Introduction
Gastrointestinal stromal tumors (GISTs) are the most
common mesenchymal tumors of the gastrointestinal tract, occurring
most frequently in the stomach (approximately 50%) and small
intestine (25-35%) and less frequently in the colorectal regions
(10-12%), omentum/mesentery (7%) and esophagus (1-5%) (1-3). The
majority of GISTs contain mutations in the gene encoding the KIT
tyrosine kinase receptor (4);
however, but <10% have mutations in the gene encoding
platelet-derived growth factor receptor A (PDGFRA) as the oncogenic
driving force(5). Imatinib
mesylate (Novartis Pharmaceuticals), a potent tyrosine kinase
inhibitor (TKI) for both KIT and PDGFRA, is the standard first-line
therapy and can achieve a median overall survival (OS) rate of 5-6
years in patients with advanced disease (6,7).
However, disease progression, which most likely occurs due to the
development of secondary mutations (8,9), is
expected within 2 to 3 years of imatinib therapy (6,7).
Therapeutic options are limited for these patients, with sunitinib
maleate (Pfizer) as the standard second-line agent (10), and regorafenib (Bayer Schering
Pharma AG) as third-line therapy (11,12).
KIT and its downstream pathway remain a major target
of interest in TKI-refractory GISTs. Therapeutic strategies, such
as those involving the degradation of the KIT protein [by using
either a heat shock protein 90 inhibitor (13) or histone deacetylase inhibitors
(14)] or the targeting of its
downstream effectors by blocking phosphoinositide 3-kinase
(15) or mammalian target of
rapamycin (mTOR) (16), have been
shown to be effective in inhibiting GIST growth. However, only a
few of these have been successfully applied in clinical studies
(17-19). Consequently, identifying novel
therapeutic targets or strategies against GIST is a priority.
By using bioinformatics analysis, Chi et al
discovered that ETS family member E26 variant 1 (ETV1) is highly
expressed in GISTs (20). They
also demonstrated that interstitial cell of Cajal (ICC) hyperplasia
in mouse models expressing constitutively activated KIT also
strongly expressed ETV1. Their analysis revealed that genes that
were downregulated by ETV1 knockdown demonstrated a negative
correlation with genes that were upregulated in GISTs, suggesting a
key role of ETV1 in an ICC-GIST-specific transcription network.
Furthermore, their study demonstrated that the ETV1 transcriptional
program is further regulated by activated KIT and its downstream
MAP kinase, which prolongs ETV1 protein stability and cooperates
with ETV1 to promote tumorigenesis (20). In their subsequent study, Ran et
al uncovered a positive feedback circuit in which the KIT/MAP
kinase pathway stabilized the ETV1 protein and ETV1 positively
regulated KIT expression. The combined targeting of ETV1 stability
by using imatinib and MEK162 resulted in significant in
vitro and in vivo cytotoxic effects (21). This series of studies demonstrated
that ETV1 is a lineage-specific oncogenic transcription factor
required for the growth and survival of GISTs, and that the
identification of novel agents that target the ETV1 pathway may be
a strategy for the treatment of GISTs.
Lamb et al created a reference collection of
gene expression profiles from cultured human cells treated with
diverse bioactive small molecules, as well as pattern-matching
software for data mining ['The Connectivity Map' (CMAP), http://www.broad.mit.edu/cmap/] (22,23).
When any gene expression profiles of interest are uploaded to this
database, pattern-matching algorithms score each reference profile
for the direction and strength of enrichment with the query
signature. Perturbagens are then ranked by this 'connectivity
score'; those at the top ('positive') and bottom ('negative') are
functionally connected with the query state through the transitory
feature of common gene-expression changes (22). In this study, an ETV1 knockout gene
signature of GIST cell lines was uploaded, and phenothiazine was
identified as a class of ETV1-targeting agent in GISTs.
Materials and methods
Collection of expression profiles from
public datasets
Microarray and clinicopathological data from the
Gene Expression Omnibus (GEO) datasets GSE14827 [osteosarcoma
(OS)], GSE13433 [alveolar soft part sarcoma (ASPS)], GSE8167
(GIST), GSE20196 [synovial sarcoma (SynSA)], GSE20559 [liposarcoma
(LPS)] and GSE17679 [Ewing sarcoma family tumors (ESFT)] were
obtained from the NCBI website. E-MEXP-1922 [leiomyosarcoma (LMS)]
data were obtained from the ArrayExpress website. All datasets were
in the Affymetrix U133 Plus 2.0 platform. In order to reduce
intra-subtype heterogeneity, we did not include all samples. For
OS, only tumors without subsequent metastasis were selected; for
GISTs, only those with exon 11 mutations were included; for SynSA,
only tumors with SYT-SSX type 1 fusion gene and non-poorly
differentiated histology were analyzed; and for LPS, only well- and
de-differentiated tumors were chosen due to their similar genetic
background. Expression profile data of GIST cell lines with ETV1
wild-type versus knockout were also obtained from GEO (GSE19396)
(20).
Bioinformatics analysis
dChip (24,25) was the main analytic software used,
and the normalization of the expression values of individual chips
was performed according to software specifications. One-way
analysis of variance (ANOVA) was used to identify genes showing
differentiated expression between different sarcoma subtypes, as
well as different mutation status categories in GISTs. The
expression levels of individual genes were obtained using z-score
transformation, and the differences between different subtypes were
then compared using the t-test.
Chip data from GSE19396 were analyzed separately.
The dChip 'Compare Samples' function was used to explore
differentially expressed genes between wild-type and ETV1 knockdown
strains of GIST cell lines using 1.4-fold changes (with P<0.05
by the unpaired t-test) as the threshold (24) (please also refer to https://sites.google.com/site/dchipsoft/high-level-analysis/compare-samples/comparison-criteria).
Gene signature query of CMAP
The identified gene signature was first transformed
to probe set ID of Human Genome U133A. Up- and downregulated ID
lists were then uploaded to the CMAP website (http://www.broad.mit.edu/cmap/). The query
signature was then compared with each rank-ordered list to
determine whether upregulated query genes tend to appear near the
top of the list and downregulated query genes near the bottom
('positive connectivity'), or vice versa ('negative connectivity'),
yielding a 'connectivity score' ranging from +1 to −1. A null
(zero) connectivity score was assigned where the enrichment scores
for the up- and downregulated genes had the same sign. All
instances in the database were then ranked according to their
connectivity scores; those at the top were most strongly associated
with the query signature, and those at the bottom were most
strongly anti-associated (22,23).
Cell lines and mass ARRAY-based mutation
characterization
GIST cell lines were kindly provided by Dr J.A.
Fletcher, and the KIT mutation status of this line has been
previously described (13).
GIST882 is a human cell line established from an untreated GIST
with a primary imatinib-sensitive homozygous missense mutation in
KIT exon 13, encoding a K642E mutant KIT oncoprotein (26). GIST48 was established from GISTs
that had progressed, after initial clinical response, during
imatinib therapy. GIST48 has a primary, homozygous exon 11 missense
mutation (V560D) and a heterozygous secondary exon 17 (kinase
activation loop) mutation (D820A) (13).
The sequences of KIT of both cell lines were
surveyed again by Mass ARRAY-based mutation characterization before
this study. The Sequenom MassARRAY platform utilizes a homogeneous
reaction format with a single extension primer to generate
allele-specific products with distinct masses (27,28).
PCR and extension primers for the mutations were designed using
MassArray Assay Design 3.1 software (Sequenom). The mutation
alleles were manually designed by extension in either the forward
or reverse direction to have lower mass than the reference allele.
The PCR products of multiplexed reactions were spotted onto
SpectroCHIP II arrays, and DNA fragments were resolved by on the
MassARRAY Analyzer 4 System (Sequenom). Each spectrum was then
analyzed using the Typer 4.0 software (Sequenom) to call mutations.
Putative mutations were further filtered by manual review. The
results of the mutation analysis of two cell lines are shown in
Fig. S1, and their mutation
status remained the same.
Reagents
The drugs used in this study were imatinib
(ALX-270-492; Enzo Life Sciences), PD98059 (P215),
suberanilohydroxamic acid (SAHA) (SML0061), trifluoperazine (TFP)
(T8516), thioridazine (TDZ) (T9025) (all from Sigma-Aldrich) and
MEK162 (S7007; Selleckchem). The primary antibodies used for
western blot analysis included p-ERK (cat. no. 4376; 1:1,000), KIT
(cat. no. 3392; 1:1,000), p-KIT (cat. no. 3391; 1:1,000), AKT (cat.
no. 9272; 1:1,000), p-AKT (cat. no. 4058; 1:1,000), p-p70S6K (cat.
no. 9205; 1:500), LC3 (cat. no. 2775; 1:1,000), early growth
response protein 1 (EGR1) (cat. no. 4153; 1:1,000) and
poly-(ADP-ribose) polymerase (PARP) (cat. no. 9542; 1:1,000) from
Cell Signaling Technology; actin (cat. no. ab6276-100; 1:10,000)
and ETV1 (cat. no. ab184120; 1:1,000) from Abcam Biotechnology; and
ERK (cat. no. sc-135900; 1:1,000), ELK1 (cat. no. sc-355; 1:200)
and p-ELK1 (cat. no. sc-8406; 1:200) from Santa Cruz Biotechnology.
The secondary antibodies used were horse anti-mouse IgG (cat. no.
7076; 1:3,000) and goat anti-rabbit IgG (cat. no. 7074; 1:3,000),
both from Cell Signaling Technology.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
assay
Cell viability was measured using the TACS™
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
cell proliferation assay (R&D Systems) according to the
manufacturer's instructions. Briefly, the cells were plated in
96-well plates at a concentration of 2,000-20,000/100
µl/well overnight. Drugs at various concentrations were
added in triplicate. The plates were incubated for the desired time
at 37°C, pulsed with 10 µl of MTT reagent, and incubated for
an additional 4 h at 37°C. Detergent reagent at 200 µl/well
was added and mixed thoroughly to dissolve the dark blue crystals.
The absorbance of the converted dye was measured
spectrophotometrically in a Vmax microplate reader (Molecular
Devices) at wavelengths of 570 nm (test) and 650 nm. Cell survival
was calculated as the percentage of MTT inhibition as follows: %
survival = (mean experimental absorbance/mean control absorbance)
×100 (29).
The synergistic effect of the applied drug
combination was measured through a combination index (CI), which
was calculated using CalcuSyn software (Biosoft) (30). CI >1 was defined as antagonism,
CI =1 as additivity, and CI <1 as synergy; the experiment was
performed in triplicate.
Western blot analysis
Monolayers of cultured cells were rinsed in
phosphate-buffered saline (PBS) and scraped into lysis buffer [25
nM Tris•HCl pH 7.6, 150 nM NaCl, 1% NP-40, 1% sodium deoxycholate,
0.1% SDS (Thermo Fisher Scientific)] containing the Protease and
Phosphatase Inhibitor Cocktail (1:100 dilution; Thermo Fisher
Scientific). Lysates were incubated for 30 min at 4°C and then
clarified by centrifugation for 30 min at 13,200 rpm at 4°C.
Protein concentrations were determined with the Pierce BCA Protein
Assay kit (Thermo Fisher Scientific). Protein extracts (20-50
µg per lane) were electrophoretically separated on sodium
dodecyl sulfate-polyacrylamide gel electrophoresis gels (8-12%
depending on the molecular weights of proteins), transferred to
polyvinylidene difluoride membranes (PerkinElmer), and blotted with
specific antibodies. For primary antibodies, the sample was
incubated at 4°C overnight; for secondary antibody, the sample was
incubated at room temperature for 1 h. Immunoreactive bands were
detected using enhanced chemiluminescence (Millipore) and X-ray
film.
Measurement of caspase activity
Caspase activity was detected using the
Caspase-Glo® 3/7 assay kit (Promega) according to the
manufacturer's instructions. Briefly, GIST cells (104
cells/well) were seeded in a luminometer plate and incubated for 24
h at 37°C. GIST cells were treated with DMSO (control vehicle) or
with various concentrations of drugs for 72 h. Moreover, 100
µl of caspase-3/7 reagents was added to each well and mixed
gently using a plate shaker. The cells were then incubated for
0.5-1 h at room temperature, and the luminescence of each well was
measured.
Apoptosis assessment by Annexin V
staining
The cells were plated at a density of 350,000
cells/well or 1,000,000 cells/well in 6-well plates. The following
day, the cells were treated with TFP (0, 30 and 40 µM) or
TDZ (0, 25 and 35 µM) for 72 h, and then washed with 1X PBS
for twice and resuspended in 100 µl staining solution
containing Annexin V-APC (BD Pharmingen) and propidium iodide in
Annexin V-binding buffer. The cells were then incubated at room
temperature for 15 min, and the cells were then diluted in 400
µl 1X Annexin V-binding buffer. The percentages of apoptotic
cells were then measured using a flow cytometer (Canto II; BD
Biosciences).
Detection and quantification of
autophagic cells by staining with acridine orange
Cells were seeded in 6-well tissue culture dishes
and treated with TFP (GIST882, 20 µM; GIST48, 10 µM)
for 72 h. The cells were then incubated at room temperature with
medium containing acridine orange (100 mg/ml) for 15-20 min, washed
once with PBS, and fresh media were added. Fluorescent micrographs
were then acquired using an inverted fluorescence microscope (IX51;
Olympus). All images presented are at the same magnification
(×200). The number of cells with increased acidic vesicular
organelles was determined by flow cytometry. The cells were
trypsinized and harvested. Red fluorescence emission was measured
through a FACSCalibur from BD Biosciences using CellQuest
software.
Statistical analysis
As stated above, dChip 'Compare Samples' function
with 1.4-fold changes set as the threshold (with P<0.05 by the
unpaired t-test) was used to identify differentially expressed
genes between control (n=4) and ETV1 knockdown strains (n=8) of
GIST cell lines. The differences of expression level of individual
genes between GISTs and non-GIST subtypes, and the difference of
percentage of viability (assessed by MTT cell proliferation assay)
and the percentage of apoptotic cells (assessed by apoptosis assay)
between the control and drug treatment (all in triplicate), were
compared using ANOVA, and the Bonferroni test was used as a post
hoc test (with P<0.05 considered to indicate a statistically
significant difference).
Results
Confirmation of the overexpression of
ETV1 in GIST in comparison with other sarcomas
Eight different types of sarcoma were analyzed:
GIST, OS, ASPS, SynSA, well-differentiated LPS (WD LPS),
de-differentiated LPS (DD LPS), ESFT and LMS. Comparisons of the
expression levels of three well-known GIST-specific expression
genes, namely KIT, DOG1 and PRKCQ and ETV1, were made. As shown in
Fig. 1, KIT, DOG1, PRKCQ and ETV1
were all significantly highly expressed in the GISTs in comparison
with the other sarcomas.
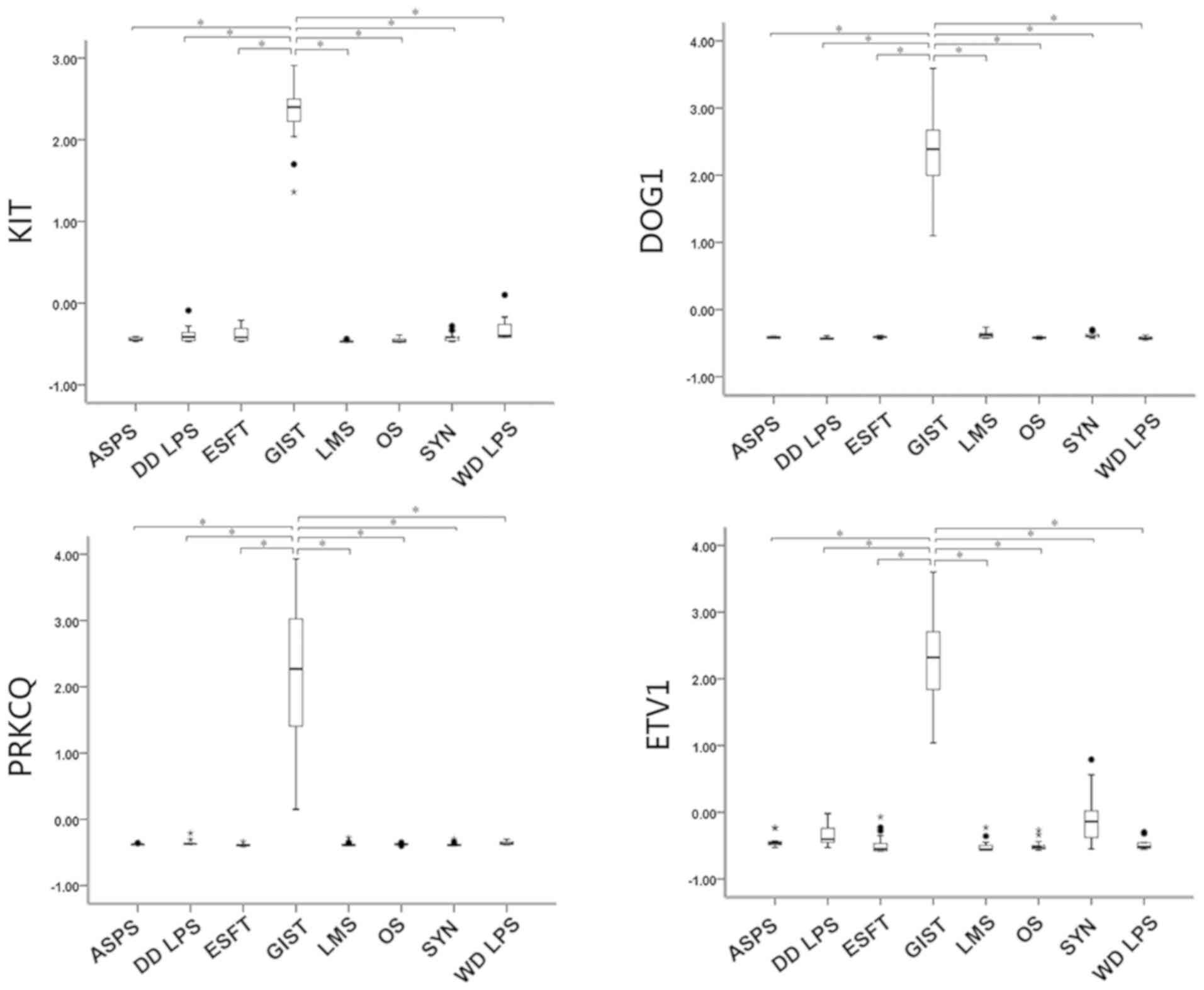 | Figure 1Differential expression of genes in
GISTs and non-GIST sarcoma. The expression levels of individual
genes were obtained using z-score transformation, and the
differences between GISTs and non-GIST subtypes were then compared
using the ANOVA (with Bonferroni test as a post hoc test). KIT,
DOG1, PKCQ and ETV1 all showed significantly high expression levels
in GISTs in comparison with other non-GIST sarcomas
(*P<0.00001). GISTs, gastrointestinal stromal tumors;
ASPS, alveolar soft part sarcoma; DD LPS, de-differentiated
liposarcoma; ESFT, Ewing sarcoma family tumors; LMS,
leiomyosarcoma; OS, osteosarcoma; WD LPS, well-differentiated
liposarcoma. |
Identification of novel agents targeting
the ETV1 pathway in GIST by CMAP
We downloaded the expression profile information
from the GEO database (GSE19396) created by Chi et al
(20) and compared the expression
profiles of wild-type GISTs and ETV1-knockdown GISTs. We obtained a
total of 207 probes of genes with differential expression between
these two types of cell lines (Tables
SI and SII). We then uploaded this set of genes onto CMAP to
search for possible agents. The five drugs that had the most
significant P-values following permutation are presented in
Table I. Prestwick-1080 is
unavailable. SAHA and trichostatin are HDACIs, of which anti-GIST
activity has been previously demonstrated (14). However, TFP and TDZ, two drugs of
the phenothiazine class, have not been reported to date to have
cytotoxic activity against GISTs, at least to the best of our
knowledge. Therefore, we focused on these two drugs in this
study.
 | Table ITop 5 agents identified by CMAP after
uploading ETV1 knockdown vs. wild-type expression profiles. |
Table I
Top 5 agents identified by CMAP after
uploading ETV1 knockdown vs. wild-type expression profiles.
| Rank | CMAP name | No. | P-value |
|---|
| 1 | Prestwick-1080 | 4 | <0.00001 |
| 2 |
Suberanilohydroxamic acid | 12 | <0.00001 |
| 3 | Trichostatin A | 182 | <0.00001 |
| 4 |
Trifluoperazine | 16 | <0.00001 |
| 5 | Thioridazine | 20 | <0.00001 |
Confirmation of the targeting of SAHA,
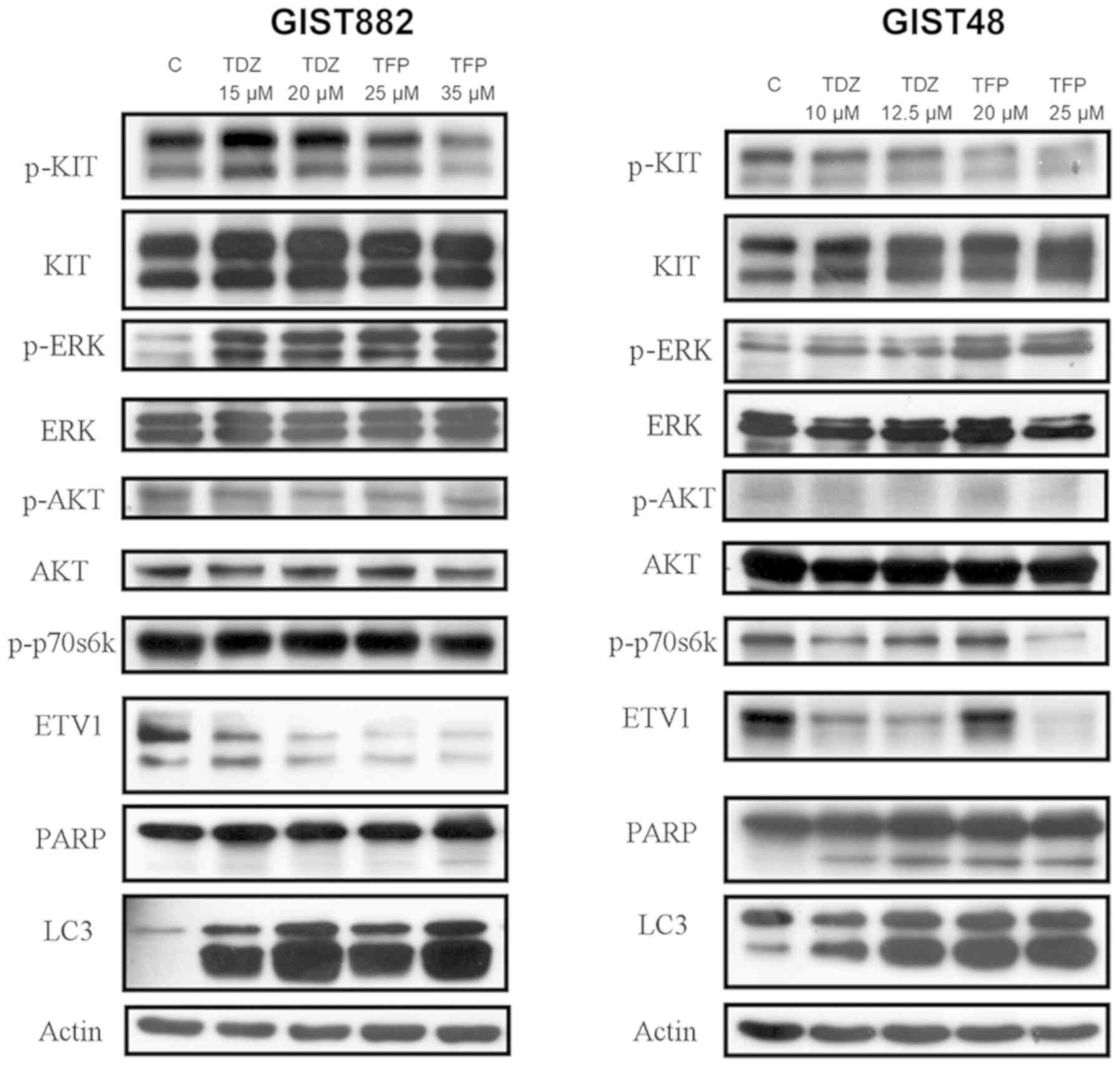
TFP and TDZ on KIT-ETV1 in GIST cell lines
Subsequently, we examined the targeting effect of
TFP and TDZ, as well as other agents, on the KIT/ETV1 pathway.
Representative western blots of GISTS882 and GIST48 following
treatment with different agents are shown in Fig. 2. The inhibition of KIT signaling by
imatinib (2 µM, 12 h) in the imatinib-sensitive GIST882
cells resulted in MEK pathway inhibition, as well as in ETV1
protein downregulation. The imatinib-resistant GIST48 cells
exhibited changes to relevant pathways following treatment with
imatinib at a higher concentration (20 µM, 12 h). By
contrast, both GIST cell lines treated with the MEK inhibitor,
PD98059 (100 nM, 12 h), exhibited an inhibition of ERK
phosphorylation and the loss of ETV1. Previous research has
demonstrated that KIT activity and expression, and the activation
of downstream pathways in GISTs are strongly inhibited by HDACI
(14). In this study, we also
found that SAHA (10 µM, 12 h), an HDACI, degraded KIT,
resulting in a subsequent decrease in ETV1 expression (Fig. 2).
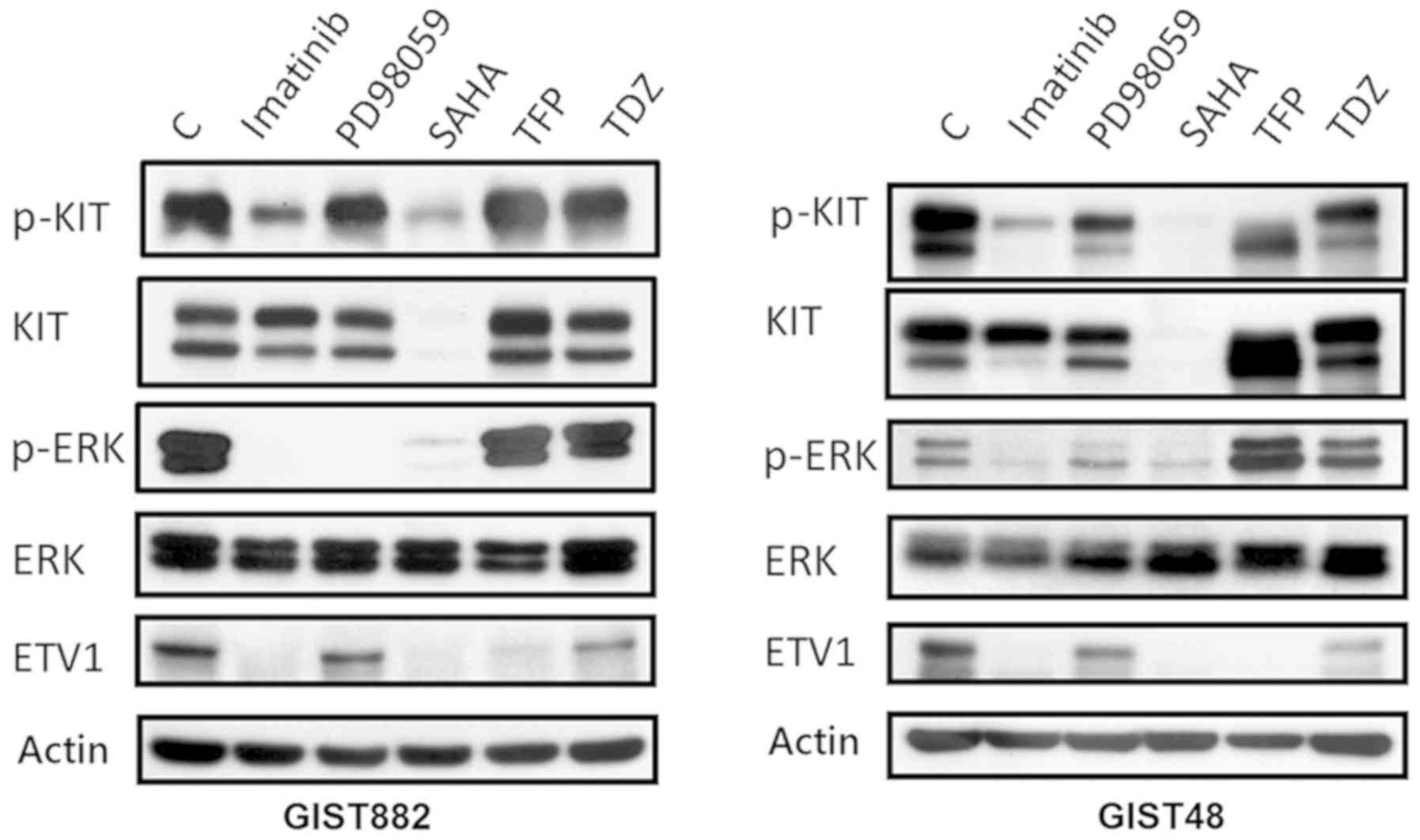 | Figure 2Western blot analysis of GISTS882 and
GIST48 cells following treatment with various agents. Imatinib (2
µM, 12 h for GIST882 and 20 µM, 12 h for GIST48)
inhibited KIT and downstream ERK, resulting in ETV1 downregulation.
The MEK inhibitor, PD98059 (100 nM, 12 h), inhibited ERK with
subsequent ETV1 downregulation. SAHA (10 µM, 12 h), an
HDACI, degraded KIT with the subsequent inhibition of ERK and the
decreased expression of ETV1. TFP (35 µM, 72 h for GIST882;
25 µM, 72 h for GIST48) and TDZ (20 µM, 72 h for
GIST882; 12.5 µM, 72 h for GIST48) downregulated ETV1, with
little change in AKT and little change in, or even the paradoxical
activation of, the ERK pathway. C, control; SAHA,
suberanilohydroxamic acid; TFP, trifluoperazine; TDZ,
thioridazine. |
We then further explored the targeting effects of
TFP (35 µM, 72 h for GIST882; 25 µM, 72 h for GIST48)
and TDZ (20 µM, 72 h for GIST882; 12.5 µM, 72 h for
GIST48) on GIST cell lines. As shown in Fig. 2, ETV1 in the GIST882 and GIST48
cells was downregulated by TFP and TDZ. However, the expression
levels of both total and activated KIT were not markedly affected
by these two drugs. This result confirms that both TFP and TDZ have
distinct targeting effects on the ETV1 pathway, compared with other
drugs.
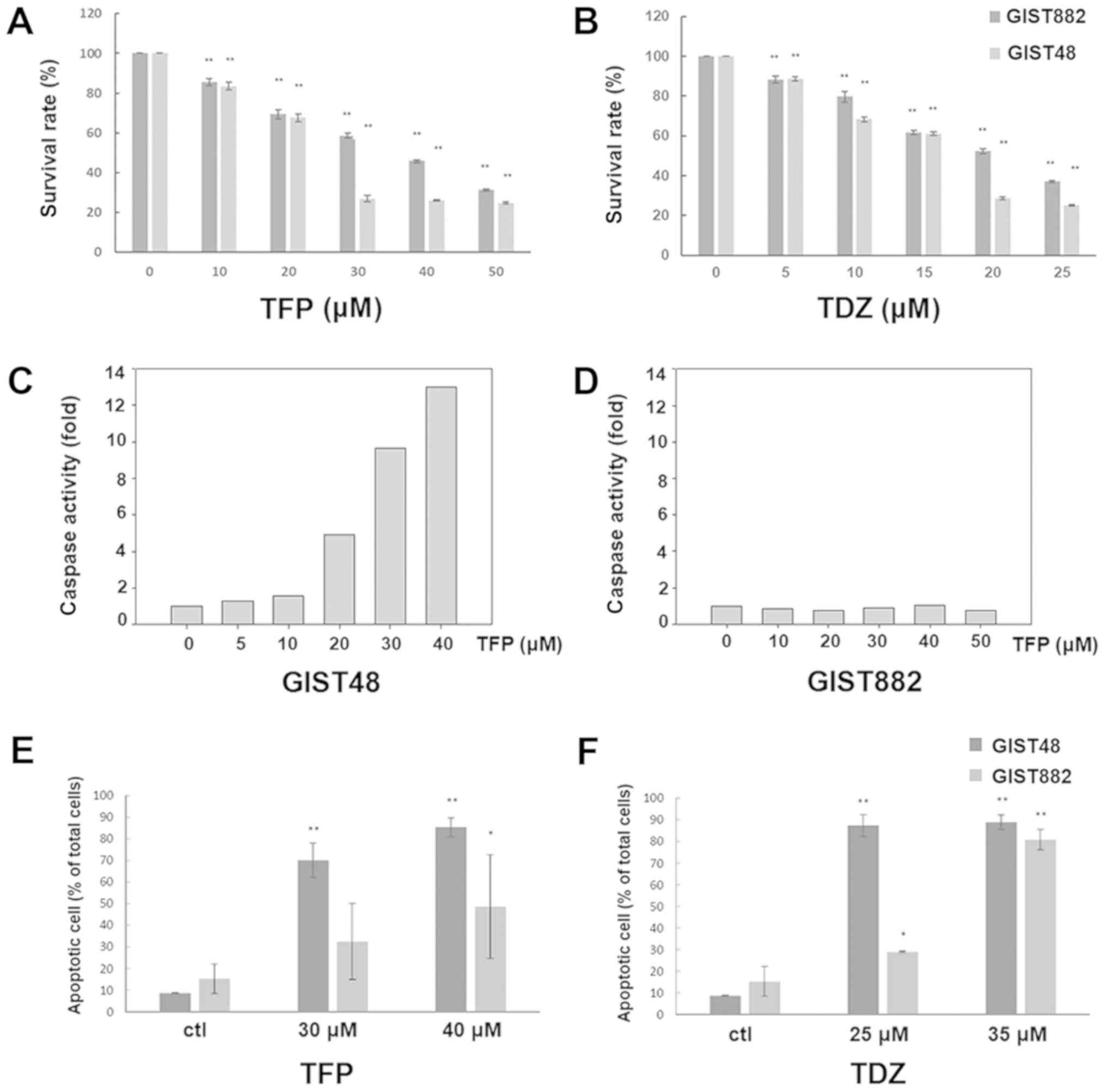
Both TFP and TDZ exert cytotoxic effects
against GISTs
We then evaluated the cytotoxic effects of TFP on
GIST cell lines. As shown in Fig. 3A
and B, both TFP and TDZ exerted significant anti-proliferative
effects on the GIST48 and GIST882 cells in a dose-dependent manner.
However, TFP induced caspase activation in the GIST48 cells, but
not in the GIST882 cells (Fig. 3C and
D). A significant increase in the number of apoptotic cells, as
assessed by Annexin V staining, was easily observed in the GIST48
cells treated with both agents. However, this could only be seen in
the GIST882 cells at relatively higher concentrations of
phenothiazine-class drugs (Fig. 3E and
F). These results indicate that both TFP and TDZ exert a
cytotoxic effect against GISTs, but not always through
apoptosis.
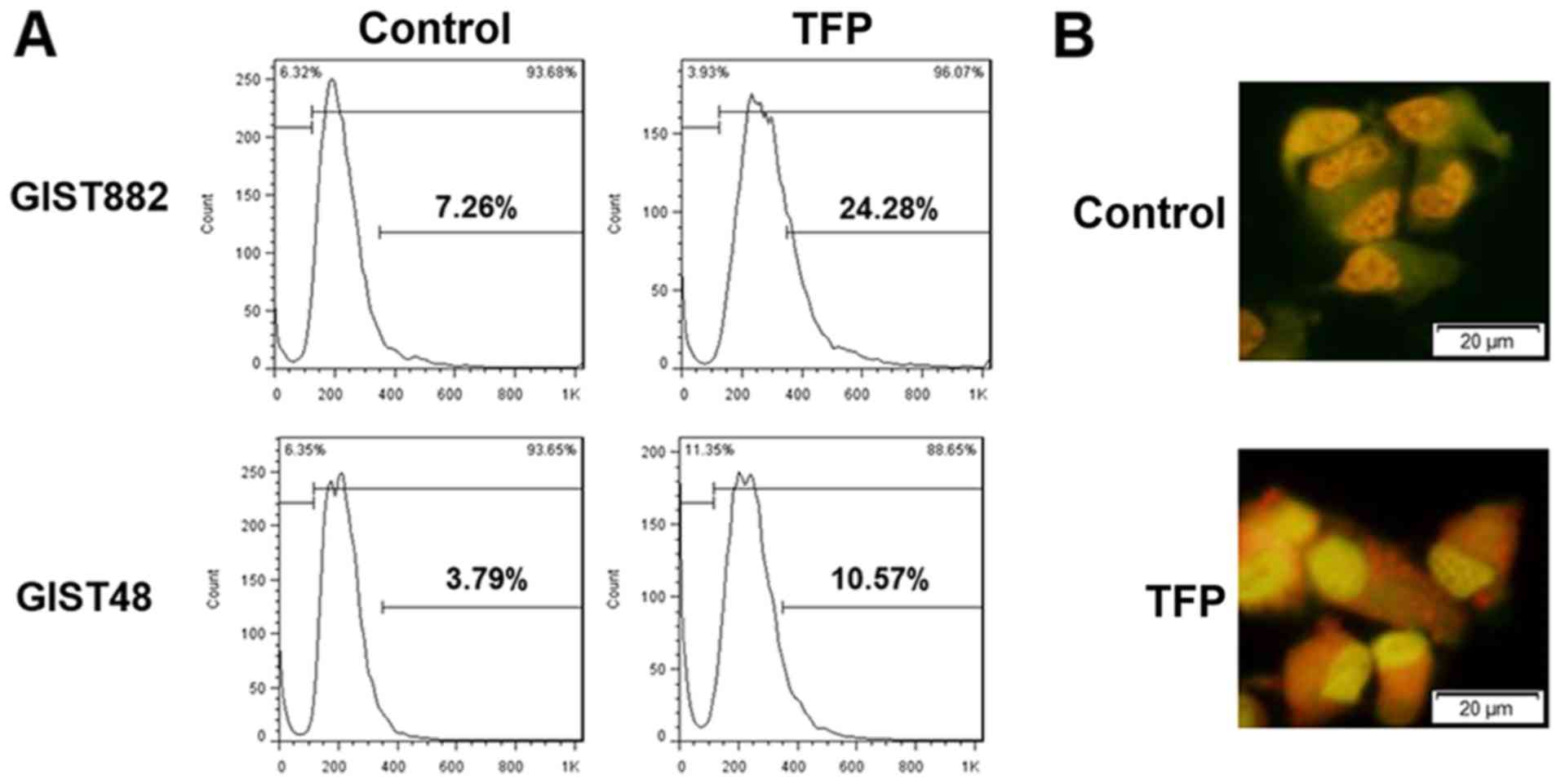
TFP and TZD induce autophagy and the
downregulation of ETV1 with the paradoxical upregulation of ERK in
GIST cell lines
Phenothiazine drugs are a well-known class of drugs
that induce autophagy (31). In
this study, we evaluated the possibility of autophagy induction in
GIST cells following drug treatment. As illustrated in Fig. 4, both TFP and TDZ induced autophagy
in GISTs, as demonstrated by an increased LC3-II expression.
Quantitative fluorescence-activated cell sorting (FACS) analysis
revealed an increase in lysosomes following drug treatment
(Fig. 5). These results indicate
that TFP and TDZ can induce autophagy in GIST cell lines. PARP
cleavage, an indicator of apoptosis, was also observed in the
GIST48 and, to a lesser extent, in the GIST882 cells. This is
compatible with the distinct pattern of caspase activation observed
in both cell lines following treatment with TFP.
Chi et al revealed that ETV1 was a downstream
effector of MEK and that MEK inhibition causes ETV1 down-regulation
(20). Similar findings were also
noted in this study (Fig. 2).
However, the GIST cell lines treated with TFP or TDZ exhibited an
ETV1 downregulation with a paradoxical ERK activation (Fig. 4), indicating a MEK-independent
mechanism in ETV1 degradation. TFP induced the suppression of the
AKT (indicated by p-AKT)/mTOR (indicated by p-p70s6k) signaling
pathway in the GIST48 cells at a relatively high concentration (25
µM). In addition, the downregulation of p-KTI was observed
in the TFP-treated GIST48 (20 and 25 µM) and GIST882 (35
µM) cells. No marked changes in the KIT/AKT/mTOR signaling
were observed in the TDZ-treated GIST cells.
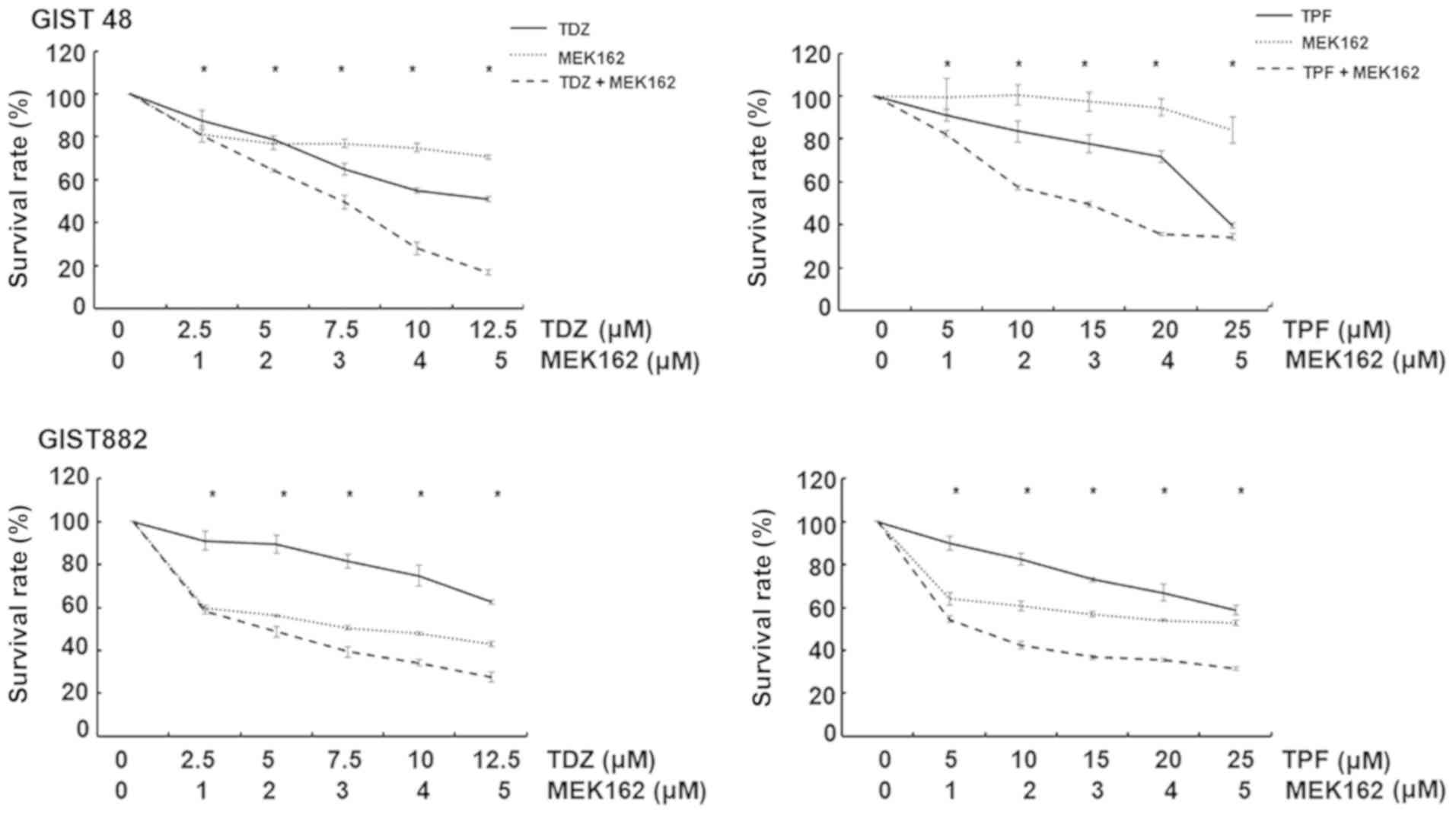
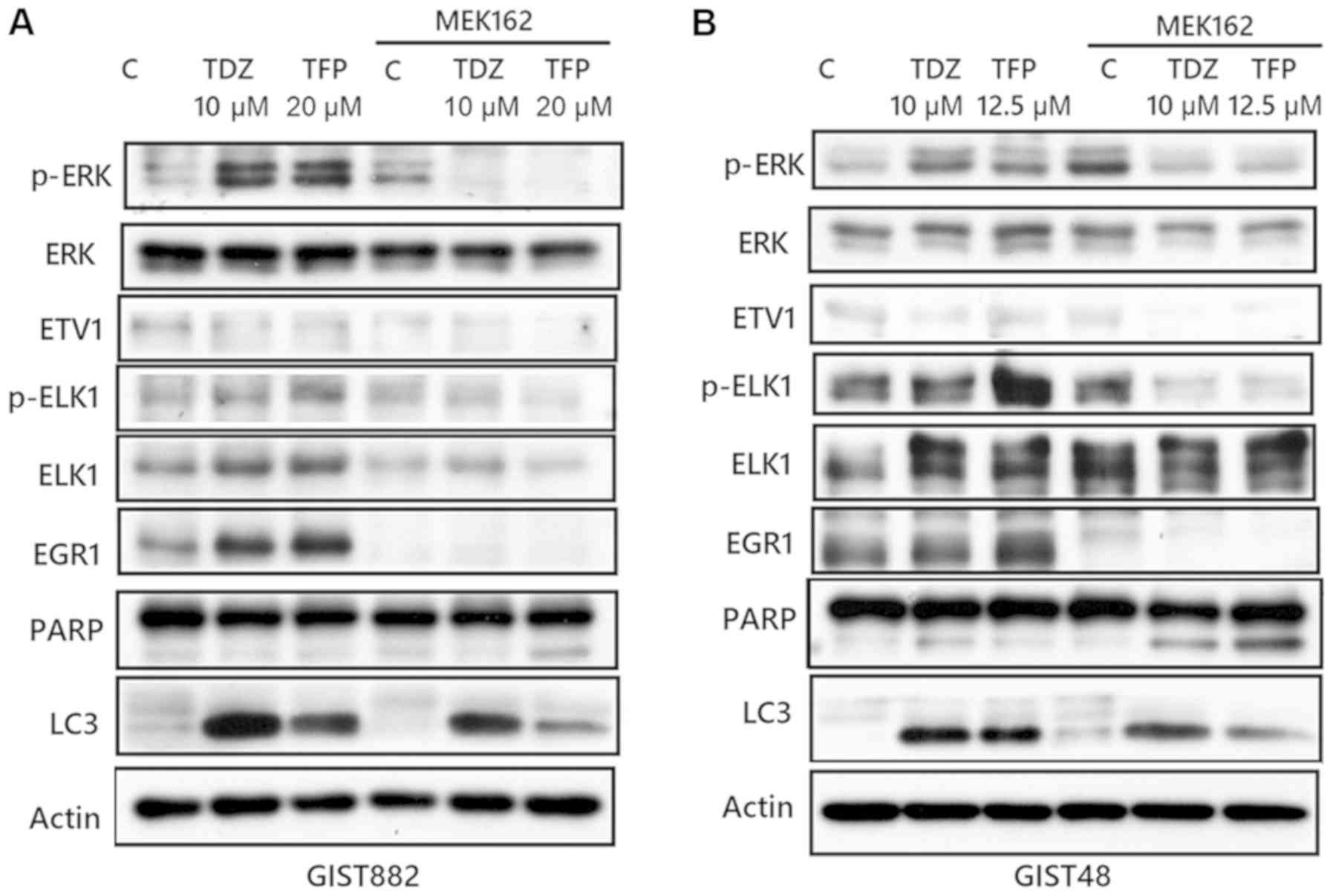
TFP or TDZ act synergistically with the
MEK inhibitor, MEK162, to exert a cytotoxic effect on GIST cell
lines and the combination of phenothiazine with MEK162 induces
apoptosis with diminished autophagy and the downregulation of ERK
and downstream ELK1 and EGR1
As we observed paradoxical ERK activation following
phenothiazine treatment, we then evaluated the possibility of
combining phenothiazine with an MEK inhibitor to treat GISTs. As
shown in Fig. 6, TFP or TDZ acted
synergistically with the MEK inhibitor, MEK162, in inducing
cytotoxic effects in the GIST48 and GIST882 cells.
The mechanisms of autophagy induction by
phenothiazine are unclear. EGR1 is a member of the EGR family of
C2H2-type zinc finger proteins. It is a nuclear protein and
functions as a transcriptional regulator, and it is also known to
be responsible for environmental stress-induced autophagy, such as
that caused by smoking (32).
Additionally, EGR1 expression can be induced by ELK1, which is a
member of the Ets family of transcription factors and of the
ternary complex factor (TCF) subfamily, as well as a known nuclear
target for the RAS/RAF/MAPK signaling cascade (33). Consequently, we considered that
phenothiazine-induced autophagy may occur through ERK activation of
the ELK1/EGR1 pathway. As shown in Fig. 7, TFP or TDZ induced autophagy with
concomitant ERK, ELK1 and EGR1 activation. However, the combination
of MEK162 and phenothiazine downregulated ERK, and downstream ELK1
and EGR1 with a resultant decrease in autophagy. Apoptosis, based
on PARP cleavage, seemed more apparent in cells treated with the
MEK162 and TFP combination. This result indicates that
phenothiazine-induced autophagy may occur through the ERK/ELK1/EGR1
pathway, and the combination of phenothiazine with MEK inhibitor is
a potential strategy for the treatment of GISTs.
Discussion
In this study, by uploading the differential
expression gene sets of wild-type versus knockdown ETV1 to CMAP, we
identified several agents with therapeutic potential. Among these,
phenothiazine had not been reported previously, at least to the
best of our knowledge. Phenothiazine-derived drugs were found to
exert cytotoxicity and could induce apoptosis and autophagy in
GISTs. Phenothiazine had little effect on the KIT/AKT/mTOR pathway,
but instead paradoxically upregulated ERK activity. Phenothiazine
may induce autophagy through the activation of the
MEK/ERK/ELK1/EGR1 pathway, and the combination of phenothiazine and
a MEK inhibitor had a synergistic cytotoxic effect on GISTs.
The important role of ETV1 in GIST was first
revealed by Chi et al (20). Their study demonstrated that ETV1
was regulated by the KIT/MAP kinase pathway, and plays a key role
in an ICC-GIST-specific transcription network (20). Furthermore, a positive feedback
circuit between the KIT/MAP kinase pathway and ETV1 was found, and
a combination therapy of imatinib and MEK162 to target ETV1
stability resulted in considerable tumor suppressive effects
(21). Their studies led us to
explore the agents with ETV1-targeting potentials in GIST
treatment.
In this study, through the analysis of a
public-domain database, we found that ETV1, along with several
known genes such as KIT, DOG1, and PKCθ, was significantly
overexpressed in GISTs in comparison with other sarcomas. This is
consistent with previous reports (20,34),
again demonstrating the uniqueness and importance of ETV1 in GISTs.
Subsequently, we uploaded an ETV1 knockout gene signature of GIST
cell lines to CMAP, and we identified several ETV1 targeting
agents. CMAP is a genome-wide transcriptional expression database
generated from cultured human cells treated with bioactive small
molecules. It applies a simple pattern-matching algorithm to
discover the functional connections between drugs, genes, and
diseases through common gene-expression changes. This database has
been widely used in the discovery of novel agents and in
deciphering previously unknown molecular mechanisms (35-38).
In this study, through CMAP analysis of an ETV1 knockout gene
signature of GIST cell lines, we discovered SAHA and trichostatin
(two HDACIs) and TFP and TDZ (two drugs of phenothiazine class) as
ETV1-targeting agents.
As expected, HDACIs were on the list, as a previous
study has already shown their activity in downregulating KIT
(14). In this study, we also
demonstrated their activity by western blot analysis (Fig. 2). However, phenothiazine has not
been previously reported to have anti-GIST activity, at least to
the best of our knowledge. Phenothiazine has been identified as a
class of potential anticancer drugs in previous publications.
Possible mechanisms include inhibiting the PDK1/AKT pathway
(39,40), Wnt/b-catenin pathway (41), dopamine receptors (42), HSP70 (43) and DNA repair (44). In this study, we did not observe a
significant effect of phenothiazine on the KIT/AKT/mTOR pathway.
However, paradoxical ERK activation was found following treatment
with the drugs.
The mechanisms of paradoxical ERK activation
following phenothiazine or MEK162 treatment are not clear. ERK
pathway activity is regulated by negative feedback at multiple
levels, including the transcriptional activation of DUSP proteins
that negatively regulate the pathway. ERK also phosphorylates and
thus regulates upstream CRAF, SOS and MEK activity directly
(45). One possible hypothesis is
that these feedback inhibitory pathways may be temporarily shut
down after phenothiazine- or MEK162- induced ERK inhibition, with
resultant re-activation of ERK, and complete inhibition could only
be achieved by combination of different agents targeting different
levels. However, the specific underlying mechanisms warrant further
investigation.
Although phenothiazine has been demonstrated to be a
class of autophagy-inducing agents (31), the associated mechanism is still
unclear. Chen et al discovered the role of EGR1 in autophagy
by showing that cigarette smoke extract (CSE) could reduce HDAC
activity, which resulted in increased binding of EGR1 and E2F
factors to the LC3B promoter with subsequently increased LC3B
expression in human pulmonary epithelial cells (32). Additionally, Guha et al
revealed that lipopolysaccharides induced ELK1 phosphorylation
through the MEK/ERK1/2 pathway with the subsequent induction of
EGR1 expression in monocytes (33). As we observed ERK activation
following phenothiazine treatment, we then explored the role of ERK
activation in autophagy. In this study, we revealed that a
combination of phenothiazine and a MEK inhibitor had a synergistic
cytotoxic effect on GISTs. Western blot analysis indicated that MEK
inhibition diminished autophagy and, together with phenothiazine,
induced apoptosis. We also demonstrated that both ELK1 and EGR1
were activated/upregulated following TDZ and TFP treatment, but
both were downregulated with a concomitant decrease in LC3-II
expression after MEK inhibition. This result indicates that
phenothiazine-induced autophagy may be mediated through the
MEK/ERK/ELK1/EGR1 pathway and that the combination of phenothiazine
with a MEK inhibitor may be a potential strategy for the treatment
of GISTs.
In conclusion, in this study, by using
bioinformatics analysis through CMAP, we identified TFP and TDZ,
two drugs of phenothiazine class, as potential ETV1-targeting
agents in GIST treatment. Phenothiazine-derived drugs induced
apoptosis and autophagy in GISTs. Phenothiazine treatment was found
to paradoxically upregulate ERK activity, but with little effect on
the KIT/AKT/mTOR pathway. Phenothiazine may induce autophagy in
GISTs through the MEK/ERK/ELK1/EGR1 pathway. A combination of
phenothiazine and a MEK inhibitor exerted a synergistic cytotoxic
effect on GISTs. Additional in vivo or clinical studies are
warranted for further confirmation.
Supplementary Data
Acknowledgments
This manuscript was edited by Wallace Academic
Editing.
Funding
This study was jointly supported by the grants from
the Department of Health in Taiwan (Center of Excellence for Cancer
Research at Taipei Veterans General Hospital, grant nos.
DOH99-TD-C-111-007 and DOH100-TD-C-111-007, and the National
Research Program for Biopharmaceuticals, grant no.
DOH100-TD-PB-111-TM026), National Science Council (NSC
100-2314-B-075-081 and NSC 101-2314-B-075-029), Ministry of Science
and Technology, Taiwan, (MOST 103-2314-B-075-066, MOST
105-2314-B-075-059, MOST 106-2314-B-075 -065), Taipei Veterans
General Hospital (V102E8-003, V103E8-001, V101C-133, V102C-034,
V103C-188, V104C-099, V104E16-001-MY3-1, V104E16-001-MY3-2,
V104D16-001-MY3-3, V105C-094, V106C-160, V107C-085,
V107D32-001-MY2-2, V108C-108) and from the Yen Tjing Ling Medical
Foundation (grant no. CI-100-19, CI-103-6, CI-105-4) designated to
CCY. This study was also supported by the Taiwan Clinical Oncology
Research Foundation, and Chong Hin Loon Memorial Cancer and
Biotherapy Research Center of National Yang-Ming University.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JAF, CCY, LTC and CFL were responsible for the
design and conception of the study; WYC, YCL, CHY and YCC were
responsible for data acquisition and interpretation; SCC, MHY and
YC were responsible for the data analysis and drafting the work.
All of the authors have read and approved the final manuscript for
publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Review on morphology, molecular pathology,
prognosis, and differential diagnosis. Arch Pathol Lab Med.
130:1466–1478. 2006.PubMed/NCBI
|
|
2
|
Reith JD, Goldblum JR, Lyles RH and Weiss
SW: Extra-gastrointestinal (soft tissue) stromal tumors: An
analysis of 48 cases with emphasis on histologic predictors of
outcome. Mod Pathol. 13:577–585. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tran T, Davila JA and El-Serag HB: The
epidemiology of malignant gastrointestinal stromal tumors: An
analysis of 1,458 cases from 1992 to 2000. Am J Gastroenterol.
100:162–168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blanke CD, Demetri GD, von Mehren M,
Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD,
Roberts PJ, Heinz D, et al: Long-term results from a randomized
phase II trial of standard- versus higher-dose imatinib mesylate
for patients with unresectable or metastatic gastrointestinal
stromal tumors expressing KIT. J Clin Oncol. 26:620–625. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yeh CN, Chen YY, Tseng JH, Chen JS, Chen
TW, Tsai CY, Cheng CT, Jan YY and Chen MF: Imatinib mesylate
forpatients with recurrent or metastatic gastrointestinal stromal
tumors expressing KIT: A decade experience from Taiwan. Transl
Oncol. 4:328–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heinrich MC, Corless CL, Blanke CD,
Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M,
Fletcher CD, Sandau K, et al: Molecular correlates of imatinib
resistance in gastrointestinal stromal tumors. J Clin Oncol.
24:4764–4774. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heinrich MC, Maki RG, Corless CL,
Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB,
Fletcher JA, et al: Primary and secondary kinase genotypes
correlate with the biological and clinical activity of sunitinib in
imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol.
26:5352–5359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Demetri GD, van Oosterom AT, Garrett CR,
Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich
MC, Morgan JA, et al: Efficacy and safety of sunitinib in patients
with advanced gastrointestinal stromal tumour after failure of
imatinib: A randomised controlled trial. Lancet. 368:1329–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Demetri GD, Reichardt P, Kang YK, Blay JY,
Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M,
Joensuu H, et al GRID study investigators: Efficacy and safety of
regorafenib for advanced gastrointestinal stromal tumours after
failure of imatinib and sunitinib (GRID): An international,
multicentre, randomised, placebo-controlled, phase 3 trial. Lancet.
381:295–302. 2013. View Article : Google Scholar
|
|
12
|
Yeh CN, Chen MH, Chen YY, Yang CY, Yen CC,
Tzen CY, Chen LT and Chen JS: A phase II trial of regorafenib in
patients with metastatic and/or a unresectable gastrointestinal
stromal tumor harboring secondary mutations of exon 17. Oncotarget.
8:44121–44130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bauer S, Yu LK, Demetri GD and Fletcher
JA: Heat shock protein 90 inhibition in imatinib-resistant
gastrointestinal stromal tumor. Cancer Res. 66:9153–9161. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mühlenberg T, Zhang Y, Wagner AJ,
Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M,
Fletcher JA, et al: Inhibitors of deacetylases suppress oncogenic
KIT signaling, acetylate HSP90, and induce apoptosis in
gastrointestinal stromal tumors. Cancer Res. 69:6941–6950. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bauer S, Duensing A, Demetri GD and
Fletcher JA: KIT oncogenic signaling mechanisms in
imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT
is a crucial survival pathway. Oncogene. 26:7560–7568. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pantaleo MA, Nicoletti G, Nanni C, Gnocchi
C, Landuzzi L, Quarta C, Boschi S, Nannini M, Di Battista M,
Castellucci P, et al: Preclinical evaluation of KIT/PDGFRA and mTOR
inhibitors in gastrointestinal stromal tumors using small animal
FDG PET. J Exp Clin Cancer Res. 29:1732010. View Article : Google Scholar
|
|
17
|
Bendell JC, Bauer TM, Lamar R, Joseph M,
Penley W, Thompson DS, Spigel DR, Owera R, Lane CM, Earwood C, et
al: A phase 2 study of the Hsp90 inhibitor AUY922 as treatment for
patients with refractory gastrointestinal stromal tumors. Cancer
Invest. 34:265–270. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deming DA, Ninan J, Bailey HH, Kolesar JM,
Eickhoff J, Reid JM, Ames MM, McGovern RM, Alberti D, Marnocha R,
et al: A Phase I study of intermittently dosed vorinostat in
combination with bortezomib in patients with advanced solid tumors.
Invest New Drugs. 32:323–329. 2014. View Article : Google Scholar :
|
|
19
|
Schöffski P, Reichardt P, Blay JY, Dumez
H, Morgan JA, Ray-Coquard I, Hollaender N, Jappe A and Demetri GD:
A phase I-II study of everolimus (RAD001) in combination with
imatinib in patients with imatinib-resistant gastrointestinal
stromal tumors. Ann Oncol. 21:1990–1998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chi P, Chen Y, Zhang L, Guo X, Wongvipat
J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, et al: ETV1 is
a lineage survival factor that cooperates with KIT in
gastrointestinal stromal tumours. Nature. 467:849–853. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ran L, Sirota I, Cao Z, Murphy D, Chen Y,
Shukla S, Xie Y, Kaufmann MC, Gao D, Zhu S, et al: Combined
inhibition of MAP kinase and KIT signaling synergistically
destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov.
5:304–315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lamb J, Crawford ED, Peck D, Modell JW,
Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et
al: The Connectivity Map: Using gene-expression signatures to
connect small molecules, genes, and disease. Science.
313:1929–1935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lamb J: The Connectivity Map: A new tool
for biomedical research. Nat Rev Cancer. 7:54–60. 2007. View Article : Google Scholar
|
|
24
|
Li C and Hung WW: Model-based analysis of
oligonucleotide arrays: model validation, design issues and
standard error application. Genome Biol.
2:RESEARCH00322001.PubMed/NCBI
|
|
25
|
Li C and Wong WH: Model-based analysis of
oligonucleotide arrays: Expression index computation and outlier
detection. Proc Natl Acad Sci USA. 98:31–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tuveson DA, Willis NA, Jacks T, Griffin
JD, Singer S, Fletcher CD, Fletcher JA and Demetri GD: STI571
inactivation of the gastrointestinal stromal tumor c-KIT
oncoprotein: Biological and clinical implications. Oncogene.
20:5054–5058. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun X, Hung K, Wu L, Sidransky D and Guo
B: Detection of tumor mutations in the presence of excess amounts
of normal DNA. Nat Biotechnol. 20:186–189. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ross P, Hall L, Smirnov I and Haff L: High
level multiplex genotyping by MALDI-TOF mass spectrometry. Nat
Biotechnol. 16:1347–1351. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W,
Zhu H, Yu AD, Xie X, Ma D, et al: Small molecule regulators of
autophagy identified by an image-based high-throughput screen. Proc
Natl Acad Sci USA. 104:19023–19028. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen ZH, Kim HP, Sciurba FC, Lee SJ,
Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ,
Yousem SA, et al: Egr-1 regulates autophagy in cigarette
smoke-induced chronic obstructive pulmonary disease. PLoS One.
3:e33162008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guha M, O'Connell MA, Pawlinski R, Hollis
A, McGovern P, Yan SF, Stern D and Mackman N: Lipopolysaccharide
activation of the MEK-ERK1/2 pathway in human monocytic cells
mediates tissue factor and tumor necrosis factor alpha expression
by inducing Elk-1 phosphorylation and Egr-1 expression. Blood.
98:1429–1439. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jang BG, Lee HE and Kim WH: ETV1 mRNA is
specifically expressed in gastrointestinal stromal tumors. Virchows
Arch. 467:393–403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang SE, Xiang B, Guix M, Olivares MG,
Parker J, Chung CH, Pandiella A and Arteaga CL: Transforming growth
factor beta engages TACE and ErbB3 to activate
phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast
cancer and desen-sitizes cells to trastuzumab. Mol Cell Biol.
28:5605–5620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hieronymus H, Lamb J, Ross KN, Peng XP,
Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, et al:
Gene expression signature-based chemical genomic prediction
identifies a novel class of HSP90 pathway modulators. Cancer Cell.
10:321–330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wei G, Twomey D, Lamb J, Schlis K, Agarwal
J, Stam RW, Opferman JT, Sallan SE, den Boer ML, Pieters R, et al:
Gene expression-based chemical genomics identifies rapamycin as a
modulator of MCL1 and glucocorticoid resistance. Cancer Cell.
10:331–342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garman KS, Acharya CR, Edelman E, Grade M,
Gaedcke J, Sud S, Barry W, Diehl AM, Provenzale D, Ginsburg GS, et
al: A genomic approach to colon cancer risk stratification yields
biologic insights into therapeutic opportunities. Proc Natl Acad
Sci USA. 105:19432–19437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi JH, Yang YR, Lee SK, Kim SH, Kim YH,
Cha JY, Oh SW, Ha JR, Ryu SH and Suh PG: Potential inhibition of
PDK1/Akt signaling by phenothiazines suppresses cancer cell
proliferation and survival. Ann NY Acad Sci. 1138:393–403. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rho SB, Kim BR and Kang S: A gene
signature-based approach identifies thioridazine as an inhibitor of
phosphati-dylinositol-3′-kinase (PI3K)/AKT pathway in ovarian
cancer cells. Gynecol Oncol. 120:121–127. 2011. View Article : Google Scholar
|
|
41
|
Yeh CT, Wu AT, Chang PM, Chen KY, Yang CN,
Yang SC, Ho CC, Chen CC, Kuo YL, Lee PY, et al: Trifluoperazine, an
antipsychotic agent, inhibits cancer stem cell growth and overcomes
drug resistance of lung cancer. Am J Respir Crit Care Med.
186:1180–1188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sachlos E, Risueño RM, Laronde S,
Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn
A, Graham M, et al: Identification of drugs including a dopamine
receptor antagonist that selectively target cancer stem cells.
Cell. 149:1284–1297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Koren J III, Jinwal UK, Jin Y, O'Leary J,
Jones JR, Johnson AG, Blair LJ, Abisambra JF, Chang L, Miyata Y, et
al: Facilitating Akt clearance via manipulation of Hsp70 activity
and levels. J Biol Chem. 285:2498–2505. 2010. View Article : Google Scholar :
|
|
44
|
Polischouk AG, Holgersson A, Zong D,
Stenerlöw B, Karlsson HL, Möller L, Viktorsson K and Lewensohn R:
The antipsychotic drug trifluoperazine inhibits DNA repair and
sensitizes non small cell lung carcinoma cells to DNA double-strand
break induced cell death. Mol Cancer Ther. 6:2303–2309. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu F, Yang X, Geng M and Huang M:
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer
therapy. Acta Pharm Sin B. 8:552–562. 2018. View Article : Google Scholar : PubMed/NCBI
|