Introduction
Breast cancer is the most common malignant tumor
affecting women (1), and ~70% of
breast cancer cases are considered estrogen receptor-positive
(ER+) (2-4). In patients with ER+ breast
cancer, tumor growth and survival are primarily dependent on the
activation of the ER signaling pathway (5-7).
Tamoxifen (TAM), which prevents the activation of ER signaling and
its downstream pathways by blocking estrogen binding to ER, is the
most frequently used endocrine drug in clinical practice (8-10).
However, ~30-40% of responsive tumors eventually acquire TAM
resistance, which remains a major clinical challenge (11-15).
However, other endocrine therapies with or without
targeted therapy may be recommended for patients who develop TAM
resistance (16,17). Chemotherapy can control the
progression of breast cancer and may prolong the survival times of
patients that exhibit rapid disease progression or visceral crisis
(18). In a previous study by the
authors, and in other studies, it was demonstrated that tumor cells
usually exhibit more rapid growth rates and increased invasiveness
following the development of TAM resistance (19-21),
which may explain why some patients may experience rapid tumor
progression and multiple visceral metastases during TAM treatment
and, thus, require chemotherapy. However, to date, at least to the
best of our knowledge, there are only a few studies available
investigating the responses of TAM-resistant patients to
chemotherapeutic drugs, and there are no guidelines for the
recommended chemotherapeutic regimen.
c-MYC is one of the most upregulated oncogenes in
several different types of cancer, and has been reported to play
varying roles in different molecular subtypes of breast cancer
(22,23). In the present study, TAM resistance
and the sensitivities of TAM-resistant cells to various
chemotherapeutic agents were examined. The results revealed that
although c-MYC plays a pivotal role in TAM resistance, it improves
cisplatin sensitivity in ER+ breast cancer. Thus,
patients with rapid disease progression during TAM treatment may
respond favorably to cisplatin, and a high c-MYC expression may be
used as a predictive marker.
Materials and methods
Reagents and antibodies
4-Hydroxytamoxifen (TAM) was purchased from
Sigma-Aldrich; Merck KGaA. Doxorubicin, paclitaxel and cisplatin
were purchased from Beijing Solarbio Science & Technology Co.,
Ltd. Antibodies against Erα (cat. no. 8644S), human epidermal
growth factor receptor 2 (HER2) (cat. no. 2242S), GAPDH (cat. no.
5174S) and AKT (cat. no. 9272S) were purchased from Cell Signaling
Technology, Inc. Antibodies against c-MYC (cat. no. ab32072),
cyclin D1 (cat. no. ab226977, phospho (p)-AKT (Ser473) (cat. no.
ab81283) and E-cadherin (cat. no. ab133597) were purchased from
Abcam. Antibodies against p21 (cat. no. 27296-1-AP) and β-catenin
(cat. no. 51067-2-AP) were purchased from ProteinTech Group, Inc.,
and an antibody against vimentin (cat. no. BS1491) was purchased
from Bioworld Technology, Inc.
Cell lines and cell culture
The human breast cancer cell lines, MCF-7 and T47D,
were purchased from the American Type Culture Collection. The
TAM-resistant cell lines, MCF-7R and T47DR, were generated by
exposing parental MCF-7 and T47D cells to progressively increasing
concentrations of TAM (up to 5 µM) over a duration of ~8
months as previously described (24,25).
All cell lines were maintained in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin (Beyotime Institute of Biotechnology) at 37°C in an
humidified incubator with 5% CO2.
Cell viability assay
Cell viability was measured using a Cell Counting
kit-8 assay (CCK-8; Beyotime Institute of Biotechnology) according
to the manufacturer's protocol. Briefly, 5×103
cells/well were seeded in a 96-well plate in 100 µl medium.
After 24 h, the cells were treated with the different drugs (TAM:
5, 10, 11, 12, 13, 14, 15, 16, 18, 20 and 25 µM;
doxorubicin: 0.25, 0.5, 1, 2, 4, 8 and 16 µM; paclitaxel:
0.25, 0.5, 1, 2, 4, 8, 16, 32 and 64 nM; cisplatin: 2, 4, 8, 16,
32, 64, 128 and 256 µM) (5 wells for each drug) for 24-72 h.
Subsequently, 10 µl CCK-8 solution was added to the medium
and the cells were incubated for a further 2 h at 37°C. Optical
density (OD) values were measured using a digital spectrophotometer
(Bio-Rad Laboratories, Inc.) at a wavelength of 450 nm, and cell
viability rates were expressed as a percentage relative to the
corresponding control cells. The IC50 value was
determined from the concentration-effect curve. After the data were
log-transformed, the specific value was obtained through the curve
parameter equation, and the viability of the control cells was
considered as 100% (26).
Cell invasion assays
The MCF7, T47D, MCF7R or T47DR cells were seeded in
the upper chamber of a Transwell insert (3×105
cells/well; EMD Millipore) coated with Matrigel (1:7.5) in 200
µl of serum-free medium. Supplemented RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) was added to the lower chamber. The
cell lines were incubated for 24-48 h at 37°C, and the cells were
fixed with 4% paraformaldehyde (Wuhan Boster Biological Technology,
Ltd.) for 15 min at room temperature and stained with 0.5% crystal
violet (Wuhan Boster Biological Technology, Ltd.) for 5 min at room
temperature. the number of migrating cells was determined in 5
random fields at an inverted light microscope (magnification, ×200;
TE2000-U; Nikon Corp.).
Cell cycle analysis
The proportion of cells in the S phase of the cell
cycle was analyzed by flow cytometry using a standard protocol.
Briefly, the cells were seeded in 6-well plates (2×105
cells/well) and cultured for 24 h. The cells were then harvested
and washed twice with cold PBS, fixed with 70% ethanol at 4°C for 1
h, incubated with Rnase for 30 min at 37°C and stained with
propidium iodide for 30 min at room temperature (cat. no. P4170;
Sigma Aldrich; Merck KGaA). Cell cycle distribution was analyzed by
flow cytometry using a FACSVantage SE instrument (BD
Biosciences).
Reverse transcription-quantitative
PCR
Total cellular RNA was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Reverse
transcription and quantitative PCR were performed using the
PrimeScript RT Master mix kit (Takara Bio, Inc.) and SYBR Pre-mix
Ex Taq™ II (Takara Bio, Inc.) according to the manufacturer's
protocols. GAPDH was used as the reference gene, and the
thermocycling conditions were as follows: 2 min at 95°C, followed
by 39 cycles at 95°C for 30 sec, 30 sec at 56.5°C and 20 sec at
72°C. Relative gene expression was normalized to GAPDH and
calculated using the 2−∆∆Cq method (27). The sequences of primers used for
amplifying GAPDH, c-MYC, multidrug resistance-related protein
(MRP), multidrug resistance protein (MDR) and breast cancer
resistance protein (BCPR) are presented in Table SI.
Western blot analysis
Cells were lysed using RIPA lysis buffer with
phenyl-methanesulfonyl fluoride (Wuhan Boster Biological
Technology, Ltd.), and the protein concentration was determined
using a bicinchoninic acid assay (Beyotime Institute of
Biotechnology). A total of 50 µg protein was loaded on a 10%
SDS gel and resolved using SDS-PAGE. The resolved proteins were
transferred to PVDF membranes. Non-specific binding sites were
blocked by incubating the membranes with 5% non-fat milk, after
which the membranes were incubated overnight at 4°C with the
primary antibodies: ERα (1:1,000); HER2 (1:1,000); AKT (1:1,000);
p-AKT (1:1,000); c-MYC (1:1,000); cyclin D1 (1:1,000); vimentin
(1:1,000); E-cadherin (1:1,000); p21 (1:500); β-catenin (1:500);
GAPDH (1:1,000) Membranes were subsequently incubated with a
horseradish peroxidase-conjugated anti-rabbit/mouse immunoglobulin
G secondary antibody (1:1,000; cat. no. BA1075 and BA1051; Wuhan
Boster Biological Technology, Ltd.). Signals were visualized using
enhanced chemiluminescence reagent (EMD Millipore). Densitometry
analysis was performed using ImageJ and normalized to the
respective expression of GAPDH.
Small interfering (si)RNA
transfection
siRNAs targeting c-MYC mRNA and a control siRNA were
purchased from Shanghai GenePharma Co., Ltd. siRNAs were
transfected into the cells using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The efficiency of RNA-interference was assessed using
RT-qPCR 24-36 h following transfection. Cell lysates were collected
48-72 h following transfection and used for western blot analysis.
The sequences of the siRNAs targeting c-MYC mRNA are presented in
Table SII.
Patient and specimen selection
A total of 178 consecutive patients with recurrent
and/or metastatic ER+ breast cancer, treated at the
Breast Cancer Center of Chongqing at The First Affiliated Hospital
of Chongqing Medical University between July, 2012 and July, 2017,
were recruited for the present study. The outcomes of endocrine
therapy were investigated for all patients, and 122 patients with
TAM resistance were identified (Table
I). At the time of diagnosis of recurrent and/or metastatic
disease, 125 patients (70.2%) had received various chemotherapeutic
regimens, whereas the remaining 53 patients continued to receive
endocrine therapy. The Response Evaluation Criteria in Solid Tumors
(RECIST) guidelines (version 1.1) (28) were utilized to assess chemotherapy
responses as follows: Partial response (PR), a reduction of all
target lesion diameters of ≥30%; progressive disease (PD), growth
of all target lesion diameters by ≥20%; stable disease (SD),
neither a sufficient reduction to be classified as PR, nor
sufficient growth to be classified as PD (29). A total of 28 paired, archived
paraffin-embedded breast cancer specimens (primary and metastatic
breast cancer tissues) from the aforementioned TAM-resistant
patients were obtained from the Clinical Diagnostic Pathology
Center of Chongqing Medical University (Chongqing, China). Detailed
information pertaining to the specimens is presented in Table SIII. The present study was
approved by the Ethics Committee of Chongqing Medical University
and written informed consent was obtained from all patients.
 | Table IClinicopathological features of
tamoxifen-resistant patients (n=122). |
Table I
Clinicopathological features of
tamoxifen-resistant patients (n=122).
| Parameters | No. (%) |
|---|
| Age (years) | |
| <30 | 3 (2.5) |
| 30-40 | 36 (29.5) |
| 40-50 | 69 (56.6) |
| ≥50 | 14 (11.4) |
| Time of metastasis
(months) | |
| <24 | 59 (48.4) |
| ≥24 | 63 (51.6) |
| Surgical
procedure | |
| Mastectomy | 103 (84.4) |
|
Breast-conserving | 19 (15.6) |
| Subtypes of
Cancer | |
| Ductal | 115 (94.4) |
| Lobular | 4 (3.2) |
| Others | 3 (2.4) |
| TNM staging | |
| I | 10 (8.2) |
| IIA | 29 (23.8) |
| IIB | 42 (34.4) |
| IIIA | 21 (17.2) |
| IIIB | 10 (8.2) |
| IIIC | 2 (1.6) |
| Unknown | 8 (6.6) |
| Recurrent or/and
metastatic site | |
| Breast | 9 (7.4) |
| Chest wall | 27 (22.1) |
| Lymph nodea | 21 (17.2) |
| Boneb | 26 (21.3) |
| Brainc | 3 (2.5) |
| Liverc | 13 (10.7) |
| Lungc | 16 (13.1) |
| Multi-visceral
metastasisd | 7 (5.7) |
Immunohistochemistry (IHC)
IHC staining was performed as previously described
(19). For histochemical analysis,
Image-Pro Plus version 6.0 (Media Cybernetics, Inc.) was used to
assess the percentage of positive cells. The detailed steps were as
follows: Deparaffinized specimens were then sectioned
(4-µm-thick slices). Following antigen repair and blocking
for non-specific binding, the slices were incubated with specific
primary antibodies against c-MYC (1:200; cat. no. ab32072, Abcam)
overnight at 4°C. Subsequently, the sections were treated with
HRP-conjugated goat anti-rabbit IgG secondary antibody (1:200; cat.
no. TA140003; OriGene Technologies, Inc.) for 30 min at room
temperature. After staining with diaminobenzidine (OriGene
Technologies, Inc.) and hematoxylin for 5 sec at room temperature,
images were captured using a Nikon Eclipse 80i microscope
(magnification, ×200; Nikon Corp.). The mean OD in 5 randomly
selected areas (MOD=IOD/area) was used to evaluate the levels of
protein expression. The c-MYC staining intensities in tumor tissues
were scored as follows: 0, no staining; 1, weak staining; 2,
intermediate staining; 3, strong staining; and 4, very strong
staining. Finally, the IHC data were quantified by multiplying the
staining intensity by the proportion of positive cells as
previously described (30).
Gene Expression Omnibus (GEO) and The
Cancer Genome Atlas (TCGA)
The microarray analysis data of MCF-7 cells and
TAM-resistant cell lines were downloaded from GEO (accession no.
GSE26459). c-MYC expression in breast cancer tissues,
para-cancerous tissues and in different molecular subtypes of
breast cancer were obtained from TCGA, and survival curves based on
c-MYC expression in breast cancers were obtained from Kaplan-Meier
plotter: c-MYC expression data in tumors and follow-up information
for patients with ER-positive breast cancer treated with adjuvant
TAM were also downloaded from GEO (accession nos. GSE6532, n=87;
and GSE9195, n=77), and the log-rank test was then used to assess
the association between c-MYC expression and survival in breast
cancer patients.
Statistical analysis
SPSS version 22.0 (SPSS Inc.) was used for the data
analysis. Data are presented as the means ± standard deviation of 3
repeats. A one-way ANOVA followed by Tukey's post hoc test was used
to compare differences among multiple groups. A χ2 test
was used to evaluate clinical response to chemotherapy in patients
with recurrent and/or metastatic breast cancer. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Proliferation and invasion are increased
in TAM-resistant cells
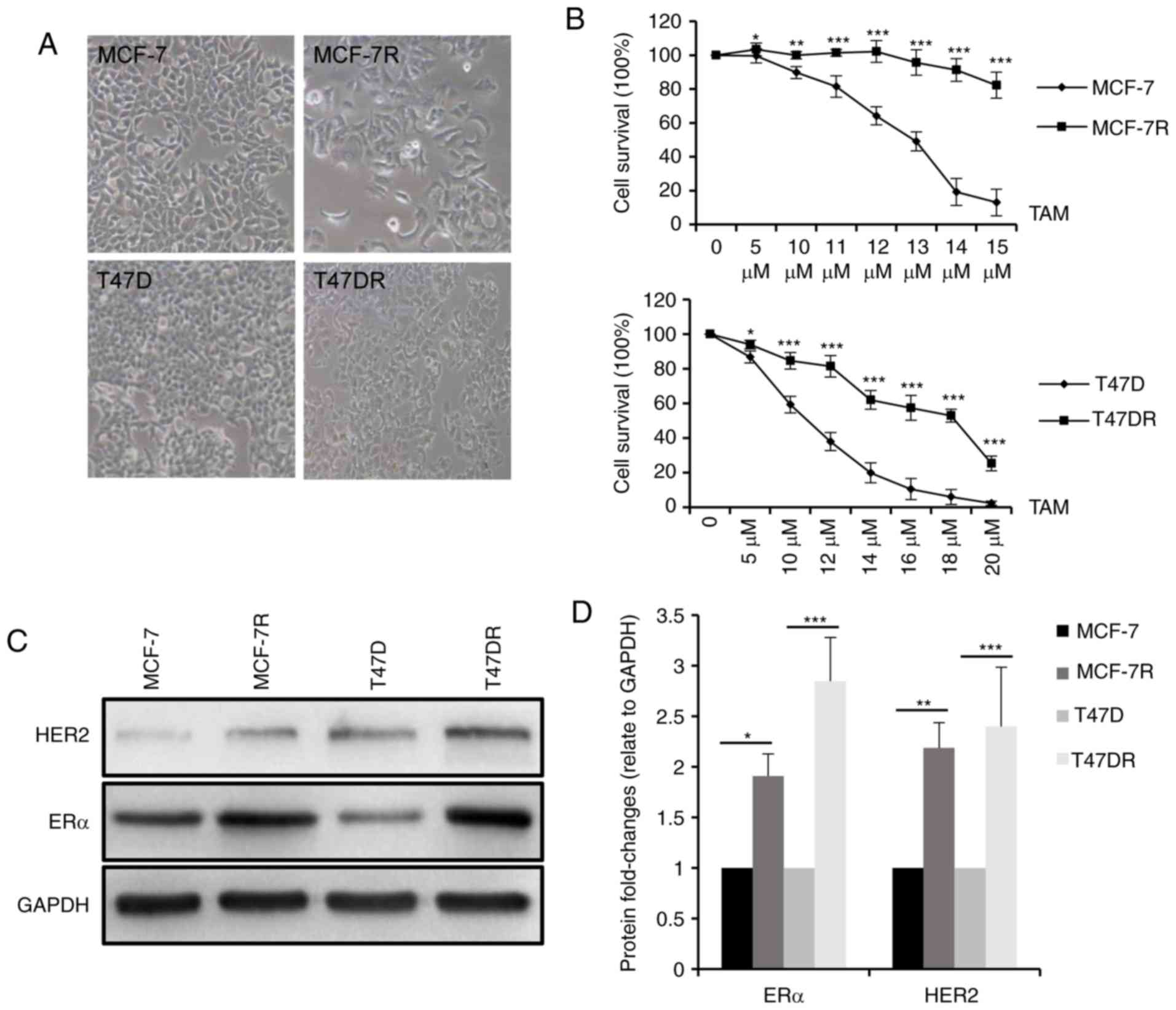
In agreement with a previous study (19) by the authors, the morphology of the
MCF-7R and T47DR cells exhibited a flatter and more polygonal shape
compared with that of the parental control cells, and the cells
were more mesenchymal phenotypically (Fig. 1A). To validate TAM resistance in
these established cells, MCF-7R and T47DR cells, and their parental
cells were treated with various concentrations of TAM and the cell
survival rates were measured compared with a negative control. As
shown in Fig. 1B, survival was
higher in the MCF-7R and T47DR cells compared with the respective
parental cells treated with the same concentration of TAM. The
IC50 values of the 4 cell lines to TAM were 12.53±0.95
µM for the MCF-7, 26.93±1.76 µM for the MCF-7R,
9.29±1.23 µM for the T47D and 17.27±1.16 µM for the
T47D-7R cells (data not shown). In addition, the protein expression
levels of ERα and HER2 were significantly higher in the
TAM-resistant cells (Fig. 1C and
D), consistent with previous preclinical models and clinical
observations (24,31-33).
To evaluate the proliferation of the TAM-resistant
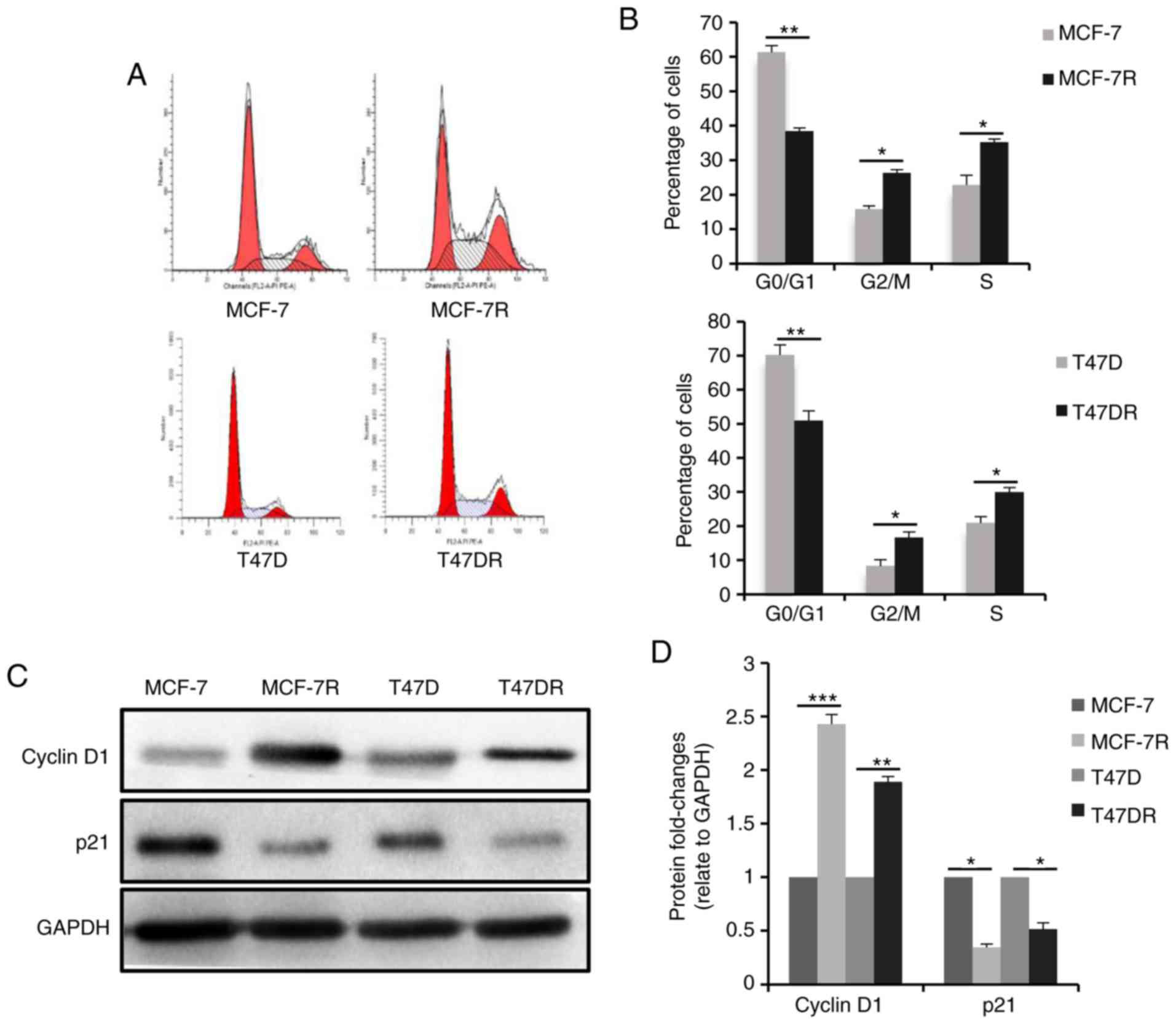
cells, cell cycle progression was analyzed by flow cytometry, and
the results revealed that the percentage of cells in the G0/G1
phase was decreased and the percentage of cells in the S phase was
increased significantly in the TAM-resistant cells (Fig. 2A and B), suggesting that
proliferation of the TAM-resistant cells was increased.
Consistently, the expression of the cell cycle
checkpoint-associated protein, p21waf1, decreased
significantly in the TAM-resistant cells, whereas the expression of
cyclin D1 was increased (Fig. 2C and
D). Furthermore, the TAM-resistant cells were significantly
more invasive compared with the parental cells (Figs. 2E and F, and S1A and B). E-cadherin protein expression
was decreased and vimentin expression was increased in the
TAM-resistant cells compared with the respective parental cells
(Figs. 2G and H, and S1C and D), suggesting the cells has
undergone epithelial-mesenchymal transition, consistent with the
morphology of resistant cells.
c-MYC expression is increased in
TAM-resistant cells and c-MYC knockdown enhances sensitivity to
TAM
To explore the potential mechanisms through which
TAM resistance is acquired, microarray data from TAM-resistant
cells were downloaded from GEO (accession no. GSE26459) and used to
identify TAM resistance-associated genes. A heatmap depicting the
expression of proliferation-related genes in the TAM-resistant
cells relative to the parental controls is presented in Fig. 3A. c-MYC expression was found to be
abnormally high in the TAM-resistant cells (P<0.001, Fig. 3B). c-MYC is one of the primary
target genes of the ER signaling pathway and possesses similar
functions to ER (34-36). The expression of c-MYC was observed
in the TAM-resistant cells used in the present study (Fig. 3C and D). Consistently, the
activation of PI3K/AKT and β-catenin signaling in TAM-resistant
cells was also confirmed in the cells used (Fig. 3C and D). To further examine the
role of c-MYC in TAM resistance, the siRNA-mediated knockdown of
c-MYC in the MCF-7R cells (Fig.
3E-G) was shown to enhance the sensitivity of MCF-7R cells to
TAM (Fig. 3H). The TAM
IC50 values of the MCF-7R cells prior to and following
the knockdown of c-MYC were 20.26±0.76 µM for the MCF-7R,
18.88±0.69 µM for the MCF-7R/NC,13.95±1.82 µM for the
MCF-7R/siRNA1 and 13.71±1.63 µM for the MCF-7R/siRNA2 cells
(data not shown). Taken together, these data demonstrate that c-MYC
plays a critical role in TAM resistance in vitro.
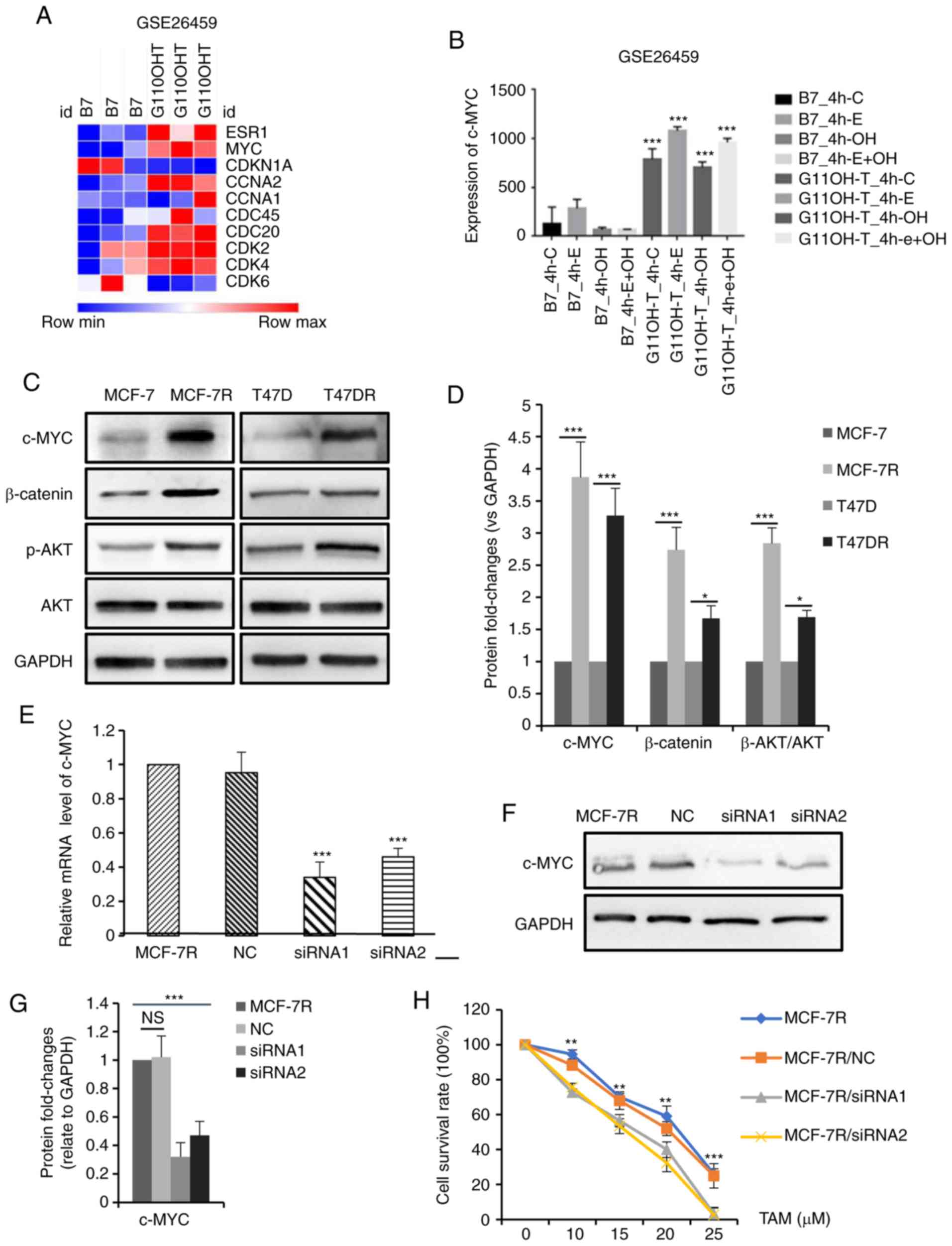 | Figure 3c-MYC expression is increased in
TAM-resistant breast cancer cells, and the knockdown of c-MYC
increases TAM sensitivity. (A) Heatmap depicting the fold-changes
of proliferation-related genes in TAM-sensitive and TAM-resistant
cells. (B) Bar graph representing the expression levels of c-MYC in
TAM-sensitive and TAM-resistant cells. (C) Western blot analysis
and (D) densitometric analysis of c-MYC, β-catenin, p-AKT and AKT
expression in TAM-sensitive and TAM-resistant cells. Efficiency of
siRNA in knocking down c-MYC expression in MCF-7R cells was
evaluated by (E) reverse transcription-quantitative PCR and (F and
G) western blot analysis. (H) Cellular viability was evaluated in
MCF-7R cells transfected with siRNA and treated with TAM for 24 h
(**P<0.01, ***P<0.001, compared to
siRNA). Data are presented as the means ± standard deviation of
mean of 3 repeats. *P<0.05, **P<0.01,
***P<0.001. TAM, tamoxifen; siRNA, small interfering
RNA; NC, negative control; p-, phospho-. |
c-MYC expression is associated with TAM
resistance in patients with breast cancer
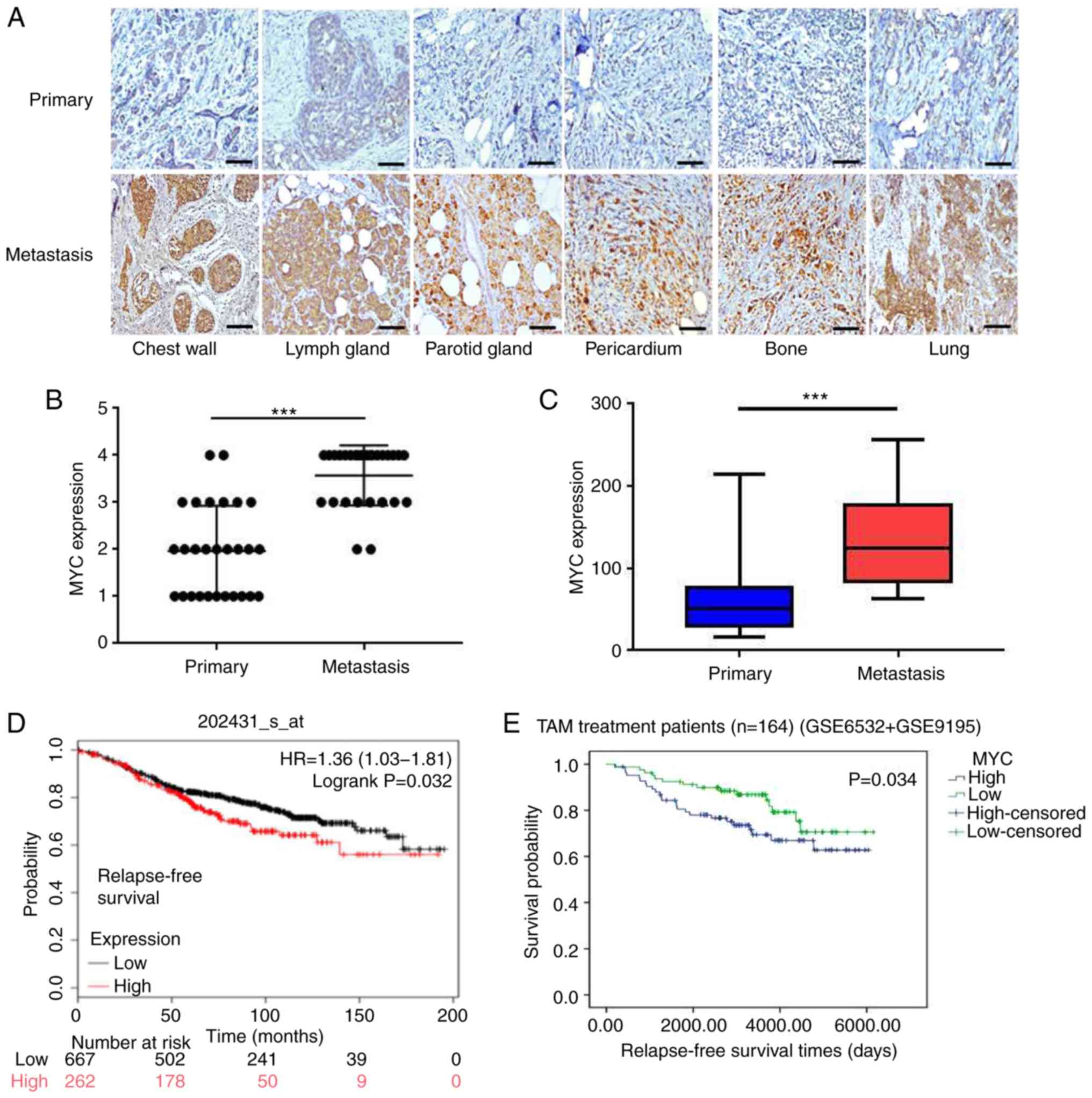
To further confirm the association between c-MYC and
clinical TAM-resistance in patients with breast cancer, 28 pairs of
primary and recurrent/metastatic tumor specimens from the same
patients who underwent adjuvant TAM treatment were collected. As
shown in Fig. 4A, c-MYC expression
in the primary tumors was weaker, but was increased significantly
in the metastatic lesions following the development of TAM in the
same patients (all P<0.001; Fig. 4B
and C), consistent with the results observed in TAM-resistant
cell lines.
To assess c-MYC expression in breast cancer, TCGA
was used for bioinformatics analysis. As shown in Fig. S2A and B, c-MYC expression was
relatively low in breast cancer tissues compared with normal
tissues (P<0.05); however, c-MYC was highly expressed in basal
subtypes (endocrine therapy-resistant), which indicated an
association with the therapeutic efficacy of TAM. Furthermore,
Kaplan-Meier survival analysis revealed that a higher c-MYC
expression was associated with a shorter distant metastasis-free
survival in patients with breast cancer (P=0.011; Fig. S2C), and its expression was also
associated with a poor relapse-free survival in patients with
breast cancer that received endocrine therapy (P=0.032; Fig. 4D). In addition, a high c-MYC
expression predicted an improved post-progression survival of
patients with ER+ breast cancer (P=0.023), but not of
the patients with ER-negative breast cancer (P=0.27; Fig. S2D and E).
To determine whether c-MYC expression may serve as a
direct predictor of favorable outcomes following adjuvant TAM
therapy, tumor gene expression levels and corresponding follow-up
data from patients with ER+ breast cancer treated with
TAM as an adjuvant were downloaded from GEO (accession nos. GSE6532
and GSE9195). The results revealed that an increased c-MYC
expression was associated with a reduced relapse-free survival
(P=0.034; Fig. 4E), suggesting
that c-MYC expression was closely associated with clinical TAM
resistance.
c-MYC affects the sensitivity of
cisplatin by regulating cell cycle progression
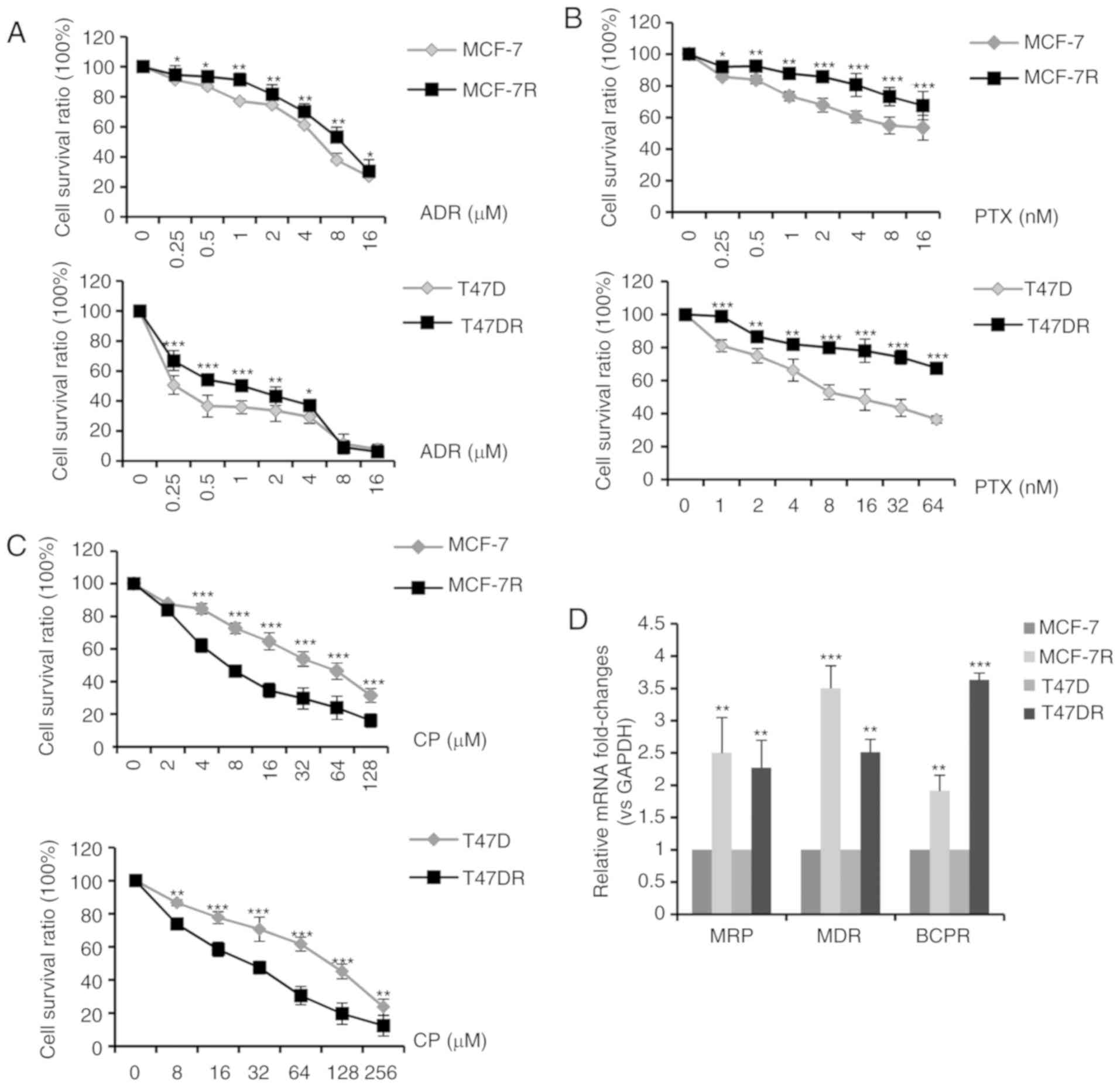
According to standard clinical chemotherapy, 3 major
chemotherapeutic drugs (doxorubicin, paclitaxel and cisplatin) were
evaluated to determine the alterations in chemosensitivity
following the development of TAM resistance. As shown in Fig. 5A-C, the MCF-7R and T47DR cells
exhibited an increased resistance to doxorubicin and paclitaxel;
however, they exhibited a greater sensitivity to cisplatin, in
comparison with the parental cells. The IC50 values of
ADR, PTX and CP to the MCF-7, MCF-7R, T47D and T47DR cells were
5.30±0.85, 8.37±1.05, 1.36±0.36, 1.94±0.26 µM for ADR,
14.10±1.63, 57.78±3.85, 16.20±2.37, 102.89±5.37 nM for PTX, and
42.37±3.35, 20.76±2.86, 84.32±4.35, 25.76±3.24 µM for CP,
respectively (data not shown). RT-qPCR assays revealed that the
mRNA expression levels of genes associated with multidrug
resistance (MRP, MDR and BCPR) were significantly higher in the
TAM-resistant cells compared with the parental cells (Fig. 5D), which may partially explain the
increase in resistance to doxorubicin and paclitaxel.
Thus, the mechanisms underlying increased cisplatin
sensitivity were examined. It is hypothesized that
cisplatin-induced DNA lesions are most cytotoxic in the S phase of
the cell cycle (37-39) and that c-MYC accelerates the G1/S
phase transition (40,41). In the present study, it was
hypothesized that a high c-MYC expression increased cisplatin
sensitivity by increasing the proportion of cells in the S phase.
To test this hypothesis, the MCF-7R cells were treated with a
medium concentration of cisplatin for 24 h, and the dead cells were
washed away. Western blot analysis was performed to evaluate c-MYC
protein expression levels in the residual adherent cells. As shown
in Fig. 6A and B, c-MYC protein
expression significantly decreased in the surviving cells following
cisplatin treatment, indicating that the cells with a high c-MYC
expression were sensitized to cisplatin-induced cell death.
Furthermore, the percentage of MCF-7R cells in the S phase
decreased significantly following the knockdown of c-MYC expression
(Fig. 6C and D), and c-MYC
knockdown desensitized the MCF-7R cells to cisplatin (Fig. 6E). The IC50 values of
the MCF-7R cells to cisplatin prior to and following knockdown of
c-MYC were 24.07±2.25 µM for the MCF-7R, 28.89±3.02
µM for the MCF-7R/NC and 54.23±3.62 µM for the
MCF-7R/siRNA cells (data not shown). Collectively, these results
suggest that a high c-MYC expression in TAM-resistant cells
increased cisplatin sensitivity by regulating the cell cycle.
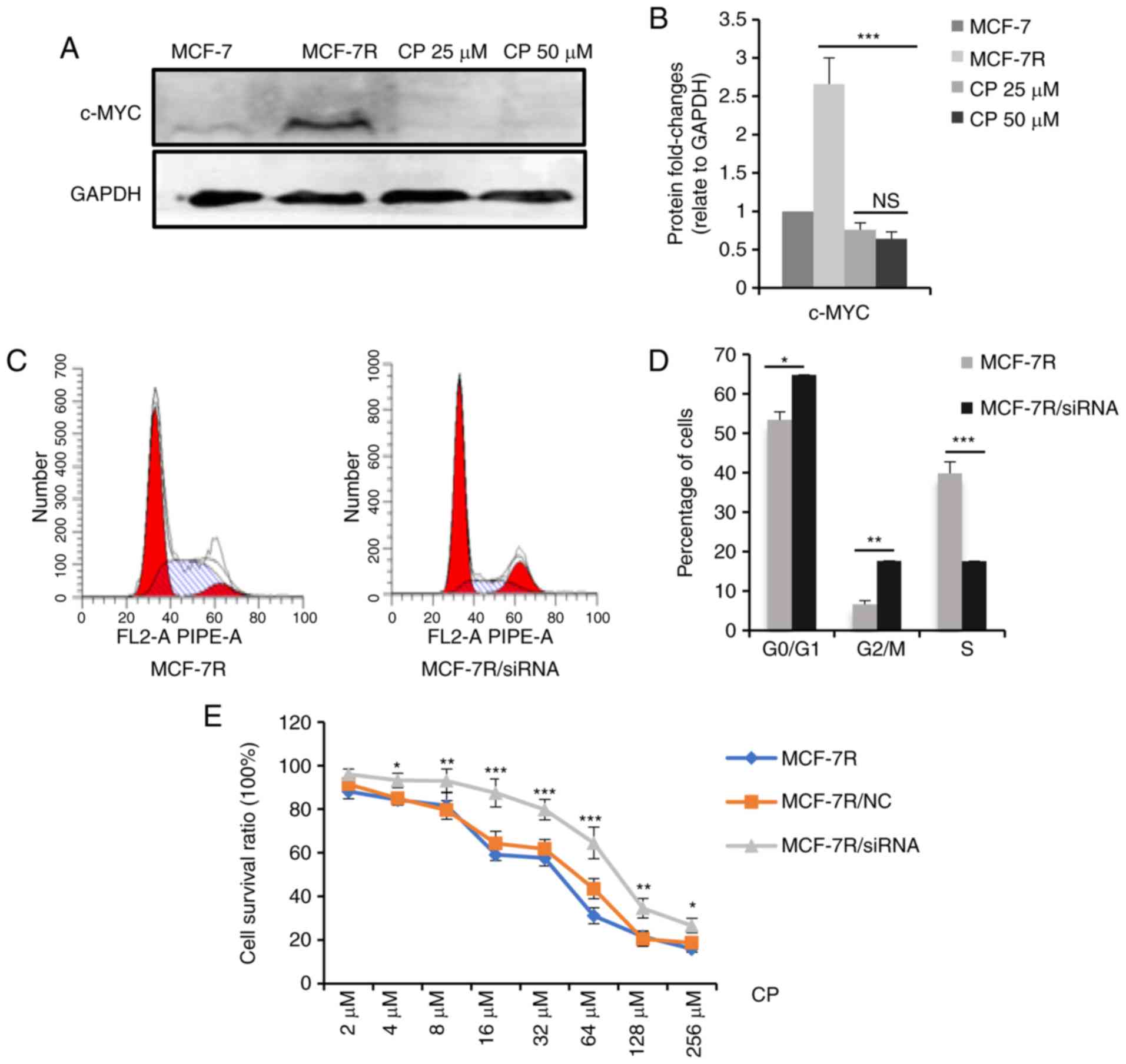 | Figure 6c-MYC affects the sensitivity of
cisplatin through regulating the cell cycle. (A) MCF-7R cells were
treated with 0, 25 or 50 µM cisplatin for 24 h. After
removing the dead cells, cells were lysed. c-MYC expression levels
in the indicated cells were determined by western blot analysis.
(B) Densitometric analysis of c-MYC in the western blots shown in
(A). (C) c-MYC expression was knocked down in MCF-7R cells and flow
cytometric analysis was performed to evaluate proportion of cells
in the S-phase. (D) Bar chart representing the percentage of MCF-7R
cells in the G0/G1, G2/M or S phase, prior to and following
c-MYC/siRNA transfection. (E) Cell-viability of MCF-7R with c-MYC
knocked down compared with MCF-7R and MCF-7R/NC cells, after
treatment of cells with different doses of cisplatin for 24 h
(*P<0.05, **P<0.01,
***P<0.001, compared to NC only). Data are presented
as the means ± standard deviation of the mean of 3 repeats.
*P<0.05, **P<0.01,
***P<0.001. TAM, tamoxifen; siRNA, small interfering;
NC, negative control. |
Patients with TAM-resistant breast cancer
exhibit higher remission rates with cisplatin-based
chemotherapy
A total of 122 patients were shown to possess
TAM-resistant breast cancer among the 178 patients with recurrent
and/or metastatic ER+ breast cancer based on their
endocrine therapy regimen (Fig.
7A). The median age of the patients at the time of
recurrence/metastatic disease was 42.2 years (range, 25-65 years),
and the median time from TAM therapy to recurrence and metastasis
was 36.0 months (range, 5-132 months). In addition, 23 patients
(18.9%) received treatment with TAM for >5 years. The baseline
characteristics of the TAM-resistant patients are presented in
Table I.
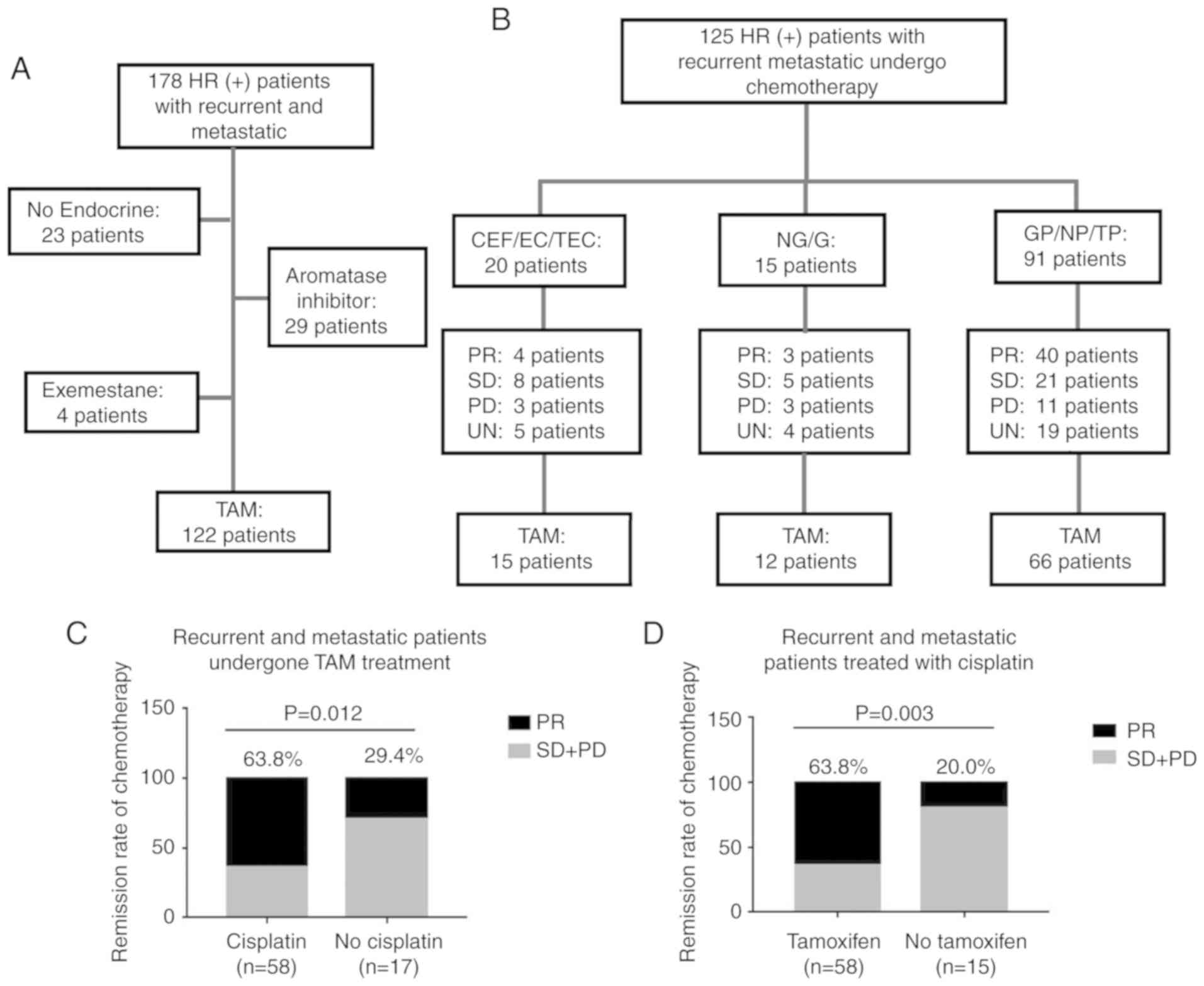 | Figure 7Patients with TAM-resistance have
favorable remission rates in response to cisplatin-based
chemotherapy. (A) Summary of endocrine therapies administered to
178 patients with ER+ recurrent and metastatic breast
cancer. A total of 122 patients were determined to exhibit TAM
resistance. (B) Chemotherapy regimens and responses in 125 patients
with ER+ recurrent and metastatic breast cancer.
Clinical responses were evaluated in comparison with alteration of
target lesions during chemotherapy. (C) Clinical remission rates of
TAM-resistant patients administered different chemotherapy
regimens. (D) Clinical remission rates of patients administered
cisplatin-based chemotherapy following treatment with different
endocrine-therapy regimens. C, cyclophosphamide; E, epirubicin; F,
fluorouracil; T, paclitaxel; N, vinorelbine; G, gemcitabine; P,
cisplatin; PR, partial response; SD, stable disease; PD,
progressive disease; UN, unknown; ER+, estrogen
receptor-positive; TAM, tamoxifen. |
Following diagnosis with tumor recurrence and
metastasis, 125 patients (93 with TAM resistance) received
chemotherapy. The chemotherapeutic regimens and therapeutic
responses are detailed in Fig. 7B.
Following the analysis of the clinical remission efficiencies of
patients that had undergone different chemotherapy regimens, it was
determined that the TAM-resistant patients who received
cisplatin-based chemotherapy exhibited improved clinical responses
compared with those who received other regimens (P=0.012; Fig. 7C). Furthermore, the patients
treated with TAM exhibited higher clinical remission rates when
treated with cisplatin-based chemotherapy, compared with patients
that had undergone other endocrine treatments (P=0.003; Fig. 7D).
Discussion
Despite the fact that aromatase inhibitors and other
endocrine therapy drugs have been successfully used in the
treatment of breast cancer (42,43),
TAM remains the most widely used therapeutic in clinical practice
(9,10,44).
A major obstacle of using TAM is that a substantial proportion of
patients develop drug resistance, leading to relapse and
progression (12,15,45).
For these patients, chemotherapy is recommended to control the
disease when visceral crisis and rapid tumor progression occurs
(18,46). However, in contrast to the large
number of studies on the mechanisms underlying the development of
TAM resistance (13-15,47,48),
to the best of our knowledge, few studies to date have examined the
effect of endocrine resistance on sensitivity to
chemotherapeutics.
As an oncogene, c-MYC has been shown to play an
important role in multiple cellular functions, including
proliferation, migration, angiogenesis and differentiation through
binding with nuclear DNA (41,49).
It has been reported that c-MYC may partially replace the function
of ER in estrogen-deprived breast cancer (34,37,50)
and that c-MYC signaling may predict responses to endocrine therapy
(51,52). Thus, c-MYC is hypothesized to play
an important role in TAM-resistant breast cancer. In agreement with
the findings of previous studies, the present study also confirmed
that c-MYC expression was significantly increased in TAM-resistant
cells and clinical tumor samples, and the inhibition of c-MYC
expression in MCF-7R cells partly restored TAM sensitivity.
Subsequently, alterations in the sensitivities of
cells to various chemotherapeutic agents following the occurrence
of TAM resistance was determined, and the results revealed that
TAM-resistant cells were significantly more sensitive to
cisplatin-induced cell death compared with the parental cell line,
consistent with previous literature (53-55).
Moreover, previous clinical findings have demonstrated that a high
c-MYC expression predicts more favorable clinical responses in
patients with cervical cancer treated with cisplatin-based
chemotherapy (56), and
TAM-resistant triple-negative breast cancer exhibits an increased
sensitivity to cisplatin; in addition, c-MYC amplification is the
most frequently reported aberration in these tumors (55,57).
Therefore, it was hypothesized that c-MYC may enhance sensitivity
to cisplatin by regulating the proportion of cells in the S phase
in TAM-resistant cells, and the results of the present study
confirmed the above hypothesis. The clinical data generated in the
present study also demonstrated that TAM-resistant breast cancer
patients treated with cisplatin-based chemotherapy had higher
clinical remission rates compared with those treated with other
chemotherapeutic regimens, further confirming the findings of the
in vitro experiments. To the best of our knowledge, the
present study is the first to demonstrate that c-MYC plays
different roles in treatment with endocrine therapy and
chemotherapy in ER+ breast cancer. Furthermore, it is
also the first time that the changes of chemosensitivity have been
investigated in clinical TAM-resistant patients. In addition, it is
noteworthy that the c-MYC downstream genes, CCNA2 and CDK4, also
exhibit a high expression in TAM resistance, and it has been
reported that both CCNA2 (58) and
CDK4 (59) play an important role
in TAM resistance. Therefore, the high expression of CCNA2 and CDK4
may also be involved in the sensitivity of TAM-resistant cells
cisplatin; further research is warranted to confirm the above
mechanisms.
In the present study, TAM-resistant cells also
exhibited resistance to doxorubicin and paclitaxel; however, they
exhibited an increased sensitivity to cisplatin, in agreement with
the findings of a previous study (60). A possible reason underlying this
phenomenon may be that upregulated expression levels of
multidrug-resistance-associated genes in TAM-resistant cells are
involved in the doxorubicin and paclitaxel efflux; thus, the cycle
change caused by c-MYC may not increase the sensitivity of these
two drugs to TAM-resistant cells (58). In addition, these proteins
exhibited less of an effect on cisplatin efflux, as the limited
intracellular accumulation of cisplatin is usually the result of
reduced uptake (61-63). Furthermore, p21 has been identified
as an important effector of c-MYC activity in the cell cycle, and
its expression was significantly reduced in the TAM-resistant cells
in the present study. Previous studies have revealed that the
inhibition of p21 expression sensitized tumor cells to cisplatin
(64,65). Thus, it was hypothesized that c-MYC
may affect the cell cycle and increase cisplatin sensitivity by
regulating p21 expression in TAM-resistant cells, and further
experiments are required to examine this hypothesis. In addition, a
recent study demonstrated that TAM-resistant breast cancer cells
are resistant to DNA-damaging chemotherapy (66), which was inconsistent with the
findings of the present and previous studies (53-55,60).
The different results observed in the various studies may reflect
the diversity in the cell models used, and further research aimed
at determining the underlying mechanisms is required due to the
complexity of drug resistance.
In conclusion, the results of the present study
suggest that although c-MYC participates in TAM resistance, it
improves cisplatin sensitivity in ER+ breast cancer.
TAM-resistant patients may respond favorably to cisplatin, and the
upregulated expression of c-MYC expression may be used as a
predictive marker. However, further research and prospective
clinical trials are required to confirm the observations of the
present study.
Supplementary Data
Funding
This study was funded by National Natural Science
Foundation of China (grant nos. 81472658, NSFC 81772979, NSFC
81472476 and NSFC 31671481).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
RC designed the study, performed the in vitro
experiments and drafted the manuscript. SG and CY participated in
the development of methodology of the study and performed the
bioinformatics analysis. LS, BZ, KL and LL obtained the clinical
data and performed the analysis. GT performed the analysis and
interpretation of data. ML and SL participated in design of the
study and revised the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
committee of Chongqing Medical University (Chongqing, China).
Written informed consent was obtained from all patients.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Anderson WF, Katki HA and Rosenberg PS:
Incidence of breast cancer in the United States: Current and future
trends. J Natl Cancer Inst. 103:1397–1402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marshall E: Breast cancer. Dare to do less
Science. 343:1454–1456. 2014.
|
|
4
|
Vargo-Gogola T and Rosen JM: Modelling
breast cancer: One size does not fit all. Nat Rev Cancer.
7:659–672. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Green KA and Carroll JS:
Oestrogen-receptor-mediated transcription and the influence of
co-factors and chromatin state. Nat Rev Cancer. 7:713–722. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patani N and Martin LA: Understanding
response and resistance to oestrogen deprivation in ER-positive
breast cancer. Mol Cell Endocrinol. 382:683–694. 2014. View Article : Google Scholar
|
|
8
|
Cuzick J, Sestak I, Pinder SE, Ellis IO,
Forsyth S, Bundred NJ, Forbes JF, Bishop H, Fentiman IS and George
WD: Effect of tamoxifen and radiotherapy in women with locally
excised ductal carcinoma in situ: Long-term results from the UK/ANZ
DCIS trial. Lancet Oncol. 12:21–29. 2011. View Article : Google Scholar :
|
|
9
|
Jordan VC: Tamoxifen as the first targeted
long-term adjuvant therapy for breast cancer. Endocr Relat Cancer.
21:R235–R246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Briest S and Stearns V: Tamoxifen
metabolism and its effect on endocrine treatment of breast cancer.
Clin Adv Hematol Oncol. 7:185–192. 2009.PubMed/NCBI
|
|
11
|
Lønning PE: Adjuvant endocrine treatment
of early breast cancer. Hematol Oncol Clin North Am. 21:223–238.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arimidex, Tamoxifen, Alone or in
Combination (ATAC) Trialists' Group; Forbes JF, Cuzick J, Buzdar A,
Howell A, Tobias JS and Baum M: Effect of anastrozole and tamoxifen
as adjuvant treatment for early-stage breast cancer: 100-month
analysis of the ATAC trial. Lancet Oncol. 9:45–53. 2008. View Article : Google Scholar
|
|
13
|
Raha P, Thomas S and Munster PN:
Epigenetic modulation: A novel therapeutic target for overcoming
hormonal therapy resistance. Epigenomics. 3:451–470. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thewes V, Simon R, Schroeter P, Schlotter
M, Anzeneder T, Büttner R, Benes V, Sauter G, Burwinkel B,
Nicholson RI, et al: Reprogramming of the ERRalpha and ERalpha
target gene landscape triggers tamoxifen resistance in breast
cancer. Cancer Res. 75:720–731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baneshi MR, Warner P, Anderson N, Edwards
J, Cooke TG and Bartlett JM: Tamoxifen resistance in early breast
cancer: Statistical modelling of tissue markers to improve risk
prediction. Br J Cancer. 102:1503–1510. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baselga J, Campone M, Piccart M, Burris HA
III, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun
F, et al: Everolimus in postmenopausal hormone-receptor-positive
advanced breast cancer. N Engl J Med. 366:520–529. 2012. View Article : Google Scholar
|
|
17
|
Turner NC, Ro J, André F, Loi S, Verma S,
Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, et al:
Palbociclib in hormone-receptor-positive advanced breast cancer. N
Engl J Med. 373:209–219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zagouri F, Liakou P, Bartsch R, Peccatori
FA, Tsigginou A, Dimitrakakis C, Zografos GC, Dimopoulos MA and
Azim HA Jr: Discrepancies between ESMO and NCCN breast cancer
guidelines: An appraisal. Breast. 24:513–523. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen
M, Cheng H, Luo H, Fu W, Li Z and Yang G: Acquisition of
epithelial-mesenchymal transition phenotype in the
tamoxifen-resistant breast cancer cell: A new role for G
protein-coupled estrogen receptor in mediating tamoxifen resistance
through cancer-associated fibroblast-derived fibronectin and
β1-integrin signaling pathway in tumor cells. Breast Cancer Res.
17:692015. View Article : Google Scholar
|
|
20
|
Bui QT, Im JH, Jeong SB, Kim YM, Lim SC,
Kim B and Kang KW: Essential role of Notch4/STAT3 signaling in
epithelial-mesenchymal transition of tamoxifen-resistant human
breast cancer. Cancer Lett. 390:115–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ward A, Balwierz A, Zhang JD, Küblbeck M,
Pawitan Y, Hielscher T, Wiemann S and Sahin Ö: Re-expression of
microRNA-375 reverses both tamoxifen resistance and accompanying
EMT-like properties in breast cancer. Oncogene. 32:1173–1182. 2013.
View Article : Google Scholar
|
|
22
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fallah Y, Brundage J, Allegakoen P and
Shajahan-Haq AN: MYC-driven pathways in breast cancer subtypes.
Biomolecules. 7:E532017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Raha P, Thomas S, Thurn KT, Park J and
Munster PN: Combined histone deacetylase inhibition and tamoxifen
induces apoptosis in tamoxifen-resistant breast cancer models, by
reversing Bcl-2 overexpression. Breast Cancer Res. 17:262015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qi H, Jiang Z, Wang C, Yang Y, Li L, He H
and Yu Z: Sensitization of tamoxifen-resistant breast cancer cells
by Z-ligustilide through inhibiting autophagy and accumulating DNA
damages. Oncotarget. 8:29300–29317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sebaugh JL: Guidelines for accurate
EC50/IC50 estimation. Pharm Stat. 10:128–134.
2011. View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S,
Mooney M, et al: New response evaluation criteria in solid tumours:
Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247.
2009. View Article : Google Scholar
|
|
29
|
Yuan JQ, Wang SM, Tang LL, Mao J, Wu YH,
Hai J, Luo SY, Ou HY, Guo L, Liao LQ, et al: Relative dose
intensity and therapy efficacy in different breast cancer molecular
subtypes: A retrospective study of early stage breast cancer
patients treated with neoadjuvant chemotherapy. Breast Cancer Res
Treat. 151:405–413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han C, Yang L, Choi HH, Baddour J, Achreja
A, Liu Y, Li Y, Li J, Wan G, Huang C, et al: Amplification of USP13
drives ovarian cancer metabolism. Nat Commun. 7:135252016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lindström LS, Karlsson E, Wilking UM,
Johansson U, Hartman J, Lidbrink EK, Hatschek T, Skoog L and Bergh
J: Clinically used breast cancer markers such as estrogen receptor,
progesterone receptor, and human epidermal growth factor receptor 2
are unstable throughout tumor progression. J Clin Oncol.
30:2601–2608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cui J, Germer K, Wu T, Wang J, Luo J, Wang
SC, Wang Q and Zhang X: Cross-talk between HER2 and MED1 regulates
tamoxifen resistance of human breast cancer cells. Cancer Res.
72:5625–5634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gonzalez-Malerva L, Park J, Zou L, Hu Y,
Moradpour Z, Pearlberg J, Sawyer J, Stevens H, Harlow E and LaBaer
J: High-throughput ectopic expression screen for tamoxifen
resistance identifies an atypical kinase that blocks autophagy.
Proc Natl Acad Sci USA. 108:2058–2063. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng R, Liu YJ, Cui JW, Yang M, Liu XL,
Li P, Wang Z, Zhu LZ, Lu SY, Zou L, et al: Aspirin regulation of
c-myc and cyclinD1 proteins to overcome tamoxifen resistance in
estrogen receptor-positive breast cancer cells. Oncotarget.
8:30252–30264. 2017.PubMed/NCBI
|
|
35
|
Cheng AS, Jin VX, Fan M, Smith LT,
Liyanarachchi S, Yan PS, Leu YW, Chan MW, Plass C, Nephew KP, et
al: Combinatorial analysis of transcription factor partners reveals
recruitment of c-MYC to estrogen receptor-alpha responsive
promoters. Mol Cell. 21:393–404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Musgrove EA, Sergio CM, Loi S, Inman CK,
Anderson LR, Alles MC, Pinese M, Caldon CE, Schütte J,
Gardiner-Garden M, et al: Identification of functional networks of
estrogen- and c-Myc-responsive genes and their relationship to
response to tamoxifen therapy in breast cancer. PLoS One.
3:e29872008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cepeda V, Fuertes MA, Castilla J, Alonso
C, Quevedo C and Pérez JM: Biochemical mechanisms of cisplatin
cytotoxicity. Anticancer Agents Med Chem. 7:3–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dobbelstein M and Sørensen CS: Exploiting
replicative stress to treat cancer. Nat Rev Drug Discov.
14:405–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
40
|
Wang Z, Yang B, Zhang M, Guo W, Wu Z, Wang
Y, Jia L, Li S; Cancer Genome Atlas Research Network; Xie W and
Yang D: lncRNA epigenetic landscape analysis identifies EPIC1 as an
oncogenic lncRNA that interacts with MYC and promotes cell-cycle
progression in cancer. Cancer Cell. 33:706–720.e9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Graham J, Pitz M, Gordon V, Grenier D,
Amir E and Niraula S: Clinical predictors of benefit from
fulvestrant in advanced breast cancer: A Meta-analysis of
randomized controlled trials. Cancer Treat Rev. 45:1–6. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma CX, Reinert T, Chmielewska I and Ellis
MJ: Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer.
15:261–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Coates AS, Winer EP, Goldhirsch A, Gelber
RD, Gnant M, Piccart-Gebhart M, Thürlimann B and Senn HJ: Tailoring
therapies-improving the management of early breast cancer: St
Gallen International Expert Consensus on the primary therapy of
early breast cancer 2015. Ann Oncol. 26:1533–1546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
van Agthoven T, Sieuwerts AM, Meijer-van
Gelder ME, Look MP, Smid M, Veldscholte J, Sleijfer S, Foekens JA
and Dorssers LC: Relevance of breast cancer antiestrogen resistance
genes in human breast cancer progression and tamoxifen resistance.
J Clin Oncol. 27:542–549. 2009. View Article : Google Scholar
|
|
46
|
Cardoso F, Costa A, Senkus E, Aapro M,
André F, Barrios CH, Bergh J, Bhattacharyya G, Biganzoli L, Cardoso
MJ, et al: 3rd ESO-ESMO International Consensus Guidelines for
advanced breast cancer (ABC 3). Ann Oncol. 28:31112017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jeselsohn R, Buchwalter G, De Angelis C,
Brown M and Schiff R: ESR1 mutations-a mechanism for acquired
endocrine resistance in breast cancer. Nat Rev Clin Oncol.
12:573–583. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar
|
|
49
|
Lin CY, Lovén J, Rahl PB, Paranal RM,
Burge CB, Bradner JE, Lee TI and Young RA: Transcriptional
amplification in tumor cells with elevated c-Myc. Cell. 151:56–67.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jin K, Park S, Teo WW, Korangath P, Cho
SS, Yoshida T, Győrffy B, Goswami CP, Nakshatri H, Cruz LA, et al:
HOXB7 is an ERα cofactor in the activation of HER2 and multiple ER
target genes leading to endocrine resistance. Cancer Discov.
5:944–959. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Manso L, Mourón S, Tress M, Gómez-López G,
Morente M, Ciruelos E, Rubio-Camarillo M, Rodriguez-Peralto JL,
Pujana MA, Pisano DG and Quintela-Fandino M: Analysis of paired
primary-metastatic hormone-receptor positive breast tumors (HRPBC)
uncovers potential novel drivers of hormonal resistance. PLoS One.
11:pp. e01558402016, View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Miller TW, Balko JM, Ghazoui Z, Dunbier A,
Anderson H, Dowsett M, González-Angulo AM, Mills GB, Miller WR, Wu
H, et al: A gene expression signature from human breast cancer
cells with acquired hormone independence identifies MYC as a
mediator of antiestrogen resistance. Clin Cancer Res. 17:2024–2034.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yin S, Rishi AK and Reddy KB:
Anti-estrogenresistant breast cancer cells are sensitive to
cisplatin plus TRAIL treatment. Oncol Rep. 33:1475–1480. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yde CW, Gyrd-Hansen M, Lykkesfeldt AE,
Issinger OG and Stenvang J: Breast cancer cells with acquired
antiestrogen resistance are sensitized to cisplatin-induced cell
death. Mol Cancer Ther. 6:1869–1876. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Leung EY, Kim JE, Askarian-Amiri M, Joseph
WR, McKeage MJ and Baguley BC: Hormone resistance in Two MCF-7
breast cancer cell lines is associated with reduced mTOR signaling,
decreased glycolysis, and increased sensitivity to cytotoxic drugs.
Front Oncol. 4:2212014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhu H, Wu J, Zhang W, Luo H, Shen Z, Cheng
H and Zhu X: PKM2 enhances chemosensitivity to cisplatin through
interaction with the mTOR pathway in cervical cancer. Sci Rep.
6:307882016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gao T, Han Y, Yu L, Ao S, Li Z and Ji J:
CCNA2 is a prognostic biomarker for ER+ breast cancer and tamoxifen
resistance. PLoS One. 9:pp. e917712014, View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gao A, Sun T, Ma G, Cao J, Hu Q, Chen L,
Wang Y, Wang Q, Sun J, Wu R, et al: LEM4 confers tamoxifen
resistance to breast cancer cells by activating cyclin D-CDK4/6-Rb
and ERα pathway. Nat Commun. 9:41802018. View Article : Google Scholar
|
|
60
|
Kangaspeska S, Hultsch S, Jaiswal A,
Edgren H, Mpindi JP, Eldfors S, Brück O, Aittokallio T and
Kallioniemi O: Systematic drug screening reveals specific
vulnerabilities and co-resistance patterns in endocrine-resistant
breast cancer. BMC Cancer. 16:3782016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jiang D, Sui M, Zhong W, Huang Y and Fan
W: Different administration strategies with paclitaxel induce
distinct phenotypes of multidrug resistance in breast cancer cells.
Cancer Lett. 335:404–411. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Baekelandt MM, Holm R, Nesland JM, Tropé
CG and Kristensen GB: P-glycoprotein expression is a marker for
chemotherapy resistance and prognosis in advanced ovarian cancer.
Anticancer Res. 20:1061–1067. 2000.PubMed/NCBI
|
|
63
|
Wada H, Saikawa Y, Niida Y, Nishimura R,
Noguchi T, Matsukawa H, Ichihara T and Koizumi S: Selectively
induced high MRP gene expression in multidrug-resistant human HL60
leukemia cells. Exp Hematol. 27:99–109. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jacobsen C and Honecker F: Cisplatin
resistance in germ cell tumours: Models and mechanisms. Andrology.
3:111–121. 2015. View Article : Google Scholar
|
|
65
|
Beuvink I, Boulay A, Fumagalli S,
Zilbermann F, Ruetz S, O'Reilly T, Natt F, Hall J, Lane HA and
Thomas G: The mTOR inhibitor RAD001 sensitizes tumor cells to
DNA-damaged induced apoptosis through inhibition of p21
translation. Cell. 120:747–759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhu Y, Liu Y, Zhang C, Chu J, Wu Y, Li Y,
Liu J, Li Q, Li S, Shi Q, et al: Tamoxifen-resistant breast cancer
cells are resistant to DNA-damaging chemotherapy because of
upregulated BARD1 and BRCA1. Nat Commun. 9:15952018. View Article : Google Scholar : PubMed/NCBI
|