Introduction
The dysregulation of protein synthesis by oncogenes
and tumor suppressor genes is a common event in cancer (1) The therapeutic targeting of the
translational machinery has thus become an area of substantive
interest for cancer therapy. In this respect, a number of small
molecules/compounds have been discovered to target various
components of the translational apparatus (2).
Mitogen-activated protein kinase (MAPK)-interacting
serine/threonine kinases (MKNKs) 1/2 (MKNK1/MKNK2) control
translational initiation through the phosphorylation of eukaryotic
initiation factor 4E (eIF4E) (3,4).
MKNK1 is recruited to eIF4E through the binding of eIF4G (5). When phosphorylated, eIF4E binds to
other components within the initiation complex to begin translation
(6). Of note, fragile X mental
retardation protein (FMR1) has been shown to regulate the
translation of specific mRNAs through its binding to eIF4E
(7). Higher levels of
phosphorylated eIF4E have also been reported to increase the rate
of development and progression of various types of cancer (6). Upstream events that activate ERK/MAPK
and p38 kinases have been shown to phosphorylate residues Thr209
and Thr214 in MKNK1 and Thr244 and Thr249 in MKNK2 (4,8,9).
While the phosphorylation and activation of eIF4E are necessary for
oncogenic transformation, knockout experiments have revealed that
both MKNK1 and MKNK2 are dispensable for normal development
(10). Targeting eIF4E may
therefore be an effective strategy for cancer therapy with minimal
side-effects (11).
Friend virus-induced Leukemia-1 (FLI1) transcription
factor (TF) is a potent oncogene that drives the initiation and
progression of leukemia and other types of cancer (12-15).
During oncogenesis, FLI1 alters a number of hallmarks of cancer,
including proliferation, apoptosis, differentiation, angiogenesis,
genomic stability and inflammation (15-17).
Thus, the therapeutic targeting of FLI1 is an attractive strategy.
Accordingly, several classes of compounds with a potent anti-FLI1
activity have recently been identified (18-22).
Among these, two flavagline-like compounds exhibiting potent FLI1
inhibition were also identified (22). These compounds suppress
c-Raf-MEK-MAPK/ERK signaling, resulting in the reduced
phosphorylation of eIF4E and the inhibition of FLI1 protein
synthesis.
The present study, at least to the best of our
knowledge, demonstrates for the first time that FLI1 is capable of
binding to the promoter of MKNK1, directly activating MKNK1
transcription and subsequently, the phosphorylation of eIF4E. Thus,
these results highlight the use of FLI1 inhibitors for the
suppression of protein translation for the treatment of cancers
overexpressing this TF.
Materials and methods
Cell lines and drug treatment
Mycoplasma negative erythroleukemia cell lines
(human HEL [ATCC-TIB-180] and K562 [ATCC-CCL-243]; mouse KH16, CB7
and DP17 (generated by the authors and others) (13,18)
were cultured and maintained in Dulbecco’s Modified Eagle Medium
supplemented with 5% fetal bovine serum (HyClone, GE
Healthcare).
For drug treatment, the HEL cells were treated with
3 µM of A1544, A1545 (22)
and CGP57380 and 24 h later subjected to either western blot
analysis or RT-qPCR. The drug CGP57380 was obtained from
Selleckchem. The drugs were dissolved to a stock solution of 2 mM
in dimethyl sulfoxide (DMSO), diluted to concentrations indicated
in the text and used in treatments. DMSO was also used as a vehicle
control.
The K562-fli-1 inducible cell line was generated by
the authors, as previously described (18). To induce FLI1, 106 cells
were treated for 24 h with 10 nM of doxycycline (Cat. no. D8960-5g,
Solarbio) and used in the experiments described below.
Gene cloning, transfections and
luciferase activity
To clone the MKNK1 promoter, various regions
of the MKNK1 promoter (for details please see Fig. 2B) were isolated by qPCR (the list
of primers is presented in Table
SI) and cloned into the luciferase reporter vector PGL3
(Promega), as previously described (21). These promoter vectors (1 µg)
with either MigR1 (1 µg) or MigR1-FLI1 (1 µg) were
transfected into 293T cells (ATCC-CRL-3216) using Lipofectamine
2000 (Life Technologies; Thermo Fisher Scientific) following the
manufacturer’s protocol. Renilla luciferase was used in
transfection as an internal control to examine the transfection
efficiency, according to the manufacturer’s recommendations
(Promega). The transfected cells were then plated 8×103
cells/well into 96-well plates and luciferase activity was
determined, as previously described (21).
The DP17 (1×106) cells were transfected
with MigR1-FLI1 (2 µg) and MigR1 (2 µG) vector, using
Lipofectamine 2000, and cells were then selected for neomycin
resistance by growing the cells in medium containing G418 (0.8
mg/ml; Gibco; Thermo-Fisher Scientific).
Chromatin immunoprecipitation (ChIP)
analysis
For ChIP assay, CB7 cells were washed, crosslinked
with formaldehyde, resuspended in lysis buffer Magna Chip G kits
(Millipore) and fixed cells sonicated using the Sonics Vibra VCX150
(Scientz Biotechnology). At this stage, a portion of chromatin
aliquot was removed for input control. To the isolated chromatin
protein G sepharose beads was added followed by incubation for 1 h
at room temperature. Immunoprecipitations were performed overnight
at 4̊C with 1 µg of FLI1 antibody (Cat. no. ab15289, Abcam)
or non-specific normal rabbit immunoglobulin G (IgG; Cat. no. 2729,
Cell Signaling Technology) antibodies. Precipitates were then
washed and reverse crosslinked, using the instructions provided
with the Magna Chip G kits (Millipore). Precipitated chromatin was
then incubated with proteinase K at 50°C for 2 h, DNA-purified with
phenol chloroform extraction and resuspended in TE buffer. RT-qPCR
was performed to amplify the MKNK1 promoter regions
containing FLI1 binding site 1 (position -482 to -205) and for
negative ChI control (position −730 to -453). The sequences of the
ChIP primers are presented in Table
SII. The percentage of input was calculated by RT-qPCR based
upon the intensity of the amplified FLI1 DNA divided by the
amplified input DNA. Amplified DNA was also resolved on a 2%
agarose gel and illustrated in Fig.
3E (right panel).
RNA preparations and RT-qPCR
Total RNA was extracted from the growing culture of
HEL cells using TRIzol reagent (Life Technologies; Thermo Fisher
Scientific) according to the manufacturer’s protocol. A NanoDrop
2000 spectrophotometer (Thermo Fisher Scientific) was used to
determine the RNA concentration. To generate cDNA, the reverse
transcription reaction was performed using the PrimeScript RT
Reagent kit (Takara). qPCR was performed using FastStart Universal
SYBR-Green Master (Roche) and the Step One Plus Real-time PCR
system (Applied Biosystems). The expression was normalized to the
β-actin level. The primer sequences are presented in Table SII. Three biological triplicates
were used for all RT-qPCRs, each in triplicate (n=3). The primer
efficiency was calculated and is summarized in Table SII.
shRNA and siRNA transfection
The sh-FLI1 expression construct (FLI1-shRNA) was as
previously described (18). The
Mknk1 siRNAs (Mknk1-si1-si3) and control scrambled plasmids were
purchased from (GenePharma). The sequences are presented in
Table SII. The transfection of
these siRNAs into the HEL cells was performed using Lipofectamine
2000 according to the manufacturer’s instructions (Invitrogen;
Thermo Fisher Scientific), and as previously described (18).
Western blot analysis and inhibitor
drugs
The procedure used for western blot analysis was as
previously described (18,23). Polyclonal rabbit antibodies against
MKNK2 (Cat. no. ab84345), eIF4E (Cat. no. ab33766), phospho-eIF4E
(Cat. no. ab76256), cMYC (Cat. no. ab39688) and FLI1 (Cat. no.
ab133485) were all purchased from Abcam; MKNK1 (Cat. no. 2195) and
survivin (Cat. no. 2808) antibodies were obtained from Cell
Signaling Technology); GAPDH (Cat. no. G9545) antibody was obtained
from Sigma-Aldrich; β-actin antibody (Cat. no. 20536-1-AP) was
obtained from Proto-Technology (Protein-Tech); goat-anti-mouse and
goat anti-rabbit HRP-conjugated antibodies were obtained from Cell
Signaling Technology (Cat. nos. 5470s and 5151s, respectively).
Primary antibodies were added to the filters and incubated
overnight at 4̊C. After washing, secondary antibodies were added
for 1 h at room temperature. Antibody dilution was according to the
manufacturer’s instructions. The Odyssey system (LI-COR
Biosciences) was used to image proteins in western blot
analysis.
The inhibitor of MKNK1 (CGP57380) was obtained from
Selleckchem. The Fli-1 inhibitors, A1544 and A1545, were used as
previously described (22).
Statistical analysis
Statistical analysis was carried out using the
two-tailed Student t-test with significance considered at
P<0.05, and by one-way ANOVA with Tukey’s post-hoc test, using
Origin 3.5 software (Microcal Software). The results were expressed
as the means ± standard deviation from at least 3 independent
experiments.
Results
FLI1 expression is associated with MKNK1
expression in leukemic cell lines
The authors have previously reported that the
anti-FLI1 A1544 and A1545 compounds downregulate the
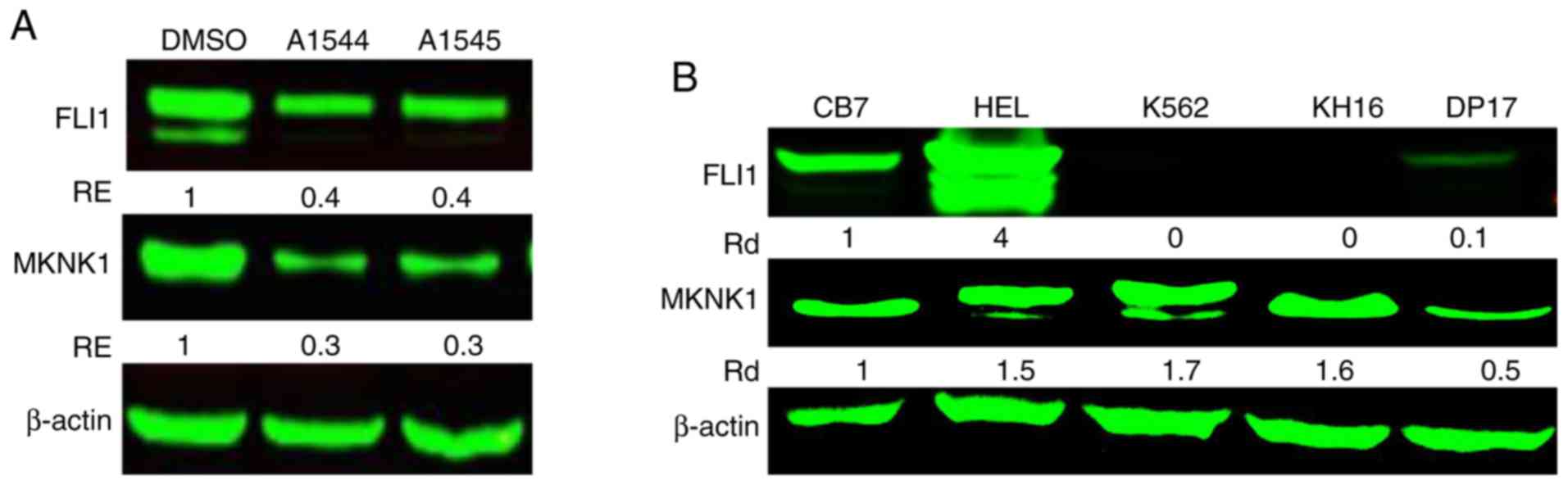
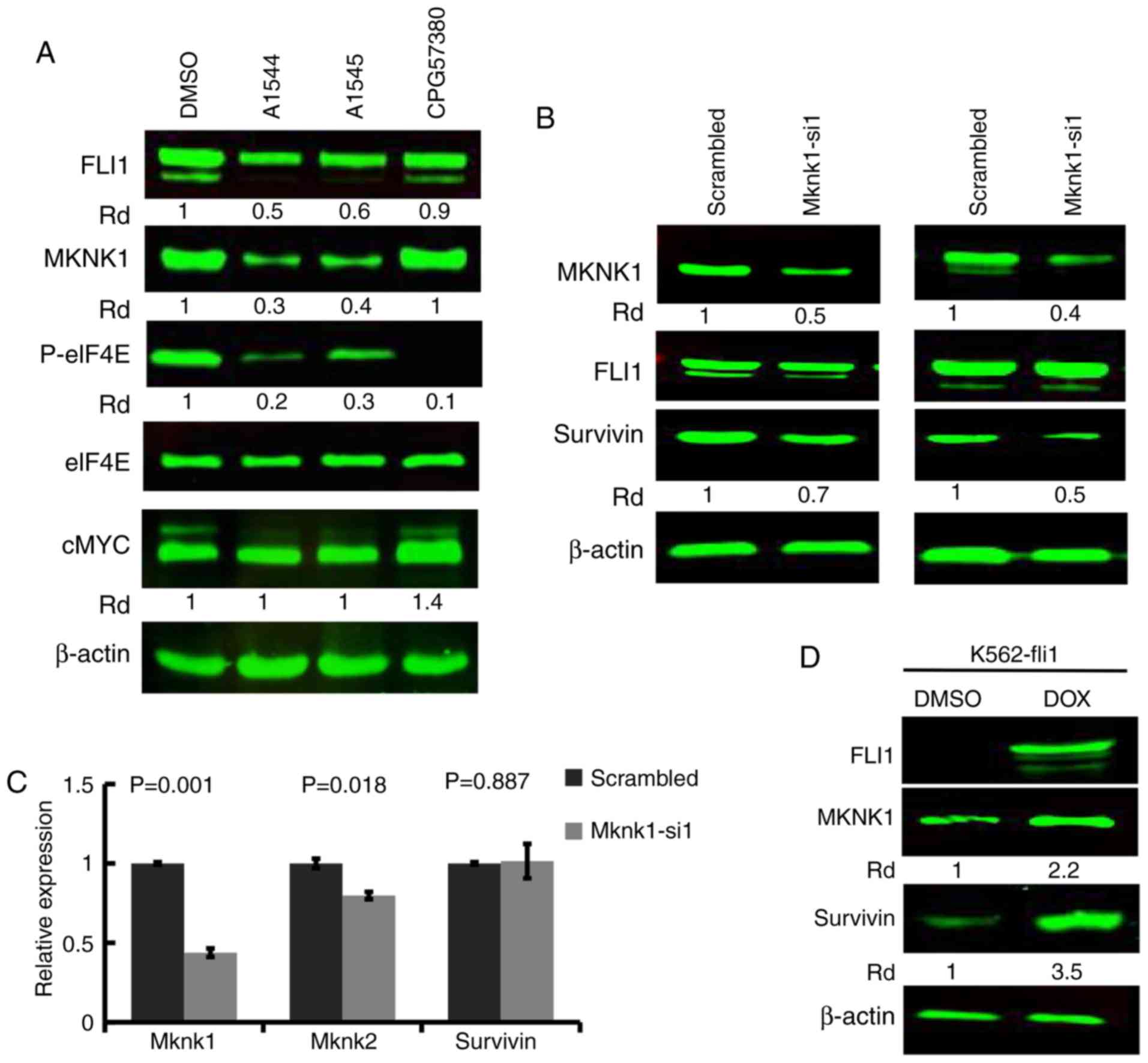
phosphorylation of eIF4E in leukemic cell lines (22). Accordingly, in this study, the
expression of MKNK1, an upstream regulator of eIF4E, was found to
be lower in the HEL cells treated with the A1544 and A1545
compounds (3 µM), compared to the control DMSO-treated cells
(Fig. 1A). This and related
analyses raised the possibility that FLI1 may regulate MKNK1
expression. Indeed, in the cell lines expressing FLI1 (HEL, CB7 and
DP17), the expression of this oncogene was positively associated
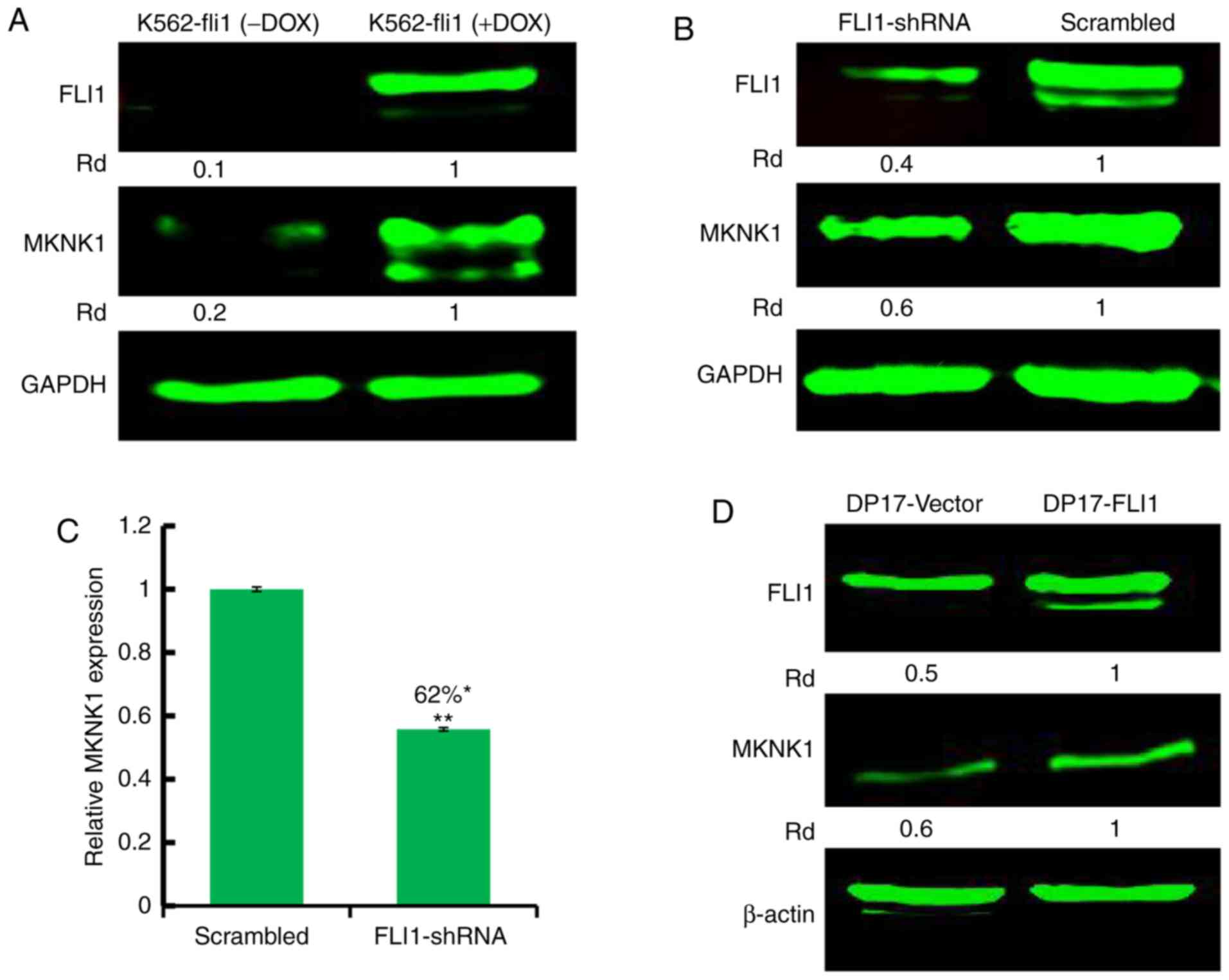
with MKNK1 expression (Fig. 1B).
In the FLI1-negative cell lines, K562 and KH16, MKNK1 expression
was relatively high, indicating different mechanisms of gene
regulation. However, when FLI1 expression was induced by
doxycycline in the K562 cells transfected with a doxycycline
inducible promoter (18), the
increased FLI1 expression resulted in a higher MKNK1 protein
(Fig. 2A) and mRNA (Fig. S1) expression compared to the
non-induced control cells (Fig.
2A). In HEL cells expressing high levels of FLI1, the
anti-FLI1 shRNA-mediated silencing resulted in a lower MKNK1
protein abundance (Fig. 2B) and
mRNA (Fig. 2C) expression.
Moreover, in murine DP17 cells with a lower FLI1 expression, the
exogenous expression of FLI1 markedly increased MKNK1 expression
(Fig. 2D). Overall, these data
strongly suggest that FLI1 regulates MKNK1 expression in both
murine and human cells.
FLI1 binds to the murine Mknk1 promoter
and positively regulates its expression
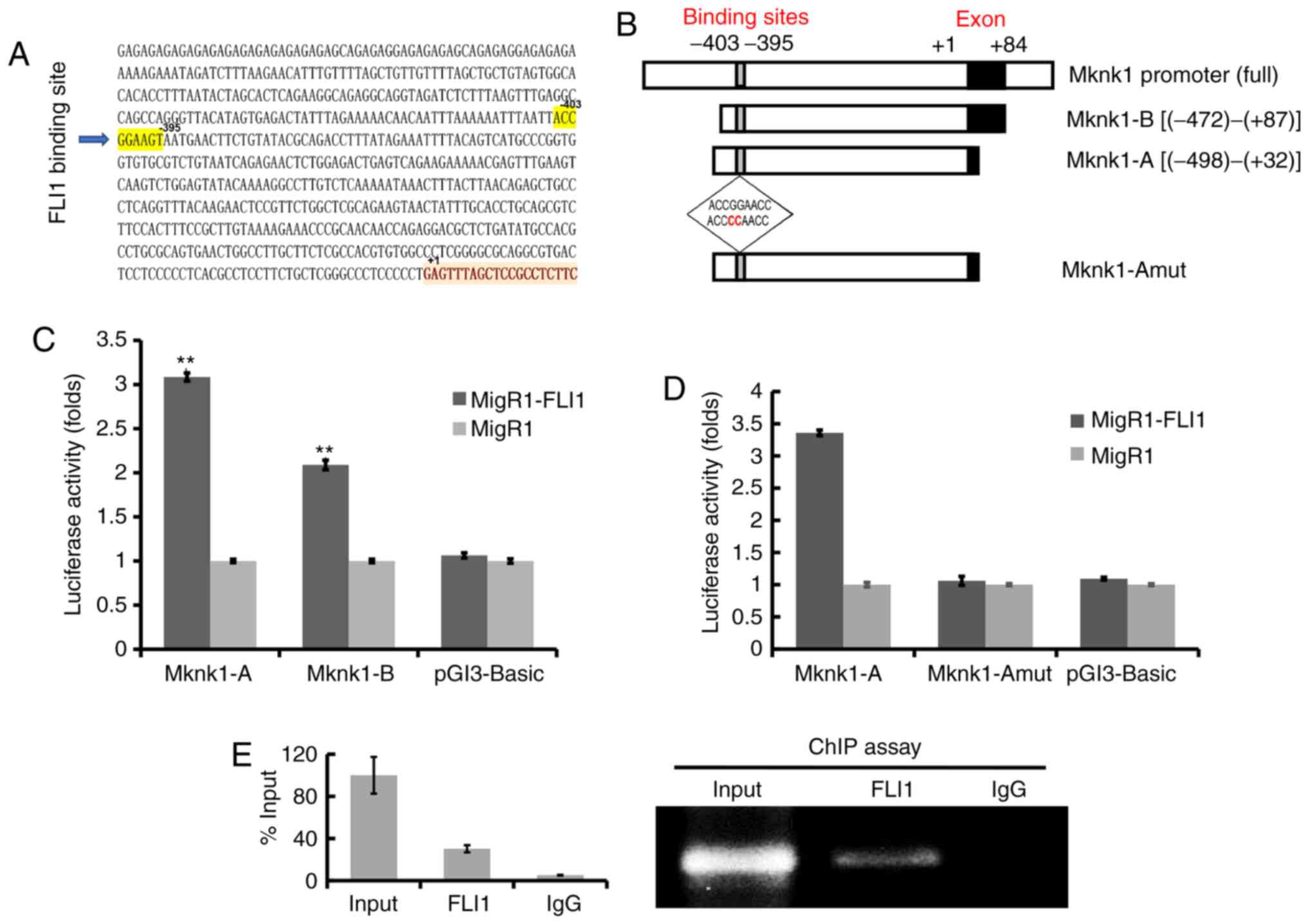
The promoter of murine Mknk1 contains a
putative FLI1 binding site (ACCGGAAGT) at position -403 to -395
(Fig. 3A). Fragments of the
Mknk1 promoter regions (designated as Mknk-A and Mknk-B)
were subcloned upstream of the luciferase reporter plasmid PGL3
(Fig. 3B). These promoters were
transfected with FLI1 (MigR1-FLI1) or control (MigR1)
expression vectors into 293T cells, examined for FLI1 expression
(Fig. S2) and later for
luciferase activity. The Mknk-A promoter fragment displayed a
significantly higher luciferase activity than the other constructs
(Fig. 3C). The putative FLI1
binding site within Mknk-A was subsequently mutated (Fig. 3B, bottom insert) and tested for
luciferase activity. The mutation of the FLI1 binding site
(ACCGGAAGT to ACCCCAAGT) within Mknk-A completely blocked promoter
activity (Fig. 3D). ChIP assay
using FLI1 antibody and control IgG was performed on chromatin
fragments of the mknk1 promoter isolated from sheared
genomic DNA of mouse CB7 cells. This analysis revealed significant
binding of FLI1 to the Mknk1 promoter having a Fli-1 binding
site (FBS), relative to input (Fig.
3E). This binding was not observed by ChIP analysis in the
immediate upstream region of the Mknk1 promoter (designated
FBS-NC), lacking FLI1 binding site (Fig. S3 and Table SII). These results demonstrate
that the murine Mknk1 promoter is a direct target of
FLI1.
siRNA-mediated MKNK1 silencing suppress
the proliferation of leukemic cells in culture
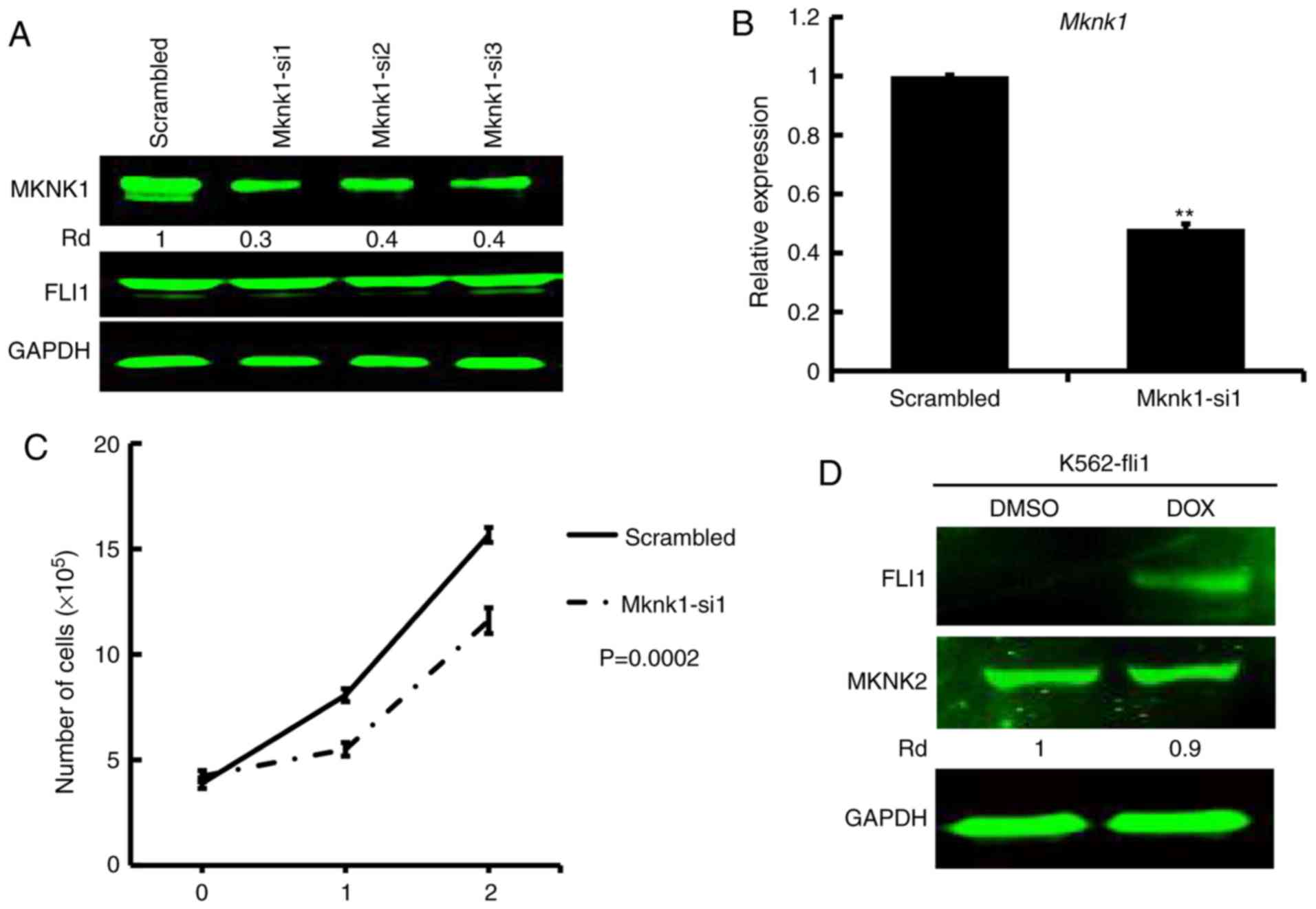
To examine the effects of MKNK1 expression on
proliferation, siRNA was used to silence the gene in HEL cells. All
3 siRNAs (Mknk1-si1, Mknk1-si2 and Mknk1-si3) markedly suppressed
MKNK1 expression (Fig. 4A);
Mknk1-si1 was used in the subsequent experiments. The
siRNA-mediated downregulation of MKNK1, as detected by
western blot analysis and RT-qPCR (Fig. 4A and B), resulted in a significant
suppression of proliferation compared to the scrambled
control-transfected cells (Fig.
4C). Of note, the induction of FLI1 in K562-fli1 inducible
cells did not increase the level of MKNK2 (Fig. 4D). Since MKNK1 was induced by FLI1
in this system (Fig. 2A) and
downregulated by shFLI1 (Fig. 2B),
this result excluded MKNK2 as a target of FLI1 (Fig. 4D). Thus, while MKNK1 downregulation
clearly inhibited cell proliferation, FLI1 altered cell
proliferation in part through the specific upregulation of this
gene.
Regulation of survivin (BIRC5) by FLI1
through the MKNK1 pathway
Previous studies have suggested that MYC
(cMYC) expression is downstream of the MKNK1 pathway (22,24).
However, in this study, while the anti-FLI1 compounds, A1544 and
A1545, and the specific MKNK1 inhibitor, CGP57380, markedly
inhibited MKNK1 and downstream phospho-eIF4A expression, the level
of MYC remained unaltered by treatment with A1544 and A1545
(Fig. 5A). Indeed, a significant
increase in MYC expression was observed in the CGP57380-treated HEL
cells. This observation excluded MYC translational
regulation by MKNK1 in these cells.
The FLI1 inhibitors, A1544 and A1545, have
previously shown to inhibit anti-apoptotic protein BIRC5 expression
through the suppression of eIF4E (22), in which the high expression of this
protein in leukemia cells facilitates cell growth (22,25).
In this study, since eiF4E phosphorylation and its activity were
regulated by MKNK1, the si1-mediated silencing of MKNK1 in 2
independent experiments resulted in a marked decrease in the
expression of BIRC5 protein (Fig.
5B), but not its mRNA expression (Fig. 5C). Notably, in this study,
MKNK2 expression was slightly downregulated, although the
level of MKNK1 was significantly decreased (Fig. 5C). As predicted, induction of FLI1
in K562 inducible fli1 cells by doxycycline significantly increased
survivin expression (Fig. 5D).
Moreover, the upregulation of survivin expression following
induction by doxycycline in K562-fli1 cells was partially blocked
through the si1-mediated downregulation of Mknk1 (Fig. S4). These results demonstrate the
regulation of BIRC5 by the FLI1/MKNK/eIF4E pathway.
Association between miR-145, FLI1 and
MKNK1 in regulating protein translation in leukemic cells
Previous studies by the authors, as well as others
have revealed a negative regulation of the miR-145 promoter by FLI1
(21,26-29),
and the inhibition of FLI1 mRNA translation by miR-145 (26), establishing a negative regulatory
loop (21,26). The anti-FLI1 compounds A1544 and
A1545 have been previously shown to target the proto-oncogene
serine/threonine-protein kinase c-RAF for inhibition, leading to
suppression of MAPK/ERK and inhibition of FLI1 translation
(22). However, the mechanisms
through which MAPK/ERK activation controls FLI1 translation remain
unknown. In this study, it was demonstrated that both the A1544 and
A1545 compounds significantly increased the level of miR-145 in the
HEL cells (Fig. 6A). Since
MAPK/ERK activation blocks miR-145 expression (30), A1544 and A1545 likely suppress FLI1
translation and subsequently MKNK1 expression through miR145
upregulation (Fig. 6B). These
results emphasize the important role played by FLI1 in the
regulation of translation through both miR-145 and
MKNK1.
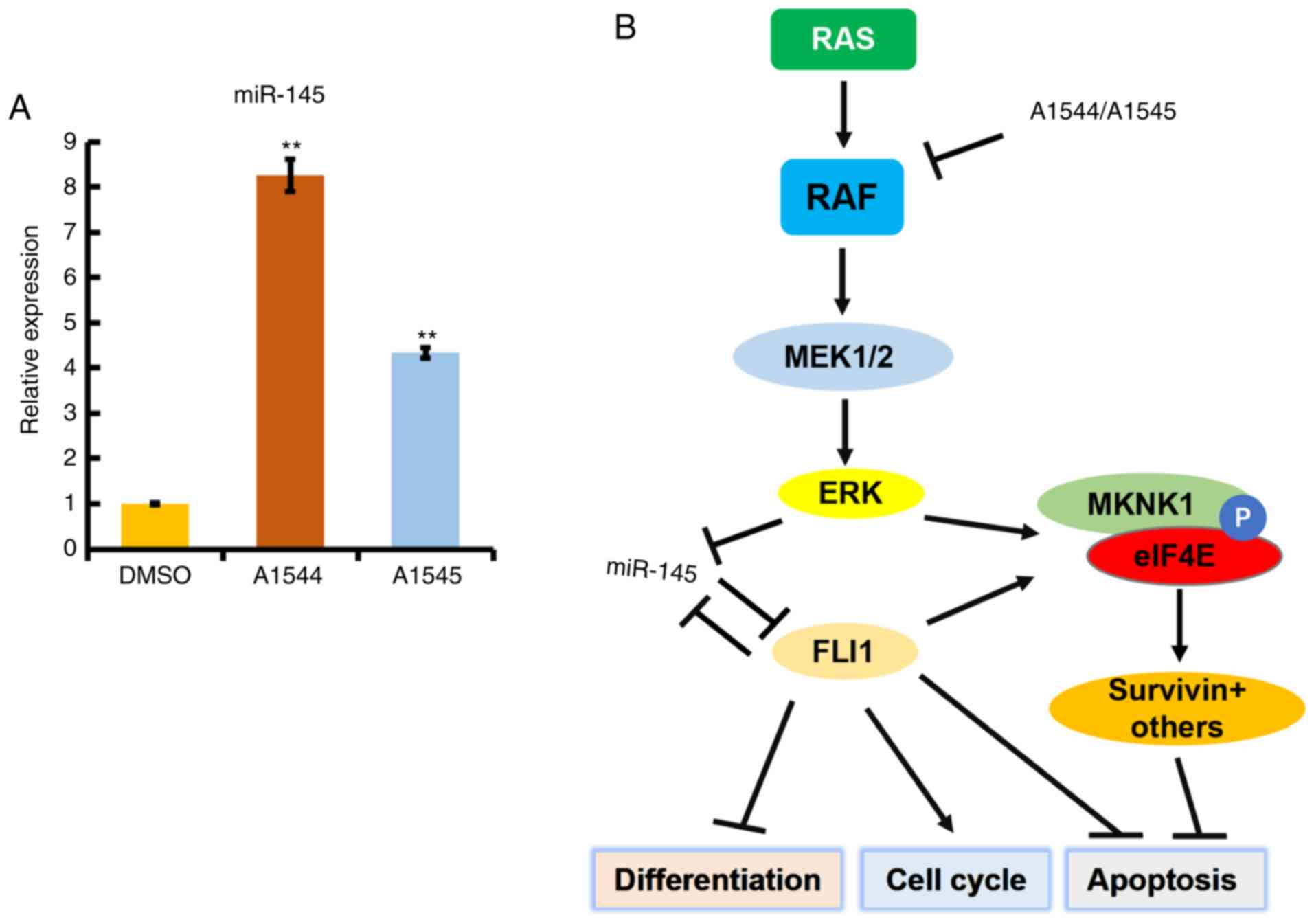 | Figure 6Anti-FLI1 compounds inhibit FLI1
expression through the upregulation of miR-145. (A) Treatment of
HEL cells with the indicated drugs (3 µM) resulted in
upregulation of miR145, as detected by RT-qPCR.
**P<0.005. (B) A model of FLI1 regulation and its
downstream effectors. The inhibition of RAF by anti-FLI1 compounds
A1544 and A1545 resulted in the suppression of MAPK/ERK, as well as
FLI1 protein expression. MAPK/ERK inhibition by the compounds
upregulate miR-145 whose expression negatively regulates FLI1
protein expression. FLI1 downregulation, as well as suppression of
MAPK/ERK, both then reduce expression of MKNK1, the phosphorylation
of eIF4E and downstreamsurvivin. FLI1 downregulation by
A1544 and A1545 compounds alters the expression of various target
genes associated with the induction of differentiation, cell cycle
and apoptosis, as previously described (15). FLI1, friend leukemia integration 1;
MKNK, mitogen-activated protein kinase (MAPK)-interacting
serine/threonine kinase; MAPK, mitogen-activated protein kinase;
ERK, extracellular signal-regulated kinase. |
Discussion
FLI1 has emerged as a potent oncogene in the
development of various types of cancer through the regulation of
multiple downstream pathways. Although protein translation has been
implicated in FLI1 tumorigenesis, the underlying mechanisms have
not yet been elucidated. To the best of our knowledge, this study
identified for the first time the translation initiator regulator
gene MKNK1 as a target of FLI1. This study demonstrates that
FLI1 binds a specific site in the Mknk1 promoter to activate
its transcription. This in turn confers proliferative advantages to
leukemic cells. Survivin (BIRC5) was identified as a
downstream target of the MKNK1/eIF4E pathway, the expression of
which is known to be involved in cell survival and proliferation
(31). These data further
emphasize the importance of targeting FLI1 or its downstream
genes for cancer therapy.
The overexpression of FLI1 in both human and murine
cells clearly resulted in a higher MKNK1 expression. While a
canonical FLI1 binding site was identified in the vicinity of the
transcription start site in the mouse Mknk1 promoter, the
location of this binding site in humans remains to be determined in
future studies. However, in human leukemic cells, the
downregulation of FLI1 by shRNA, drugs or its overexpression,
resulted in the significant reduction or increase in the expression
of MKNK1, respectively. These results strongly suggest a direct
regulation of the Mknk1 gene by FLI1 in both mice and
humans.
In gene knockout experiments, both Mknk1 and
Mknk2 have been found to be dispensable for normal
development, although higher levels of both genes in various cancer
cells are associated with growth properties (10,11).
While MKNK2 is not a direct target of FLI1, the loss or
overexpression of MKNK1 appears to have small or marginal
effect on the expression of MKNK2, respectively. Moreover,
the inhibition of MKNK1 expression by either a FLI1
inhibitor (22) or siRNA
significantly suppressed leukemic cell proliferation. These results
demonstrate that targeting MKNK1/eIF4E or its upstream regulators,
such as the MAPK pathway and FLI1, may be a potent approach for
cancer therapy, particularly for cancers which are driven by
FLI1.
A previous study by the authors demonstrated the
suppression of phospho-eIF4E by anti-FLI1 compounds through both
the inhibition of MAPK/ERK and the downregulation of FLI1
(22). MYC and cyclin D1
(CCND1) are two genes reported to be regulated by
MKNK1/eIF4E in multiple myelomas (22,24)
However, the findings of this study clearly revealed that the
expression of MYC was not altered through the suppression of
MKNK1/eIF4E activity. However, BIRC5 protein expression, but not
its transcription, was found to be downregulated when MKNK1
expression was suppressed either by anti-FLI1 compounds or siRNA;
or upregulated when FLI1 was overexpressed in leukemic cells.
Survivin is a member of the inhibitor of apoptosis (IAP) family of
proteins that play a critical role in suppressing apoptosis and
regulating the cell cycle (31).
Due to these properties, survivin has been identified as an
important target for cancer therapy (32). The identification of other genes
that are directly regulated by the MKNK1/eIF4E pathway will further
increase our understanding of tumor progression by FLI1 and
MKNK1.
In conclusion, in this study, MKNK1 was
identified as a downstream target of FLI1, which can alter
the translation of specific downstream targets, leading to tumor
progression. Survivin was identified as a potential target of
MKNK1; however, the existence of other factors that further
orchestrate protein synthesis and cancer progression by FLI1 is
also possible and thus further studies are warranted to fully
investigate this matter.
Supplementary Data
Funding
This study was supported by the Science and
Technology Department of Guizhou Province innovation and project
grants (6012-4001), research grants from the Thousand Talent
Program of China (WQ20135200171), the 100 Leading Talents of
Guizhou Province and The Natural National Science Foundation of
China (21867009 and U1812403) to YBD.
Availability of data and materials
All data and materials are available without
restriction. Researchers can obtain data by contacting the
corresponding authors.
Authors’ contributions
CW, WL, YY, BG, XH, EZ and JS contributed to the
conception, design of the study, as well as data acquisition and
interpretation. PK and KMS were involved in data and statistical
analysis. CW drafted the manuscript. YBD, XH, PK, EZ and JS
reviewed the manuscript critically. YBD supervised and also
conceived and designed the study. All authors contributed to the
interpretation of the findings, and reviewed, edited and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
MAPK
|
mitogen-activated protein kinase
|
|
FLI1
|
friend leukemia integration 1
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
eIF4E
|
eukaryotic initiation factor 4E
|
|
FMR1
|
fragile X mental retardation
protein
|
|
MKNK
|
mitogen-activated protein kinase
(MAPK)-interacting serine/threonine kinase
|
|
ChIP
|
chromatin immunoprecipitation
|
Acknowledgments
Not applicable.
References
|
1
|
Bhat M, Robichaud N, Hulea L, Sonenberg N,
Pelletier J and Topisirovic I: Targeting the translation machinery
in cancer. Nat Rev Drug Discov. 14:261–278. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chu J and Pelletier J: Therapeutic
opportunities in eukaryotic translation. Cold Spring Harb Perspect
Biol. 10:pp. a0329952018, View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Waskiewicz AJ, Johnson JC, Penn B,
Mahalingam M, Kimball SR and Cooper JA: Phosphorylation of the
cap-binding protein eukaryotic translation initiation factor 4E by
protein kinase Mnk1 in vivo. Mol Cell Biol. 19:1871–1880. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waskiewicz AJ, Flynn A, Proud CG and
Cooper JA: Mitogen-activated protein kinases activate the
serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16:1909–1920. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pyronnet S, Imataka H, Gingras AC,
Fukunaga R, Hunter T and Sonenberg N: Human eukaryotic translation
initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E.
EMBO J. 18:270–279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siddiqui N and Sonenberg N: Signalling to
eIF4E in cancer. Biochem Soc Trans. 43:763–772. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Napoli I, Mercaldo V, Boyl PP, Eleuteri B,
Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, et
al: The fragile X syndrome protein represses activity-dependent
translation through CYFIP1, a new 4E-BP. Cell. 134:1042–1054. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jauch R, Jäkel S, Netter C, Schreiter K,
Aicher B, Jäckle H and Wahl MC: Crystal structures of the Mnk2
kinase domain reveal an inhibitory conformation and a zinc binding
site. Structure. 13:1559–1568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jauch R, Cho MK, Jäkel S, Netter C,
Schreiter K, Aicher B, Zweckstetter M, Jäckle H and Wahl MC:
Mitogen-activated protein kinases interacting kinases are
autoinhibited by a reprogrammed activation segment. EMBO J.
25:4020–4032. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ueda T, Watanabe-Fukunaga R, Fukuyama H,
Nagata S and Fukunaga R: Mnk2 and Mnk1 are essential for
constitutive and inducible phosphorylation of eukaryotic initiation
factor 4E but not for cell growth or development. Mol Cell Biol.
24:6539–6549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Diab S, Kumarasiri M, Yu M, Teo T, Proud
C, Milne R and Wang S: MAP kinase-interacting kinases-emerging
targets against cancer. Chem Biol. 21:441–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ben-David Y and Bernstein A: Friend
virus-induced erythro-leukemia and the multistage nature of cancer.
Cell. 66:831–834. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ben-David Y, Giddens EB and Bernstein A:
Identification and mapping of a common proviral integration site
Fli-1 in erythro-leukemia cells induced by Friend murine leukemia
virus. Proc Natl Acad Sci USA. 87:1332–1336. 1990. View Article : Google Scholar
|
|
14
|
Ben-David Y, Giddens EB, Letwin K and
Bernstein A: Erythroleukemia induction by Friend murine leukemia
virus: Insertional activation of a new member of the ets gene
family, Fli-1, closely linked to c-ets-1. Genes Dev. 5:908–918.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Luo H, Liu T, Zacksenhaus E and
Ben-David Y: The ets transcription factor Fli-1 in development,
cancer and disease. Oncogene. 34:2022–2031. 2015. View Article : Google Scholar
|
|
16
|
Lou N, Lennard Richard ML, Yu J, Kindy M
and Zhang XK: The Fli-1 transcription factor is a critical
regulator for controlling the expression of chemokine C-X-C motif
ligand 2 (CXCL2). Mol Immunol. 81:59–66. 2017. View Article : Google Scholar
|
|
17
|
Sato S and Zhang XK: The Friend leukaemia
virus integration 1 (Fli-1) transcription factor affects lupus
nephritis development by regulating inflammatory cell infiltration
into the kidney. Clin Exp Immunol. 177:102–109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu T, Yao Y, Zhang G, Wang Y, Deng B,
Song J, Li X, Han F, Xiao X, Yang J, et al: A screen for Fli-1
transcriptional modulators identifies PKC agonists that induce
erythroid to megakaryocytic differentiation and suppress
leukemogenesis. Oncotarget. 8:16728–16743. 2017.PubMed/NCBI
|
|
19
|
Cui JW, Vecchiarelli-Federico LM, Li YJ,
Wang GJ and Ben-David Y: Continuous Fli-1 expression plays an
essential role in the proliferation and survival of F-MuLV-induced
erythroleukemia and human erythroleukemia. Leukemia. 23:1311–1319.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li YJ, Zhao X, Vecchiarelli-Federico LM,
Li Y, Datti A, Cheng Y and Ben-David Y: Drug-mediated inhibition of
Fli-1 for the treatment of leukemia. Blood Cancer J. 2:e542012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu T, Xia L, Yao Y, Yan C, Fan Y,
Gajendran B, Yang J, Li YJ, Chen J, Filmus J, et al: Identification
of diterpenoid compounds that interfere with Fli-1 DNA binding to
suppress leukemogen-esis. Cell Death Dis. 10:1172019. View Article : Google Scholar
|
|
22
|
Song J, Yuan C, Yang J, Liu T, Yao Y, Xiao
X, Gajendran B, Xu D, Li YJ, Wang C, et al: Novel flavagline-like
compounds with potent Fli-1 inhibitory activity suppress diverse
types of leukemia. FEBS J. 285:4631–4645. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vecchiarelli-Federico LM, Liu T, Yao Y,
Gao Y, Li Y, Li YJ and Ben-David Y: Fli-1 overexpression in
erythroleukemic cells promotes erythroid de-differentiation while
Spi-1/PU.1 exerts the opposite effect. Int J Oncol. 51:456–466.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Benedetti A and Graff JR: eIF-4E
expression and its role in malignancies and metastases. Oncogene.
23:3189–3199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cong XL and Han ZC: Survivin and leukemia.
Int J Hematol. 80:232–238. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ban J, Jug G, Mestdagh P, Schwentner R,
Kauer M, Aryee DN, Schaefer KL, Nakatani F, Scotlandi K, Reiter M,
et al: Hsa-mir-145 is the top EWS-FLI1-repressed microRNA involved
in a positive feedback loop in Ewing’s sarcoma. Oncogene.
30:2173–2180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu P, Liang J, Yu F, Zhou Z, Tang J and Li
K: miR-145 promotes osteosarcoma growth by reducing expression of
the transcription factor friend leukemia virus integration 1.
Oncotarget. 7:42241–42251. 2016.PubMed/NCBI
|
|
28
|
Zhang J, Guo H, Zhang H, Wang H, Qian G,
Fan X, Hoffman AR, Hu JF and Ge S: Putative tumor suppressor
miR-145 inhibits colon cancer cell growth by targeting oncogene
Friend leukemia virus integration 1 gene. Cancer. 117:86–95. 2011.
View Article : Google Scholar
|
|
29
|
Larsson E, Fredlund Fuchs P, Heldin J,
Barkefors I, Bondjers C, Genové G, Arrondel C, Gerwins P, Kurschat
C, Schermer B, et al: Discovery of microvascular miRNAs using
public gene expression data: miR-145 is expressed in pericytes and
is a regulator of Fli1. Genome Med. 1:1082009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang S, Liu JC, Ju Y, Pellecchia G, Voisin
V, Wang DY, Leha LR, Ben-David Y, Bader GD and Zacksenhaus E:
microRNA-143/145 loss induces Ras signaling to promote aggressive
Pten-deficient basal-like breast cancer. JCI Insight. 2:933132017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wheatley SP and Altieri DC: Survivin at a
glance. J Cell Sci. 132:jcs2238262019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garg H, Suri P, Gupta JC, Talwar GP and
Dubey S: Survivin: A unique target for tumor therapy. Cancer Cell
Int. 16:492016. View Article : Google Scholar : PubMed/NCBI
|