Introduction
Multiple ankyrin repeats single KH domain (MASK),
consisting of multiple ankyrin repeat and single KH domains, was
first identified in Drosophila eyes and found to play a role
in cell proliferation, differentiation and survival (1). Ankyrin repeat and KH
domain-containing 1 (ANKHD1), the corresponding orthologous human
protein, was first reported to be expressed in the prostate cancer
cell line LNCap (2). The ankyrin
repeat structure enables its function as a scaffold protein,
mediating protein-protein interactions and regulating gene
transcription, cell cycle, cell survival, and cell signaling
(3,4). For example, the KH domain enables
ANKHD1 to mediate protein-nucleic acid interactions (5), and drives cell proliferation via
specific miRNA interactions (6).
ANKHD1 also interacts with Src homology 2 domain-containing
phosphatase 2 (SHP2) to affect the malignant phenotype of leukemic
cells (7). Importantly, the
expression level of ANKHD1 was reported to correlate with patient
prognosis, with lower expression levels predicting better prognosis
(8). It was recently revealed that
ANKHD1 functions as a potential member of the Hippo signaling
pathway (9), and is involved in
organ growth and maintenance of tissue homeostasis (10). In humans, vital molecules of the
Hippo signaling pathway include yes-associated protein (YAP), large
tumor suppressors 1 and 2 (LATS1/2), mammalian STE-20 kinases 1 and
2 (MST1/2), and Msp-one-binder 1, which are highly conserved and
act as suppressors of tumorigenesis (11,12).
YAP can enter the nucleus and act as a transcriptional activator
via binding to multiple transcriptional factors, including ErbB4,
TEAD1-4 and p73, to regulate gene expression (13-17).
YAP phosphorylation results in its degradation in the cytoplasm,
thereby activating the Hippo pathway (12,18).
Notably, ANKHD1 was found to play a crucial role in the
YAP-mediated Hippo pathway in humans (9,19).
In prostate cancer cells, ANKHD1 expression promotes proliferation
and cell cycle progression by modulating the expression of cyclin
A, followed by activation of YAP (20).
The aim of the present study was to investigate the
role and expression levels of ANKHD1 in non-small-cell lung cancer
(NSCLC) and normal tissues and to determine whether ANKHD1 affects
the proliferation and invasion of NSCLC cells and to elucidate the
underlying mechanism.
Materials and methods
Patients and specimens
A total of 170 tumor specimens, including NSCLC
tissues and 170 paired non-tumor tissues (>5 cm from the edge of
the primary tumor), were collected between January 1999 and
December 2006 at the First Affiliated Hospital of China Medical
University. Written informed consent was obtained from all the
patients, and the procedures were approved by the Institutional
Research Ethics Committee of China Medical University. All
specimens were obtained during surgical resection from patients who
had not received chemotherapy or radiotherapy prior to surgery.
According to the World Health Organization 2015 classification
criteria for lung cancer (21), 93
and 77 patients presented with adenocarcinoma and squamous cell
carcinoma, respectively. According to the International Union of
Cancer 2010 tumor-node-metastasis (TNM) staging standards (22), 73 tumors were classified as stage
I/II and 97 as stage III/IV.
Immunohistochemistry
All tissue blocks were cut into 4-µm sections,
deparaffinized, rehydrated, stained overnight at 4˚C using the
Ultrasensitive TM S-P system (KIT-9710, MaiXin), and incubated with
antibodies against ANKHD1 (1:100, cat. no. ab199164; Abcam) and YAP
(1:100, cat. no. 14074; Cell Signaling Technology, Inc.). The
tissue sections were incubated with secondary antibody labeled with
biotin at 37˚C for 30 min (Ultrasensitive TM S-P, MaiXin).
Diaminobenzidine tetrahy-drochloride substrate (MaiXin) was used as
the chromogen. The intensity of ANKHD1 staining was scored as
follows: 0 (no staining), 1 (weak), 2 (moderate) and 3 (strong).
Percentage scores were assigned as follows: 1 (1-25%), 2 (26-50%),
3 (51-75%) and 4 (76-100%). The scores of each tumor sample were
multiplied to give a final score of 0-12, and positive expression
for tumor samples was defined as scores ≥4; scores 1-4 were
categorized as weak expression, whereas tumors with a score of 0
were considered as negative. Phosphate-buffered saline (PBS) and
goat serum were used as negative controls.
Cell lines
The human bronchial epithelium (HBE) cell line was
obtained from the American Type Culture Collection. The LK2 cell
line was obtained from the Japanese Collection of Research
Bioresources Cell Bank. The PG-LH7 (LH7) cell line was a gift from
Dr Jie Zheng (Department of Pathology, Peking University). The
A549, H1299, BE1, H292 and H460 cell lines were obtained from the
Shanghai Cell Bank. All cells were cultured in RPMI-1640
(Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific,
Inc.), 100 IU/ml penicillin (Sigma-Aldrich; Merck KGaA), and 100
µg/ml streptomycin (Sigma-Aldrich; Merck KGaA) at 37˚C in 5%
CO2.
Western blotting
Cells were harvested during the exponential phase.
Total protein from cells was extracted in lysis buffer (Pierce;
Thermo Fisher Scientific, Inc.) and quantified using the Bradford
method. Equal amounts (50 µg) of total protein extracts were
subjected to 6-10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and then transferred to a polyvinylidene fluoride
membrane (EMD Millipore). The membrane was blocked with 5% non-fat
milk for 1 h and incubated with antibodies against ANKHD1 (1:500,
cat. no. ab117788); LATS1 (1:100, cat. no. sc398560; Santa Cruz
Biotechnology, Inc.) or LATS1 (1:500, cat. no. 9153; Cell Signaling
Technology, Inc.); phosphorylated (p)-LATS1 (1:500, cat. no. 9157;
Cell Signaling Technology, Inc.); YAP (1:500, cat. no. 14074; Cell
Signaling Technology, Inc.); p-YAP (Ser127) (1:500, cat. no. 4911;
Cell Signaling Technology); MST1 (1:500, cat. no. 3682; Cell
Signaling Technology, Inc.); p-MST1 (1:500, cat. no. 49332; Cell
Signaling Technology, Inc.); connective tissue growth factor (CTGF;
1:100, cat. no. sc14940; Santa Cruz Biotechnology, Inc.); cyclin D1
(1:500, cat. no. 2922; Cell Signaling Technology, Inc.); or GAPDH
(1:2,000, cat. no. G8795; Sigma-Aldrich; Merck KGaA) overnight at
4˚C. The membrane was then incubated with goat anti-mouse or
anti-rabbit secondary antibody at 37˚C for 2 h. Protein bands were
visualized with enhanced chemiluminescence and detected using a
bioimaging system (UVP, LLC). All experiments were performed in
triplicate.
Plasmids and transfection
Plasmid pCMV6-Myc/DDK-ANKHD1 (no. RC221886) was
purchased from OriGene Technologies, Inc. Small interfering RNA
(siRNA)-ANKHD1 (sc-92073), ANKHD1 short hairpin RNA (shRNA) plasmid
(sc-92073-SH), and siRNA-YAP (sc-38637) were purchased from Santa
Cruz Biotechnology, Inc. The plasmids or siRNAs mentioned above
were transfected into cells using Lipofectamine 3000 transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
Immunofluorescence staining
The cells were fixed for 30 min at room temperature
in 4% paraformaldehyde in PBS, permeabilized with Triton X-100, and
then blocked with 1% bovine serum albumin for 1 h at room
temperature. The cells were incubated with antibody against ANKHD1
(1:100, cat. no. ab199164; Abcam) or YAP (1:100, cat. no. 14074;
Cell Signaling Technology, Inc.). After washing three times (5 min
per wash) at room temperature with PBS, the cells were incubated
with fluorescein-isothiocyanate-conjugated secondary antibody (cat.
no. ZF-0311; ZsBio Technology). The nuclei were counterstained at
25˚C for 5 min with DAPI (cat. no. C1005; Beyotime Institute of
Biotechnology).
MTT assay
At 24 h after transfection, the cells were plated in
a 96-well plate in medium containing 10% FBS at ~3,000 cells/well,
and viability was quantified by the MTT assay. MTT (5 mg/ml
solution, 20 µl) was added to each well and incubated for 4 h at
37˚C; subsequently, the medium was removed from each well, and the
resultant MTT formazan was solubilized in 150 µl dimethyl
sulfoxide. All experiments were performed in triplicate. The
results were quantified spectrophotometrically using a test
wavelength of 490 nm.
Matrigel invasion assay
A 24-well Transwell chamber was used with a pore
size of 8 µm (Costar; Corning, Inc.), and the inserts were coated
with Matrigel (BD Biosciences) in serum-free medium. At 48 h after
transfection, cells (100 µl) were trypsinized and transferred to
the upper Matrigel chamber in serum-free medium containing
5x105 cells and incubated for 16 h. The non-invading
cells on the upper membrane surface were removed, and the cells
that passed through the filter were fixed with 4% paraformaldehyde
for 15 min and stained for 10 min with hematoxylin at room
temperature. The number of invading cells was counted in 10
randomly selected high-power fields (magnification, x400) under an
Olympus IX73 inverted microscope (Olympus Corporation). The data
are representative of three individual wells.
Colony formation assay
At 48 h after transfection, cells were plated in
6-cm cell culture dishes (1,000 cells/dish) and incubated for 14
days. The cells were then stained for 20 min at room temperature
with Giemsa and the number of colonies (>50 cells) was
determined.
Immunoprecipitation
For immunoprecipitation, sufficient antibody was
added to 200 mg protein and gently rotated overnight at 4˚C. The
immunocomplex was captured by adding 25 ml protein A/G agarose
beads (Beyotime Institute of Biotechnology) and gently rotating for
3 h at 4˚C. Following centrifugation at 1,500 x g for 5 min at 4˚C,
the supernatant was discarded. The precipitate was washed three
times with ice-cold radioimmunoprecipitation assay buffer,
resuspended in sample buffer, and boiled for 5 min to dissociate
the immu-nocomplex from the beads. The supernatant was then
collected by centrifugation at 10,000 x g for 10 min at 4˚C and
subjected to western blot analysis.
RNA extraction and quantitative PCR
(qPCR) analysis
Total RNA was extracted from cells using RNeasy Plus
Mini Kit (Qiagen GmbH), and 1 µg RNA was reverse-transcribed (37˚C
for 15 min, 85˚C for 5 sec and 4˚C for 5 min) using the Prime
Script TM RT Master Mix (Takara Biotechnology Co., Ltd.). qPCR was
performed with a 20-µl reaction mixture using SYBR Green PCR Master
Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.) on an
ABI7900 system (Applied Biosystems; Thermo Fisher Scientific, Inc.)
as follows: 50˚C for 2 min, 95˚C for 10 min, and 95˚C for 40 sec
(40 cycles), and then 60˚C for 60 sec. GAPDH was used as an
internal control, and the mRNA values were normalized to GAPDH; all
experiments were performed in triplicate.
The following specific primer sequences were used:
ANKHD1: Forward (F): 5'-AGACCAATCGGAACACGGCT CT-3' and reverse (R):
5'-CAGAAGCTGCTTCCATCAAG GG-3'; YAP: F: 5'-TGTCCCAGATGAACGTCACAGC-3'
and R: 5'-TGGTGGCTGTTTCACTGGAGCA-3'; and GAPDH: F:
5'-GTCTCCTCTGACTTCAACAGCG-3' and R: 5'-ACCACC
CTGTTGCTGTAGCCAA-3'.
Xenograft model of tumorigenesis
All experiments with nude mice were performed
according to the guidelines of China Medical University for the use
of experimental animals. The study was approved by the
Institutional Animal Research Committee of China Medical
University. A total of 12 4-week-old female BALB/c nude mice were
purchased from Charles River Laboratories (Beijing, China). The
mice were kept in a sterile laboratory at a constant temperature of
25±1˚C and at a constant humidity of 45-50% and were fed inside the
laminar air flow rack. Each mouse was subcutaneously inoculated
with 5x106 tumor cells in 0.2 ml sterile PBS in the
axillary area. Six weeks after inoculation, the mice were
euthanized and autopsied to examine tumor growth and
dissemination.
Statistical analysis
SPSS version 22.0 (IBM Corp.) was used for all
analyses. The χ2 test was used to assess possible
associations between ANKHD1 expression and clinicopathological
factors. The Kaplan-Meier method was used to estimate the
probability of patient survival. The Cox regression model was used
for multivariate analysis. Differences between two groups were
assessed with Student's t-test. All P-values were based on a
two-sided statistical analysis, and P<0.05 was considered to
indicate statistically significant differences.
Results
ANKHD1 is upregulated in NSCLC tissues
and cells
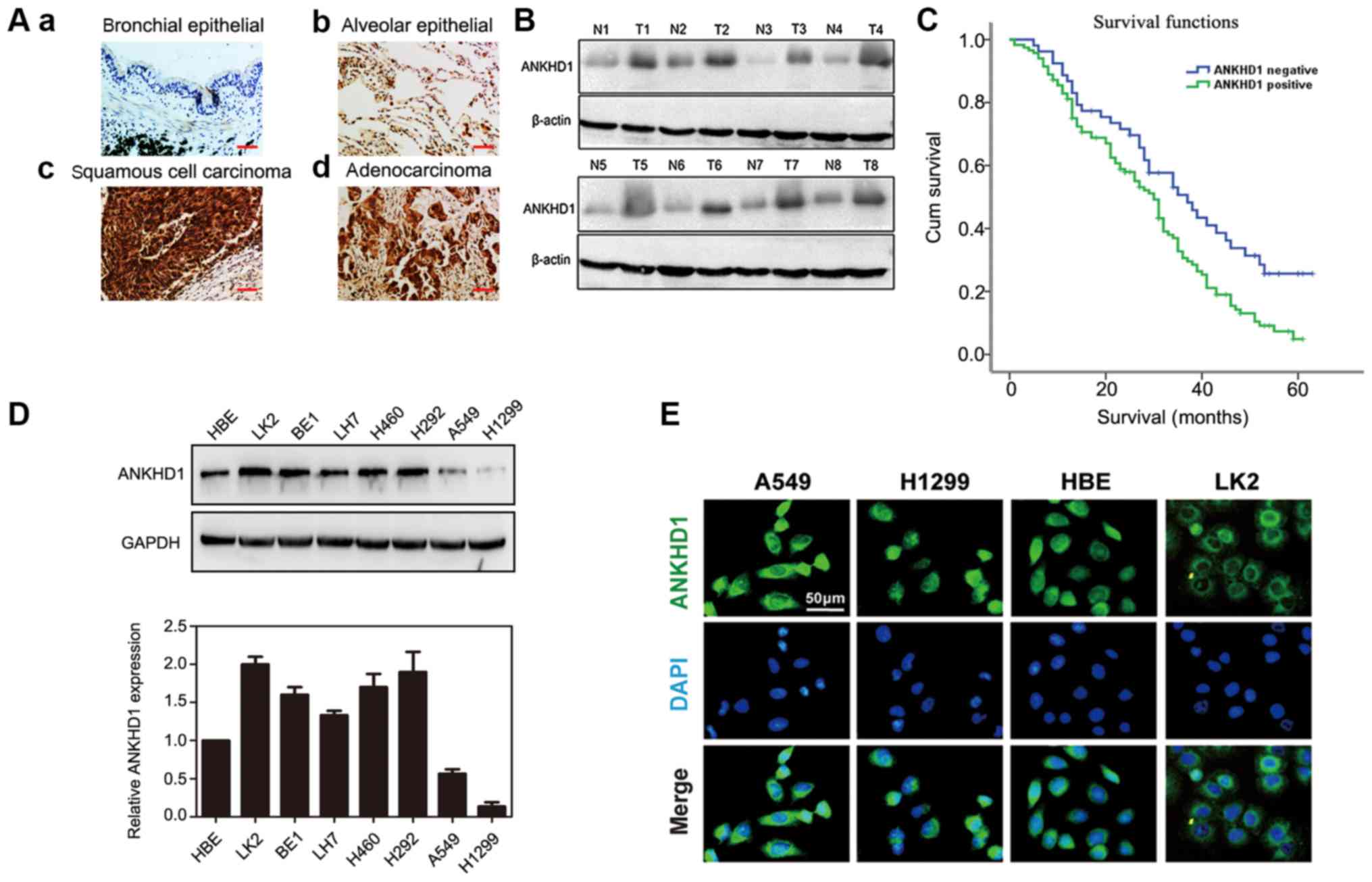
Immunohistochemical staining demonstrated that the
expression of ANKHD1 was significantly higher in NSCLC specimens
compared with that in normal tissues (117/170 vs. 59/170,
respectively; P=0.000; Table I).
In the bronchial and alveolar cells of normal tissue, ANKHD1 was
negative or weakly expressed [Fig. 1A
(a and b)]. By contrast, ANKHD1 was strongly expressed in the
cytoplasm and nuclei of NSCLC tissues [Fig. 1A (c and d)]. Consistently, a higher
ANKHD1 protein level was also observed in tumor tissues compared
with adjacent normal tissues (n=8, Fig. 1B). ANKHD1 transcript and protein
levels were also higher in NSCLC cell lines compared with HBE cells
(LK2, BE1, LH7, H460, H292, A549 and H1299; Fig. 1D). To assess the localization of
ANKHD1 in cells, immunofluorescence staining was utilized to
visualize ANKHD1 in A549, H1299, HBE and LK2 cells. Consequently,
ANKHD1 protein was shown to be clearly expressed and localized in
the cytoplasm and nuclei (Fig.
1E).
 | Table IExpression pattern of ANKHD1 in
normal lung and NSCLC tissues. |
Table I
Expression pattern of ANKHD1 in
normal lung and NSCLC tissues.
| N | ANKHD1, n(%)
| P-value |
|---|
| Negative | Positive |
|---|
| Normal | 170 | 111 (65.3) | 59 (34.7) | 0.000 |
| Tumor | 170 | 53 (31.2) | 117 (68 .8) | |
Expression of ANKHD1 is associated with
clinical factors
The association between ANKHD1 expression and
clinicopathological factors was investigated in patients with
NSCLC. The expression of ANKHD1 was found to be significantly
associated with advanced pathological TNM (pTNM) stage and the
presence of lymph node metastasis, but not with age, sex,
histological type, or degree of differentiation and tumor size
(Table II). Moreover,
Kaplan-Meier survival analysis revealed that the overall survival
rate of the ANKHD1-positive group was significantly lower compared
with that of the ANKHD1-negative group, suggesting that a high
level of ANKHD1 may be associated with poor prognosis (P=0.037,
log-rank test; Fig. 1C).
Univariate analysis was then performed, which demonstrated that
ANKHD1 expression and lymph node metastasis were significant
prognostic factors for NSCLC (positive ANKHD1 expression: Hazard
ratio 1.698, P=0.008; lymph node metastasis: Hazard ratio 1.797,
P=0.001). Multivariate analysis using a Cox regression model also
indicated that ANKHD1 and lymph node metastasis were independent
prognostic factors in patients with NSCLC (Table III).
| Table IIAssociation of ANKHD1 expression with
clinical and pathological factors in NSCLC. |
Table II
Association of ANKHD1 expression with
clinical and pathological factors in NSCLC.
| Clinicopathological
factors | N | ANKHD1, n (%)
| P-value |
|---|
| Negative | Positive |
|---|
| Total | 170 | 53 (31.2) | 117(68.8) | |
| Age, years | | | | 0.615 |
| ≤55 | 66 | 19 (11.2) | 47 (27.6) | |
| >55 | 104 | 34 (20.0) | 70 (41.2) | |
| Sex | | | | 0.740 |
| Male | 94 | 28 (16.5) | 66 (38 .8) | |
| Female | 76 | 25 (14.7) | 51 (30.0) | |
| Histological
type | | | | 0.743 |
| Squamous cell
carcinoma | 77 | 20(11.8) | 57 (33.5) | |
|
Adenocarcinoma | 93 | 33 (19.4) | 60 (35.3) | |
| Grade | | | | 0.188 |
| Well
differentiated | 44 | 10 (5.9) | 34 (20.0) | |
| Moderately/poorly
differentiated | 126 | 43 (25.3) | 83 (48.8) | |
| TNM stage | | | | 0.019 |
| I and II | 73 | 30 (17.6) | 43 (25.3) | |
| III and IV | 97 | 23 (13.5) | 74 (43.5) | |
| Tumor size, cm | | | | 0.862 |
| <3 | 58 | 19 (11.2) | 39 (22.9) | |
| ≥3 | 112 | 34 (20.0) | 78 (45.9) | |
| Lymph node
metastasis | | | | 0.020 |
| Yes | 81 | 17 (10.0) | 64 (37.6) | |
| No | 89 | 36 (21.2) | 53 (31.2) | |
| Table IIIUnivariate and multivariate analysis
for predictive factors in patients with NSCLC. |
Table III
Univariate and multivariate analysis
for predictive factors in patients with NSCLC.
| Clinicopathological
characteristics | Hazard ratio (95%
Cl) | P-value |
|---|
| Univariate
analysis |
| Age >55
years | 0.929
(0.652-1.323) | 0.682 |
| Male sex | 0.941
(0.665-1.331) | 0.730 |
|
Adenocarcinoma |
1.238(0.875-1.752) | 0.227 |
| Poor
differentiation |
1.155(0.786-1.697) | 0.464 |
| High TNM
stage |
0.993(0.701-1.406) | 0.968 |
| Positive lymph
node metastasis | 1.797
(1.271-2.540) | 0.001 |
| Positive ANKHD1
expression |
1.698(1.149-2.509) | 0.008 |
| Multivariate
analysis |
| Positive lymph
node metastasis | 2.299
(1.449-3.648) | 0.000 |
| Positive ANKHD1
expression |
1.564(1.032-2.370) | 0.035 |
ANKHD1 promotes the proliferation and
invasion of NSCLC cells
To further test our hypothesis, exogenous expression
vectors were transfected to achieve ANKHD1 overexpression in A549
cells, and RNA interference was used to deplete endogenous ANKHD1
in LK2 cells. Exogenous ANKHD1 significantly promoted colony
formation in A549 cells (ANKHD1 vs. control: 284±28 vs. 140±20,
respectively; P<0.05). Conversely, depleting endogenous ANKHD1
in LK2 cells reduced colony formation (negative siRNA vs.
siRNA-ANKHD1: 123±9 vs. 60±3, respectively; P<0.05; Fig. 2A). In the Transwell assay, the
invasive ability of ANKHD1-overexpressing A549 cells was
significantly enhanced compared with that of the control group
(ANKHD1 vs. control: 134±5 vs. 43±6, respectively; P<0.05). By
contrast, knockdown of ANKHD1 reduced the invasive ability of LK2
cells (negative siRNA vs. si-ANKHD1: 144±8 vs. 76±4, respectively;
P<0.05; Fig. 2B).
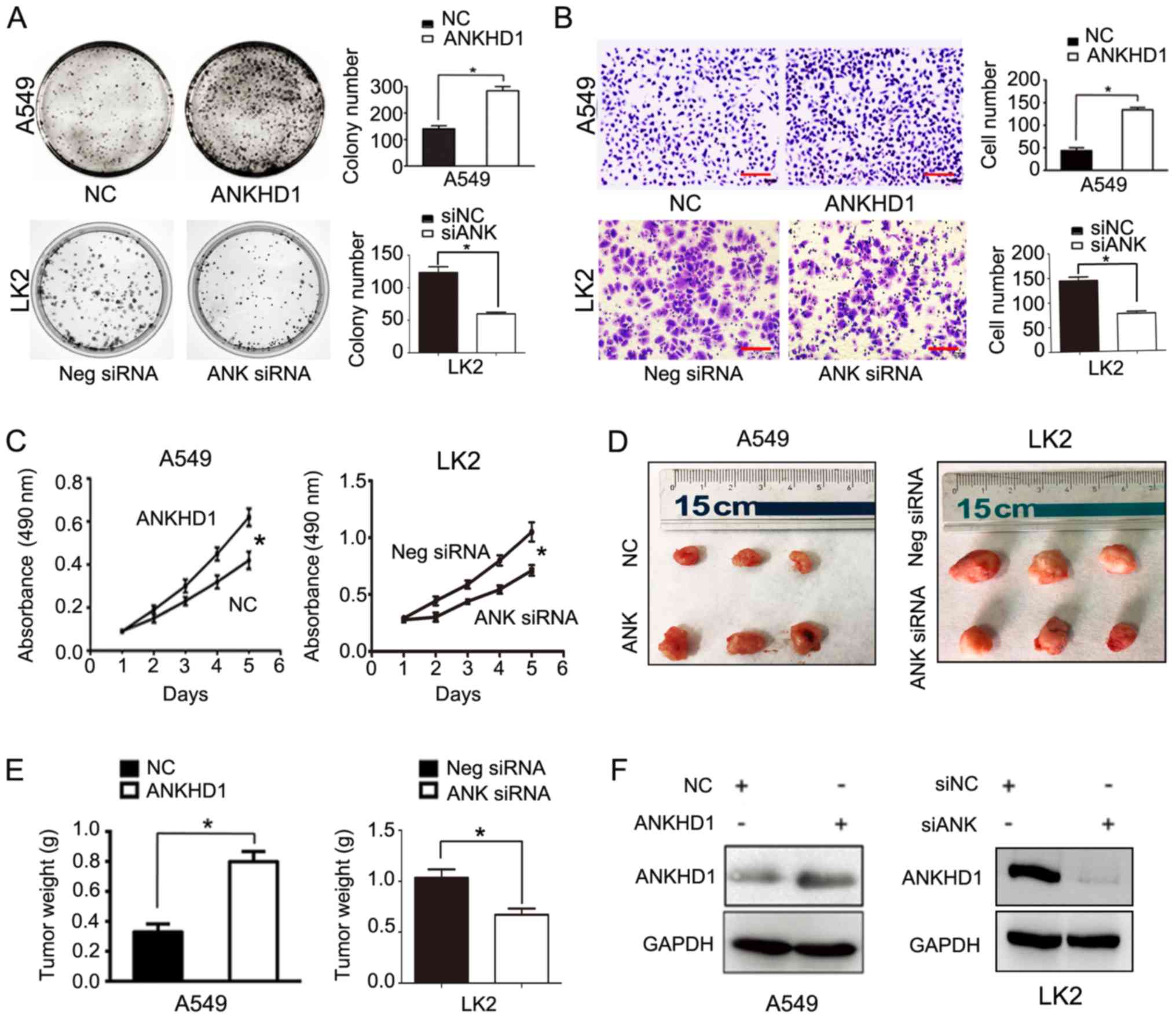 | Figure 2Role of ANKHD1 in NSCLC cell
proliferation and invasion; *P<0.05. (A) Colony
formation assay demonstrated that ANKHD1 overexpression in A549
cells promoted colony formation, whereas ANKHD1 depletion inhibited
colony formation in LK2 cells. (B) Transwell assay demonstrated
that ANKHD1 over-expression in A549 cells promoted invasion,
whereas ANKHD1 depletion inhibited invasion in LK2 cells
(magnification, x100; scale bar, 100 µm). (C) MTT assay also
demonstrated that ANKHD1 overexpression in A549 cells increased
proliferation, whereas ANKHD1 depletion decreased proliferation in
LK2 cells. (D) ANKHD1 regulated NSCLC growth in vivo. Mice
that received A549 cells stably expressing ANKHD1 (bottom row, G418
screening) exhibited an increase in tumor weight compared with the
control group (upper row), whereas animals that received LK2 cells
transduced with lentiviral shRNA-ANKHD1 (upper row, G418 screening)
exhibited a reduction in tumor weight compared with the control
group (bottom row). (E) ANKHD1-expressing A549 cells exhibited more
progressive tumor growth in the nude mice (n=3) compared with the
control group (n=3). Consistently, the ANKHD1-depleted LK2 cell
inoculation resulted in lower proliferative ability in the nude
mice (n=3) compared with the control group (n=3). (F) Transfection
of plasmid pCMV6-Myc/DDK-ANKHD1 for ANKHD1 overexpression in A549
cells, compared with negative plasmid. siRNA-ANKHD1 to knockdown
the expression of ANKHD1 in LK2 cells, compared with negative
plasmid. The transfection efficiency was determined by western
blotting. ANKHD1, ankyrin repeat and KH domain-containing 1; NSCLC,
non-small-cell lung cancer. |
Similarly, the MTT test demonstrated that the
proliferation rate of the ANKHD1-expressing A549 cells was
significantly higher compared with that of the control group
(ANKHD1 vs. control: 0.613±0.077 vs. 0.394±0.048, respectively;
P<0.05). Consistently, knockdown of ANKHD1 decreased the
proliferation rate of LK2 cells (negative siRNA vs. siRNA-ANKHD1:
1.050±0.084 vs. 0.712±0.04, respectively; P<0.05; Fig. 2C). The effects of ANKHD1 on the
growth of NSCLC cells were also determined in vivo using a
nude mouse xenograft NSCLC model. A549 cells stably expressing
ANKHD1 were generated and subcutaneously injected into nude mice.
Six weeks later, ANKHD1-expressing A549 cells exhibited more
progressive tumor growth in the nude mice compared with the control
group (ANKHD1 vs. control group: 0.798±0.068 vs. 0.330±0.052,
respectively; P<0.05; n=3). Consistently, the ANKHD1-depleted
LK2 cells exhibited a lower proliferation ability in nude mice
compared with the control group (negative siRNA vs. siRNA-ANKHD1:
1.34±0.048 vs. 0.671±0.035, respectively; P<0.05; n=3).
Collectively, these results indicate that ANKHD1 regulates the
proliferation of NSCLC cells in vivo (Fig. 2D and E). The ANKHD1 protein levels
are shown in Fig. 2F.
ANKHD1 inactivates the Hippo pathway by
upregulating YAP expression and inhibiting its phosphorylation in
NSCLC cells
To elucidate the mechanism underlying the
ANKHD1-dependent regulation of NSCLC cell proliferation, downstream
molecules involved with ANKHD1 in A549 or LK2 cells were
investigated. The CTGF and cyclin D1 proteins, which are both
involved in the Hippo signaling pathway, were found to be
downregulated in LK2 cells and upregulated in ANKHD1-overexpressing
A549 cells (Fig. 3A), suggesting
that ANKHD1 is involved in the Hippo signaling pathway. Notably,
during activation of the Hippo pathway, the phosphorylated kinase
MST1/2 and LATS1/2 are sequentially activated, and the latter can
phosphorylate YAP to p-YAP, resulting in its cytoplasmic retention
and degradation (23,24). When Hippo signaling is lost,
non-phosphorylated YAP enters the nucleus to act as a synergistic
transcription factor. Therefore, we sought to determine whether
ANHKD1 is involved in YAP/p-YAP during Hippo signaling. In A549
cells, it was observed that the YAP protein was upregulated by
ANKHD1 overexpression, whereas it was decreased in the absence of
endogenous ANKHD1 (Fig. 3A). By
contrast, p-YAP was decreased (Fig.
3B) and YAP was increased in the presence of ANKHD1
overexpression, suggesting that ANKHD1 regulates the transition
between YAP and p-YAP. When endogenous ANKHD1 was depleted, it was
consistently observed that the YAP protein was downregulated in the
LK2 cells, whereas the levels of p-YAP were increased. However, no
significant changes in LATS1, p-LATS1, MST, or p-MST were detected
(Fig. 3B). Thus, these results
suggest that ANKHD1 inactivates the Hippo signaling pathway via
YAP.
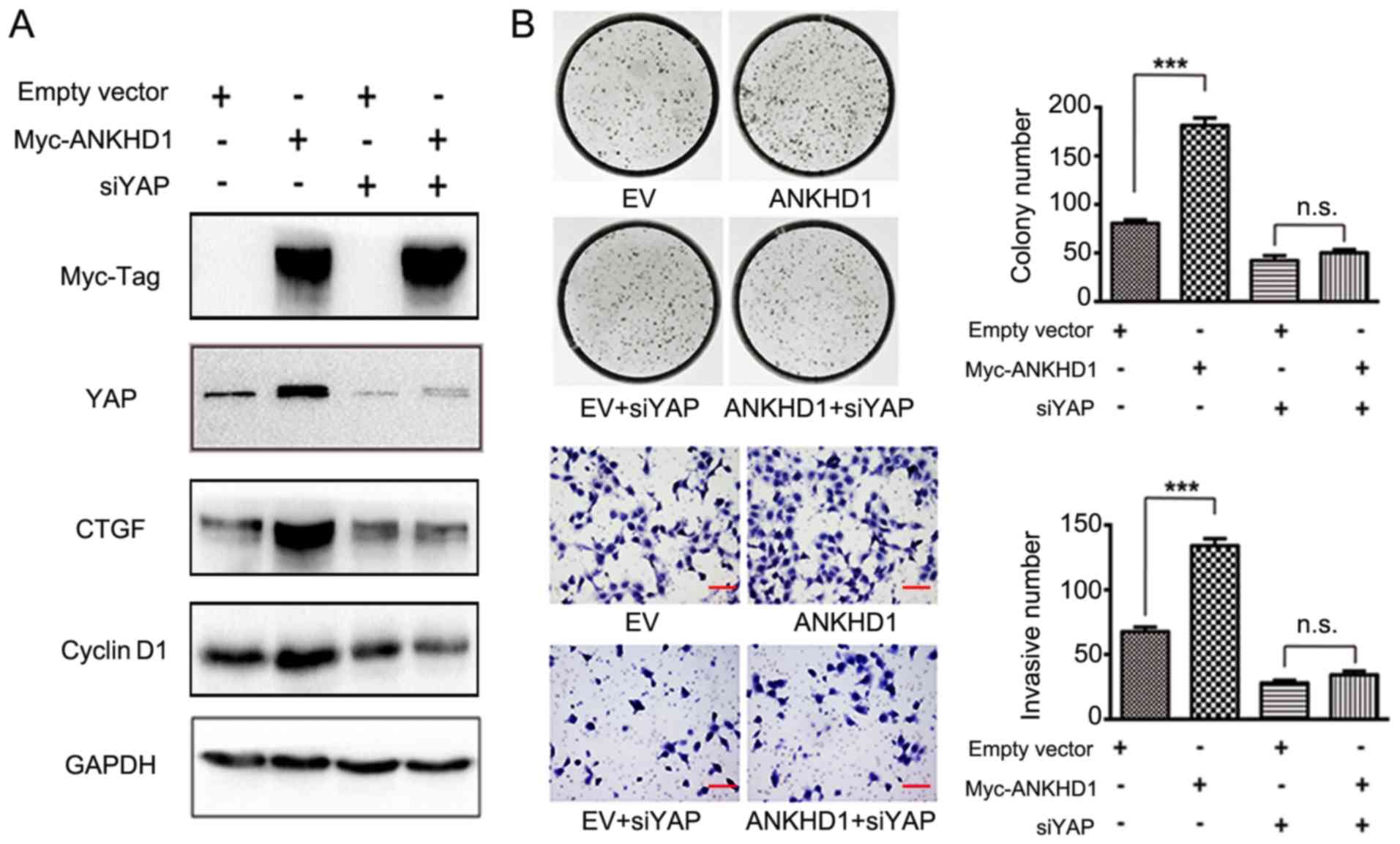
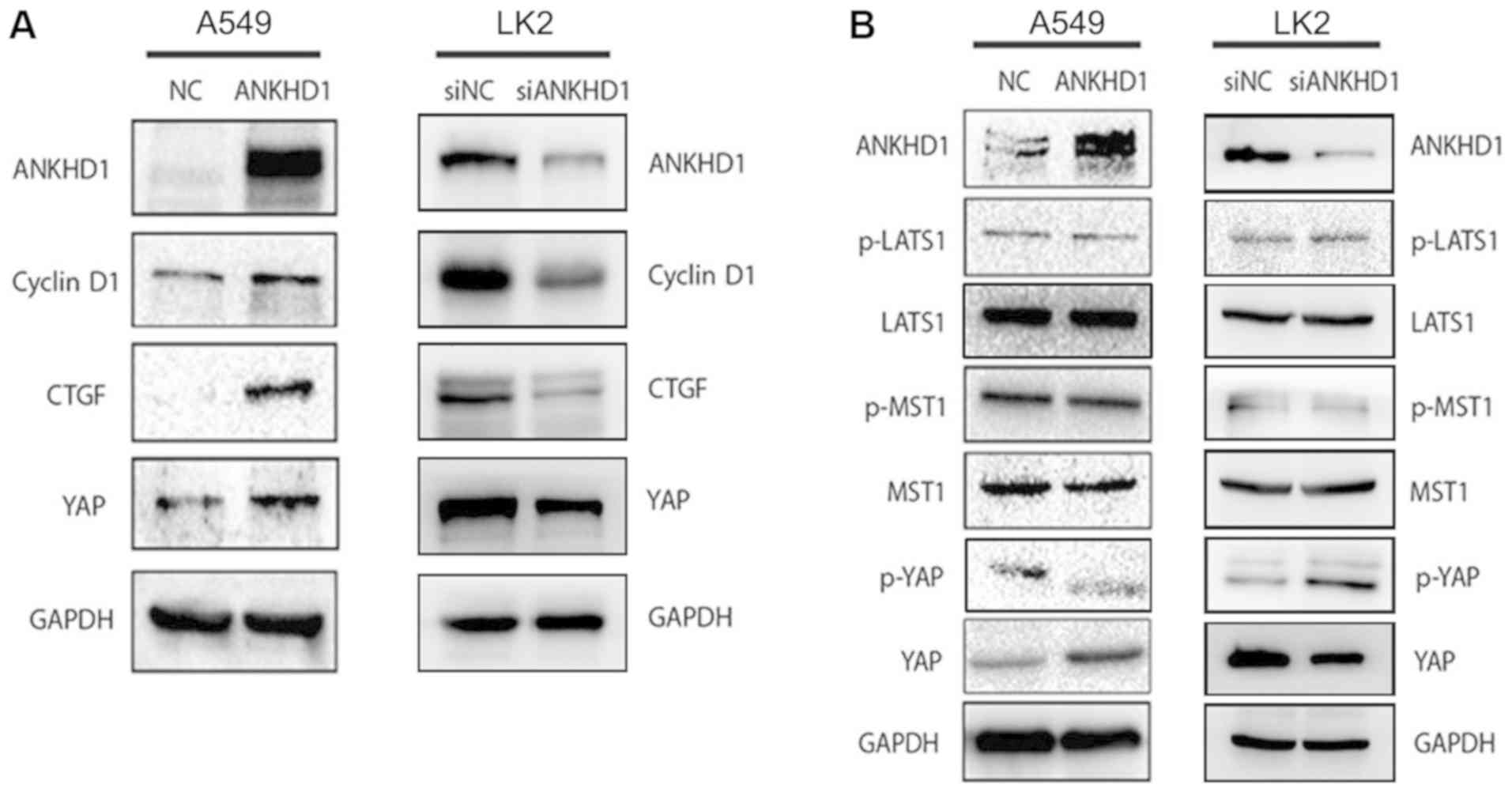 | Figure 3ANKHD1 regulates the activity of the
Hippo pathway. (A) Overexpression of ANKHD1 in A549 cells resulted
in increased expression of total YAP and the downstream effectors
CTGF and cyclin D1. Depletion of ANKHD1 in LK2 cells resulted in a
decrease in total YAP, as well as CTGF and cyclin D1.(B) YAP
expression was upregulated in A549 cells after ANKHD1
overexpression. Conversely, p-YAP expression was downregulated. YAP
expression was downregulated after knockdown of ANKHD1 in LK2 cells
but p-YAP expression was upregulated. There were no significant
changes in LATS1, p-LATS1, MST, or p-MST in A549 or LK2 cells
following either overexpression or knockdown of ANKHD1. ANKHD1,
ankyrin repeat and KH domain-containing 1; YAP, yes-associated
protein; CTGF, connective tissue growth factor; LATS, large tumor
suppressor; MST, mammalian STE-20 kinase. |
Subsequently, it was examined whether ANKHD1
inactivates Hippo signaling via directing p-YAP to YAP transition.
CTGF and cyclin D1 are downstream effectors of the Hippo signaling
pathway (25). YAP was knocked
down in the presence of ANKHD1 in A549 cells, and it was observed
that depletion of YAP resulted in decreased levels of the CTGF and
cyclin D1 proteins only in the presence of ANKHD1 (Fig. 4A). However, in the absence of
ANKHD1, there were no effects of YAP silencing, thereby supporting
the hypothesis that the effect of ANKHD1 on the Hippo signaling
pathway depends on YAP. In the colony formation and Transwell
assays, in the presence of ANKHD1, knockdown of YAP in H1299 cells
(empty vector vs. ANKHD1: 84±4 vs. 180±9, respectively; P<0.001;
empty vector + siYAP vs. ANKHD1 + siYAP: 43±7 vs. 56±5,
respectively; P>0.05; empty vector vs. ANKHD1: 68±4 vs. 134±6,
respectively; P<0.05; empty vector + siYAP vs. ANKHD1 + siYAP:
28±2 vs. 34±2, respectively; P>0.05) consistently alleviated the
effects of ANKHD1 on cell proliferation and invasion (Fig. 4B), suggesting that YAP is necessary
for ANKHD1. Taken together, these results suggest that the
ANKHD1-induced proliferation and invasion of NSCLC cells is
mediated by YAP through Hippo signaling.
ANKHD1 may lead to increased YAP
transcription
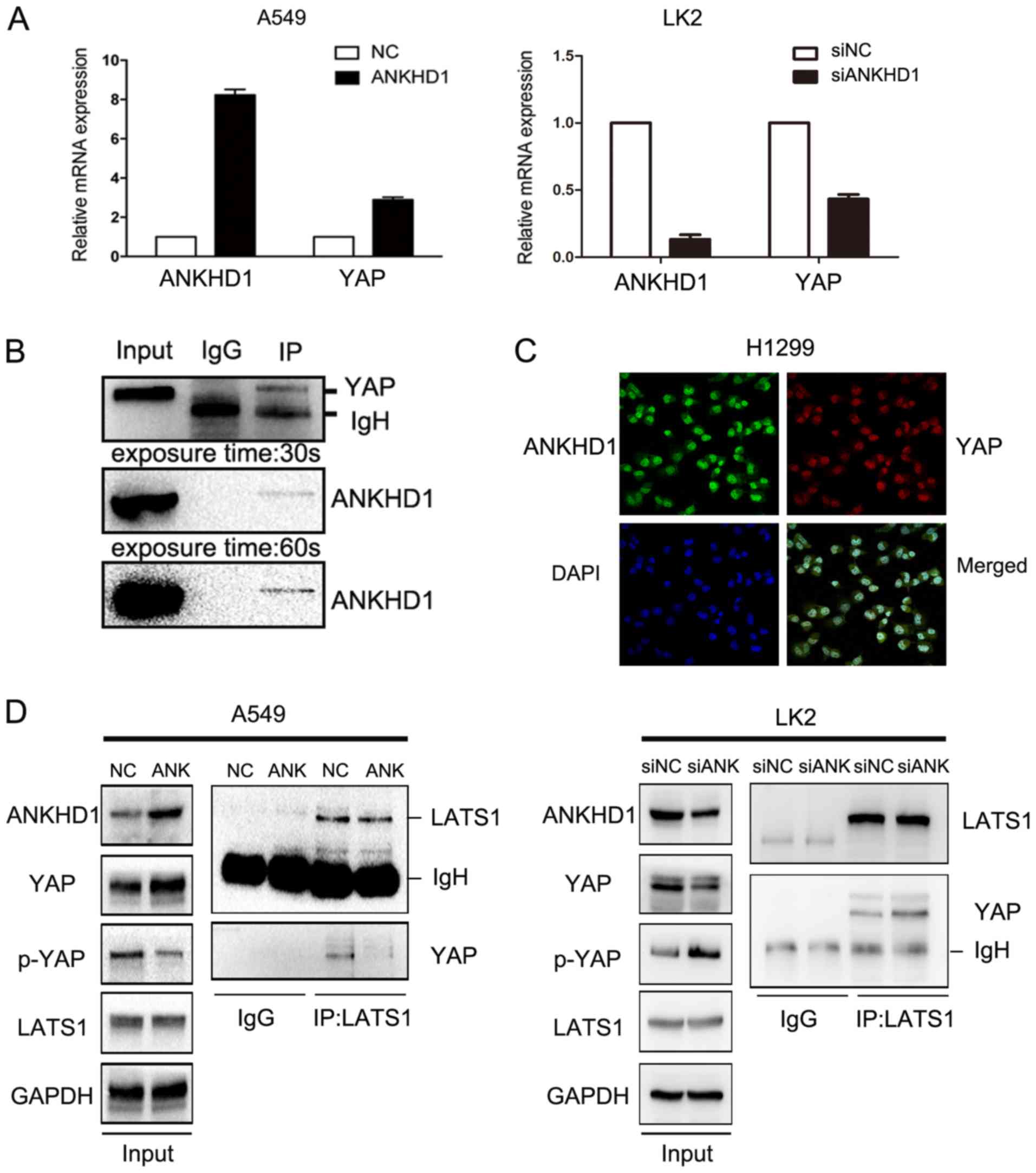
To further investigate the potential mechanism
through which ANKHD1 increased the levels of YAP protein, YAP mRNA
expression was examined in the presence or absence of ANKHD1. In
A549 cells, we observed a significant YAP mRNA upregulation
compared with the control group (P<0.05) in the presence of
ANKHD1. By contrast, in the LK2 cell line, YAP mRNA was
downregulated when endogenous ANKHD1 was knocked down (P<0.05;
Fig. 5A). To confirm these
results, immunohistochemical staining was performed and revealed
that the levels of the ANKHD1 protein in NSCLC specimens were
correlated with YAP protein levels (117/170 vs. 114/170,
respectively; P=0.000; Table IV,
Fig. S1). These findings suggest
that ANKHD1 increased YAP expression.
| Table IVCorrelation between the expression of
ANKHD1 and YAP in NSCLC. |
Table IV
Correlation between the expression of
ANKHD1 and YAP in NSCLC.
| ANKHD1 | YAP
| Spearman's
correlation | P-value
(two-tailed) |
|---|
| Negative | Positive | Total |
|---|
| Negative | 28 | 25 | 53 | | |
| Positive | 28 | 89 | 117 | 0.285 | 0.000 |
| Total | 56 | 114 | 170 | | |
ANKHD1 may inhibit phosphorylation of the
YAP protein
The previous results clearly demonstrated that
ANKHD1 inhibited YAP phosphorylation. Therefore, to explore the
mechanism of action of ANKHD1, an immunoprecipitation assay was
performed, and it was observed that ANKHD1 bound to YAP (Fig. 5B). Immunofluorescence staining
demonstrated that ANKHD1 co-localized with YAP, and this
association was predominantly observed in the nuclei (Fig. 5C). We also investigated whether
ANKHD1 affected the interaction between LATS1 and YAP, and observed
that ANKHD1 decreased this binding between LATS1 and YAP, despite
higher levels of YAP protein being induced by ANKHD1 overexpression
(Fig. 5D). Consistently, knockdown
of ANKHD1 promoted binding of LATS1 to YAP, despite the lower
levels of YAP. Therefore, ANKHD1 may inactivate the Hippo pathway
by regulating YAP binding with LATS1.
Discussion
The Hippo pathway controls organ size in several
species, and deregulation of this pathway is involved in a broad
range of human cancers (10,11).
Over the last few decades, the role of the Hippo pathway in cancer
has attracted considerable attention. Previous studies have
demonstrated that the ANKHD1 protein is highly expressed in the
stomach, small intestine and liver, but its expression is low in
the spleen, lung and kidney (7).
Furthermore, the expression of ANKHD1 is high in the blood cells of
patients with multiple myeloma and leukemia (7,20,26-28).
However, only few studies have investigated the role of ANKHD1 in
solid cancers to date (21).
In the present study, the expression of ANKHD1 was
first determined in NSCLC tissues, and significantly higher
expression of ANKHD1 was observed in NSCLC tissues compared with
that in adjacent normal tissues. Immunohistochemical staining also
revealed that the expression of ANKHD1 in tumor tissues was
significantly higher compared with that in normal tissues. Further
analysis of the clinicopatho-logical characteristics demonstrated
that the upregulation of ANKHD1 in NSCLC was significantly
associated with higher pathological stage and lymph node
metastasis. Positive expression of ANKHD1 was found to be
associated with poor prognosis, which is consistent with a previous
study of MASK1 gene expression in breast cancer patients, with low
expression levels indicating a better prognosis (8). Hence, it was hypothesized that ANKHD1
may play a role in NSCLC progression. Previous studies reported
that ANKHD1 was highly expressed in a leukemic cell line, prostate
cancer cells and myeloma cells (7,20,26-28).
Similarly, our data demonstrated that the ANKHD1 mRNA and protein
levels were also significantly upregulated in NSCLC cells. These
results were supported by the findings of immunohistochemical
staining in NSCLC specimens. Moreover, silencing ANKHD1 inhibited
the proliferation and invasion of NSCLC cells in vitro and
in vivo, in accordance with previous findings (20). Furthermore, overexpression of
ANKHD1 promoted the proliferation and invasion of NSCLC cells in
vitro, and their proliferation ability was also increased in
vivo.
It has previously been reported that ANKHD1 and/or
its ortholog protein, MASK, are involved in the Hippo, SHP2 and
JAK2/STAT signaling pathways (7-9,19,28-30).
The present study demonstrated that the expression of ANKHD1 was
upregulated in NSCLC, in which the Hippo signaling pathway was
inactivated. To investigate the impact of ANKHD1 on the Hippo
signaling pathway, the expression levels of its core effectors were
determined. There were no obvious changes in the expression levels
of MST1, p-MST1, LATS1, or p-LATS1, whereas upregulation of ANKHD1
increased the levels of YAP and simultaneously decreased the levels
of p-YAP. In mammals, CTGF is a direct target gene of YAP, and it
is associated with cell proliferation (31). ANKHD1 increases the levels of the
Hippo pathway downstream target proteins cyclin D1 and CTGF. It was
hypothesized that the effects of ANKHD1 on the Hippo signaling
pathway may be due to its effect on the YAP protein, a potential
cancer-promoting factor in several types of cancers, including lung
cancer (32-37). YAP is highly expressed in the
nucleus and can promote cell proliferation, resulting in
inactivation of the Hippo pathway. In leukemia cells, YAP is
expressed at low or undetectable levels; thus, the effects of
ANKHD1 silencing on leukemia cell growth and clonogenicity may not
be associated with Hippo (38,39).
ANKHD1 has been reported to play an oncogenic role in a prostate
cancer cell line and breast cancer, which is mediated by YAP
(20). We herein investigated
whether ANKHD1 activity in the Hippo signaling pathway was
dependent on YAP in NSCLC. First, ANKHD1 was overexpressed with and
without knockdown of YAP. Overexpression of ANKHD1 was sufficient
to induce cell proliferation and invasion, but in combination with
YAP knockdown, proliferation and invasion were inhibited. In
addition, the expression of downstream genes of the Hippo signaling
pathway following regulation of ANKHD1 was also decreased. The
mechanism underlying ANKHD1-dependent improvement of the Hippo
signaling pathway through YAP remains unclear, although the
expression levels of YAP and p-YAP were both affected by changes in
ANKHD1 expression. YAP mRNA expression was examined by qPCR
following ANKHD1 transfection or depletion, and the results
suggested that ANKHD1 regulated YAP protein expression via
regulating YAP mRNA transcription; i.e., ANKHD1 upregulated YAP at
the transcriptional level. As regards the simultaneous inhibition
of YAP phosphorylation by ANKHD1, we hypothesized that, as reported
by Machado-Neto et al (20), the binding of ANKHD1 and YAP may
inhibit the phosphorylation of YAP by other kinases, including
LATS1. To confirm this hypothesis, an immunoprecipitation assay was
performed in A549 cells overexpressing ANKHD1 and in LK2 cells with
ANKHD1 depletion. The results demonstrated that the binding of
LATS1 and YAP was reduced in A549 cells overexpressing ANKHD1,
whereas the binding of LATS1 and YAP was increased in LK2 cells
with ANKHD1 depletion. These findings indicate that the
upregulation and binding of ANKHD1 and YAP affect the binding of
YAP and LATS1, which inhibits the phosphorylation of YAP.
In conclusion, the expression of ANKHD1 was found to
be upregulated in NSCLC cell lines and tissues, and was associated
with advanced pTNM stage, lymph node metastasis and poor prognosis
in patients with NSCLC. ANKHD1 may affect the proliferation and
invasion of NSCLC cells through upregu-lating YAP expression,
inhibiting YAP phosphorylation, and inactivating the Hippo
signaling pathway.
Supplementary Data
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81401885 and
81301837), the Basic Scientific Project of Liaoning University,
China (grant no. LQNK201705), and the Liaoning Provincial Natural
Science Foundation (grant no. 20170541007).
Availability of data and materials
The datasets analyzed during the current study are
not publicly available, as they will be used in our further
studies, but they are available from the corresponding author on
reasonable request.
Authors' contributions
XFL, QH and XYL acquired the data and created a
draft of the manuscript. XFL, QH and XZR collected clinical samples
and performed the in vitro and in vivo assays. XFL,
XZR and MY analyzed and interpreted the data, and performed
statistical analysis. YCH, JHY and XYL reviewed the manuscript,
figures and tables. All authors have read and approved the final
version of the manuscript for publication.
Ethics approval and consent to
participate
Written informed consent was obtained from all the
patients, and the procedures were approved by the Institutional
Research Ethics Committee of China Medical University. All
experiments with nude mice were performed according to the
guidelines of China Medical University for the use of experimental
animals. The experimental protocols were approved by the
Institutional Animal Research Committee of China Medical
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors would like to thank all our laboratory
members for their contribution to this study. We would also like to
thank Dr Guoxin Liang for critical reading and editing of our
manuscript.
References
|
1
|
Smith RK, Carroll PM, Allard JD and Simon
MA: MASK, a large ankyrin repeat and KH domain-containing protein
involved in Drosophila receptor tyrosine kinase signaling.
Development. 129:71–82. 2002.PubMed/NCBI
|
|
2
|
Poulin F, Brueschke A and Sonenberg N:
Gene fusion and overlapping reading frames in the mammalian genes
for 4E-BP3 and MASK. J Biol Chem. 278:52290–52297. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li J, Mahajan A and Tsai MD: Ankyrin
repeat: A unique motif mediating protein-protein interactions.
Biochemistry. 45:15168–15178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sedgwick SG and Smerdon SJ: The ankyrin
repeat: A diversity of interactions on a common structural
framework. Trends Biochem Sci. 24:311–316. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valverde R, Edwards L and Regan L:
Structure and function of KH domains. FEBS J. 275:2712–2726. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fragiadaki M and Zeidler MP: Ankyrin
repeat and single KH domain 1 (ANKHD1) drives renal cancer cell
proliferation via binding to and altering a subset of miRNAs. J
Biol Chem. 293:9570–9579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Traina F, Favaro PM, Medina Sde S, Duarte
Ada S, Winnischofer SM, Costa FF and Saad ST: ANKHD1, ankyrin
repeat and KH domain containing 1, is overexpressed in acute
leukemias and is associated with SHP2 in K562 cells. Biochim
Biophys Acta. 1762:828–834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sansores-Garcia L, Atkins M, Moya IM,
Shahmoradgoli M, Tao C, Mills GB and Halder G: Mask is required for
the activity of the Hippo pathway effector Yki/YAP. Curr Biol.
23:229–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Du Toit A: Cell signalling: A new Hippo
pathway component. Nat Rev Mol Cell Biol. 14:1962013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao B, Tumaneng K and Guan KL: The Hippo
pathway in organ size control, tissue regeneration and stem cell
self-renewal. Nat Cell Biol. 13:877–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bao Y, Hata Y, Ikeda M and Withanage K:
Mammalian Hippo pathway: From development to cancer and beyond. J
Biochem. 149:361–379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan SW, Lim CJ, Chen L, Chong YF, Huang
C, Song H and Hong W: The Hippo pathway in biological control and
cancer development. J Cell Physiol. 226:928–939. 2011. View Article : Google Scholar
|
|
13
|
Espanel X and Sudol M: Yes-associated
protein and p53-binding protein-2 interact through their WW and SH3
domains. J Biol Chem. 276:14514–14523. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Komuro A, Nagai M, Navin NE and Sudol M:
WW domain-containing protein YAP associates with ErbB-4 and acts as
a co-transcriptional activator for the carboxyl-terminal fragment
of ErbB-4 that translocates to the nucleus. J Biol Chem.
278:33334–33341. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lapi E, Di Agostino S, Donzelli S, Gal H,
Domany E, Rechavi G, Pandolfi PP, Givol D, Strano S, Lu X and
Blandino G: PML, YAP, and p73 are components of a proapoptotic
autoregulatory feedback loop. Mol Cell. 32:803–814. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zaidi SK, Sullivan AJ, Medina R, Ito Y,
van Wijnen AJ, Stein JL, Lian JB and Stein GS: Tyrosine
phosphorylation controls Runx2-mediated subnuclear targeting of YAP
to repress transcription. EMBO J. 23:790–799. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Ren F, Zhang Q, Chen Y, Wang B
and Jiang J: The TEAD/TEF family of transcription factor Scalloped
mediates Hippo signaling in organ size control. Dev Cell.
14:377–387. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu FX, Zhao B, Panupinthu N, Jewell JL,
Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al:
Regulation of the Hippo-YAP pathway by G-protein-coupled receptor
signaling. Cell. 150:780–791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Enderle L and McNeill H: Hippo gains
weight: Added insights and complexity to pathway control. Sci
Signal. 6:re72013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Machado-Neto JA, Lazarini M, Favaro P,
Franchi GC Jr, Nowill AE, Saad ST and Traina F: ANKHD1, a novel
component of the Hippo signaling pathway, promotes YAP1 activation
and cell cycle progression in prostate cancer cells. Exp Cell Res.
324:137–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Travis WD, Brambilla E, Nicholson AG,
Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E,
Flieder DB, et al: The 2015 World Health Organization
Classification of Lung Tumors: Impact of genetic, clinical and
radiologic advances since the 2004 classification. J Thorac Oncol.
10:1243–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goldstraw P: Updated staging system for
lung cancer. Surg Oncol Clin N Am. 20:655–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishioka N, Inoue K, Adachi K, Kiyonari H,
Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N,
et al: The Hippo signaling pathway components Lats and Yap pattern
Tead4 activity to distinguish mouse trophectoderm from inner cell
mass. Dev Cell. 16:398–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xin M, Kim Y, Sutherland LB, Murakami M,
Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et
al: Hippo pathway effector Yap promotes cardiac regeneration. Proc
Natl Acad Sci USA. 110:13839–13844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang Y, Fu XR, Cui M, Li WM, Zhang L, Li
X, Li L, Sun ZC, Zhang XD, Li ZM, et al: Activated hippo signal
pathway inhibits cell proliferation and promotes apoptosis in NK/T
cell lymphoma cells. Cancer Med. 8:3892–3904. 2019.PubMed/NCBI
|
|
26
|
Dhyani A, Duarte AS, Machado-Neto JA,
Favaro P, Ortega MM and Olalla Saad ST: ANKHD1 regulates cell cycle
progression and proliferation in multiple myeloma cells. FEBS Lett.
586:4311–4318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dhyani A, Machado-Neto JA, Favaro P and
Saad ST: ANKHD1 represses p21 (WAF1/CIP1) promoter and promotes
multiple myeloma cell growth. Eur J Cancer. 51:252–259. 2015.
View Article : Google Scholar
|
|
28
|
Machado-Neto JA, Lazarini M, Favaro P, de
Melo Campos P, Scopim-Ribeiro R, Franchi Junior GC, Nowill AE, Lima
PR, Costa FF, Benichou S, et al: ANKHD1 silencing inhibits Stathmin
1 activity, cell proliferation and migration of leukemia cells.
Biochim Biophys Acta. 1853:583–593. 2015. View Article : Google Scholar
|
|
29
|
Mosavi LK, Cammett TJ, Desrosiers DC and
Peng ZY: The ankyrin repeat as molecular architecture for protein
recognition. Protein Sci. 13:1435–1448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sidor CM, Brain R and Thompson BJ: Mask
proteins are cofactors of Yorkie/YAP in the Hippo pathway. Curr
Biol. 23:223–228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu
J, Lin JD, Wang CY, Chinnaiyan AM, et al: TEAD mediates
YAP-dependent gene induction and growth control. Genes Dev.
22:1962–1971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hao Y, Chun A, Cheung K, Rashidi B and
Yang X: Tumor suppressor LATS1 is a negative regulator of oncogene
YAP. J Biol Chem. 283:5496–5509. 2008. View Article : Google Scholar
|
|
33
|
Oka T, Mazack V and Sudol M: Mst2 and Lats
kinases regulate apoptotic function of Yes kinase-associated
protein (YAP). J Biol Chem. 283:27534–27546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Remue E: TAZ interacts with zonula
occludens-1 and -2 proteins in a PDZ-1 dependent manner. FEBS Lett.
584:175–180. 2010. View Article : Google Scholar
|
|
35
|
Ren F, Zhang L and Jiang J: Hippo
signaling regulates Yorkie nuclear localization and activity
through 14-3-3 dependent and independent mechanisms. Dev Biol.
337:303–312. 2010. View Article : Google Scholar :
|
|
36
|
Wu S, Liu Y, Zheng Y, Dong J and Pan D:
The TEAD/TEF family protein Scalloped mediates transcriptional
output of the Hippo growth-regulatory pathway. Dev Cell.
14:388–398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao B, Li L, Tumaneng K, Wang CY and Guan
KL: A coordinated phosphorylation by Lats and CK1 regulates YAP
stability through SCF(beta-TRCP). Genes Dev. 24:72–85. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cottini F, Hideshima T, Xu C, Sattler M,
Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri
A, et al: Rescue of Hippo coactivator YAP1 triggers DNA
damage-induced apop-tosis in hematological cancers. Nat Med.
20:599–606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Machado-Neto JA, de Melo Campos P, Olalla
Saad ST and Traina F: YAP1 expression in myelodysplastic syndromes
and acute leukemias. Leuk Lymphoma. 55:2413–2415. 2014. View Article : Google Scholar : PubMed/NCBI
|