair PMA-induced
differentiation of U937 cells
The U937 cell line is widely employed as a model of
monocyte/macrophage differentiation following treatment with PMA,
which is a typical inducer of differentiation. Following treatment
with PMA for 96 h, WT, MT+/− and MT−/− cell
lines were examined by Wright-Giemsa staining to detect morphology,
and by flow cytometry to detect CD11b expression; CD11b is a
pan-macrophage marker that is highly expressed in monocytes and
macrophages. The majority of cells differentiated into macrophages
and adhered to the plates. WT cells (WTblk and WT1) were larger and
richer in cytoplasm compared with MT cells (MT2 and MT11) (Fig. 3A). As determined by flow cytometry,
there were fewer CD11b-positive cells in the MT+/− and
MT−/− groups compared with in the WT group following PMA
treatment (Fig. 3B and C). These
results indicated that ASXL1-mutated U937 cells were less
responsive to PMA-induced differentiation.
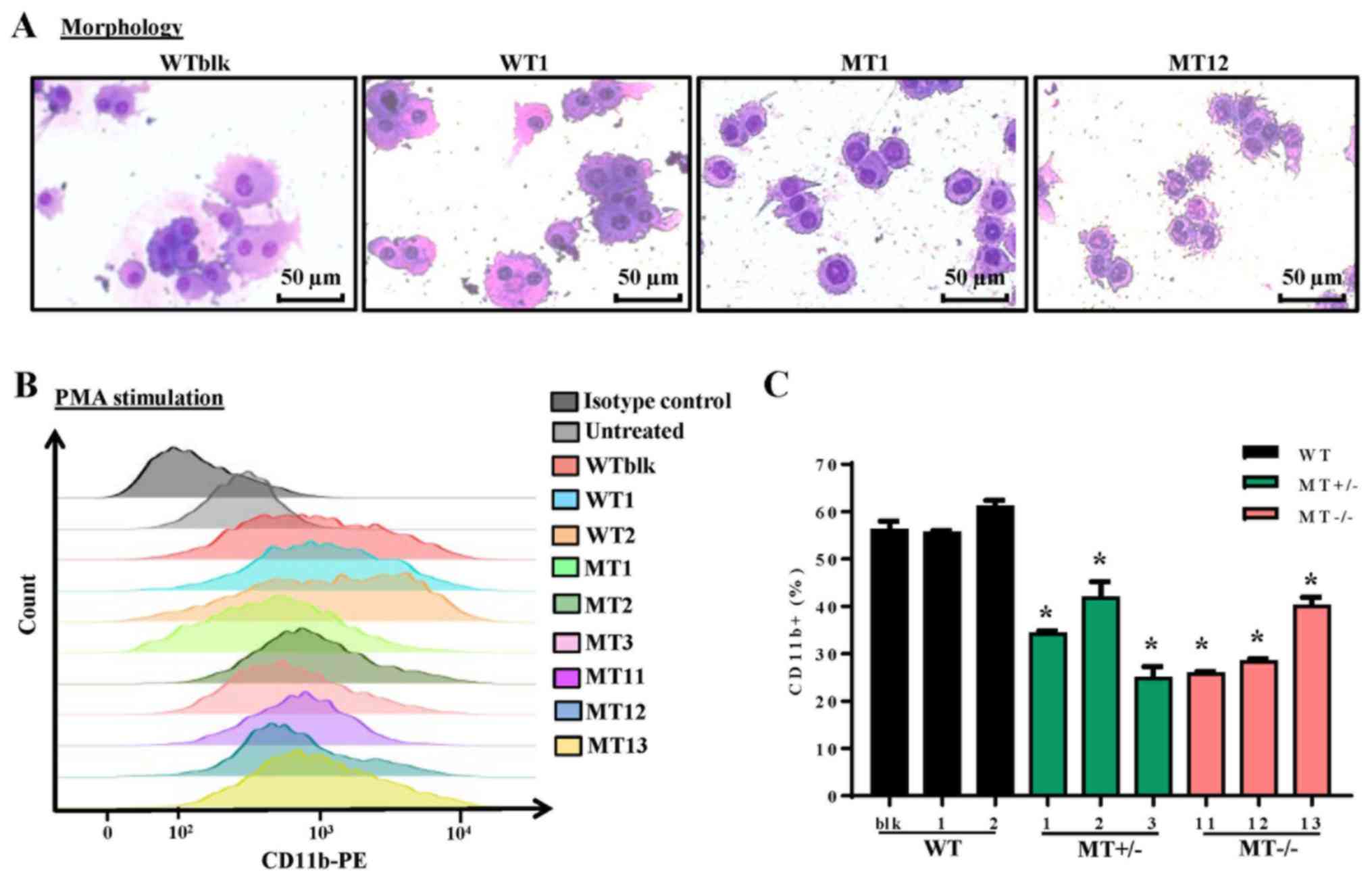 | Figure 3Effects of ASXL1 mutations on
the differentiation of PMA-treated U937 cells. Following treatment
with various concentrations of PMA, cells were harvested at 96 h.
(A) Representative images of Wright-Giemsa-stained WT and
ASXL1-mutated U937 cell lines, which attached to chamber
slides following 200 ng/ml PMA-induced differentiation. (B) CD11b
expression in cells harvested from slide chambers, as determined by
flow cytometric analysis. (C) Percentages of CD11b-positive cells
in WT and MT U937 cells calculated using flow cytometry data. Data
are presented as the means ± standard error of the mean.
*P<0.05 vs. the WT group (WTblk, WT1 and WT2),
one-way analysis of variance. ASXL1, additional sex combs-like 1;
CD11b, cluster of differentiation 11b; PE, phycoerythrin; PMA,
phorbol 12-myristate 13-acetate; MT, mutated; WT, wild-type; WTblk,
wild-type bulk parental. |
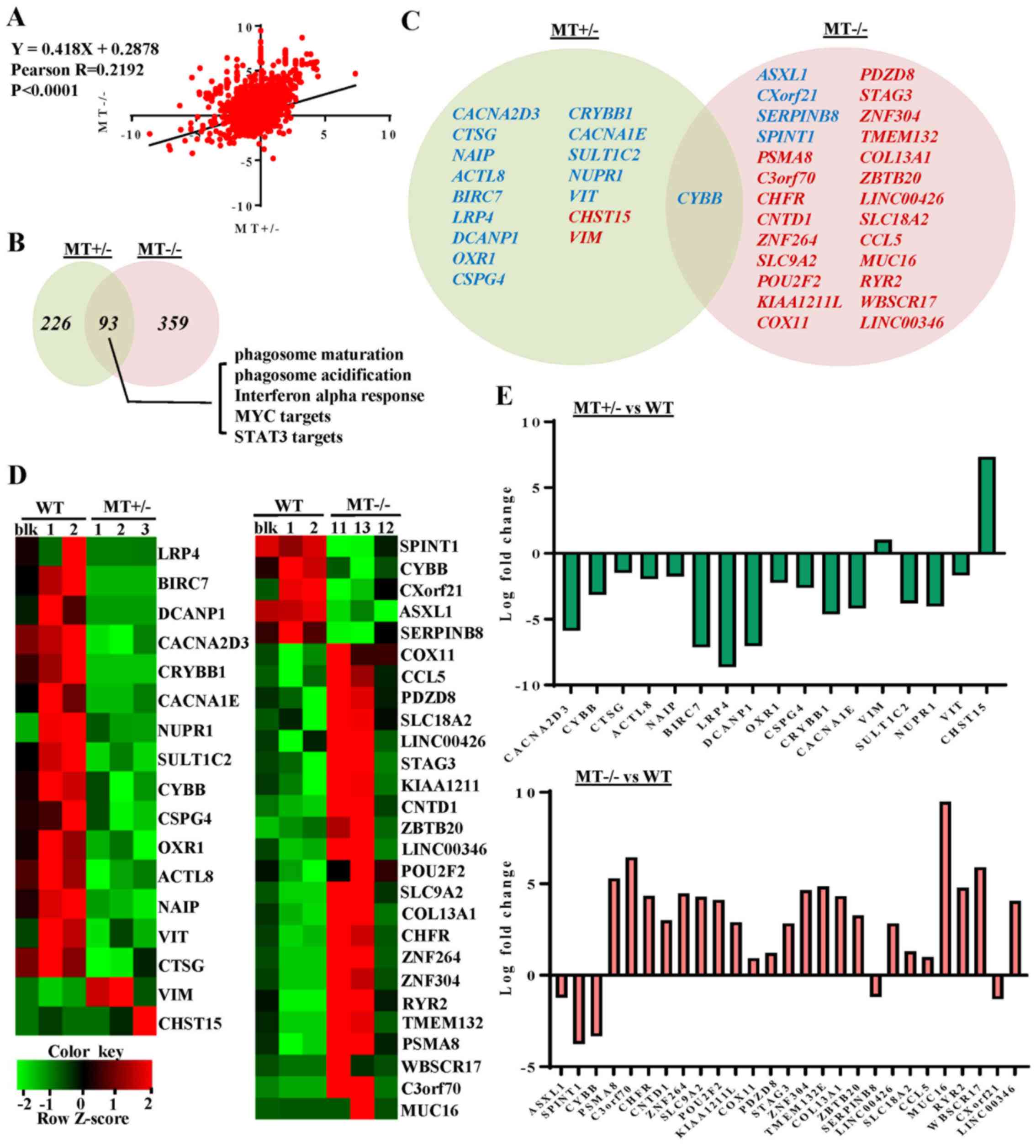
RNA-seq reveals dysregulated gene
expression profiles in ASXL1-mutated U937 cell lines
To characterize the molecular mechanisms underlying
differentiation defects, and to obtain dysregulation profiles of
ASXL1-mutated U937 cells, RNA-seq of WT and
ASXL1-mutated U937 cell lines was performed. When compared
with the WT group (WTblk, WT1 and WT2), with criteria of >2-fold
change and P<0.05, 107 upregulated and 54 downregulated genes
were identified in the MT group (including MT1-MT3 and MT11-MT13;
data not shown). At an FDR-corrected P<0.05, CYBB was the
only differentially expressed gene in the MT group compared with
the WT group. Considering likely variations in gene expression
within the MT group, the present study analyzed the differentially
expressed genes in the MT+/− (MT1-MT3) and
MT−/− (MT11-MT13) groups relative to the WT group, with
cutoff values of P<0.05, fold change >2 and FDR-corrected
P<0.20 (Fig. 4). By determining
the fold changes of individual genes in the MT+/− and
MT−/− groups relative to the WT group, a linear
correlation (P<0.0001 and Pearson R-value, 0.2192) was observed
between the MT+/− and MT−/− groups (Fig. 4A). When compared with the WT group,
15 downregulated and 2 upregulated genes were detected in the
MT+/− group, whereas 5 downregulated and 22 upregulated
genes were identified in the MT−/− group. In addition,
only CYBB was differentially expressed (downregulated) in
both groups relative to WT (Fig. 4C
and D). Log fold changes of the differentially expressed genes
ranged between -10 and 10 (Fig.
4E).
ASXL1 mutations dysregulate
transcriptional programs associated with U937 cell differentiation
and survival
To further explore the effects of ASXL1
mutations on the molecular machinery of U937 cells, GSEA was
performed in MT+/− (MT1-MT3) and MT−/−
(MT11-MT13) groups, and the results were compared to the WT (WTblk,
WT1 and WT2) group (Fig. 5). There
were 93 common skewed gene sets shared by MT+/− and
MT−/−, whereas in total, MT−/− cells harbored
133 more skewed gene sets than MT+/− cells (Fig. 4B). A gene set associated with the
negative regulation of myeloid differentiation was upregulated in
the MT+/− group while the macrophage differentiation
pathway was specifically downregulated in the MT−/−
group (Fig. 5A and B). Apoptosis
gene sets exhibited no significant alterations in the
MT+/− and MT−/− groups compared with in the
WT group; this finding is consistent with the results of the
functional assay (Fig. 5F). Two
genes (CYBB and CLEC5A) that were revealed to be
highly associated with myeloid cell differentiation were
downregulated in the MT groups, as determined by gene set analysis
of RNAseq data (Fig. 5B and C).
Gene sets associated with monocyte functions, including phagosome
maturation and phagosome acidification, were also downregulated in
the MT groups (Fig. 5D and E).
Other gene sets associated with various cell functions were
downregulated in the MT+/− and MT−/− cells:
Monocyte functions (such as ATP synthesis, oxidative
phosphorylation and respiratory chain), cell development and
survival, cell growth and death (such as interferon α response, MYC
targets, and signal transducer and activator of transcription 3
targets), and cell-to-cell signaling and interaction (Fig. 4B).
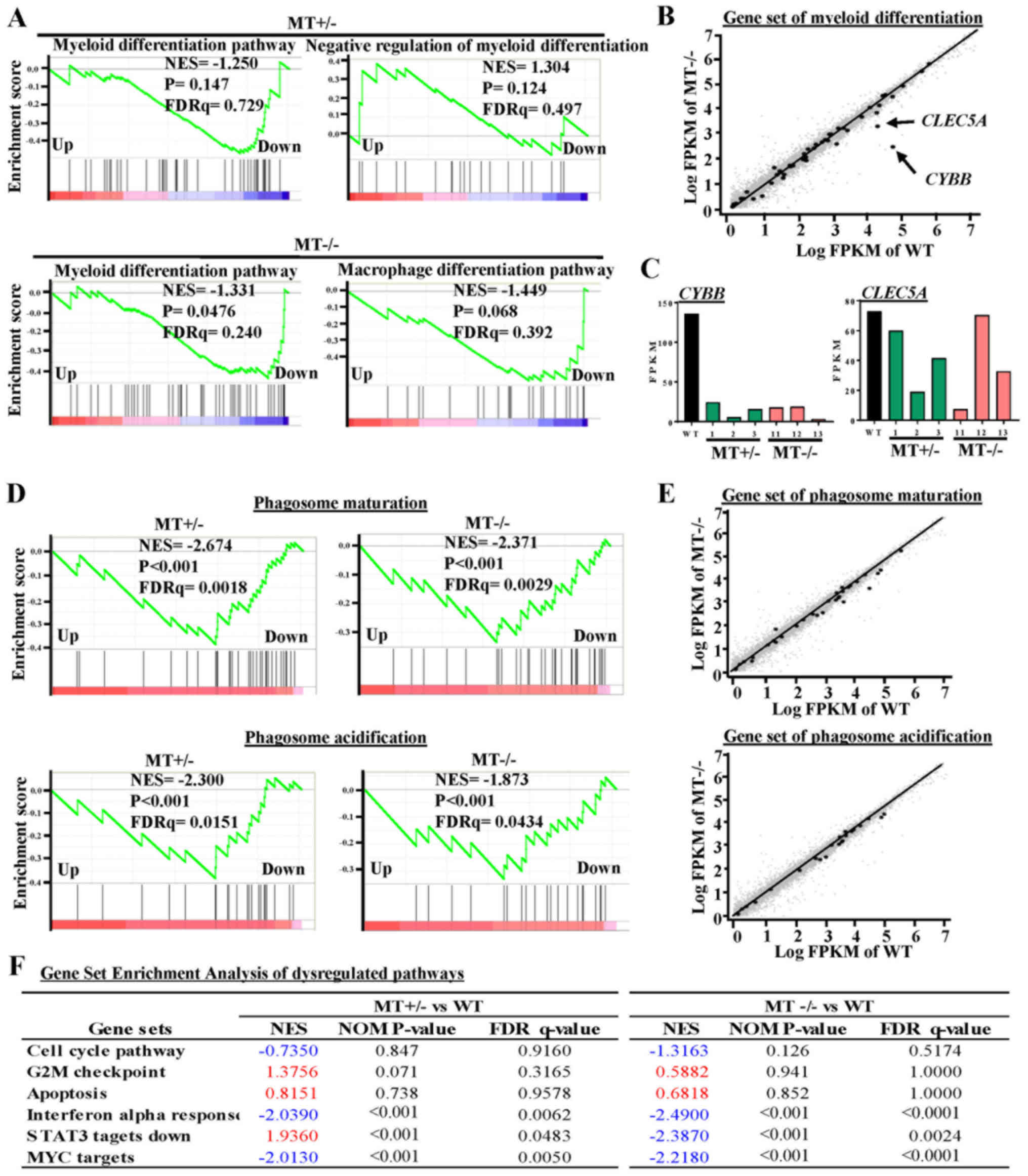 | Figure 5GSEA of dysregulated pathways in
MT+/− and MT−/− U937 cell lines. (A) GSEA
plots represent decreased expression of gene sets involved in
myeloid differentiation-related pathways in MT+/−
(MT1-MT3) and MT−/− (MT11-MT13) groups relative to the
WT (WT bulk parental, WT1 and WT2) group. NES, P-value and FDRq
value (FDR-corrected P-value) are presented. (B) Scatter plot of
genes in the myeloid differentiation pathway, as constructed by
comparing Log FPKM values between the MT−/− and the WT
groups. Black or small grey dots represent genes in the myeloid
differentiation pathway and all genes detected by RNA sequencing,
respectively; CYBB and CLEC5A are indicated with arrows. (C) FPKM
values of CYBB and CLEC5A in RNA-seq data. (D) GSEA
of phagosome maturation and phagosome acidification gene sets for
the MT+/− and MT−/− groups compared with the
WT group. (E) Scatter plots of phagosome maturation (top panel) and
phagosome acidification (bottom panel) gene sets for the
MT−/− group in which x- and y-axes refer to the Log FPKM
of MT−/− and Log FPKM of WT groups, respectively. (F)
Six representative pathways associated with the cell cycle,
apoptosis and other signaling pathways. NES are highlighted in blue
(downregulated) and red (upregulated). CLEC5A, C-type lectin
domain family 5, member A; CYBB, cytochrome B-245 β chain;
FDR, false discovery rate; FPKM, fragments per kilobase of
transcript per million mapped reads; GSEA, Gene Set Enrichment
Analysis; MT, mutated; NES, normalized enrichment score; WT,
wild-type. |
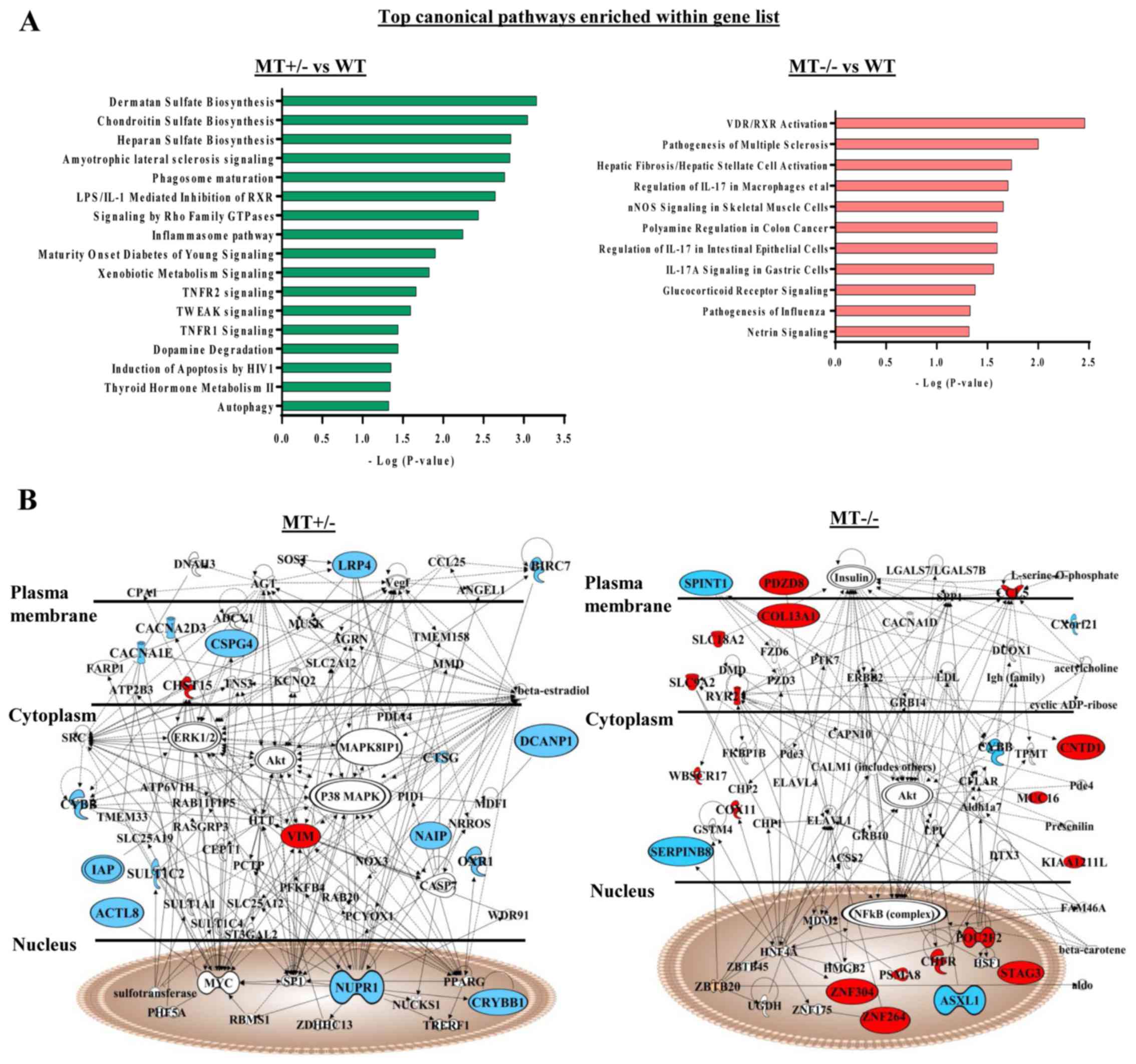
IPA of differentially expressed genes was performed
in the MT+/− and MT−/− groups compared with
in the WT group. Some of these genes were revealed to be involved
in the phagocyte development signaling pathway, which is consistent
with the GSEA data of defects in monocyte/macrophage
differentiation of ASXL1-mutated cells (Fig. 6A). In addition, protein products
from differentially expressed genes in the MT+/− group
were interconnected, and were involved in cell death and survival
signaling [such as tumor necrosis factor (TNF) receptor (TNFR)1,
TNFR2 and TNF-related weak inducer of apoptosis]; these proteins
were components of a comprehensive network, with mitogen-activated
protein kinase (MAPK), protein kinase B (AKT), extracellular
signal-regulated kinase, nuclear factor (NF)-κB, caspase and MYC as
molecular hubs (Fig. 6B).
To validate altered gene expression detected by
RNA-seq analysis, RT-qPCR, immunoblotting and flow cytometry were
performed. RT-qPCR confirmed that the expression levels of genes
essential for myeloid differentiation were significantly reduced,
including CYBB and CLEC5A, and that genes involved in
cell death and survival, such as NAIP, CACNA2D3,
ACTL8, CTSG, OXR1 and CSPG4, were
decreased in ASXL1-mutated cells compared with in WT cells
(Fig. 7A). ASXL1 expression
was also assessed by RT-qPCR using two different primer sets (set
1, nt799-865; set 2, nt1151-1248); the results indicated that
ASXL1 expression was reduced in MT+/− and
MT−/− cells compared with in WT cells (Fig. 7B), which are in agreement with the
RNA-seq results. For comparison, the FPKM values of ASXL1
from RNA-seq of the WT and MT cell lines are also shown in Fig. 7B. Reduced ASXL1 expression
in ASXL1-mutated cells may be attributed to premature stop
codons created by single nucleotide insertion, since degradation of
transcripts containing premature stop codons is the evolutionarily
conserved mRNA quality control system in all eukaryotes (30). Consistent with gene expression
levels, CYBB (Fig. 7C and D) and
CLEC5A (Fig. 7D) protein
expression levels were reduced in MT+/− and
MT−/− cells compared with in WT cells. These results
indicated that ASXL1 mutations markedly affected the
molecular machinery of U937 cells by dysregulating the expression
of genes essential for myeloid differentiation; notable effects
were detected on gene sets associated with cell death and
survival.
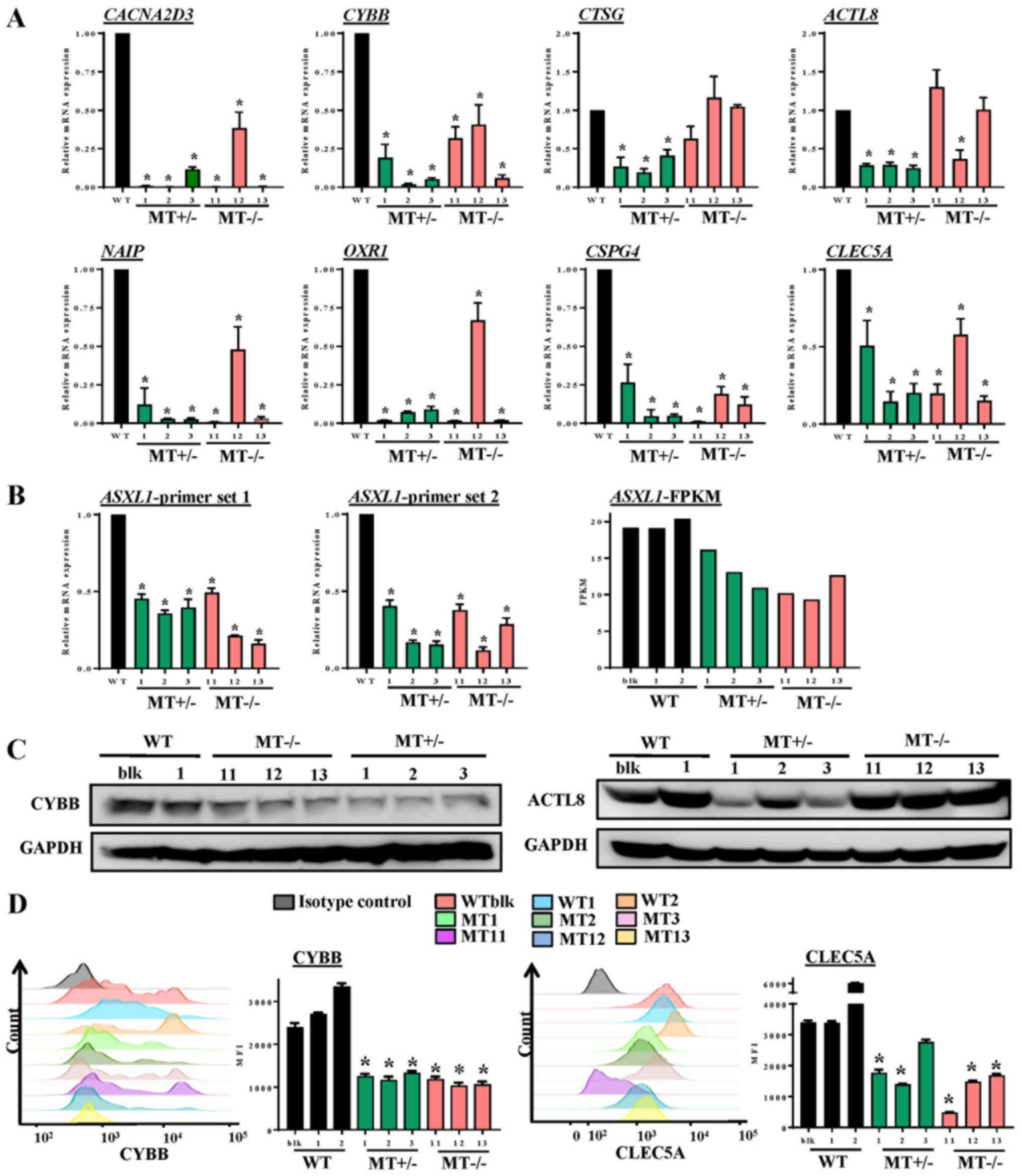 | Figure 7Validation of RNA-seq data by RT-qPCR
and immunoblotting. (A) Expression levels of genes in WT and
ASXL1-mutated U937 cells were validated using RT-qPCR. Data
are presented as the means ± standard error of the mean
(*P<0.05 vs. the WT group, one-way analysis of
variance). (B) ASXL1 expression was also validated by
RT-qPCR using two primer sets targeted before (ASXL1-probe
1) and after (ASXL1-probe 2) the mutation point. FPKM values
from RNA-seq data obtained from individual WT and
ASXL1-mutated cell lines are also shown (ASXL1-FPKM).
(C) Proteins extracted from WT, MT+/− and
MT−/− cells were subjected to immunoblotting to measure
CYBB and ACTL8 protein expression levels using GAPDH as a loading
control. (D) Histograms of CYBB expression and MFI of CYBB (left
panel), and histograms of CLEC5A expression and MFI of CLEC5A
(right panel) in WT and MT U937 cells. Data are presented as the
means ± standard error of the mean. *P<0.05 vs. the
WT group (WTblk, WT1 and WT2), one-way analysis of variance. ACTL8,
actin-like 8; ASXL1, additional sex combs-like 1;
CACNA2D3, calcium voltage-gated channel auxiliary subunit
α2δ3; CLEC5A, C-type lectin domain family 5, member A; CTSG,
cathepsin G; CYBB, cytochrome B-245 β chain; FPKM, fragments per
kilobase of transcript per million mapped reads; MFI, mean
fluorescence intensity; NAIP, NLR family apoptosis
inhibitory protein; OXR1, oxidation resistance 1; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; MT, mutated;
WT, wild-type; WTblk, wild-type bulk parental. |
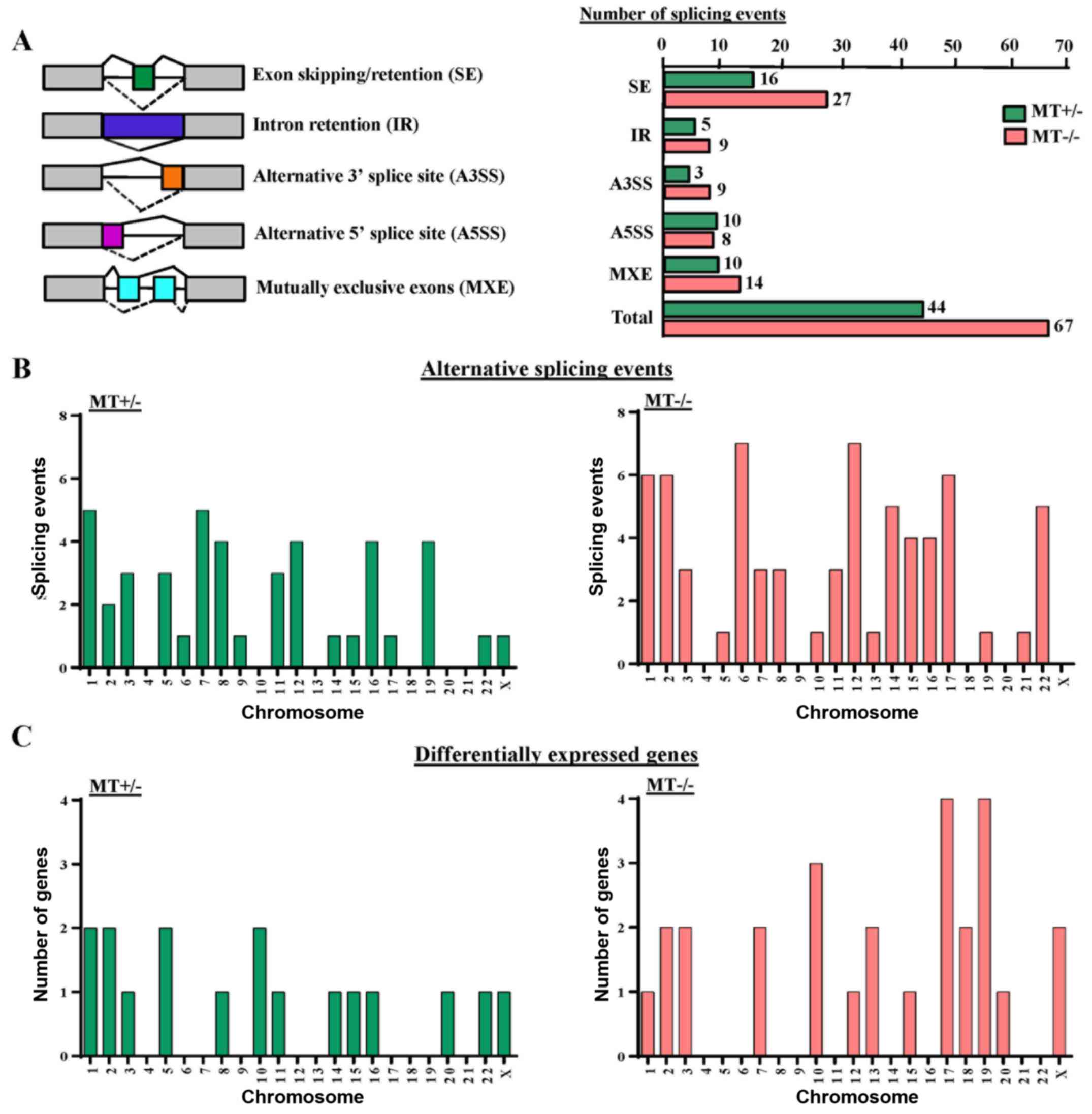
ASXL1 mutations cause altered gene
splicing in U937 cells
GSEA analysis revealed that gene sets associated
with mRNA splicing were downregulated in the MT cell lines (data
not shown). Therefore, the present study analyzed alternative
splicing in ASXL1-mutated U937 cells. In total, 44 and 67
differential splicing events were observed in the MT+/−
and MT−/− cell lines compared with the WT group,
respectively. These events could be categorized into five different
patterns of alternative splicing, among which 16 (36.4%) and 27
(40.3%) genes represented abnormal exon inclusion and/or retention
in MT+/− and MT−/− cells, respectively
(Fig. 8A). Calculation of
differential splicing events on each chromosome revealed that
ASXL1 mutations affected alternative splicing on individual
chromosomes randomly, with no preference for any specific
chromosomes (Fig. 8B). In
addition, the affected chromosomes containing differentially
expressed genes were analyzed in MT+/− and
MT−/− cells; however, there was no preference for
chromosomes containing differentially expressed genes (Fig. 8C). Notably, differentially spliced
genes in the MT cell lines were highly associated with gene
expression regulation, cell cycle, apoptosis, DNA/RNA/protein
synthesis and even RNA splicing itself, including the spliceosomal
gene LUC7-like, small nuclear ribonucleoprotein D3 polypeptide (a
core component of the spliceosome) and pre-mRNA processing factor
40 homolog A (data not shown). Alternative splicing of solute
carrier family 25 member 37 (SLC25A37), synoviolin 1
(SYVN1), enhancer of zeste 1 polycomb repressive complex 2
subunit (EZH1), zinc finger protein 227 (ZNF227) and
ADP-dependent glucokinase (ADPGK) were detected in
ASXL1-mutated cell lines (data not shown).
Discussion
To determine the molecular and cellular consequences
of ASXL1 mutations, the present study generated
ASXL1-MT+/− and ASXL1-MT−/− U937 cell
lines. These mutated cells produced short, truncated ASXL1 proteins
lacking most of the C-terminus domain, thus mimicking the majority
of ASXL1 mutations in myeloid malignancies, in which
reported point or frameshift mutations result in alterations, or
partial or complete loss, of the C-terminus PHD domain.
ASXL1-mutated U937 cells displayed comparable cell growth
and cell cycle progression as WT cells; however, defects were
observed in PMA-induced monocyte/macrophage differentiation.
RNA-seq revealed that ASXL1 mutations led to dysregulation
of genes involved in myeloid differentiation, including
downregulation of CYBB and CLEC5A. Gene sets
associated with various cellular functions, such as cell growth and
cell death were also impaired in ASXL1-mutated cells.
Furthermore, gene splicing analysis implicated ASXL1
mutations in altered mRNA splicing.
Previous studies have investigated the effects of
ASXL1 knockdown on cell survival, with variable results.
ASXL1 knockdown did not affect proliferation or apoptosis of
human CD34+ cells (31), whereas ASXL1-mutated KBM5
cells derived from chronic myelogenous leukemia cells exhibited a
growth advantage following ASXL1-mutation correction
(32). Enforced expression of
ASXL1 in murine leukemic cells resulted in growth
suppression (16); similarly,
hematopoietic-specific ASXL1 knockdown manifested as
impaired hematopoiesis, increased apoptosis and altered cell cycle
regulation of mouse hematopoietic stem and progenitor cells
(17). In the present study,
significant abnormalities in cell growth, cell cycle progression
and apoptosis were not observed in ASXL1-mutated cells
compared with in the WT cells, with the exception of some variation
among individual clones, which may be associated with clonal
expansion and stochastic selection of cells with enhanced
proliferative capacity in vitro.
In response to various stimuli, U937 cells become
adherent to substrates, exhibit slow proliferation and display cell
surface antigen characteristics of monocytes/mature macrophages
(33,34). U937 cells are used to investigate
the mechanisms underlying monocyte/macrophage differentiation and
monocyte-endothelium attachment. In the present study, upon
stimulation with PMA, ASXL1-mutated U937 cells displayed
less CD11b-positive cells and low CD11b intensity compared with in
the WT cells, thus indicating a decreased response to PMA and
thereby inefficient PMA-induced monocyte/macrophage
differentiation. According to the two-hit model of AML development
(35), these results are
supportive of ASXL1 as a class II gene, in which mutations
impair cell differentiation.
Due to the variability of RNA-seq data among
single-cell derived cell lines, less stringent statistical criteria
(FDR-corrected P<0.20, instead of 0.05) were applied for
analysis of RNA-seq data, in order to screen differentially
expressed genes. RT-qPCR data also displayed variability of gene
expression among the cell lines, indicating biological
heterogeneity among individual cell lines rather than technical
irreproducibility. Among the differentially expressed genes,
CYBB and CLEC5A, which are involved in myeloid
differentiation, were downregulated in the MT+/− and
MT−/− groups, and CYBB was the most consistently
downregulated gene in the present study. CYBB encodes the
gp91-phox component of the phagocyte oxidase enzyme complex, and
its expression is restricted to terminally differentiating myeloid
cells beyond the promyelocyte stage (36). CYBB downregulation decreases
production of reactive oxygen species (ROS), which are involved in
cell signaling associated with differentiation, cell cycle and
apoptosis. ROS prime Drosophila hematopoietic progenitors
for differentiation (37), and ROS
levels are low in mammalian hematopoietic stem cells but high in
common myeloid progenitors (38).
CLEC5A is a cell surface receptor strongly associated with
myeloid maturation (39). In the
mouse 32Dcl3 cell line, ASXL1 mutations suppressed
methylation of histone H3K27 located near the transcription start
site of microRNA (miR)-125a, which led to suppression of miR-125,
which in turn repressed Clec5a expression, directly
inhibiting cellular differentiation (17). CLEC5A mRNA expression is
also known to be low in primary MDS and AML patient samples
compared with in samples from healthy donors (17,40).
The downregulation of CYBB and CLEC5A may be related,
in part, to the diminished response of ASXL1-mutated U937
cells to PMA.
Differentially expressed genes in
ASXL1-mutated U937 cells were interconnected and scattered
within a comprehensive network, the key molecular hubs of which
included classical pathways of cell functions, such as MAPK,
AKT, NF-κB and MYC. Among the differentially
expressed genes validated by RT-qPCR, NAIP, CACNA2D3
and CSPG4 were consistently downregulated in individual
MT+/− and MT−/− cell lines. These genes are
involved in cell apoptosis and/or tumorigenicity (41-45).
In addition, the three MT+/− cell lines displayed
downregulated ACTL8, CTSG and OXR1 expression
compared with in the WT cells, whereas some of the MT−/−
cell lines exhibited different expression (downregulated or not
significantly different). These three genes are involved in various
aspects of cellular mechanisms, including hematopoietic cell
survival (46,47). There were more aberrant gene sets
identified in the MT−/− cell lines than in the
MT+/− cells, thus suggesting that complete loss of
ASXL1 appeared to have more complex transcriptional
consequences than were observed in the MT+/− cell lines
with a partial loss. No dose-effects were observed with regards to
cell functions, perhaps due to lethality (a much lower yield of
MT−/− single cell clones was observed than of
MT+/− following CRISPR/Cas9 editing); therefore, milder
defects may have been detected in MT−/− clones as a
result of clone selection. Alternatively, gain-of-function or
dominant-negative roles of truncated ASXL1 have been
suggested by other studies (48-51).
More than 50% of patients with MDS harbor mutations
in genes encoding proteins involved in pre-mRNA splicing (52). ASXL1 mutations appear to
cooperate with mutations in genes encoding splicing factors
[including splicing factor 3b subunit 1 (SF3B1), U2 small
nuclear RNA auxiliary factor 1 (U2AF1), and serine and
arginine rich splicing factor 2] in MDS. For example, ASXL1
mutations are more frequent in patients with U2AF35-mutated
MDS compared with in patients with U2AF35 WT MDS (53,54).
The present study observed alternative splicing events in
ASXL1-mutated U937 cell lines, which affected genes related
to cell survival and the regulation of gene expression. However,
there is no direct evidence to suggest that alternative splicing is
the mechanism responsible for altered gene expression. Several
alternatively spliced genes (SLC25A37, SYVN1,
EZH1, ZNF227 and ADPGK) detected in the
ASXL1-mutated cell lines in the present study were also
shown to exhibit differential usage of exons in patients with MDS
with SF3B1 mutations (55,56).
Ankyrin repeat and MYND domain containing 1 and chromosome 17 open
reading frame 62, which were found to be differentially spliced in
the present study (data not shown), were reported to be associated
with U2AF1 mutations in leukemic cell lines (57). Further investigation is required to
better define the effects of mutated ASXL1 on splicing
patterns, and to elucidate the relationship between ASXL1
and commonly mutated spliceosome genes in clinical samples.
Using CRISPR-Cas9 genome editing, ASXL1
mutations were introduced into U937 cells by electroporation of a
plasmid and gRNA, instead of viral infection, in order to avoid
continued gene expression from the CRISPR-Cas9 machinery. The
present study has the following limitations: The use of a cell line
and the application of less strict criteria (FDR <0.20) to
screen differentially expressed genes for discovery purposes. A
more applicable model for myeloid malignancies is the use of
primary cells; however, gene editing of CD34+ cells is
technically challenging, due to the low efficiency of mutation
induction, very limited survival of cells in vitro, and
their heterogeneity and propensity to mature in cell culture.
Single cell clones in the present study manifested larger
variations than anticipated from data derived from bulk cell
samples. CRISPR/Cas9 introduces gene mutations with low efficiency
(<10% in the present study), and various types of mutations are
introduced into targeted exons of individual cells by homologous
recombination following double strand breaks. Therefore, the
single-cell-clone method was adopted and three clones with
identical mutations were used in the MT+/− and
MT−/− groups, and two transfected single cell clones
with WT ASXL1, as well as U937 WTblk cells comprised the WT
group. Despite variations among the clones and a less stringent
threshold for expression screening, nine differentially expressed
genes were validated by qPCR and/or immunoblotting. In addition,
variability of these differentially expressed genes among the
ASXL1-mutated cell lines may be attributed to heterogeneity
of individual MT cell lines. A total of 20 loci most likely to
occur as off-site targets were predicted using the online MIT
CRISPR sgRNA design tool (http://crispr.mit.edu/) and were assessed by RNA
sequencing; no off-target mutations were observed in any of the
cell lines used in the present study (data not shown).
In conclusion, ASXL1 mutations, which are
potential class II gene mutations in leukemogenesis, disrupted
monocyte/macrophage differentiation in U937 cells, which is a
hallmark of myeloid malignancies, by downregulating genes essential
to myeloid differentiation, including CYBB and
CLEC5A. In addition, ASXL1 mutations affected
numerous gene sets involved in cell survival, and appeared to
induce altered splicing in mutated cell lines.
ASXL1-MT+/− mutations in the present study better
mimicked abnormality in patients, whereas homozygous mutations
likely exhibited more pronounced dysregulated biological behavior
and gene expression. ASXL1-mutated U937 cell lines may
therefore be considered helpful to further elucidate the molecular
mechanisms of ASXL1 in hematopoiesis.
Acknowledgments
The authors would like to thank Dr Valentina
Giudice (Hematology Branch, National Heart, Lung and Blood
Institute, National Institutes of Health, Bethesda, MD, USA), for
her advice and helpful discussion.
Notes
[1]
Funding
This study was supported by the Intramural Research
Program of the NIH, National Heart, Lung and Blood Institute.
[2] Availability
of data and materials
The datasets generated and/or analyzed during the
present study are available in the Gene Expression Omnibus
repository, (https://www.ncbi.nlm.nih.gov/geo/); accession no.
GSE98104 (available May 1, 2018).
[3] Authors'
contributions
ZJW, XZ, LGB, SK and NSY participated in the design
of the present study. ZJW conceptualized, conducted experiments,
analyzed data, interpreted results and drafted the manuscript. FGR
and DQR conducted experiments and data acquisition. KK conducted
cell sorting. SGG conducted the bioinformatics analysis. XZ, DQR,
and SK edited the manuscript. NSY was involved in
conceptualization, interim discussions and interpretation of
results, and edited the manuscript. All authors critically reviewed
the manuscript content and agree with the submission of the final
manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Rocquain J, Carbuccia N, Trouplin V,
Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N,
Birnbaum D, et al: Combined mutations of ASXL1, CBL, FLT3, IDH1,
IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in
myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer.
10:4012010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gelsi-Boyer V, Trouplin V, Adélaïde J,
Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M,
Sainty D, et al: Mutations of polycomb-associated gene ASXL1 in
myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br
J Haematol. 145:788–800. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boultwood J, Perry J, Pellagatti A,
Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, Larrayoz
MJ, Garcia-Delgado M, Giagounidis A, Malcovati L, et al: Frequent
mutation of the polycomb-associated gene ASXL1 in the
myelodysplastic syndromes and in acute myeloid leukemia. Leukemia.
24:1062–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thol F, Friesen I, Damm F, Yun H,
Weissinger EM, Krauter J, Wagner K, Chaturvedi A, Sharma A,
Wichmann M, et al: Prognostic significance of ASXL1 mutations in
patients with myelodysplastic syndromes. J Clin Oncol.
29:2499–2506. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gelsi-Boyer V, Trouplin V, Roquain J,
Adélaïde J, Carbuccia N, Esterni B, Finetti P, Murati A, Arnoulet
C, Zerazhi H, et al: ASXL1 mutation is associated with poor
prognosis and acute transformation in chronic myelomonocytic
leukaemia. Br J Haematol. 151:365–375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue D, Kitaura J, Matsui H, Hou HA, Chou
WC, Nagamachi A, Kawabata KC, Togami K, Nagase R, Horikawa S, et
al: SETBP1 mutations drive leukemic transformation in ASXL1-mutated
MDS. Leukemia. 29:847–857. 2015. View Article : Google Scholar :
|
|
7
|
Schnittger S, Eder C, Jeromin S, Alpermann
T, Fasan A, Grossmann V, Kohlmann A, Illig T, Klopp N, Wichmann HE,
et al: ASXL1 exon 12 mutations are frequent in AML with
intermediate risk karyotype and are independently associated with
an adverse outcome. Leukemia. 27:82–91. 2013. View Article : Google Scholar
|
|
8
|
Zong X, Yao H, Wen L, Ma L, Wang Q, Yang
Z, Zhang T, Chen S and Depei W: ASXL1 mutations are frequent in de
novo AML with trisomy 8 and confer an unfavorable prognosis. Leuk
Lymphoma. 58:204–206. 2017. View Article : Google Scholar
|
|
9
|
Alpermann T, Haferlach C, Eder C,
Nadarajah N, Meggendorfer M, Kern W, Haferlach T and Schnittger S:
AML with gain of chromosome 8 as the sole chromosomal abnormality
(+8 sole) is associated with a specific molecular mutation pattern
including ASXL1 mutations in 46.8% of the patients. Leuk Res.
39:265–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshizato T, Dumitriu B, Hosokawa K,
Makishima H, Yoshida K, Townsley D, Sato-Otsubo A, Sato Y, Liu D,
Suzuki H, et al: Somatic Mutations and Clonal Hematopoiesis in
Aplastic Anemia. N Engl J Med. 373:35–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fisher CL, Lee I, Bloyer S, Bozza S,
Chevalier J, Dahl A, Bodner C, Helgason CD, Hess JL, Humphries RK,
et al: Additional sex combs-like 1 belongs to the enhancer of
trithorax and polycomb group and genetically interacts with Cbx2 in
mice. Dev Biol. 337:9–15. 2010. View Article : Google Scholar :
|
|
12
|
Park UH, Yoon SK, Park T, Kim EJ and Um
SJ: Additional sex comb-like (ASXL) proteins 1 and 2 play opposite
roles in adipogenesis via reciprocal regulation of peroxisome
proliferator-activated receptor {gamma}. J Biol Chem.
286:1354–1363. 2011. View Article : Google Scholar
|
|
13
|
Cho YS, Kim EJ, Park UH, Sin HS and Um SJ:
Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts
as a ligand-dependent coactivator for retinoic acid receptor. J
Biol Chem. 281:17588–17598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Katoh M: Functional and cancer genomics of
ASXL family members. Br J Cancer. 109:299–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scheuermann JC, de Ayala Alonso AG, Oktaba
K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW and Müller
J: Histone H2A deubiquitinase activity of the Polycomb repressive
complex PR-DUB. Nature. 465:243–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abdel-Wahab O, Gao J, Adli M, Dey A,
Trimarchi T, Chung YR, Kuscu C, Hricik T, Ndiaye-Lobry D, LaFave
LM, et al: Deletion of Asxl1 results in myelodysplasia and severe
developmental defects in vivo. J Exp Med. 210:2641–2659. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inoue D, Kitaura J, Togami K, Nishimura K,
Enomoto Y, Uchida T, Kagiyama Y, Kawabata KC, Nakahara F, Izawa K,
et al: Myelodysplastic syndromes are induced by histone
methylation-altering ASXL1 mutations. J Clin Invest. 123:4627–4640.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Li Z, He Y, Pan F, Chen S, Rhodes
S, Nguyen L, Yuan J, Jiang L, Yang X, et al: Loss of Asxl1 leads to
myelodysplastic syndrome-like disease in mice. Blood. 123:541–553.
2014. View Article : Google Scholar :
|
|
19
|
Carbuccia N, Murati A, Trouplin V,
Brecqueville M, Adélaïde J, Rey J, Vainchenker W, Bernard OA,
Chaffanet M, Vey N, et al: Mutations of ASXL1 gene in
myeloproliferative neoplasms. Leukemia. 23:2183–2186. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carbuccia N, Trouplin V, Gelsi-Boyer V,
Murati A, Rocquain J, Adélaïde J, Olschwang S, Xerri L, Vey N,
Chaffanet M, et al: Mutual exclusion of ASXL1 and NPM1 mutations in
a series of acute myeloid leukemias. Leukemia. 24:469–473. 2010.
View Article : Google Scholar
|
|
21
|
Abdel-Wahab O, Adli M, LaFave LM, Gao J,
Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, et al:
ASXL1 mutations promote myeloid transformation through loss of
PRC2-mediated gene repression. Cancer Cell. 22:180–193. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hilgendorf S, Folkerts H, Schuringa JJ and
Vellenga E: Loss of ASXL1 triggers an apoptotic response in human
hematopoietic stem and progenitor cells. Exp Hematol.
44:1188–1196.e6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Banaszak LG, Giudice V, Zhao X, Wu Z, Gao
S, Hosokawa K, Keyvanfar K, Townsley DM, Gutierrez-Rodrigues F,
Fernandez Ibanez MP, et al: Abnormal RNA splicing and genomic
instability after induction of DNMT3A mutations by CRISPR/Cas9 gene
editing. Blood Cells Mol Dis. 69:10–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liao Y, Smyth GK and Shi W: featureCounts:
An efficient general purpose program for assigning sequence reads
to genomic features. Bioinformatics. 30:923–930. 2014. View Article : Google Scholar
|
|
25
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar
|
|
27
|
Alexa A, Rahnenfuhrer J and Lengauer T:
Improved scoring of functional groups from gene expression data by
decorrelating GO graph structure. Bioinfomatics. 22:1600–1607.
2006. View Article : Google Scholar
|
|
28
|
Katz Y, Wang ET, Airoldi EM and Burge CB:
Analysis and design of RNA sequencing experiments for identifying
isoform regulation. Nat Methods. 7:1009–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) methods. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Miller JN and Pearce DA: Nonsense-mediated
decay in genetic disease: Friend or foe. Mutat Res Rev Mutat Res.
762:52–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davies C, Yip BH, Fernandez-Mercado M,
Woll PS, Agirre X, Prosper F, Jacobsen SE, Wainscoat JS, Pellagatti
A and Boultwood J: Silencing of ASXL1 impairs the granulomonocytic
lineage potential of human CD34+ progenitor cells. Br J
Haematol. 160:842–850. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Valletta S, Dolatshad H, Bartenstein M,
Yip BH, Bello E, Gordon S, Yu Y, Shaw J, Roy S, Scifo L, et al:
ASXL1 mutation correction by CRISPR/Cas9 restores gene function in
leukemia cells and increases survival in mouse xenografts.
Oncotarget. 6:44061–44071. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harris PE, Ralph P, Litcofsky P and Moore
MA: Distinct activities of interferon-gamma, lymphokine and
cytokine differentiation-inducing factors acting on the human
monoblastic leukemia cell line U937. Cancer Res. 45:9–13.
1985.PubMed/NCBI
|
|
34
|
Valledor AF, Borràs FE, Cullell-Young M
and Celada A: Transcription factors that regulate
monocyte/macrophage differentiation. J Leukoc Biol. 63:405–417.
1988. View Article : Google Scholar
|
|
35
|
Gilliland DG: Hematologic malignancies.
Curr Opin Hematol. 8:189–191. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nauseef WM and Borregaard N: Neutrophils
at work. Nat Immunol. 15:602–611. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Owusu-Ansah E and Banerjee U: Reactive
oxygen species prime Drosophila haematopoietic progenitors for
differentiation. Nature. 461:537–541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tothova Z, Kollipara R, Huntly BJ, Lee BH,
Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams
IR, Sears C, et al: FoxOs are critical mediators of hematopoietic
stem cell resistance to physiologic oxidative stress. Cell.
128:325–339. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gingras MC, Lapillonne H and Margolin JF:
TREM-1, MDL-1, and DAP12 expression is associated with a mature
stage of myeloid development. Mol Immunol. 38:817–824. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Batliner J, Mancarelli MM, Jenal M, Reddy
VA, Fey MF, Torbett BE and Tschan MP: CLEC5A (MDL-1) is a novel
PU.1 transcriptional target during myeloid differentiation. Mol
Immunol. 48:714–719. 2011. View Article : Google Scholar :
|
|
41
|
Negoro E, Yamauchi T, Urasaki Y, Nishi R,
Hori H and Ueda T: Characterization of cytarabine-resistant
leukemic cell lines established from five different blood cell
lineages using gene expression and proteomic analyses. Int J Oncol.
38:911–919. 2011.PubMed/NCBI
|
|
42
|
Li Y, Zhu CL, Nie CJ, Li JC, Zeng T, Zhou
J, Chen J, Chen K, Fu L, Liu H, et al: Investigation of tumor
suppressing function of CACNA2D3 in esophageal squamous cell
carcinoma. PLoS One. 8:e600272013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Palmieri C, Rudraraju B, Monteverde M,
Lattanzio L, Gojis O, Brizio R, Garrone O, Merlano M, Syed N, Lo
Nigro C, et al: Methylation of the calcium channel regulatory
subunit α2δ-3 (CACNA2D3) predicts site-specific relapse in
oestrogen receptor-positive primary breast carcinomas. Br J Cancer.
107:375–381. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wanajo A, Sasaki A, Nagasaki H, Shimada S,
Otsubo T, Owaki S, Shimizu Y, Eishi Y, Kojima K, Nakajima Y, et al:
Methylation of the calcium channel-related gene, CACNA2D3, is
frequent and a poor prognostic factor in gastric cancer.
Gastroenterology. 135:580–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Price MA, Colvin Wanshura LE, Yang J,
Carlson J, Xiang B, Li G, Ferrone S, Dudek AZ, Turley EA and
McCarthy JB: CSPG4, a potential therapeutic target, facilitates
malignant progression of melanoma. Pigment Cell Melanoma Res.
24:1148–1157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jin W, Wu K, Li YZ, Yang WT, Zou B, Zhang
F, Zhang J and Wang KK: AML1-ETO targets and suppresses cathepsin
G, a serine protease, which is able to degrade AML1-ETO in t(8;21)
acute myeloid leukemia. Oncogene. 32:1978–1987. 2013. View Article : Google Scholar
|
|
47
|
Yang M, Lin X, Rowe A, Rognes T, Eide L
and Bjørås M: Transcriptome analysis of human OXR1 depleted cells
reveals its role in regulating the p53 signaling pathway. Sci Rep.
5:174092015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fisher CL, Pineault N, Brookes C, Helgason
CD, Ohta H, Bodner C, Hess JL, Humphries RK and Brock HW:
Loss-of-function Additional sex combs like 1 mutations disrupt
hematopoiesis but do not cause severe myelodysplasia or leukemia.
Blood. 115:38–46. 2010. View Article : Google Scholar :
|
|
49
|
Vainchenker W, Delhommeau F,
Constantinescu SN and Bernard OA: New mutations and pathogenesis of
myeloproliferative neoplasms. Blood. 118:1723–1735. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Inoue D, Matsumoto M, Nagase R, Saika M,
Fujino T, Nakayama KI and Kitamura T: Truncation mutants of ASXL1
observed in myeloid malignancies are expressed at detectable
protein levels. Exp Hematol. 44:172–6.e1. 2016. View Article : Google Scholar
|
|
51
|
Balasubramani A, Larjo A, Bassein JA,
Chang X, Hastie RB, Togher SM, Lähdesmäki H and Rao A:
Cancer-associated ASXL1 mutations may act as gain-of-function
mutations of the ASXL1-BAP1 complex. Nat Commun. 6:73072015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bejar R and Steensma DP: Recent
developments in myelodysplastic syndromes. Blood. 124:2793–2803.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gelsi-Boyer V, Brecqueville M, Devillier
R, Murati A, Mozziconacci MJ and Birnbaum D: Mutations in ASXL1 are
associated with poor prognosis across the spectrum of malignant
myeloid diseases. J Hematol Oncol. 5:122012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Damm F, Kosmider O, Gelsi-Boyer V,
Renneville A, Carbuccia N, Hidalgo-Curtis C, Della Valle V,
Couronne L, Scourzic L, Chesnais V, et al: Mutations affecting mRNA
splicing define distinct clinical phenotypes and correlate with
patient outcome in myelodysplastic syndromes. Blood. 119:3211–3218.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Visconte V, Rogers HJ, Singh J, Barnard J,
Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska
A, et al: SF3B1 haploinsufficiency leads to formation of ring
sideroblasts in myelodysplastic syndromes. Blood. 120:3173–3186.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dolatshad H, Pellagatti A,
Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, Przychodzen B,
Sahgal N, Kanapin AA, Lockstone H, et al: Disruption of SF3B1
results in deregulated expression and splicing of key genes and
pathways in myelodysplastic syndrome hematopoietic stem and
progenitor cells. Leukemia. 29:1092–1103. 2015. View Article : Google Scholar :
|
|
57
|
Ilagan JO, Ramakrishnan A, Hayes B, Murphy
ME, Zebari AS, Bradley P and Bradley RK: U2AF1 mutations alter
splice site recognition in hematological malignancies. Genome Res.
25:14–26. 2015. View Article : Google Scholar :
|