Introduction
The PTEN gene is a tumor suppressor that negatively
regulates the phosphoinositol-3-kinase/AKT survival pathway
(1). PTEN is important in
sustaining cell homeostasis and genomic stability, and its
inactivation almost always leads to cancer (2-4).
Recently, PTEN has been proposed to conform to the continuum model
of tumor suppression (5). Rather
than following discrete and stepwise changes in PTEN allele loss as
described in the two-hit hypothesis, increasing evidence supports
dosage-dependency of PTEN tumor suppressor function. A tight
correlation between PTEN expression level and function has been
demonstrated using a series of targeted PTEN alleles which resulted
in varying levels of PTEN expression in mice. Data from these
studies show that a subtle 20% reduction in PTEN protein levels may
already lead to deregulated signaling and subsequent development of
sporadic tumors, albeit to a lesser extent compared to mice with
heterozygous knockout of PTEN (6).
However, further downregulation may be required for the disease to
progress (7).
Quasi-insufficiency of PTEN can be caused by rare
mutations, promoter hypermethylation and antisense regulation.
Additionally, post-transcriptional regulation by oncogenic
microRNAs (miRNAs/miRs) has been shown to subtly decrease PTEN
levels by binding to the 3′untranslated region (3′UTR) (8,9). In
colorectal cancer, which has a low rate of PTEN coding region
mutations, dysregulation by miRNAs appears to be the main cause of
PTEN inactivation, accounting for ~70% of cases (4). While it may seem unlikely that these
miRNAs alone are able to downregulate PTEN levels enough to allow
cancer progression, studies have shown that these miRNAs can
simultaneously modulate the levels of transcription factors in the
nucleus, directly or indirectly, to augment downregulation of their
target proteins (10,11). Furthermore, individual miRNAs can
simultaneously downregulate several tumor suppressor mRNA targets,
leading to a single phenotypic readout (12-15).
As certain miRNAs only require 6-8 bp of perfect complementarity,
they can bind to multiple and otherwise unrelated transcripts and
form competing endogenous RNA (ceRNA) networks (16). Thus, a physiologically relevant
PTEN miRNA should be able to negatively regulate a ceRNA transcript
or network that sustains PTEN transcript levels.
Extensive investigation of the PTEN ceRNA network
has revealed transcripts that can act as potent PTEN derepressors
(17-19). The majority of these ceRNAs have
not been functionally linked to PTEN mRNA before, but are able to
act as molecular sponges that can sequester and inhibit shared
miRNAs from inactivating PTEN (16). These PTEN ceRNAs included PTEN1P,
TNRC6B, VCAN and ZEB2, all of which were able to rescue PTEN
expression upon overexpression of either their transcripts or their
isolated 3′UTR fragments (17-19).
The functional characterization of ceRNAs as derepressors of the
PTEN transcript has helped clarify the poorly-understood crosstalk
mediated by the 3′UTR of the mRNA, and the specific nuances that
PTEN expression levels manifest in cancer, either via their
synergistic or antagonistic interactions with other
PTEN-inactivating mechanisms (18).
The present study aimed to identify other PTEN
ceRNAs that may modulate its tumor suppressive function and
validate the functional relevance of the putative ceRNAs.
Materials and methods
Cell line maintenance and
transfection
HCT116 colorectal carcinoma cells were sourced from
the American Type Culture Collection. HCT116 cells were cultured in
Roswell Park Memorial Institute 1640 (RPMI-1640) medium
supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo
Fisher Scientific, Inc.), 50 U/ml penicillin/streptomycin and 2 g/l
sodium bicarbonate. All transfection experiments were performed
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer’s instructions. The
amount of plasmid and volume of Lipofectamine 2000 was optimized to
achieve 70-80% transfection efficiency for all functional and
molecular characterization experiments. A ratio of 1 ng plasmid per
100 cells was used to transfect cells that were seeded 24 h
earlier. Transfection efficiency for the overexpression constructs
was assessed through parallel transfection with pmR-ZsGreen1 empty
vector (Clontech Laboratories, Inc.) and fluorescence imaging,
while knockdown efficiency of siRNA experiments was assessed via
RT-qPCR. miRNA overexpression set-ups included pmR-ZsGreen1 empty
vector control and pmR-ZsGreen1-miR4465, whereas 3′UTR
overexpression set-ups included pmiRGLO empty vector control, and
the different 3′UTRs cloned into the pmiRGLO vector, namely:
pmiRGLO-DNMT3B, pmiRGLO-TET3, and pmiRGLO-PTEN. Daily transfection
of the siRNAs using their optimized concentrations were performed
for the knockdown experiments. Cells were maintained in a 37°C
humidified incubator with 5% CO2.
Antibodies and plasmid constructs
The rabbit polyclonal anti-PTEN (cat. no. PA5-27304;
1:1,000) and rabbit poly-clonal anti-TET methylcytosine dioxygenase
3 (TET3; cat. no. PA5-31860; 1:1,000) antibodies were from
Invitrogen (Thermo Fisher Scientific, Inc.). The mouse monoclonal
anti-GAPDH (cat. no. CB1001; 1:1,500) antibody was from EMD
Millipore. The rabbit monoclonal anti-DNA methyl-transferase 3β
(DNMT3B; cat. no. 67259; 1:1,000) antibody was from Cell Signaling
Technology, Inc. The goat anti-rabbit IgG (1:5,000; cat. no. 31460)
and goat anti-mouse IgG (H+L) (1:10,000; cat. no. 31430) secondary
antibodies conjugated with horseradish peroxidase were obtained
from Invitrogen (Thermo Fisher Scientific, Inc.).
The 3′UTR of human PTEN (NM_001304717), DNMT3B
(NM_006892.3), TET3 (NM_001287491.1) and the pre-miR-4465 gene
(NR_039675.1) were amplified by PCR from human genomic DNA using
primers listed in Table I. The
wild-type 3′UTR fragments were cloned into the pmirGLO
dual-luciferase miRNA target expression vector (Promega
Corporation), downstream of the firefly luciferase reporter. A
mutant version of the 3′UTRs was generated by site-directed
mutagenesis of the wild-type 3′UTRs in pmirGLO, in which a TG>AC
substitution within the miR-4465 seed sequence binding site
(5′-ACTACAA-3′) was introduced through
splicing-by-overlap-extension PCR using the primers listed in
Table I.
 | Table IPrimers used for generation and
site-directed mutagenesis of 3′UTR constructs. |
Table I
Primers used for generation and
site-directed mutagenesis of 3′UTR constructs.
| Primer | Primer sequence
(5′→3′) |
|---|
| DNMT3B-3′UTR-F |
ATTAAGGAGCTCGCACAAATCAGACCTGGCTGCTTG |
|
DNMT3B-3′UTR-Fmut |
TTCTCCTAAAACTTTAAAACTACAAGTAGGTAGCAACGTGGC |
| DNMT3B-3′UTR-R |
GTTCTAGAGCTCCCCAGAGGGTTCTATTG |
|
DNMT3B-3′UTR-Rmut |
GCCACGTTGCTACCTACTTGTAGTTTTAAAGTTTTAGGAG |
| TET3-3′UTR-F |
CATGGCGAGCTCTAACAATCAGATGACCGCTATAGG |
|
TET3-3′UTR-Fmut |
GCCTAGATTTAAACAGCAACTACAAAAAAAAAGTATGTTTTAAC |
| TET3-3′UTR-R |
ACTTATTCTAGACCGTGGACAGGGCACACTGGGGAG |
|
TET3-3′UTR-Rmut |
GTTAAAACATACTTTTTTTTTGTAGTTGCTGTTTAAATCTAGGC |
| PTEN-3′UTR-F |
AAGAGGGATAAAACACCATGAAAATAAACTACAATAAACTGAAAATG |
|
PTEN-3′UTR-Fmut |
TTAACTGTTAGGGAATTTTACTACAATACTGAATACATATAATG |
| PTEN-3′UTR-R |
GCGGAGCTCGAGCACATGTAGGACAATTTCTACTG |
|
PTEN-3′UTR-Rmut |
CATTATATGTATTCAGTATTGTAGTAAAATTCCCTAACAGTTAA |
The miR-4465 precursor primers were designed to
amplify a genomic DNA fragment that includes 206 bp upstream and
226 bp downstream of the pri-miR-4465 sequence. The amplicon was
cloned into the mammalian miRNA expression vector pmR-ZsGreen1
(Clontech Laboratories, Inc.).
The PCR reaction mixture contained a final
concentration of 1X Titanium® Taq PCR buffer (Clontech
Laboratories, Inc.), 0.125 µM of each deoxynucleoside
triphosphate (dNTP) (Promega Corporation), 2 µM of each
forward and reverse primers, 1X Titanium® Taq polymerase
(Clontech Laboratories, Inc.) and human genomic DNA template
extracted from HK-2 cells (ATCC® CRL-2190™) available at
the Disease Molecular Biology and Epigenetics Laboratory of the
National Institute of Molecular Biology and Biotechnology (Quezon
City, Philippines). The PCR program used was as follows: Initial
denaturation at 95°C for 5 min, followed by 25-30 cycles of
denaturation at 95°C for 30 sec, annealing at 55-65°C for 30 sec,
and extension at 72°C for 30-60 sec, with a final extension step at
72°C for 10 min. All plasmid constructs were verified by
sequencing.
RT-qPCR
HCT116 cells were seeded at a density of 200,000
cells/well in a 12-well plate and transfected after 24 h with the
appropriate amount of pmiRGLO constructs, pmRZsGreen1 constructs,
siRNA and target protectors using Lipofectamine 2000. Cells were
harvested 48 h post-transfection and total RNA was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). For first strand cDNA synthesis, 2 µg of total RNA
was mixed with 1 µl of 50 µg/µl random
hexamers, 10 µM oligo-dT and 15 µl diethyl
pyrocarbonate (DEPC)-treated sterile distilled deionized
H2O (sddH2O). This was incubated in a
thermocycler for 5 min at 70°C then immediately placed on ice. The
following were then added to make up a total volume of 25
µl: 5 µl 5X M-MLV RT buffer, 1.5 µl 10 mM
dNTPs, 0.625 µl 40 U/µl RNAsin ribonuclease
inhibitor, 1.0 µl M-MLV RT (200 U/µl; Promega
Corporation) and 1.875 µl DEPC-treated sddH2O.
The first strand cDNA samples were then used as template for
RT-qPCR.
PowerUp™ SYBR® Select Master mix
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for RT-qPCR
following the manufacturer’s protocol. For statistical analysis of
gene expression, triplicate reactions were prepared per template
per primer pair. Adjustment of the baseline and threshold values
determined the threshold cycles for the amplification curves.
Relative quantification was done with the use of generated standard
curves per gene target. Target gene expression was normalized
against cyclophilin expression, and fold-change expression relative
to the untransfected control/empty vector control was calculated
(20). Primers used for RT-qPCR
are summarized in Table II.
| Table IIRT-qPCR primers used for detecting
DNMT3B, TET3 and PTEN mRNA. |
Table II
RT-qPCR primers used for detecting
DNMT3B, TET3 and PTEN mRNA.
| Primer | Sequence
(5′→3′) |
|---|
| DNMT3B-qRT-F |
GAATTACTCACGCCCCAAGGA |
| DNMT3B-qRT-R |
ACCGTGAGATGTCCCTCTTGTC |
| PTEN-qRT-F |
CGTATTCTGTAGGTAATCTCTG |
| PTEN-qRT-R |
GGTGATGCCTTCAAGTAAC |
| TET3-qRT-F |
CAGAAGGAGAAGCTGAGCAC |
| TET3-qRT-R |
AGCACCGAGTAGCTCTCCAC |
| Cyclophilin
A-F |
CCTAAAGCATACGGGTCCTGGCATC |
| Cyclophilin
A-R |
GTGGAGGGGTGCTCTCCTGAGCTAC |
Absolute quantification using Quantigene
analysis
Quantigene miRNA Assay (Affymetrix; Thermo Fisher
Scientific, Inc.) was used for absolute quantification of miR-4465
in HCT116 cells. Since there is no required RNA extraction and cDNA
synthesis step, miRNA quantification is more reliable compared with
RT-qPCR using TaqMan probes due to minimal loss of RNA yield. This
branched DNA signal amplification technology is a
hybridization-based assay that relies on using miRNA-specific
probes for normalization.
Functionally validated pre-designed probes with high
sensitivity and specificity for miR-4465 were obtained from
eBioscience (Affymetrix; Thermo Fisher Scientific, Inc.). The
miR-4465 probe (5′-CUCAAGUAGUCUGACCAGGGGA-3′) was determined
bioinformatically by Affymetrix to be specific to human samples and
to have low cross-reactivity towards closely related miRNA family
members.
Cultured cells were lysed to release the miRNAs to
be stabilized by overnight hybridization in a 96-well plate with
capture extenders, label extenders and target-specific capture
probes from the Quantigene miRNA assay kit. Pre-amplifier molecules
were added to allow hybridization to each pair of gene-specific
probe sets for signal amplification. Hybridization of amplifier
molecules with pre-amplifiers was performed prior to probing with
alkaline phosphatase-conjugated oligo-nucleotides. Generation of a
luminescent signal proportional to the amount of target miRNA
present in each sample was achieved upon addition of a
chemilumigenic substrate to each well. Luminescent signal was
detected using the FLUOstar Omega microplate reader (BMG Labtech
GmbH). For statistical analysis, triplicates of cell lysates per
set-up were used to calculate assay precision. For absolute
quantitation, a standard curve was generated for the samples of
known miRNA concentrations, and this was used as reference to
calculate the miRNA concentration for each sample.
siRNA design and optimization
Pre-designed siRNA oligonucleotides directed against
all transcript variants of human PTEN, DNMT3B and TET3 were
obtained from Qiagen (Qiagen Sciences, Inc.) as follows: PTEN,
Product ID Hs_TET3_2, GeneGlobe cat. no. SI05784268, target
sequence: CTC GCG CGG CAT GGT ATG AAA; DNMT3B, Product ID Hs
DNMT3B_2, GeneGlobe cat. no. SI00092967, target sequence: AAG GAC
TAC TTT GCA TGT GAA; TET3, Product ID Hs_PTEN_6, GeneGlobe cat. no.
SI00301504, target sequence: AAG GCG TAT ACA GGA ACA ATA. The
siRNAs were designed to specifically target their corresponding
transcripts. Designed with asymmetrical 5′base pair stabilities,
these double-stranded oligos ensure entry of less tightly-bound
antisense strand to the RNA-induced silencing complex while
reducing risk of off-target effects by degrading the sense strand,
which can be incorrectly processed by the RNA-induced silencing
complex.
To optimize the amount of siRNA to be used for
transfections, different amounts of gene-specific siRNA or the
AllStars negative control siRNA (cat. no. 1027281; Qiagen Sciences,
Inc.) were tested. Based on the recommended optimal concentration
for knockdown, which is 5-30 nM, the concentrations of 5, 15, 30
and 60 nM were used. RNA extraction with TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.) of siRNA-trans-fected cells was performed
according to the manufacturer's protocol, and the generated
corresponding cDNA was used for RT-qPCR to validate the efficacy of
the siRNAs in decreasing individual DNMT3B, TET3 and PTEN
transcript levels. The optimum concentration for each siRNA was 30
nM.
Dual-luciferase reporter assay
HCT116 cells were seeded at a density of 10,000
cells/well in a 96-well plate and transfected after 24 h. For the
endogenous dual-luciferase assays, cells were transfected with 200
ng of empty pmirGLO vector (Promega Corporation),
pmirGLO-PTEN-3′UTR-wild type (wt)- or mutant (mut) 4465,
pmirGLO-DNMT3B-3′UTR-wt/mut4465 and pmirGLO-TET3-3′UTR-wt/mut4465.
For the miRNA co-transfection dual-luciferase assays, cells were
co-transfected with 20 ng empty pmirGLO vector,
pmirGLO-PTEN-3′UTR-wt/mut4465, pmirGLO-DNMT3B-3′UTR-wt/mut4465 or
pmirGLO-TET3-3′UTR-wt/mut4465 together with 200 ng empty
pmR-ZsGreen1 vector (Clontech Laboratories, Inc.) or
pmR-ZsGreen-miR-4465 construct. The construct preparations were
complexed with Lipofectamine 2000 for transfection of cells in
triplicate wells. Luciferase activity was measured 48 h
post-transfection using the Dual-Luciferase® Reporter
Assay System (Promega Corporation) and the FLUOstar Omega
microplate reader (BMG Labtech GmbH). The supernatant of the
centrifuged lysates was sequentially added with LARII (Luc2
substrate) and Stop and GLO reagents (Rluc substrate) with a 12-sec
timeframe of a continuous read. Fast kinetics function of the
multi-mode reader was utilized to continuously detect luminescence.
Degree of miRNA regulation was calculated by dividing the sum of
the RLUs of the Luc2 interval (0.5 to 12 sec) with that of the Rluc
interval (12.5 to 24 sec). Data are expressed as the mean values of
normalized firefly relative luciferase units (RLUs) per set-up; raw
firefly RLUs were normalized against the internal control
Renilla luciferase RLUs in each well prior to normalizing
against the empty vector control.
Target protection experiments
Target protector oligonucleotides specific to the
respective miR-4465 binding site and flanking sequences within the
3′UTR of the PTEN, DNMT3B or TET3 3′UTRs were co-transfected with
pmR-ZsGreen1-miR-4465 into HCT116 cells. The target protectors were
designed using the Qiagen miRNA Target Protector design tool
(https://www.qiagen.com/ph/shop/genes-and-pathways/custom-products/custom-assay-products/custom-mirna-products/#target-protector)
with the RefSeq ID of PTEN transcript variant 1 (NM_000268), DNMT3B
transcript variant 1 (NM_006892.3) or TET3 transcript variant 1
(NM_001287491.1) as reference templates. Selection of the 40-bp
context sequences was performed by including 20-bp sequences
flanking the respective miRNA binding site in the 3′UTRs of the
target MREs. An effective concentration of 200 nM miR-4465 target
protector co-transfected with ZsG-miR-4465 was determined through
preliminary transfection optimization experiments. Target protector
efficiency for miRNA inhibition was measured using dual luciferase
assays or RT-qPCR from transfected cells vs. control setups.
Western blotting
Seeding and transfection of HCT116 cells in 12-well
plates were performed as described for RT-qPCR. Total protein was
extracted from cells 48 h post-transfection using
radioimmunoprecipitation lysis buffer (150 mM NaCl, 0.5% sodium
deoxycholate, 0.1% SDS and 50 mM Tris pH 8.0) supplemented with
protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 5 mM EDTA
and 10 µM E64; Roche Diagnostics GmbH). Lysates were
centrifuged at 10,000 × g for 20 min at 4°C and transferred to
fresh tubes. Total protein concentration was measured using the
bicinchoninic acid assay. Protein extracts were snap-frozen in
liquid nitrogen and stored at -80°C until use. For polyacrylamide
gel electrophoresis, 30 µg of total protein per setup was
loaded into 4-15% Mini-PROTEAN® TGX Stain-Free™ Protein
Gels (Bio-Rad Laboratories, Inc.). Semi-dry electroblot transfer of
proteins onto polyvinylidene fluoride membranes was done at 2.5 mA,
25 V, 10 min using the Trans-Blot® Turbo Blotting system
(Bio-Rad Laboratories, Inc.). Blocking of membranes was performed
using 5% w/v whey protein in Tris-buffered saline with 0.1% v/v
Tween-20 (TBST) at room temperature for 1 h. Blots were probed
overnight at 4°C with the primary antibodies described above and
washed with TBST. This was followed by incubation with the
appropriate secondary antibody conjugated with horseradish
peroxidase (HRP) for 1 h at room temperature. Bands were visualized
with enhanced chemiluminescence (ECL) substrate and imaged using
the ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Inc.).
Densitometric analysis of digitized band intensities was performed
using GelQuant.NET (v1.8.2). Quantified PTEN, DNMT3B,
or TET3 gene expression was normalized against GAPDH.
Caspase 3/7 assay
HCT116 cells were seeded at a density of 100,000
cells/well in a 24-well plate and were transfected at 24 h
post-seeding. After 48 h, the cells were reseeded into a 96-well
plate. For the assay proper, trypsinized transfected cells were
seeded in two sets of triplicates corresponding to the uninduced
and induced groups per transfection set-up. Cells were treated with
5 mM sodium butyrate and incubated further for 16-20 h for the
induction of apoptosis. Following the addition of 10 ml Caspase 3/7
reagent (Promega Corporation) per well, the plate was mixed by
shaking at 300 rpm for 30 sec and incubated for 30 min at room
temperature. Luminescence was measured using a multi-mode plate
reader (FLUOstar Omega; BMG Labtech GmbH), and statistical analysis
was performed by comparing the relative luminescent units of the
induced and uninduced groups in each set-up.
Wound healing assay
HCT116 cells were seeded at 100,000 cells/well in a
96-well plate. At 24 h, 250 ng pmR-ZsGreen1 empty vector was
co-transfected with either the pmiRGLO construct or the desired
siRNA using Lipofectamine 2000 to assess the transfection
efficiency through GFP fluorescence. Upon reaching >95%
confluence at 24 h post-transfection, a thin artificial wound was
introduced to the cell monolayer using a sterile pipette tip. Cells
were washed once with 1X phosphate buffered saline (PBS) and
maintained in the appropriate media supplemented with 10 or 4%
fetal bovine serum (FBS). Wound closure was monitored at 0 and 16 h
post-scratch by capturing three fields of view per set-up at ×4
magnification using the IN Cell Analyzer 6000 High-Content Imaging
system (GE Healthcare Life Sciences). Percent open wound area was
analyzed using the TScratch software (http://www.cse-lab.ethz.ch/software/) and reported as
the mean % open wound area per set-up relative to % open wound area
upon initial scratching (t=0 h). The migration rate was calculated
as follows: [(Percentage open wound area (t=0)-(Percentage open
wound area (t=16)]/16 h.
Low serum (4%) to full serum (10%) conditions were
used in would healing assays following miRNA sequestration
experiments to allow the demonstration of the tumor suppressive
effects of PTEN as a result of its derepression. Total serum
deprivation was not performed, as no effects of oncogenic signaling
needed to be decoupled from mitogenic stimulation. Cell migration
was monitored only up to 16 h before the known doubling time of
HCT116 cells of ~21 h.
In silico analysis
CEFINDER (https://www.oncomir.umn.edu/cefinder/), a tool that
identifies mRNAs sharing miRNA binding sites, was used to rank
putative PTEN ceRNAs based on shared PTEN miRNA binding sites in
their 3′UTR. TargetScanHuman v.7.1 (http://www.targetscan.org/vert_71/) was used to
identify miRNAs that can regulate PTEN and its ceRNAs. RNAfold
(http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi)
was used to assess structural changes in the 3′UTR upon
introduction of a mutation to abrogate a miR-4465 binding site.
RNAhybrid (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid)
was used to assess miRNA affinity with 3′UTR sequence by
calculating the free energy of binding.
Statistical analysis
For statistical analysis, unpaired two-tailed
Student's t-test to measure differences between two set-ups was
performed. One-way ANOVA with post hoc Tukey's test was used to
test significant differences between multiple set-ups. Data from
all quantitative experiments are presented as the mean ± standard
deviation (SD). P<0.05 was considered to indicate a
statistically significant difference.
Results
In silico identification of novel PTEN
ceRNAs and miR-4465 as a shared microRNA
Initial bioinformatics analysis using CEFINDER
identified TET3, which is a demethylase, and DNMT3B, a de
novo methyltransferase, as two of the top scoring hits. TET3
ranked second to the validated PTEN ceRNA TNRC6B, which exhibited
the highest number of shared miRNA binding sites with PTEN. PTEN
shares 21 miRNA binding sites with TET3 and 5 with DNMT3B (Fig. 1A). By contrast, TET1, TET2 and
DNMT3A only share 7, 2 and 2 miRNA binding sites with PTEN,
respectively. DNMT3L was not included in the analysis as it had no
known regulatory miRNAs based on the analysis on TargetScanHuman
v.7.1. These findings were further qualified by mapping the shared
miRNAs among DNMT3B, TET3 and PTEN that were broadly conserved
across all vertebrates. The results demonstrated that PTEN may
theoretically share 32 miRNA binding sites with TET3 and 11 with
DNMT3B. DNMT3B and TET3 may share 11 miRNA binding sites, whereas
all three transcripts may share 6 miRNA binding sites (Fig. 1B).
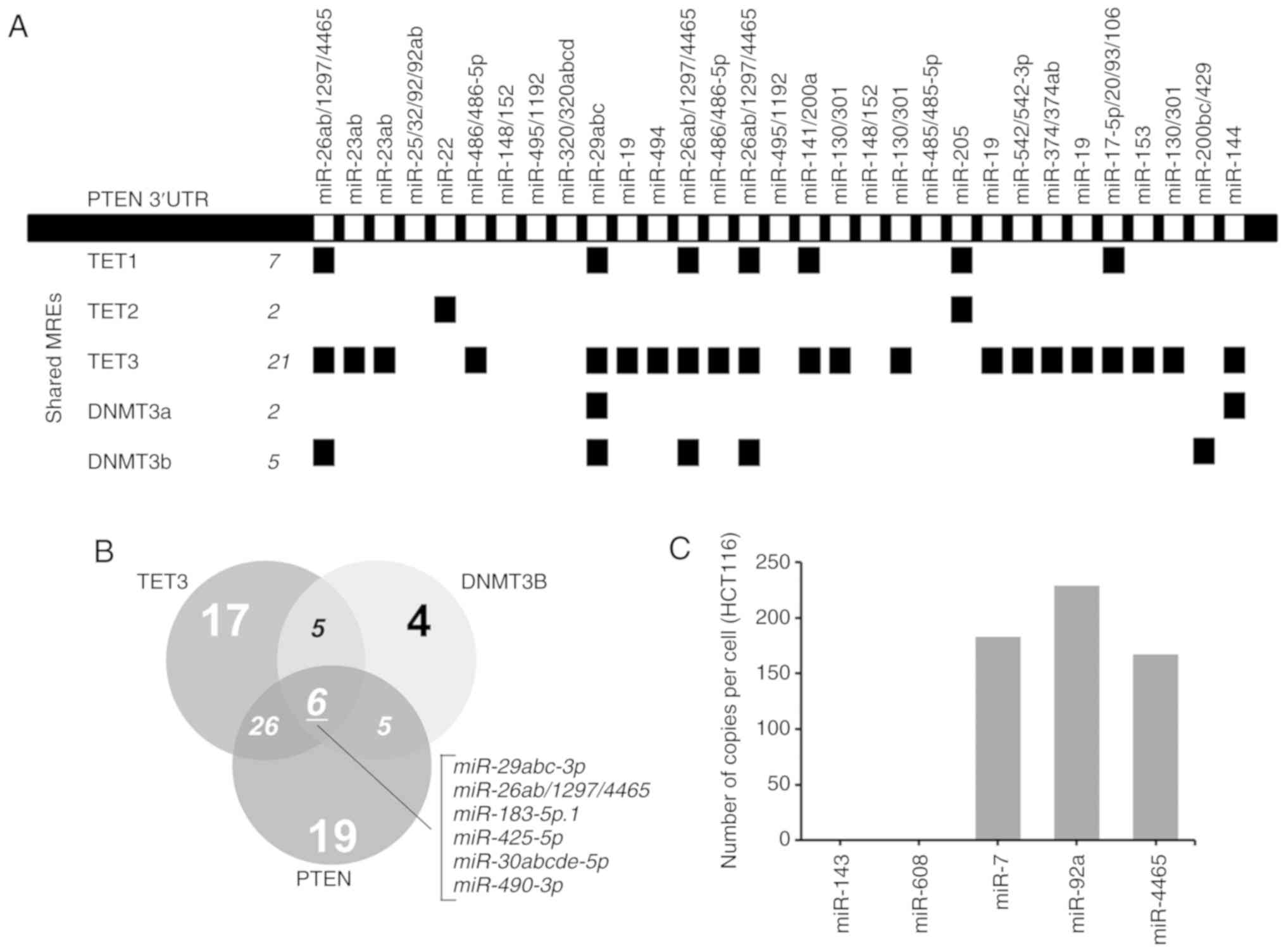 | Figure 1PTEN, TET3 and DNMT3B 3′UTRs share
multiple miRNAs. (A) Linear map of PTEN 3′UTR demonstrating the
MREs; miRNAs shared with TET1, TET2, TET3, DNMT3a and DNMT3b are
indicated below. The map was derived from ceRDB using CEFINDER. (B)
Venn diagram of the theoretical number of miRNAs shared among PTEN,
TET3 and DNMT3b 3′UTRs determined by TargetScanHuman v.7.1. (C)
Number of copies of miR-4465 per cell in HCT116 cells compared with
miR-143, miR-408, miR-7 and miR-92a. The expression levels of the
mature miRNAs were measured using the Quantigene miRNA assay.
miRNA, miR, microRNA; MREs, miRNA response elements; UTR,
untranslated region; TET, TET methylcytosine dioxygenase; DNMT3,
DNA methyltransferase 3. |
To validate the result that DNMT3B, TET3 and PTEN
form a ceRNA network, a miRNA able to negatively regulate all three
genes via their 3′UTRs needed to be identified. CEFINDER identified
only two miRNA families that could bind to all three genes,
miR-29-abc-3p and miR-26ab-5p/1297/4465. When these were
cross-referenced against the 6 shared miRNAs proposed by
TargetScanHuman v.7.1, they were also identified as the top two
miRNA families predicted to negatively regulate the three genes.
Upon further examination of the relative context scores (a metric
for predicted efficacy of repression) of the miRNAs among the three
genes, the miR-26ab-5p/1297/4465 family had the highest context
score for PTEN, but miR-29abc-3p had the highest context scores for
DNMT3B and TET3. In addition, the miR26ab-5p/1297/4465 family had a
high context score for both PTEN and TET3 and a relatively low
context score for DNMT3B. miR-4465 was chosen since it is the only
miRNA in this family the role of which in cancer has not been
functionally character-ized. PTEN and TET3 have four miR-4465
binding sites (according to TargetScanHuman v.7.1; only three
according to CEFINDER), whereas DNMT3B only has one. The results of
the bioinformatics analysis of miR-4465 binding to TET3, PTEN and
DNMT3B 3′UTRs are summarized in Table III.
| Table IIIFeatures of mir-4465 binding onto
DNMT3B, TET3 and PTEN 3′UTRs in terms of number of binding sites,
total context score and validation from previous TargetScan
publications. |
Table III
Features of mir-4465 binding onto
DNMT3B, TET3 and PTEN 3′UTRs in terms of number of binding sites,
total context score and validation from previous TargetScan
publications.
| Authors, year | Gene | No. of MREs within
3′UTR | MRE positions | Cumulative weighted
context scorea | TargetScan
publication(s) |
|---|
| Garcia et
al., 2011 | PTEN | 4 | 41-47, 1261-1268,
2619-2626, 3800-3807 | -0.76 | (47) |
| Garcia et
al., 2011; | TET3 | 4 | 1958-1964,
4662-4668, 5570-5577, 5607-5714 | -0.48 | (47,48) |
| Friedman et
al., 2009 | | | | | |
| Garcia et
al., 2011 | DNMT3B | 1 | 265-271 | -0.09 | (47) |
Previous studies have confirmed the expression of
PTEN (21), DNMT3B (22) and TET3 (23) in HCT116 cells. The expression level
of miR-4465 in HCT116 cells, however, has not been reported. The
results of the Quantigene assays in the present study demonstrated
that miR-4465 was present in HCT116 cells at 150-250 copies/cell,
which is comparable to the oncogenic miRNAs miR-7 and miR-92a
(Fig. 1C).
miR-4465 negatively regulates DNMT3B,
TET3 and PTEN via their 3′UTRs
To confirm whether miR-4465 may regulate DNMT3B,
TET3 and PTEN individually, precursor miR-4465 was cloned into the
miRNA expression vector pmR-ZsGreen1, and fragments of the 3′UTRs
of the target genes into the miRNA target expression vector
pmirGLO, downstream of a luciferase reporter. Cloned DNMT3B 3′UTR
contained one miR-4465 binding site (265-271 bp), cloned TET3 3′UTR
contained one (1,958-1,964 bp), whereas cloned PTEN 3′UTR contained
two binding sites (41-47 and 1,261-1,268 bp). Quantigene analysis
confirmed the overexpression of mature miR-4465 in HCT116 cells
transfected with the pmR-ZsGreen1-miR-4465 construct (Fig. S1). Individual overexpression of
DNMT3B, TET3 and PTEN 3′UTRs led to a decrease in the FLuc/Rluc
ratio in luciferase assays compared with the pmirGLO empty vector
set-up (Fig. 2A). These results
suggested the negative endogenous regulation of the three genes in
HCT116 cells. To implicate miR-4465 in the observed downregulation,
pmRZs-Green1-miR-4465 was co-transfected with the individual
pmirGLO constructs containing DNMT3B, TET3 and PTEN 3′UTR
fragments. Dual luciferase reporter assay revealed that miR-4465
negatively regulated DNMT3B, TET3 and PTEN 3′UTRs in a
dose-dependent manner (Fig.
2B).
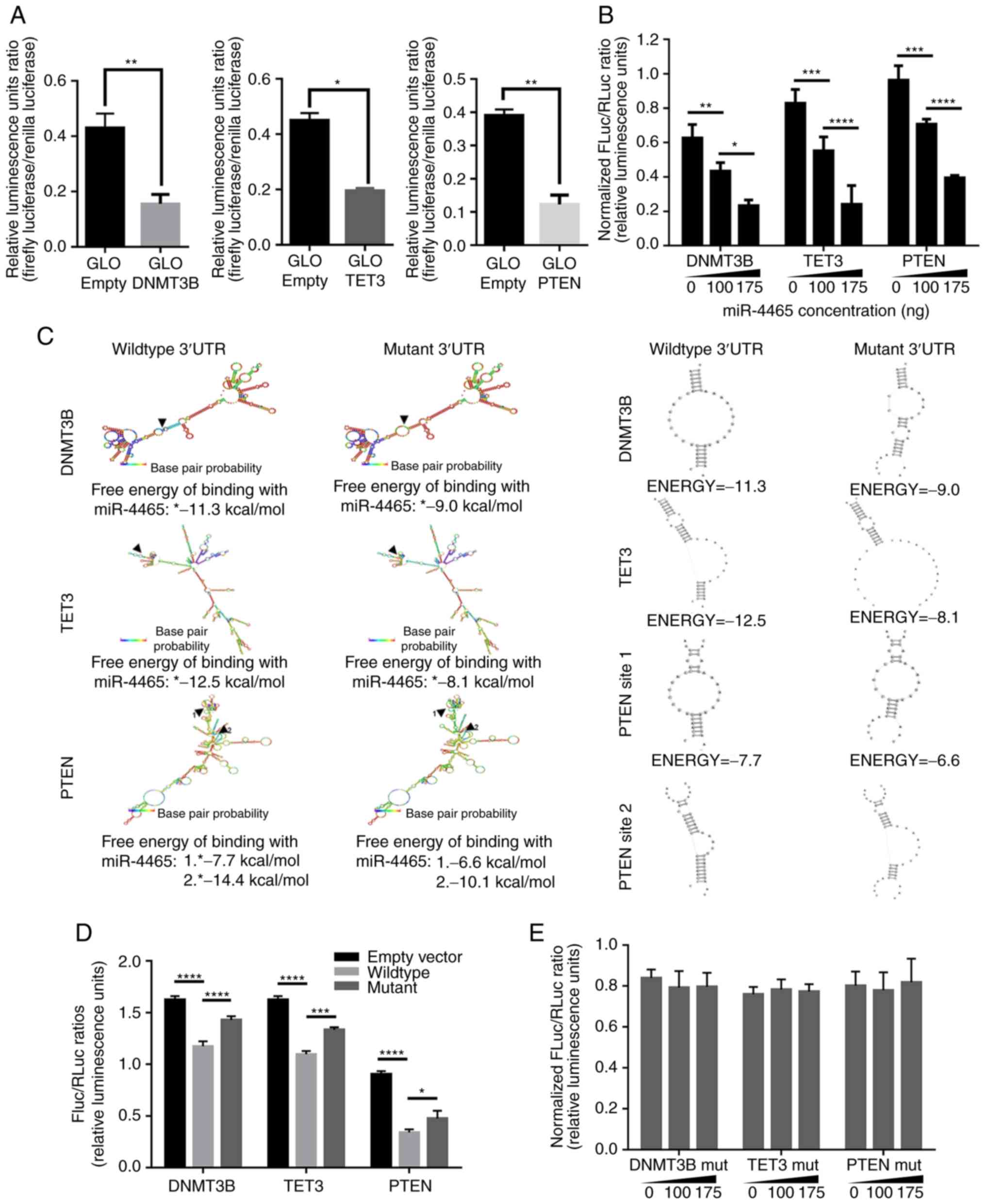 | Figure 2PTEN, TET3 and DNMT3B 3′UTRs are
negatively regulated by the shared miRNA-4465. (A) Dual-luciferase
assays of HCT1116 cells transfected with an empty vector or
pmirGLO-wt DNMT3B-, wt TET3- and wt PTEN-3′UTR demonstrated
endogenous downregulation of the transcripts. (B) Dual-luciferase
assay results of the dose-dependent downregulation exerted by
miR-4465 on DNMT3B, TET3 and PTEN 3′UTRs in HCT116 cells. (C)
Secondary structure analyses of DNMT3B, TET3 and PTEN 3′UTRs, and
changes in the free energy of binding of miR-4465 to its targets
following site-directed mutagenesis of the seed sequence
(5′-ACTTGAA-3′) with a TG>AC substitution. (D) Dual-luciferase
assay results of HCT116 cells transfected with wt or miR-4465 MRE
mutant DNMT3B-, TET3- and PTEN-3′UTRs demonstrated endogenous
derepression of the mutant transcripts. (E) Dual-luciferase assay
results on the abrogation of dose-dependent downregulation exerted
by miR-4465 in HTC116 cells co-transfected with miR-4465 MRE mutant
3′UTR constructs. All experiments were performed in cells
maintained in 10% serum. Data are representative of three
independent trials and expressed as the mean ±
SD.*P<0.05, **P<0.01, ***P<0.001 and
****P<0.0001. UTR, untranslated region; MRE, microRNA
response element; DNMT3B, DNA methyltransferase 3β; TET3, TET
methylcytosine dioxygenase 3; empty, cells transfected with an
empty pmirGLO vector; miR, microRNA; MRE, microRNA response
element; wt, wild-type. |
To further demonstrate that miR-4465 may directly
bind to the 3′UTRs of DNMT3B, TET3 and PTEN, each miR-4465 binding
site seed sequence (5′-ACTTGAA-3′) in the cloned 3′UTR
fragments was disrupted via site-directed mutagenesis with a
TG->AC substitution. In silico analysis revealed that
these mutations changed the secondary structure of each of the
3′UTRs only minimally, while making miR-4465 free energy of binding
less negative (Fig. 2C), which was
indicative of weaker pairing between the miRNA and the target. Dual
luciferase assays using cells transfected with the mut 3′UTRs
exhibited an increase in Fluc/Rluc ratio compared with the wt 3′UTR
controls (Fig. 2D). These results
suggested that the destruction of the binding site partially
derepressed the 3′UTRs from endogenous negative regulation due to
the loss of binding by miR-4465. In addition, no dose-dependent
negative regulation by miR-4465 was observed when the mutant 3′UTRs
were used for the dual luciferase assays (Fig. 2E). These results confirmed that
miR-4465 contributed to the negative regulation of DNMT3B, TET3 and
PTEN by directly binding to their 3′UTRs, since mutating the
binding site abrogated the observed dose-dependent negative
regulation exerted by miR-4465 on the three genes.
DNMT3B, TET3 and PTEN are endogenous
targets of miR-4465
To determine whether miR-4465 acted on its putative
endogenous targets, the effects of its overexpression on DNMT3B,
TET3 and PTEN transcript and protein levels in HCT116 were examined
using RT-qPCR and western blot analyses, respectively. TET3 and
PTEN transcript levels decreased, whereas DNMT3B transcript levels
were unchanged in cells overexpressing miR-4465 compared with cells
transfected with an empty vector (Fig.
3A). A decrease in the protein levels of all three genes was
also observed following miR-4465 overexpression (Fig. 3B and C). These results suggested
that miR-4465 may negatively regulate the expression of endogenous
TET3 and PTEN via transcript degradation, and DNMT3B via
translational repression.
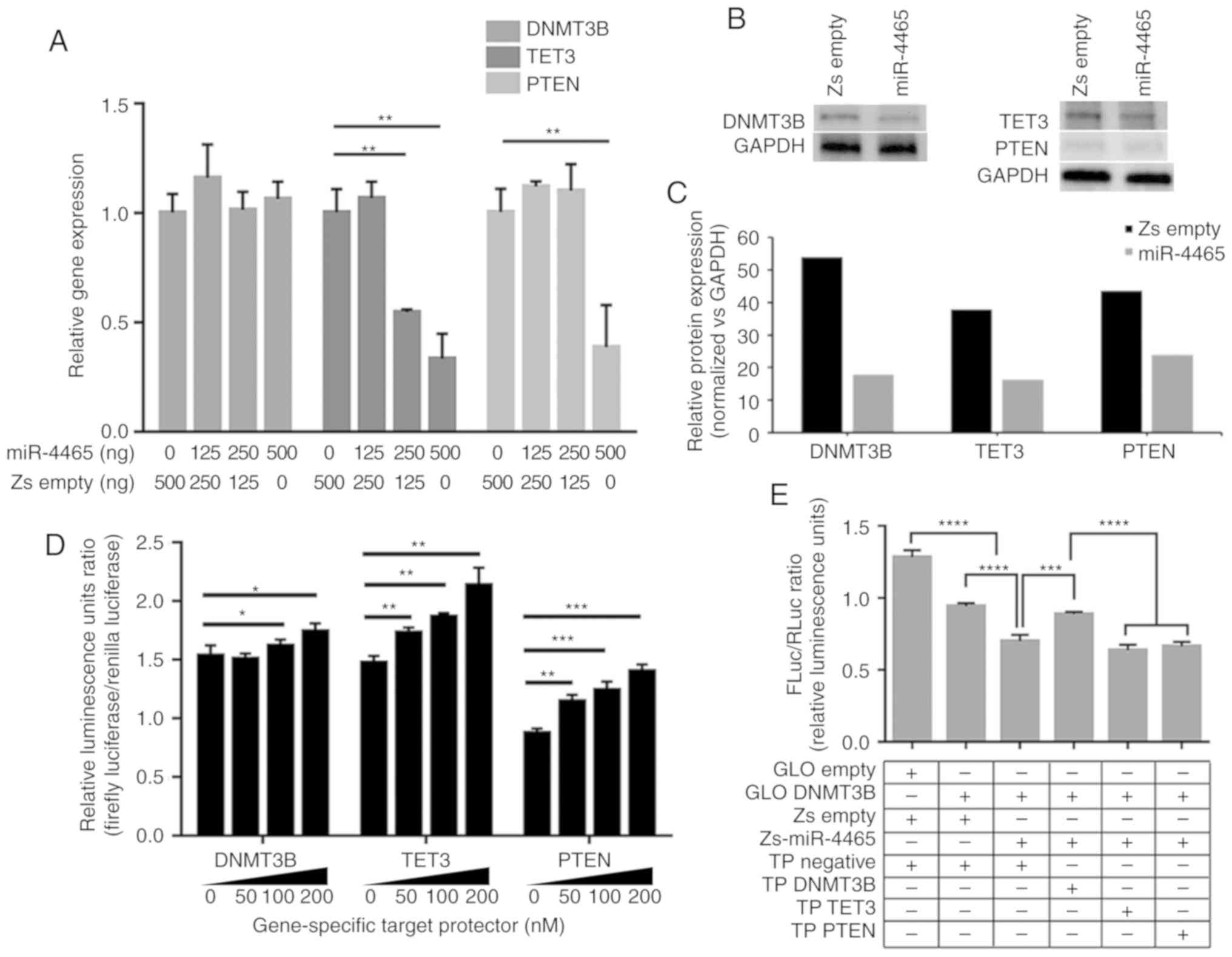 | Figure 3miR-4465 negatively regulates the
expression of endogenous DNMT3B, TET3 and PTEN. (A) RT-qPCR
demonstrated the effects of increasing amounts of transfected
miR-4465 on the endogenous transcript levels of DNMT3B, TET3 and
PTEN. (B) Western blot analyses demonstrated the effects of
miR-4465 overexpression on endogenous protein levels of DNMT4B,
TET3 and PTEN. (C) Relative quantification of the protein
expression levels of DNMT3B, TET3 and PTEN in western blots
normalized to GAPDH (n=5). (D) Dual-luciferase assays demonstrated
derepression of endogenous DNMT3B, TET3 and PTEN following
co-transfection with increasing amounts of gene-specific TPs. (E-G)
Dual-luciferase assays demonstrated direct targeting of (E)
DNMT3B-, (F) TET3- and (G) PTEN-3′UTR by miR-4465 and rescue by
gene-specific target protectors. (H) RT-qPCR demonstrated the
effects of transfected miR-4465 and co-transfected gene-specific
target TPs on the endogenous transcript levels of DNMT3B, TET3 and
PTEN. All experiments were performed in cells maintained in 10%
serum. Data presented are representative of three independent
trials and expressed as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. miR, microRNA; UTR, untranslated
region; GLO Empty, empty pmirGLO vector; Zs Empty, empty
pmR-ZsGreen1 vector; TP, target protector; RT-qPCR, reverse
transcription-quantitative PCR; DNMT3B, DNA methyltransferase 3β;
TET3, TET methylcytosine dioxygenase 3. |
To confirm that the observed endogenous
downregulation was due to direct binding of miR-4465 to its target
mRNAs, target protectors (TPs) specific to the miR-4465 binding
site of each 3′UTR were used in subsequent transfections. Unlike
anti-miRs, TPs are locked nucleic acids that can block the
interaction of a miRNA with a single target, but leaves other
targets of the same miRNA in other transcripts unaffected. Dual
luciferase assay results demonstrated derepression of the three
3′UTRs from endogenous miRNA binding upon transfection of their
respective TPs in a dose-dependent manner (Fig. 3D). In addition, co-transfection of
specific TPs with individual 3′UTR constructs and miR-4465
inhibited the repressive effect of miR-4465 (Fig. 3E-G). The target protectors were
only able to inhibit miR-4465 binding to their specific 3′UTR
targets.
Evaluation of the DNMT3B, TET3 and PTEN transcript
levels upon co-transfection of miR-4465 and the respective TPs was
performed to determine whether miR-4465 may downregulate endogenous
DNMT3B, TET3 and PTEN levels by direct binding to their 3′UTRs.
TET3 and PTEN transcripts were rescued from downregulation by
miR-4465; DNMT3B transcript levels, on the other hand, were not
affected by miR-4465 overexpression, consistent with results
presented in Fig. 3A and B,
suggesting translational repression. The DNMT3B transcript levels
remained unchanged following transfection with DNMT3B
3′UTR-specific TPs (Fig. 3H).
DNMT3B, TET3 and PTEN positively modulate
each other's expression by competitive miRNA sequestration
To demonstrate possible miRNA-mediated crosstalk
among PTEN, DNMT3B and TET3, the effects of shared miRNA
sequestration were examined via gene knockdown and 3′UTR
overexpression experiments. For knockdown experiments, 30 nM
gene-specific siRNA was used, which resulted in knockdown
efficiencies of ≥60% (Fig. S2).
Knockdown of DNMT3B led to a decrease in the transcript levels of
TET3 and PTEN, as identified by RT-qPCR (Fig. 4A). Knockdown of TET3 also led to a
decrease in the transcript levels of DNMT3B and PTEN (Fig. 4B). In addition, knockdown of PTEN
led to a decrease in the transcript levels of DNMT3B and TET3
(Fig. 4C). These results
demonstrated that in the context of HCT116 cells, knockdown of
their individual transcripts resulted in downregulated expression
of their ceRNAs.
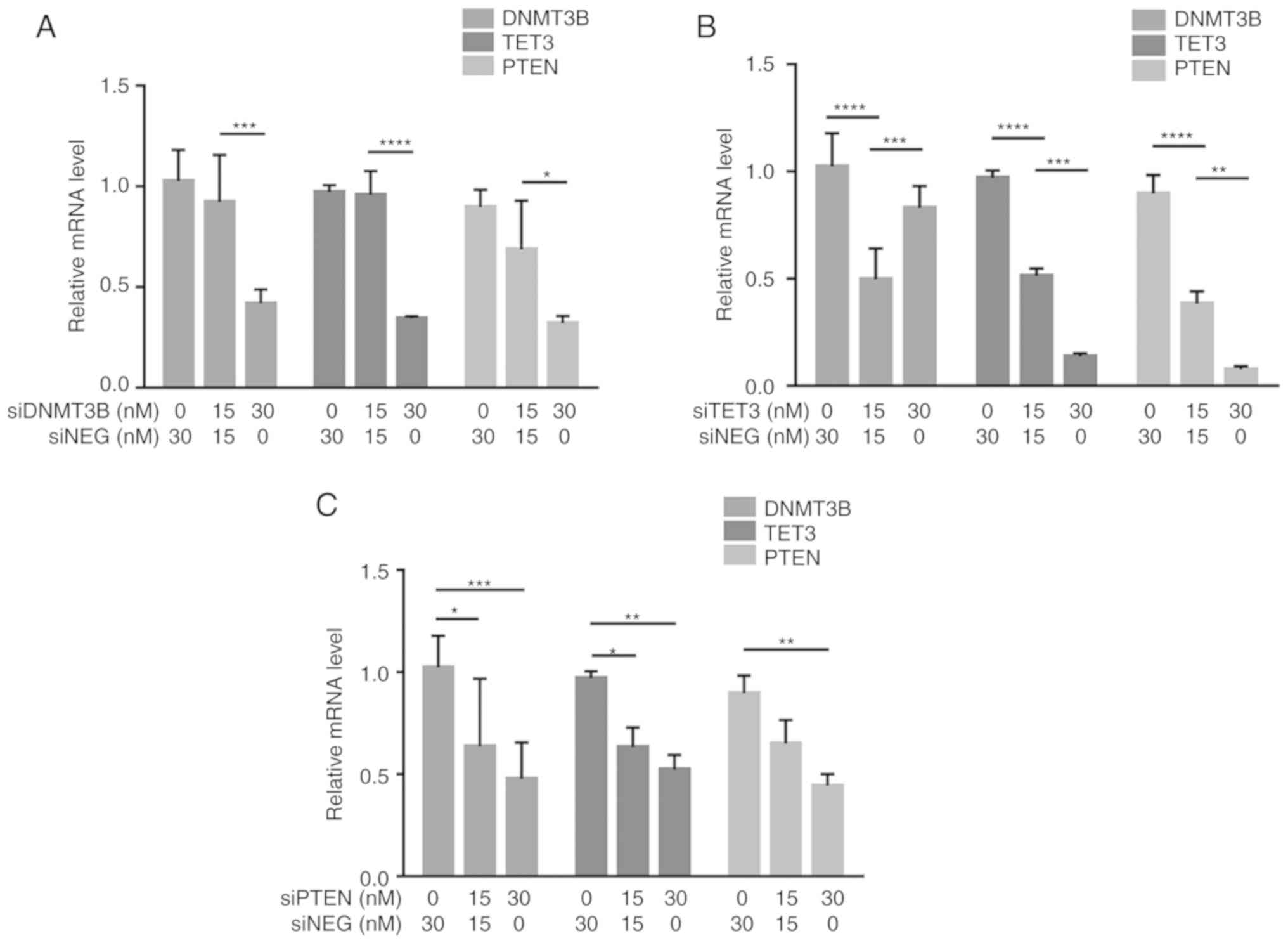 | Figure 4Crosstalk among DNMT3B, TET3 and PTEN
through shared miRNA sequestration. (A-C) RT-qPCR demonstrated the
reciprocal effects of (A) DNMT3B, (B) TET3 and (C) PTEN knockdown
on the transcript levels of their competing endogenous RNAs. (D-F)
The effects of overexpression of (D) DNMT3B- (E) TET3- and (F)
PTEN-3′UTR on the transcript levels of their competing endogenous
RNAs were also assessed by RT-qPCR. All experiments were performed
in cells maintained in 10% serum. Data are representative of three
independent trials and expressed as the mean ± SD.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. UTR,
untranslated region; GLO, pmirGLO construct; si-, small interfering
RNA; siNEG, negative control small interfering RNA; miRNA,
microRNA; DNMT3B, DNA methyltransferase 3β; TET3, TET
methylcytosine dioxygenase 3; RT-qPCR, reverse
transcription-quantitative PCR. |
To further determine if the observed downregulation
was due, at least in part, to competitive miRNA sequestration, the
3′UTR construct from each gene was overexpressed in HCT116 cells.
Overexpression of the DNMT3B 3′UTR led to an increase in the
transcript levels of TET3 and PTEN in a dose-dependent manner as
assessed by RT-qPCR (Fig. 4D).
Overexpression of the TET3 3′UTR also led to an increase in the
transcript levels of DNMT3B and PTEN compared with the control
cells (Fig. 4E). Additionally,
overexpression of the PTEN 3′UTR led to an increase in the
transcript levels of DNMT3B and TET3 (Fig. 4F). These results demonstrated that
the observed down-regulation of the endogenous competitors upon
knockdown of one gene and the converse upregulation of the
endogenous competitors when the 3′UTR of a gene is overexpressed
were at least partly miRNA-dependent and coding
region-independent.
DNMT3B and TET3 3′UTR overexpression
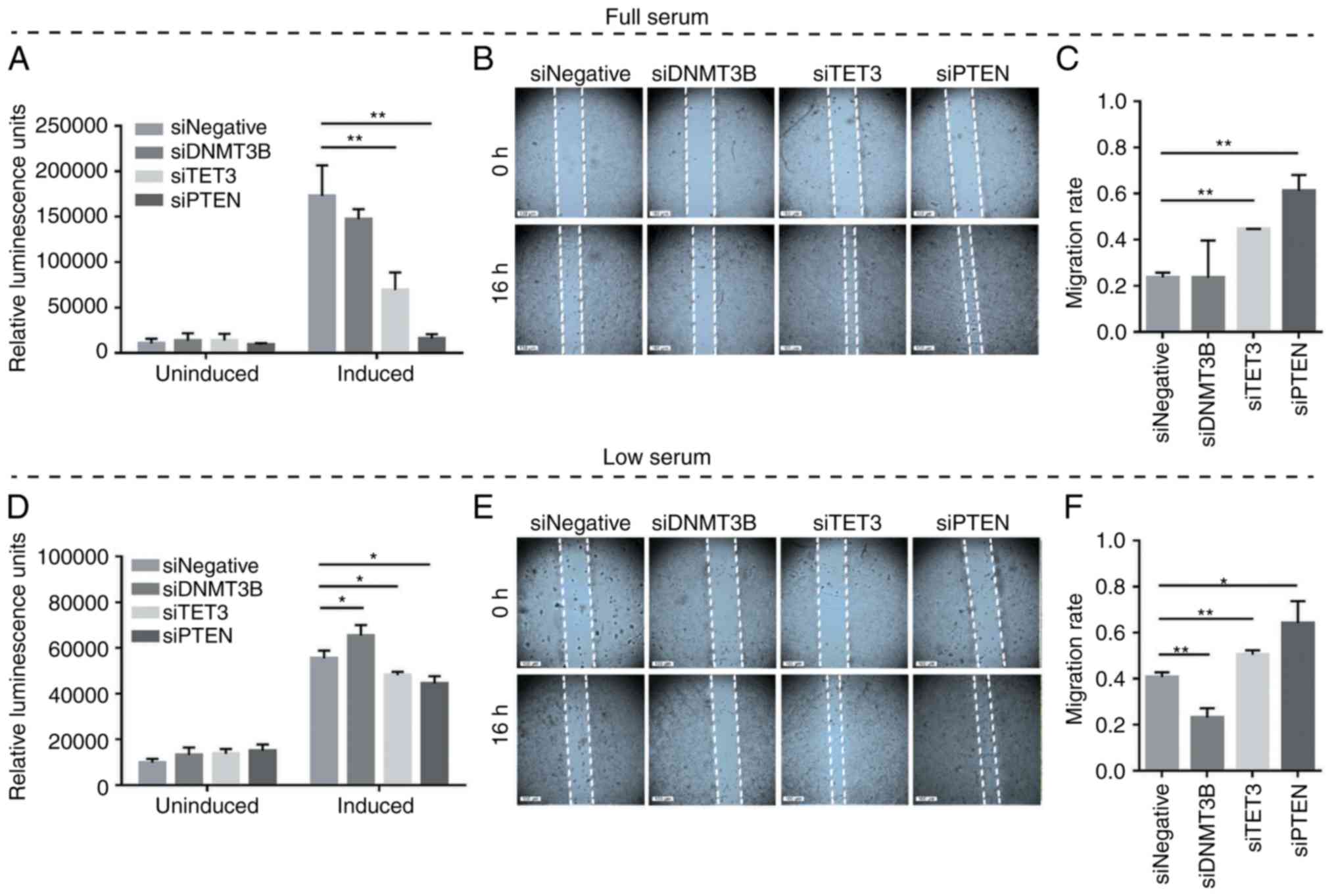
promotes apoptosis and inhibits migration
To test whether competitive miRNA sequestration and
the consequent modulation of PTEN levels may elicit phenotypic
readouts consistent with PTEN derepression, overexpression
experiments involving DNMT3B and TET3 3′UTR constructs were
performed in full-serum and low-serum conditions. Caspase-Glo 3/7
and wound healing assays were used to determine apoptosis and cell
migration, respectively. The results demonstrated the suppression
of oncogenesis by DNMT3B and TET3 3′UTRs in a coding-independent
and potentially miRNA-dependent manner, specifically by promoting
apoptosis and decreasing the migratory capacity in both full-serum
(Fig. 5A-C) and low-serum
conditions (Fig. 5D-F) compared
with the pmiRGLO empty vector control. In all migration assays, the
debris observed in the open wound areas were confirmed to be dead
cells based on fluorescence microscopy (data not shown).
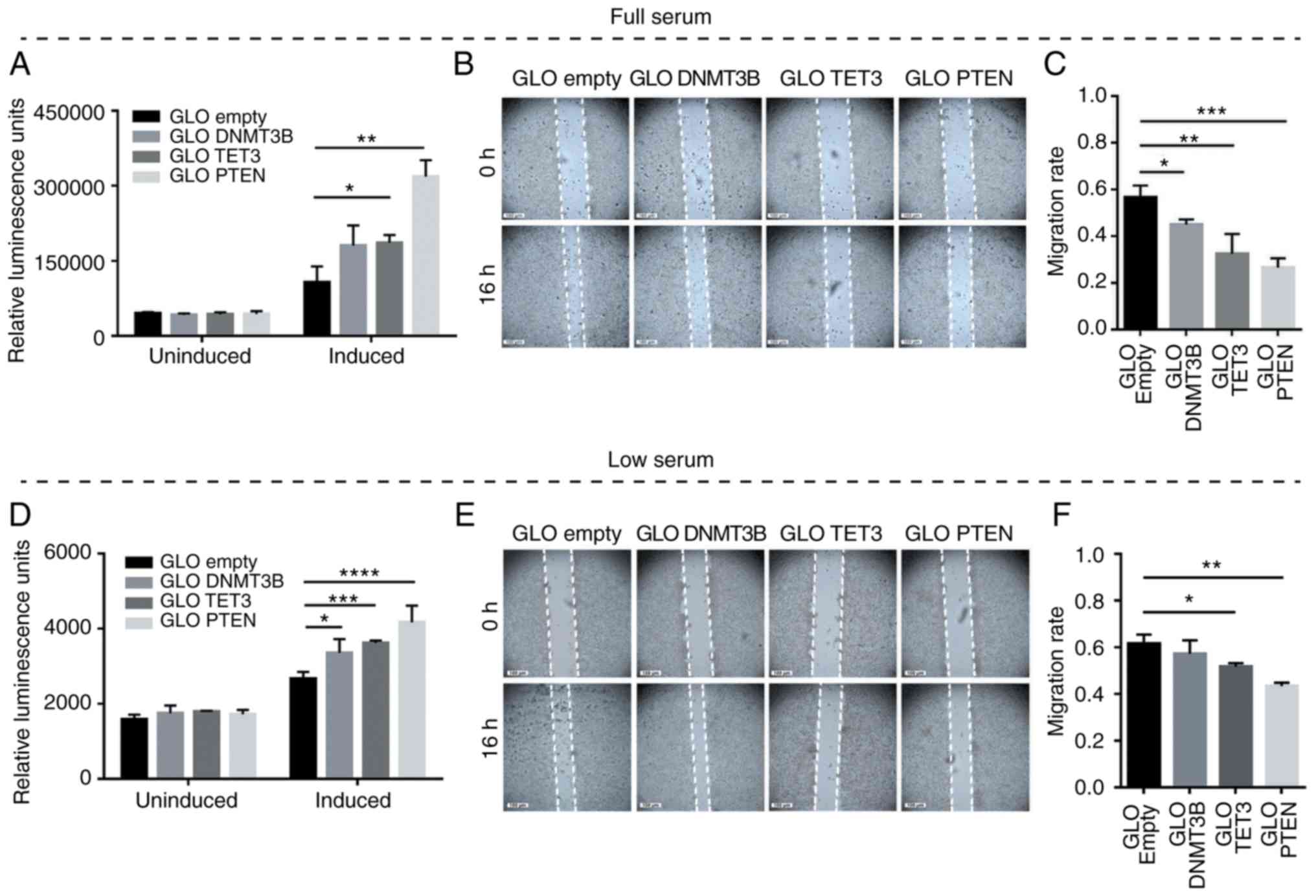 | Figure 5Effects of DNMT3B- and TET3-3′UTR
overexpression on apoptosis and cell migration in HCT116 cells. (A
and D) Caspase-Glo 3/7 and (B and E) wound healing assays were used
to monitor the effects of 3′UTR overexpression on apoptosis and
migratory capacity, respectively. (C and F) The migration rates
corresponding to (B) and (E). Set-ups A-C and D-F were performed in
full-serum (10%) and low-serum (4%) conditions, respectively. Data
are representative of three independent trials and expressed as the
mean ± SD. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. GLO, pmirGLO
construct; empty, GLO empty, pmirGLO empty vector control; DNMT3B,
DNA methyltransferase 3β; TET3, TET methylcytosine dioxygenase
3. |
Decoupling the coding-dependent from the
miRNA-dependent, coding-independent functions of TET3 and
DNMT3B
TET3 knockdown enhanced migration as well as
resistance to apoptosis in both full-serum (Fig. 6A-C) and low-serum conditions
(Fig. 6D-F) compared with the
siNEG controls. This may be the result of the abrogation of TET3
protein function as well as the redirection of shared miRNAs to
other targets such as PTEN, as demonstrated in Fig. 4B. DNMT3B knockdown, compared with
the siNEG control, exhibited pro-apoptotic and antimigratory
effects in low-serum conditions (Fig.
6D-F) and an absence of an apparent effect in full-serum
conditions (Fig. 6A-C), which may
have overridden the effects of knockdown owing to full mitogenic
stimulation. While this phenotypic readout may be attributed to the
abrogation of the DNMT3B protein function as an oncogene, the
results of DNMT3B 3′UTR overexpression (Figs. 4D and 5A-F) suggested a contribution of the
miRNA-dependent, coding-independent function of DNMT3B.
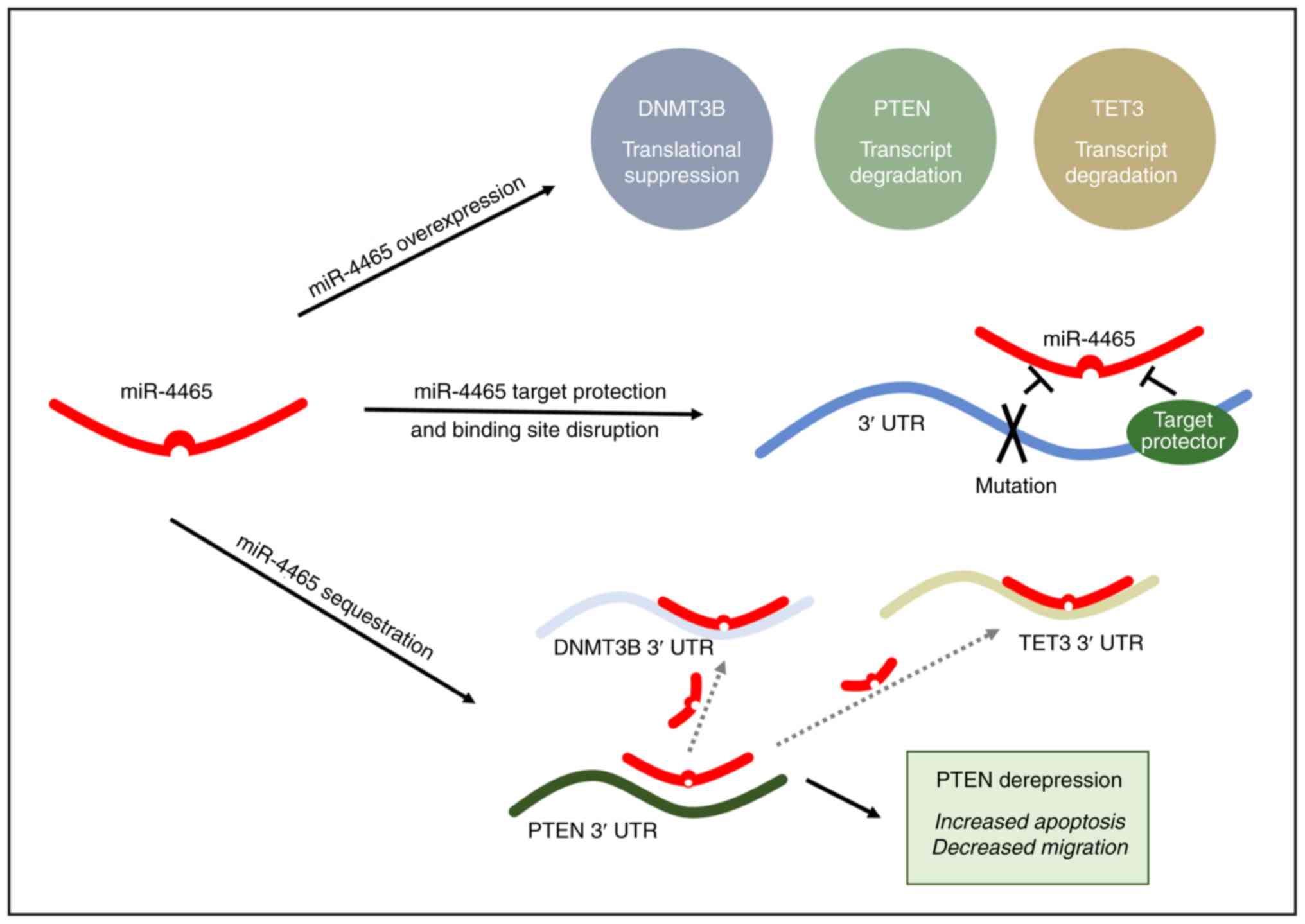
Discussion
Multiple regulatory mechanisms responsible for
downregulation of PTEN in cancer have been elucidated in the last
two decades, including negative regulation by transcription
factors, promoter hypermethylation, histone modifications,
post-translational modifications, antisense RNA and miRNA
regulation (24-29). Each one of these can have an impact
on PTEN function by affecting its abundance, catalytic activity,
protein interactions or localization (17-19).
More recently, ceRNAs have been added to this growing list of PTEN
regulators (19,30). By virtue of shared miRNA binding
sites, endogenous RNAs (mRNAs, lncRNAs and pseudogenes functioning
as molecular decoys) can sponge up miRNAs away from their primary
targets (31,32).
The in silico identification and experimental
validation of ceRNA networks in cancer are important for several
reasons: i) It has helped establish the role of miRNAs in the
regulation of oncogenes and tumor suppressors (16,31,33,34);
ii) it has revealed a novel, coding-independent and miRNA-dependent
function for endogenous mRNAs (16,19,30,31,35,36);
iii) it has assigned a molecular decoy function to pseudogenes
based on the extensive homology to and shared MREs with the
parental gene (31,37,38);
iv) it has attributed another function to 3′UTRs of mRNA targets as
trans regulatory elements that can modulate the expression
of other RNAs through the sequestration of shared miRNAs (39); v) it has led to the identification
of ceRNA hubs and higher-order ceRNA networks, as well as
cancer-specific ceRNA modules (40-43).
The present study reported the experimental
validation of two novel PTEN ceRNAs, DNMT3B and TET3, which were
identified through the MREs that they share with PTEN in their
3′UTR regions. All three transcripts have previously been
demonstrated to be subject to miRNA regulation. PTEN is regulated
by its pseudogene molecular decoy, PTENP1, via a ceRNA network in
colorectal and prostate cancer (31). In the present study, DNMT3B and
TET3 were identified as additional ceRNAs that may positively
modulate PTEN expression and enhance its tumor-suppressive
functions in HCT116 colorectal cancer cells. This is, to the best
of our knowledge, the first study to implicate both DNMT3B and TET3
in a ceRNA network. Previously, DNMT3B was only identified to be
endogenously regulated by miRNAs in breast and liver cancer
(44,45). TET3 has only been demonstrated to
be endogenously regulated by a network of miRNAs in the brain
governing epigenetic mechanisms for memory formation and
consolidation (46).
Evaluation of DNMT3B and TET3 as PTEN ceRNAs
involved a series of experiments demonstrating that these putative
ceRNAs may be regulated by shared miRNAs. miR-4465 was revealed to
directly target and regulate all three transcripts via their 3′UTRs
through a combination of luciferase reporter assays, abrogation of
miRNA binding sites, target protection with locked nucleic acids
and RT-qPCR assays. miRNA sequestration experiments including
overexpression of individual 3′UTR fragments led to derepression of
the ceRNAs. By contrast, knockdown of one of the ceRNAs through RNA
interference led to decreased expression of the other two ceRNAs.
These results suggested that indeed, PTEN, TET3 and DNMT3B may
constitute a robust ceRNA network. Of note, although overexpression
of 3′UTRs harboring miR-4465 binding sites may be largely
responsible for miRNA sequestration in the 3′UTR overexpression
experiments, knockdown by RNA interference affects the entire
length of the transcript, which harbors multiple MREs targeted by
multiple miRNAs. Thus, multiple shared miRNAs can be redirected and
sequestered by the ceRNAs.
The effects of sequestration experiments on two
cancer hallmarks, resistance to apoptosis and migratory capacity,
revealed nuances of the ceRNA crosstalk. Overexpression of DNMT3B
or TET3 3′UTR promoted apoptosis and decreased the migratory
capacity potentially, at least in part, due to the shared miRNA
sequestration and the consequent derepres-sion of PTEN. In a
physiological context, these may represent cellular conditions
where either DNMT3B or TET3 is strongly upregulated, thus providing
extra copies of the 3′UTR to sponge up miRNAs shared with PTEN.
Fig. 7 summarizes the experimental
approaches used to validate DNMT3B and TET3 as novel ceRNAs of
PTEN.
Knockdown of TET3 resulted in an increased migratory
capacity and resistance to apoptosis. The relative contribution of
shared miRNAs redirected to PTEN to this phenotypic observation
cannot easily be determined. Reduced expression of the TET3
protein, a known tumor suppressor, may have contributed to this
result. Knockdown of DNMT3B did not exhibit phenotypes indicative
of miRNA sequestration and instead promoted apoptosis and decreased
the migratory capacity. This may mean that in this context,
downregulation of DNMT3B in HCT116 colorectal cancer cells is
pheno-typically inconsequential. With fewer predicted shared MREs
between PTEN and DNMT3B, the extra pool of shared miRNA made
available by DNMT3B knockdown may not be enough to reverse the
tumor-suppressive pro-apoptotic and anti-migratory phenotypic
readout of PTEN expression. In addition, DNMT3B may participate in
other ceRNA networks, and its knockdown may dissipate its miRNA
regulators to various other target transcripts. The contribution of
the coding-dependent function of DNMT3B, however, cannot be
discounted; compared with a competitive miRNA sequestration
experiment through 3′UTR overexpression, knockdown of DNMT3B may
have simply decoupled its coding-dependent function as an oncogene
from its miRNA-dependent, coding-independent function. TET3
knockdown, on the other hand, may elicit a pro-oncogenic phenotype
by redirecting more shared miRNAs to its ceRNA PTEN, with which it
shares 32 miRNA binding sites, compared with only 11 between PTEN
and DNMT3B. However, similar to the case of DNMT3B knockdown, TET3
knockdown may have deprived the cells of a potent tumor suppressor
protein. This highlights the disadvantages of the knockdown
approach in studying potential ceRNAs; sequestration experiments
using individual 3′UTR overexpression are more instructive. The
value of using knockdown approaches lies in decoupling the
protein-coding from the coding-independent function of mRNAs. In
addition, these experiments highlight that functional relevance of
ceRNA networks is determined largely by the number of shared miRNAs
between or among ceRNAs, their relative stoichiometry and the
relative affinity of shared miRNAs with the individual targets.
Depending on these factors, redirection of shared miRNAs to
competitors may not be relevant, as demonstrated by the
dose-dependent tumor suppressive function of PTEN in the present
study. Gene regulation in cancer is very dynamic, depending on
cellular conditions at any point in time; there may or may not be
specific contexts in which ceRNAs are expected to alter phenotypic
outcomes.
In future studies, transfection experiments
involving DNMT3B and TET3 cDNA, with or without their 3′UTRs, may
help distinguish the coding-dependent and coding-independent
functions of these genes. Full-length DNMT3B and TET3
overexpression experiments, however, come with at least one caveat.
Considering the universal roles of the two genes in methylation and
demethylation, respectively, of countless oncogenes and tumor
suppressors, overexpression of their coding regions may not afford
clear-cut interpretation of results.
The existence of a ceRNA network among PTEN, DNMT3B
and TET3 may have important ramifications and merits further
investigation. With the opposing functions of DNMT3B and TET3 as a
de novo methylase and demethylase, respectively, the net
effect of miRNA-dependent competition between DNMT3B and TET3, and
among DNMT3B, TET3 and PTEN, may influence the methylation status
of the PTEN promoter, and consequently, carcinogenesis.
Supplementary Data
Funding
This work was supported by in-house funds from the
National Institute of Molecular Biology and Biotechnology,
University of the Philippines Diliman.
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RLG conceived the project and wrote the project
proposal. KARR and KMMA performed the experiments. RLG, KMMA and
KARR analyzed the data. RLG and KARR wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Ms. Marie Isabelle
Viola (Disease Molecular Biology and Epigenetics Laboratory,
National Institute of Molecular Biology and Biotechnology,
University of the Philippines Diliman) for assistance in the
preparation of figures and statistical analyses and Mr. Charles
Christopher Bataclan (Disease Molecular Biology and Epigenetics
Laboratory, National Institute of Molecular Biology and
Biotechnology, University of the Philippines Diliman) for sorting
the references.
References
|
1
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal J: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun Z, Huang C, He J, Lamb KL, Kang X, Gu
T, Shen WH and Yin Y: PTEN C-terminal deletion causes genomic
instability and tumor development. Cell Rep. 6:844–854. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Murphy SJ, Karnes RJ, Kosari F, Castellar
BE, Kipp BR, Johnson SH, Terra S, Harris FR, Halling GC, Klein JL,
et al: Integrated analysis of the genomic instability of PTEN in
clinically insignificant and significant prostate cancer. Mod
Pathol. 29:143–156. 2016. View Article : Google Scholar
|
|
4
|
Molinari F and Frattini M: Functions and
regulation of the PTEN gene in colorectal cancer. Front Oncol.
3:3262014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berger AH, Knudson AG and Pandolfi PP: A
continuum model for tumour suppression. Nature. 476:163–169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alimonti A, Carracedo A, Clohessy JG,
Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ,
Brogi E, et al: Subtle variations in Pten dose determine cancer
susceptibility. Nat Genet. 42:454–458. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trotman LC, Niki M, Dotan ZA, Koutcher JA,
Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van
Dyke T, et al: Pten dose dictates cancer progression in the
prostate. PLoS Biol. 1:E592003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garofalo M, Di Leva G, Romano G, Nuovo G,
Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P,
et al: miR-221&222 regulate TRAIL resistance and enhance
tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell.
16:498–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huse JT, Brennan C, Hambardzumyan D, Wee
B, Pena J, Rouhanifard SH, Sohn-Lee C, le Sage C, Agami R, Tuschl T
and Holland EC: The PTEN-regulating microRNA miR-26a is amplified
in high-grade glioma and facilitates gliomagenesis in vivo. Genes
Dev. 23:1327–1337. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tabach Y, Billi AC, Hayes GD, Newman MA,
Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, et al:
Identification of small RNA pathway genes using patterns of
phylogenetic conservation and divergence. Nature. 493:694–698.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mullany LE, Herrick JS, Wolff RK, Stevens
JR, Samowitz W and Slattery ML: MicroRNA-transcription factor
interactions and their combined effect on target gene expression in
colon cancer cases. Genes, Chromosom Cancer. 57:192–202. 2018.
View Article : Google Scholar
|
|
12
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Frankel LB, Christoffersen NR, Jacobsen A,
Lindow M, Krogh A and Lund AH: Programmed cell death 4 (PDCD4) is
an important functional target of the microRNA miR-21 in breast
cancer cells. J Biol Chem. 283:1026–1033. 2008. View Article : Google Scholar
|
|
14
|
Zhu S, Si ML, Wu H and Mo YY: MicroRNA-21
targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol
Chem. 282:14328–14336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chan JA, Krichevsky AM and Kosik KS:
MicroRNA-21 Is an antiapoptotic factor in human glioblastoma cells.
Cancer Res. 65:6029–6033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zarringhalam K, Tay Y, Kulkarni P, Bester
AC, Pandolfi PP and Kulkarni RV: Identification of competing
endogenous RNAs of the tumor suppressor gene PTEN: A probabilistic
approach. Sci Rep. 7:77552017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poliseno L and Pandolfi PP: PTEN ceRNA
networks in human cancer. Methods. 77-78:41–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Karreth FA, Tay Y, Perna D, Ala U, Tan SM,
Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, et al:
In vivo identification of tumor-suppressive PTEN ceRNAs in an
oncogenic BRAF-induced mouse model of melanoma. Cell. 147:382–395.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marone M, Mozzetti S, De Ritis D, Pierelli
L and Scambia G: Semiquantitative RT-PCR analysis to assess the
expression levels of multiple transcripts from the same sample.
Biol Proced Online. 3:19–25. 2001. View
Article : Google Scholar
|
|
21
|
Langlois MJ, Bergeron S, Bernatchez G,
Boudreau F, Saucier C, Perreault N, Carrier JC and Rivard N: The
PTEN phosphatase controls intestinal epithelial cell polarity and
barrier function: Role in colorectal cancer progression. PLoS One.
5:e157422010. View Article : Google Scholar
|
|
22
|
Rhee I, Bachman KE, Park BH, Jair KW, Yen
RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al:
DNMT1 and DNMT3b cooperate to silence genes in human cancer cells.
Nature. 416:552–556. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uribe-Lewis S, Stark R, Carroll T, Dunning
MJ, Bachman M, Ito Y, Stojic L, Halim S, Vowler SL, Lynch AG, et
al: 5-hydroxy-methylcytosine marks promoters in colon that resist
DNA hypermethylation in cancer. Genome Biol. 16:692015. View Article : Google Scholar
|
|
24
|
García JM, Silva J, Peña C, Garcia V,
Rodríguez R, Cruz MA, Cantos B, Provencio M, España P and Bonilla
F: Promoter methylation of the PTEN gene is a common molecular
change in breast cancer. Genes Chromosom Cancer. 41:117–124. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alvarez-Nuñez F, Bussaglia E, Mauricio D,
Ybarra J, Vilar M, Lerma E, de Leiva A and Matias-Guiu X; Thyroid
Neoplasia Study Group: PTEN promoter methylation in sporadic
thyroid carcinomas. Thyroid. 16:17–23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu J, Jeong H, Kong N, Yang Y, Carroll J,
Luo HR, Silberstein LE, Yupoma and Chai L: Stem cell factor SALL4
represses the transcriptions of PTEN and SALL1 through an
epigenetic repressor complex. PLoS One. 4:e55772009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hettinger K, Vikhanskaya F, Poh MK, Lee
MK, de Belle I, Zhang JT, Reddy SA and Sabapathy K: c-Jun promotes
cellular survival by suppression of PTEN. Cell Death Differ.
14:218–229. 2007. View Article : Google Scholar
|
|
28
|
Vasudevan KM, Gurumurthy S and Rangnekar
VM: Suppression of PTEN expression by NF-kappa B prevents
apoptosis. Mol Cell Biol. 24:1007–1021. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meng F, Henson R, Wehbe-Janek H, Ghoshal
K, Jacob ST and Patel T: MicroRNA-21 regulates expression of the
PTEN tumor suppressor gene in human hepatocellular cancer.
Gastroenterology. 133:647–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tay Y, Kats L, Salmena L, Weiss D, Tan SM,
Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al:
Coding-independent regulation of the tumor suppressor PTEN by
competing endogenous mRNAs. Cell. 147:344–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Poliseno L, Salmena L, Zhang J, Carver B,
Haveman WJ and Pandolfi PP: A coding-independent function of gene
and pseudogene mRNAs regulates tumour biology. Nature.
465:1033–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA Hypothesis: The rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamakuchi M, Lotterman CD, Bao C, Hruban
RH, Karim B, Mendell JT, Huso D and Lowenstein CJ: P53-induced
microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad
Sci USA. 107:6334–6339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang B, Hsu SH, Wang X, Kutay H, Bid HK,
Yu J, Ganju RK, Jacob ST, Yuneva M and Ghoshal K: Reciprocal
regulation of microRNA-122 and c-Myc in hepatocellular cancer: Role
of E2F1 and transcription factor dimerization partner 2.
Hepatology. 59:555–566. 2014. View Article : Google Scholar
|
|
35
|
Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer
A, Llobet-Navas D, Rajbhandari P, Bansal M, Guarnieri P, Silva J
and Califano A: An Extensive MicroRNA-mediated network of RNA-RNA
interactions regulates established oncogenic pathways in
glio-blastoma. Cell. 147:370–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jeyapalan Z, Deng Z, Shatseva T, Fang L,
He C and Yang BB: Expression of CD44 3'-untranslated region
regulates endogenous microRNA functions in tumorigenesis and
angiogenesis. Nucleic Acids Res. 39:3026–3041. 2011. View Article : Google Scholar
|
|
37
|
Rutnam ZJ, Du WW, Yang W, Yang X and Yang
BB: The pseu-dogene TUSC2P promotes TUSC2 function by binding
multiple microRNAs. Nat Commun. 5:29142014. View Article : Google Scholar
|
|
38
|
Karreth FA, Reschke M, Ruocco A, Ng C,
Chapuy B, Léopold V, Sjoberg M, Keane TM, Verma A, Ala U, et al:
The BRAF pseu-dogene functions as a competitive endogenous RNA and
induces lymphoma in vivo. Cell. 161:319–332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang L, Guo ZY, Zhang R, Xin B, Chen R,
Zhao J, Wang T, Wen WH, Jia LT, Yao LB and Yang AG: Pseudogene
OCT4-pg4 functions as a natural micro RNA sponge to regulate OCT4
expression by competing for miR-145 in hepatocellular carcinoma.
Carcinogenesis. 34:1773–1781. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sanchez-Mejias A and Tay Y: Competing
endogenous RNA networks: Tying the essential knots for cancer
biology and therapeutics. J Hematol Oncol. 8:302015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ergun S and Oztuzcu S: Oncocers:
ceRNA-mediated cross-talk by sponging miRNAs in oncogenic pathways.
Tumor Biol. 36:3129–3136. 2015. View Article : Google Scholar
|
|
42
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu J, Li Y, Lu J, Pan T, Ding N, Wang Z,
Shao T, Zhang J, Wang L and Li X: The mRNA related ceRNA-ceRNA
landscape and significance across 20 major cancer types. Nucleic
Acids Res. 43:8169–8182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen L, Zheng J, Zhang Y, Yang L, Wang J,
Ni J, Cui D, Yu C and Cai Z: Tumor-specific Expression of
MicroRNA-26a suppresses human hepatocellular carcinoma growth via
cyclin-dependent and -independent pathways. Mol Ther. 19:1521–1528.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sandhu R, Rivenbark AG and Coleman WB:
Enhancement of chemotherapeutic efficacy in hypermethylator breast
cancer cells through targeted and pharmacologic inhibition of
DNMT3b. Breast Cancer Res Treat. 131:385–399. 2012. View Article : Google Scholar
|
|
46
|
Noack F and Calegari F: MicroRNAs meet
epigenetics to make for better brains. EMBO Rep. 15:1224–1225.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garcia DM, Baek D, Shin C, Bell GW,
Grimson A and Bartel DP: Weak seed-pairing stability and high
target-site abundance decrease the proficiency of lsy-6 and other
miRNAs. Nat Struct Mol Biol. 18:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar :
|