Introduction
Colorectal cancer is a major cause of
cancer-associated mortality and is the third most common cause of
cancer-associated death in the UK (1,2).
Colorectal cancer is a multifactorial disease, with both genetic
predisposition and environmental factors (such as the microbiota
and chronic inflammation) playing important roles in its
development (3). There are a
number of regulatory pathways linking inflammation and colorectal
cancer, such as chemokines, cytokines, NF-κB, cyclooxygenase-2
(COX-2) and its metabolic product pros-taglandin E2
(PGE2) (4,5). Deregulation of the
COX-2/PGE2 pathway is considered to be an important,
relatively early event in colorectal tumorigenesis, not only
because of its role in shaping the tumour microenvironment, but
also for enabling several of the hallmarks of cancer (6).
Despite a focus on NF-κB canonical signalling in
carcinogenesis (7-10), the NF-κB homodimer subunits NF-κB1
(p50/p50) and NF-κB2 (p52/p52) have become the subject of intense
interest as they represent an alternative mode of NF-κB activation
(an atypical NF-κB pathway); reviewed in (11). Although the NF-κB homodimer
subunits (p50/p50 or p52/p52) function as transcriptional
repressors due to their lack of trans-activation domains, they can
bind transcriptional co-activators via their C-terminal ankyrin
repeats (involved in protein-protein interactions) and therefore
serve a direct role in positively or negatively regulating NF-κB
target genes (12). First
identified in a subgroup of B-cell chronic lymphocytic leukaemia
(13,14), BCL-3 selectively binds to the NF-κB
homodimers, and depending on the nature of the stimuli, can
function as either a co-activator or as an inhibitor of NF-κB by
either increasing transcriptional activation or through
stabilisation of repressive homodimeric complexes, respectively
(15-18), reviewed in (19).
BCL-3 has been shown to be widely expressed in solid
tumours (20); in particular,
elevated levels of BCL-3 and p52 homodimers have been linked to
immortalized human breast epithelial cells (21), and BCL-3 has been proposed as a
link between STAT3 signalling and NF-κB in metastatic breast cancer
(22). Furthermore, it has been
shown that BCL-3 can promote colorectal tumorigenesis by increasing
survival of colorectal cancer cells through activation of AKT
(23), through stabilizing c-MYC
protein via ERK activation, and by promoting a cancer stem cell
phenotype by enhancing β-catenin signalling (24,25).
BCL-3 expression has been reported to be
dysregulated in colorectal cancer tissues (26); high BCL-3 protein expression is
associated with a poor prognosis in patients with colorectal cancer
(24). Cytoplasmic
localisation of BCL-3 has also been suggested as a potential early
diagnostic marker in colorectal cancer (27). Furthermore, BCL-3 expression is
upregulated by key cytokines including tumour necrosis factor-α
(TNF-α) and interleukin-(IL)-1β (20,28,29),
potentially representing a novel mechanism linking inflammation and
cancer.
Although the importance of COX-2/PGE2
signalling in colorectal tumorigenesis is well established
(6,30,31),
the effect of BCL-3 on the expression of COX-2/PGE2
signalling in colorectal cancer has not been investigated before,
to the best of our knowledge. Based on results from focused arrays,
it was hypothesized that BCL-3 may be a key determinant of the
COX-2 response to inflammatory cytokines in colorectal tumours.
Therefore, the aim of the present study was to determine whether
suppressing BCL-3 expression inhibited the activity of the
COX-2/PGE2 pathway.
Materials and methods
Cell lines and cell culture
The human colorectal adeno-carcinoma-derived cell
line HCA7 was a kind gift from Dr Susan Kirkland, (Imperial
College, London); cells were mycoplasma tested on receipt
(Mycoalert Plus mycoplasma detection kit; Lonza Group Ltd.). The
human colorectal adenocarcinoma-derived cell line HT-29 and human
rectal adenocarcinoma-derived cell line SW837 were obtained from
the American Type Culture Collection (ATCC; cat. nos. HTB-38 and
CCL-235, respectively) and mycoplasma tested on receipt. All
experiments were performed within 6 passages and cells were
routinely characterised (as described below). The human colorectal
adenocarcinoma-derived HCT116, HCT15, SW480, SW620, LOVO and LS174T
cells, and the specifically rectal cell line SW1463, were all
obtained from ATCC (cat. nos. CCL-247, CCL-225, CCL-228, CCL-227,
CCL-229, CL-118 and CCL-234, respectively), cells were mycoplasma
tested on receipt and experiments were performed within 6 passages
of receipt. All cell lines were maintained as previously described
(32). Cell lines were grown in
DMEM (Gibco; Thermo Fisher Scientific, Inc.), supplemented with 10%
FBS, 2 mM glutamine (Gibco; Thermo Fisher Scientific, Inc.), 100
U/ml penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.). The human colorectal adenoma-PC/AA/C1, S/AN/C1,
S/RG/C2 and transformed adenoma-PC/AA/C1/SB10 cell lines were
derived in this laboratory and grown as described previously
(33,34). Growth medium was DMEM supplemented
with 20% FBS, 1 µg/ml hydrocortisone sodium succinate, 0.2 units/ml
insulin, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin. All cell lines were routinely assessed for microbial
contamination (including mycoplasma), and molecularly characterised
using an inhouse panel of cellular and molecular markers to check
that cell lines have not been cross contaminated (every 3-6 months;
data not shown). Stocks were securely catalogued and stored, and
passage numbers were strictly adhered to prevent phenotypic
drift.
Supplementary tissue culture
treatments
Conditioned medium (CM; 24 h) was harvested from
BCL-3 siRNA, COX-2 siRNA or negative control transfected HCA7
cells, filtered and used to culture S/RG/C2 adenoma for 72 h (CM
was refreshed every 24 h). Add back experiments were carried out
using 1 µM dimethyl PGE2 (Sigma-Aldrich; Merck KGaA) to the CM
prior to culture.
HCA7 cells were treated with 75 µM NS-398
(Sigma-Aldrich; Merck KGaA), for 24 h, using a dose selective for
COX-2 inhibition (35). Both HCA7
and HT-29 cells were treated with 100 ng/ml TNF-α (Insight
Biotechnology Ltd.), and SW837 cells were treated with 10 ng/ml
IL-1β (Insight Biotechnology Ltd.), both for up to 72 h. The
concentration of TNF-α and IL-1β used had been previously optimized
(36).
Determining percentage of floating cells
as a measure of apoptosis
Growth medium from individual flasks was collected
and the number of floating cells counted. Attached cell yield was
determined by trypsinising and counting the number of viable cells
from the same flask. The floating cells are represented as a
proportion of the total cell population (floating and attached),
and used as a measure of cell death, as described previously
(37).
RNA interference
Cells were transfected using Lipofectamine RNAiMAX
(Invitrogen, Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol, with small interfering RNAs (siRNAs final
concentration 50 nM; GE Healthcare Dharmacon, Inc.) targeting BCL-3
(AGA CAC GCC UCU CCA UAU U) or COX-2, for which four different
siRNA sequences were pooled (GGA CUU AUG GGU AAU GU UA, CAU CAA CAC
UGC CUC AAU U, GAA AUU UGA CCC AGA ACU A and GAA UUA CCC AGU UUG
UUG A) (23). Cells were incubated
overnight at 37˚C, before changing the medium. Samples were
prepared up to 72 h after transfection.
Reverse transcription (RT)
Total RNA was extracted from cells using TRI-reagent
(Sigma-Aldrich; Merck KGaA), an RNeasy mini kit (Qiagen, Inc.) was
utilised according to manufacturer's protocol with an additional
on-column DNase digestion step (RNase-Free DNase Set; Qiagen,
Inc.), prior to complementary (c)DNA synthesis. The RNA
concentration of samples was measured using a NanoDrop (Thermo
Fisher Scientific, Inc.).
cDNA was synthesised from 2 µg RNA by RT, using the
RNA-dependent DNA polymerase, Moloney murine leukaemia virus
reverse transcriptase, (Promega Corporation), at 40˚C for 1 h. A
second tube, without reverse transcriptase was used as the negative
control (no RT). The samples were diluted further giving a final
concentration of 10 ng/µl.
Quantitative-PCR (qPCR)
Following optimisation of primers and ensuring the
annealing temperature provided ~100% amplification efficiency per
cycle (data not shown), qPCR was performed as previously described
(24), using SYBR Green PCR mix
(Qiagen, Inc.) and the following Qiagen QuantiTect primers, at a
dilution of 1:10: BCL-3, cat. no. QT00040040; and COX-2, cat. no.
QT00040586; with gene expression normalised interchangeably with
both housekeeping genes TATA-binding protein (TBP; cat. no.
QT00000721) or Hypoxanthine Phosphoribosyl transferase (HPRT; cat.
no. QT00059066). The QuantiTect primer assays sets are designed to
have annealing temperatures of 55˚C. For PCR, a 40-cycle program
was performed (denaturing, 15 sec at 94˚C; annealing, 30 sec at
55˚C; and extension, 30 sec at 72˚C) using a MxPro 3005P Real-time
Thermal Cycler (Agilent Technologies, Inc.); samples were amplified
in triplicate with one no RT well per condition. Amplification data
was analysed using MxPro software version 4.10 (Agilent
Technologies, Inc.), using the 2-ΔΔCq method (38).
Immunoblotting
Whole-cell lysates were prepared in situ, on
ice, using 100 µl 1x lysis buffer (Cell Signaling Technology, Inc.)
with the addition of a Protease Inhibitor Cocktail Tablet (Roche
Diagnostics) per 10 ml lysis buffer. The cell debris were removed
by centrifugation at 1˚C for 10 min, at 18,500 x g. Protein
concentration of the cell lysate was determined using a Bio-Rad DC
Protein assay kit (Bio-Rad Laboratories, Inc.), according to the
manufacturer's protocol. Absorption was measured in duplicate at
750 nm using an iMark Microplate Absorbance Reader (Bio-Rad
Laboratories, Inc.). Samples of 100 µg total protein were prepared
in a volume of 20 µl (adjusted with distilled water), and 5 µl 5x
Laemmli buffer was added to each lysate sample and boiled for 5
min.
Mini-Protean 3 Electrophoresis Cells (Bio-Rad
Laboratories, Inc.) were used to cast 9% acrylamide resolving gels,
using 1.5 mm spacers. Gels were run at 100 V for ~15 min, allowing
the samples to move through the stacking gel, before voltage was
increased to 180 V for ~1 h, or until the blue dye front had
migrated through the gel. Following separation of the protein
samples using SDS-PAGE, the proteins were transferred onto
Immobilon-P, a PVDF membrane (EMD Millipore). The gel and membrane
were then assembled into a Transblot Cell (Bio-Rad Laboratories,
Inc.), and a voltage of 100 V was applied for 1.5 h. The membrane
was blocked in 5% (w/v) milk blocking buffer for a minimum of 1 h
at room temperature, prior to immunoblotting, as previously
described (39), using the
following antibodies diluted in 0.5% (w/v) milk dilution buffer:
Rabbit polyclonal anti-BCL-3 (1:2,000; cat. no. 23959-1-AP;
ProteinTech Group, Inc.), goat polyclonal anti-COX-1 (1:500; cat.
no. sc-1752; Santa Cruz Biotechnology, Inc.), goat polyclonal
anti-COX-2 (1:500; cat. no. sc-1745; Santa Cruz Biotechnology,
Inc.) or rabbit polyclonal anti-15-PGDH (1:2,000; cat. no. ab37148;
Abcam). The membranes were incubated overnight at 4˚C, after which
they were washed three times 10 min each in Tris-Buffered
Saline-0.1% (v/v) Tween-20 (TBS-T). Subsequently, the membranes
were incubated with an appropriate horseradish
peroxidise-conjugated secondary antibody (1:1,000, anti-mouse IgG,
cat. no. A4416; anti-rabbit IgG, cat. no. A6154, 1:30,000;
anti-goat IgG, cat. no. A9452; all from Sigma-Aldrich; Merck KGaA)
in 0.5% (w/v) milk dilution buffer. After an 1 h incubation at room
temperature on a rocker, the membranes were washed three times, 10
min each, in TBS-T, and rinsed once with distilled water prior to
visualisation of the signals using LumiGLO Peroxidase
Chemiluminescence Substrate (Kirkegaard & Perry Laboratories,
Inc.). The signal was detected using x-ray films developed in a
Compact X4 Film Processor (XOgraph Imaging Systems Ltd.). The
length of time films were exposed was dependent on the strength of
the signal (between 1-20 min). Equal loading was confirmed using a
monoclonal mouse anti-α-tubulin incubated for 2 h at room
temperature (1:10,000; cat. no. T9026; Sigma-Aldrich; Merck
KGaA).
Blots were quantified using ImageJ version 1.52p
(National Institutes of Health). Densitometry analysis was used to
measure the change in intensity of individual western blot
bands.
PGE2 assay
PGE2 released by cells into culture media
was quantified using a PGE2 enzyme immunoassay solid
well kit (Cayman Chemical Company), that used a high-affinity
PGE2 monoclonal antibody for quantification of
PGE2. Medium collected from 70% confluent HCA7 cells was
snap frozen in liquid nitrogen (-196˚C) and stored at -70˚C.
PGE2 levels were determined using an immunoassay kit
(Cayman Chemical Company), as described previously (40). Cells were pre-treated with TNF-α
(100 ng/ml, Insight Biotechnology Ltd.) for 16 h or NS-398 (75 µM,
Sigma-Aldrich; Merck KGaA) for 24 h as required. Samples were
performed in triplicate.
Statistical analysis
For statistical analysis, a one sample t-test, a
Student's t-test, or a one-way ANOVA with Tukey's post-hoc test was
used to compare groups in GraphPad Prism version 7 (GraphPad
Software, Inc.). Results are expressed as the mean ± standard error
of the mean, where a minimum of three independent experiments were
performed. Where a single experiment was performed with multiple
technical repeats, the data are presented as the mean of the
technical repeats ± standard deviation.
Results
Knockdown of BCL-3 expression decreases
COX-2 expression in colorectal cancer cells
BCL-3 consistently repressed PTGS2 expression
in a number of human colorectal cancer cell lines in focused
mini-arrays (data not shown). Therefore, to determine whether BCL-3
regulated COX-2/PGE2 signalling in colorectal cancer,
HCA7 human colorectal cancer cells, which endogenously express high
levels of COX-2 and high BCL-3 expression (Fig. 1A), were transfected with BCL-3
siRNA and the expression of COX-2 was determined (Fig. 1B). Cells were transfected with
BCL-3 siRNA or non-targeting control. To confirm the specificity of
the siRNA and to control for off-target effects, BCL-3 expression
was silenced using four separate siRNA sequences (A, B, C and D).
Transfection with all four siRNA sequences separately resulted in a
reduction in the levels of COX-2 protein (Fig. 1B). Sequence A was used in all
subsequent experiments. The protein expression levels of both BCL-3
and COX-2 in HCA7 cells were assessed 24, 48 and 72 h
post-transfection (Fig. 1C).
Western blot analysis showed that the levels of COX-2 protein were
reduced following knockdown of BCL-3 at all-time points (Fig. 1C). BCL-3 and COX-2 mRNA levels were
evaluated using RT-qPCR. Results are presented as the fold change
of the negative siRNA transfected levels (Fig. 1C). Knockdown of BCL-3 expression
resulted in >60% reduction in COX-2 mRNA levels, confirming that
knockdown of BCL-3 in colorectal cancer cells decreased both the
COX-2 mRNA and protein expression levels.
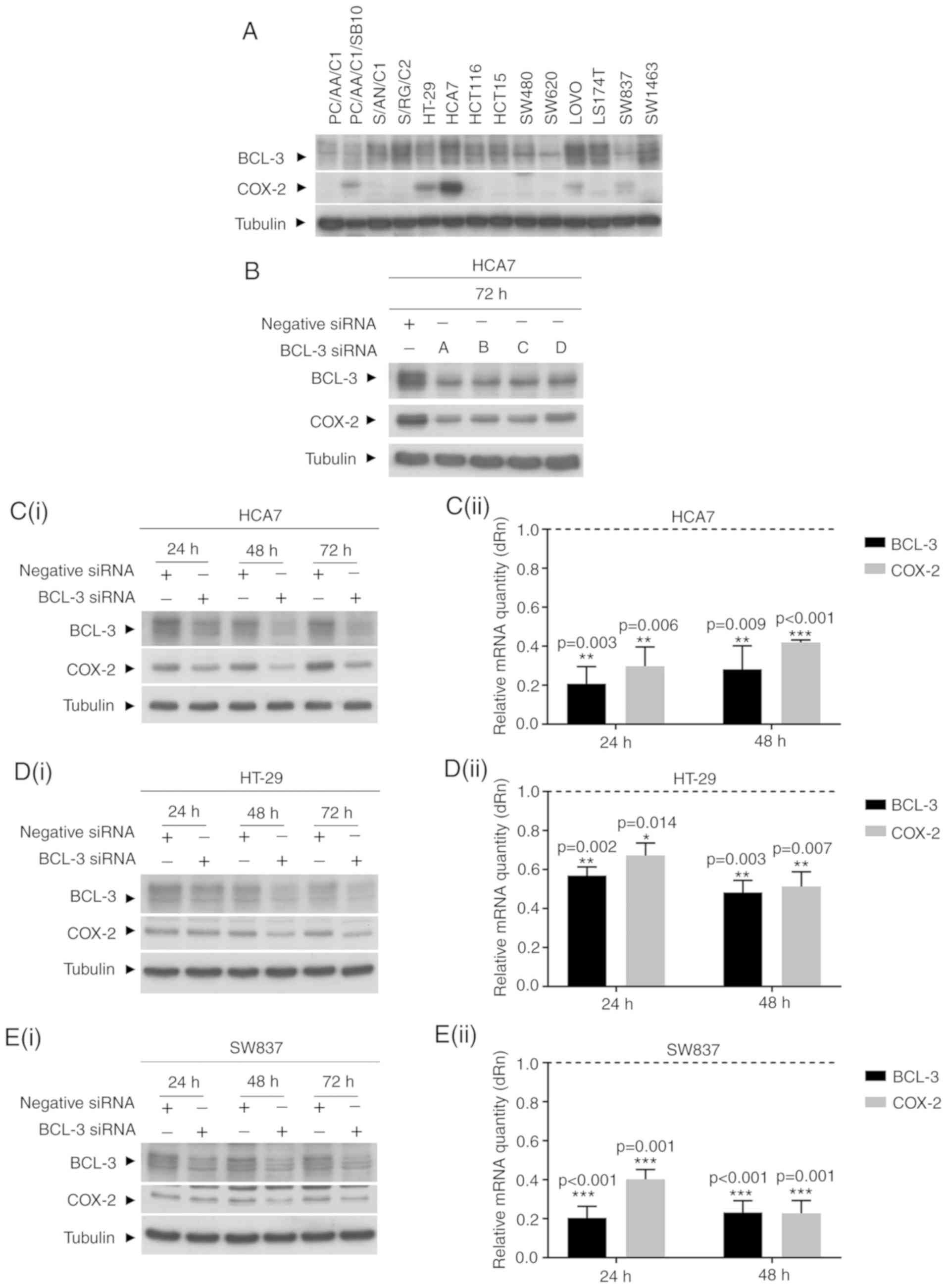 | Figure 1BCL-3 knockdown reduces COX-2
expression. (A) Endogenous levels of BCL-3 and COX-2 expression in
a panel of colorectal adenoma and carcinoma derived cell lines.
PC/AA/C1, S/AN/C1, S/RG/C2 colorectal cells, PC/AA/C1/SB10
transformed adenocarcinoma cells, HT-29, HCA7, HCT116, HCT15,
SW480, SW620, LOVO, LS174T colorectal adenocarcinoma cells, and
SW837 and SW1463 rectal adenocarcinoma cells were grown to ~70%
confluence before collection of total protein for western blot
analysis. α-tubulin was used as the loading control. (B) Western
blot of BCL-3 expression in HCA7 cells to determine the efficiency
of BCL-3 siRNA sequences. The expression levels of BCL-3 was
measured in HCA7 cells 72 h after knockdown of BCL-3 using four
separate BCL-3 siRNAs (A, B, C and D). The western blot is
representative of two independent repeats. Western blot and RT-qPCR
analysis of (C) HCA7, (D) HT-29 and (E) SW837 cells transfected
with control or BCL-3 siRNA. The visible non-specific higher weight
band is the result of a long exposure time used to detect low
endogenous expression levels of COX-2 in the SW837 cells. i) BCL-3
siRNA transfected cells were harvested, total protein was extracted
24, 48 and 72 h post-transfection, and BCL-3 and COX-2 levels were
assessed by western blotting. α-tubulin was used as the loading
control. The results are representative of three independent
experiments. ii) RT-qPCR was performed on cells from parallel
flasks 24 and 48 h post-transfection. Relative mRNA quantity of
BCL-3 and COX-2 are presented as a fold change of the control
negative siRNA, which itself was normalised to one. All mRNA values
were normalised to the housekeeping genes TBP or HPRT (n=4).
*P≤0.05, **P≤0.01 ***P≤0.001.
BCL-3, B-cell chronic lymphocytic leukaemia 3; COX-2,
cyclooxygenase 2; siRNA, small interfering RNA; dRn, baseline
corrected normalised fluorescence. |
To determine whether BCL-3 regulated COX-2
expression in other colorectal cancer cell lines, HT-29 (which has
an intermediate level of endogenous COX-2 protein expression;
Fig. 1D) and the SW837 rectal
cancer cell lines (low endogenous COX-2 protein expression;
Fig. 1E) were assessed.
Transfection with BCL-3 siRNA also resulted in downregulation of
COX-2 mRNA and protein expression levels in both cell lines.
Knockdown of BCL-3 expression decreases
COX-2/PGE2 signalling
To confirm that the decrease in COX-2 expression
following transfection with BCL-3 siRNA resulted in reduced COX-2
activity, PGE2 levels were measured in the culture
medium. HCA7 cells were used for this experiment as the high
endogenous COX-2 expression allowed PGE2 to be measured
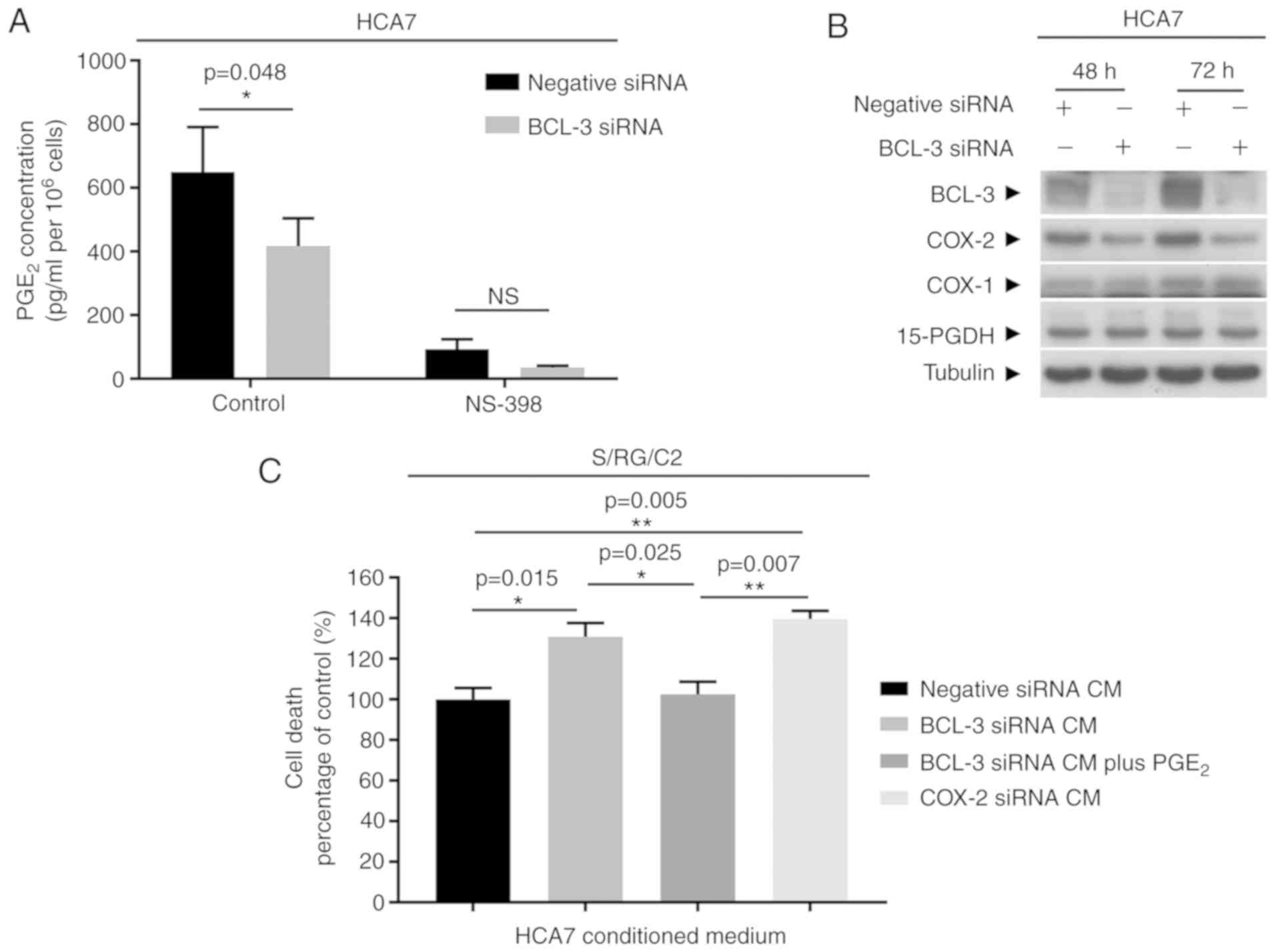
directly by ELISA (Fig. 2A). COX-1
and 15-PGDH protein expression were also assessed by western
blotting 72 h after BCL-3 siRNA transfection (Fig. 2B) to determine whether expression
of any other enzymes which regulate PGE2 levels were
altered. Furthermore, cells were treated with NS-398 to validate
that changes in PGE2 detected in the cell culture medium
were due to COX-2 and not COX-1 activity (Fig. 2A). The results showed that
PGE2 production was significantly reduced when BCL-3
expression was knocked down (P=0.048); there was a ~30% decrease in
basal PGE2 production in BCL-3 siRNA transfected cells
compared with the negative siRNA control. The levels of
PGE2 were reduced by the COX-2 selective inhibitor
NS-398 by >90% (Fig. 2A).
Finally, COX-1 and 15-PGDH levels were not altered by transfection
with the BCL-3 siRNA (Fig. 2B).
Taken together, these findings show that the observed decrease in
PGE2 production in the BCL-3 siRNA transfected cells was
due to a loss of COX-2 activity.
Subsequently, whether the regulation of
PGE2 observed by suppression of BCL-3 affected survival
of other tumour cells was determined. CM exchange experiments were
performed using a PGE2 sensitive adenoma derived cell
line (S/RG/C2). It was previously shown that PGE2
promoted the growth of S/RG/C2 (41). CM from the BCL-3 siRNA, COX-2 siRNA
or negative control transfected HCA7 cells were harvested and used
to culture the S/RG/C2 adenoma derived cell lines. The number of
HCA7 cells were not affected by BCL-3 knockdown or COX-2
downregulation during the preparation of CM over the 72 h period,
(Fig. S1A). Treating S/RG/C2
cells with CM from the BCL-3 siRNA transfected HCA7 cells resulted
in a significant increase in floating cells (P=0.015). Cell death
was rescued by addition of dimethyl PGE2 to the CM prior
to culture. In addition, the degree of cell death was similar to
that observed in cells grown in CM from HCA7 cells transfected with
the COX-2 siRNA (Fig. 2C). These
findings suggest that the changes in PGE2 secretion
caused by knockdown of BCL-3 affected the survival of
PGE2 sensitive tumour cells.
Together these results showed that knockdown of
BCL-3 expression downregulated expression of COX-2 and
significantly reduced the quantity of potentially pro-tumorigenic
PGE2 produced by the cancer cells.
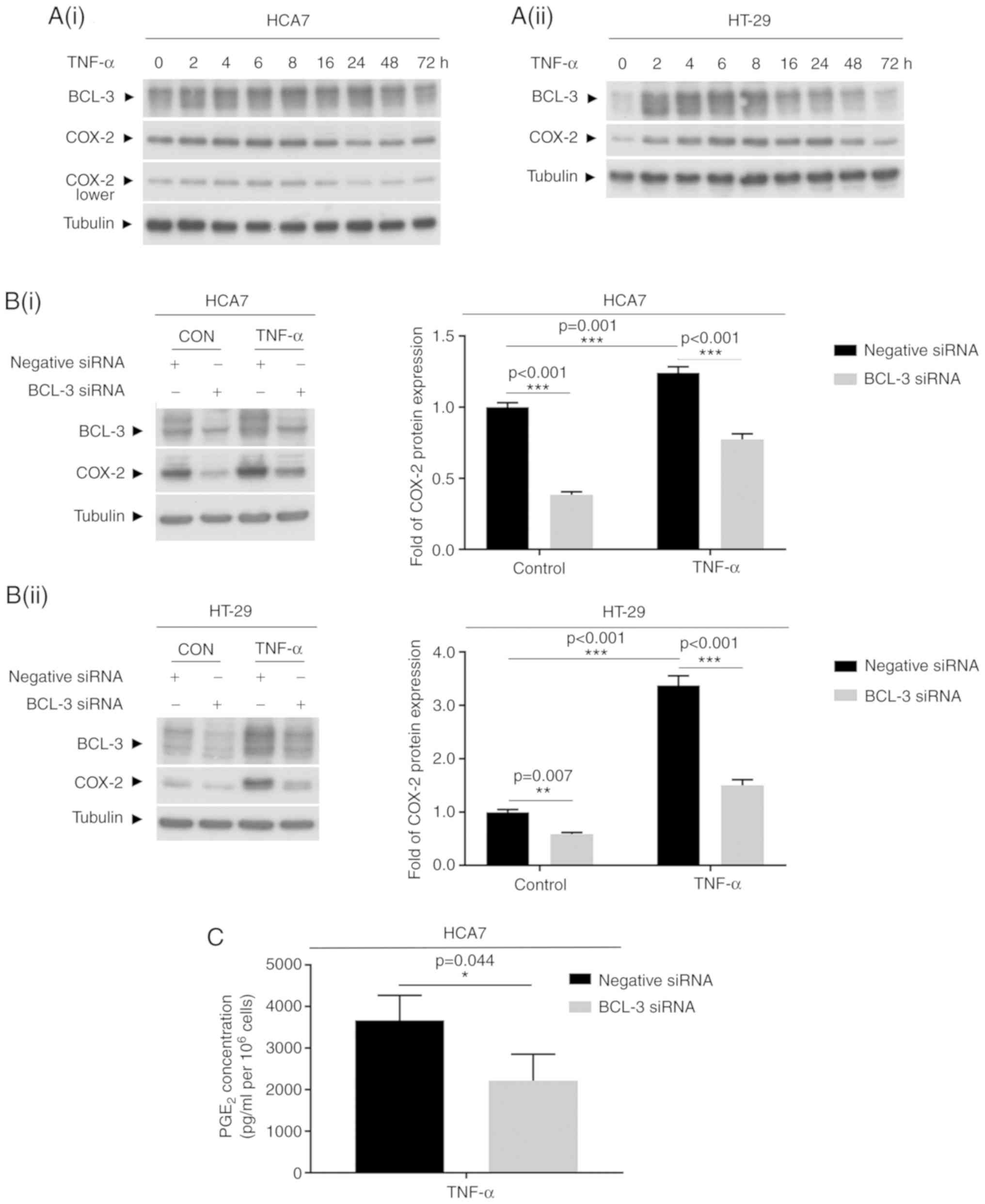
Induction of COX-2 expression by TNF-α or
IL-1β is partially blocked by knockdown of BCL-3
COX-2 protein has been identified as an
immediate-early response gene that although normally absent from
most cells, may be induced at sites of inflammation in response to
cytokines such as TNF-α and IL-1β (31). These cytokines have also been shown
to increase COX-2 expression in colorectal cancer cells (4,42,43).
To determine whether targeting BCL-3 reduced the induction of COX-2
in response to inflammatory cytokines, HCA7 and HT-29 cells were
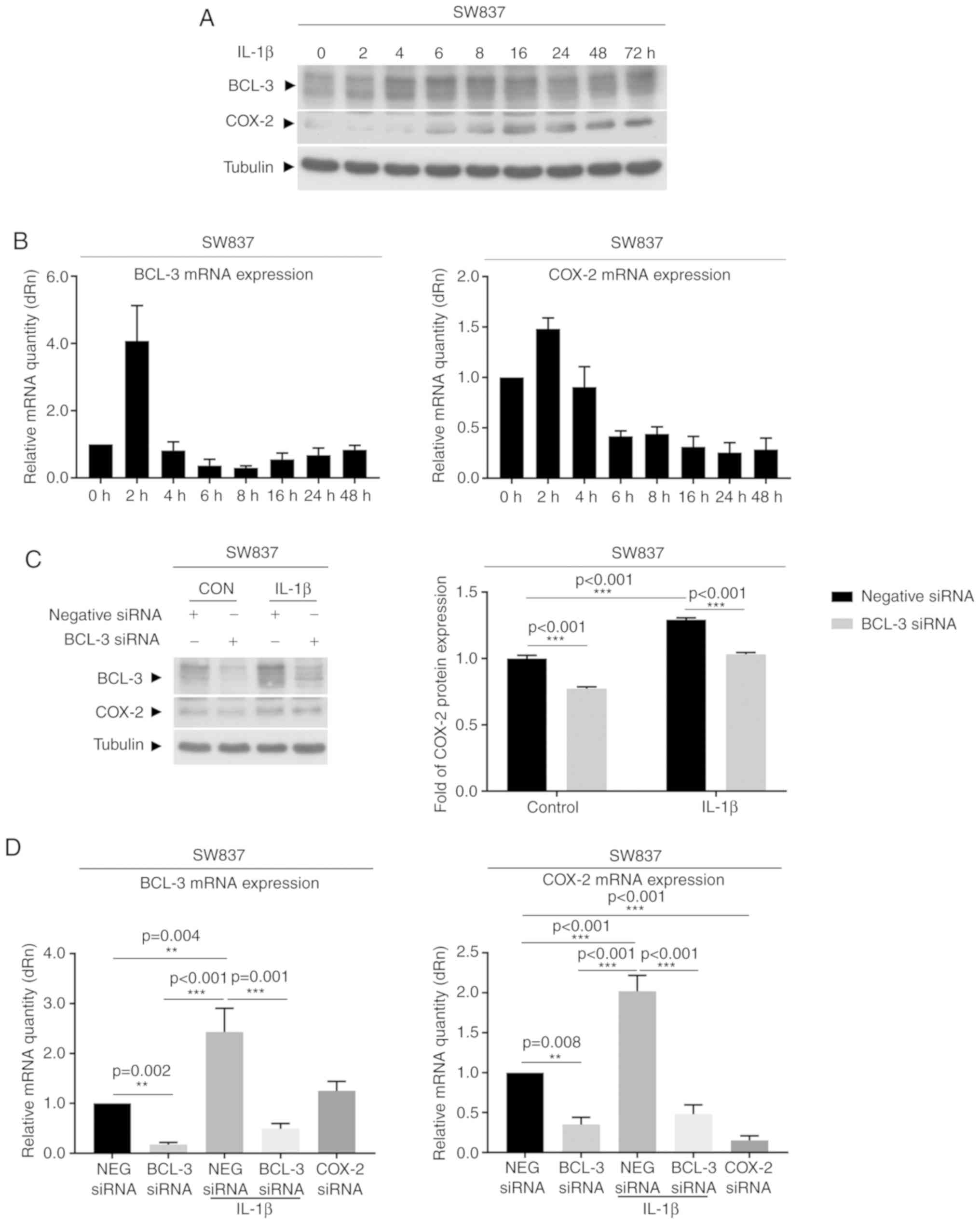
treated with 100 ng/ml TNF-α for up to 72 h (Fig. 3), and SW837 were treated with 10
ng/ml IL-1β (Fig. 4) as SW837
cells are more responsive to IL-1β compared with TNF-α. BCL-3 and
COX-2 protein expression were measured over 72 h. BCL-3 protein
levels increased after 2 h of treatment with TNF-α (Fig. 3A), reaching peak levels after 4-8 h
of treatment and returning to basal levels by 72 h. COX-2
expression followed a similar pattern to BCL-3 induction; COX-2 was
induced after 4-8 h, and remained high for 24-48 h when treated
with TNF-α, consistent with its regulation with BCL-3 (Fig. 3A). Notably, the increased
expression of COX-2 in response to TNF-α treatment (as shown in
Fig. 3B at 4 h) was found to be
partially blocked following the knockdown of BCL-3 in both HCA7 and
HT-29 cell lines. Suppression of BCL-3 also reduced PGE2
production by the HCA7 cells in response to TNF-α treatment
(Fig. 3C). Similar results were
observed in the SW837 cells treated with 10 ng/ml IL-1β (Fig. 4). Due to the low endogenous levels
of COX-2 protein expression in the SW837 cells, expression of both
BCL-3 and COX-2 were also measured using RT-qPCR (Fig. 4B and D). There was a rapid
induction of BCL-3 (within 2 h) following treatment with IL-1β,
followed by a more prolonged induction of COX-2 (Fig. 4A and B). In addition, the increase
in COX-2 expression 2-4 h after IL-1β treatment was partially
blocked following knockdown of BCL-3 (Fig. 4C and D). These data show that
inhibition of BCL-3 expression not only reduced basal COX-2
expression levels but also signifi-cantly supressed cytokine
induction mediated by COX-2. These results suggest that expression
of BCL-3 may be an important determinant in the COX-2-mediated
response to inflammatory cytokines.
Discussion
Discovered as an oncogene in leukaemia, BCL-3 has
been shown to be overexpressed in a range of solid tumours
including breast (21), prostate
(44), endometrial (45) and nasopharyngeal carcinoma
(46). The importance of BCL-3 in
colorectal carcinogenesis is supported by both clinical and
mechanistic studies. Puvvada et al (26) were the first to show that high
levels of nuclear BCL-3 were correlated with a poor prognosis in
patients with colorectal cancer, and Saamarthy et al
(27) proposed BCL-3 cellular
localisation as a marker for early diagnosis in colorectal cancer.
Both studies reported that at least 30% of colorectal tumours had
increased nuclear expression of BCL-3 (26,27).
More recently high BCL-3 expression has been shown to be associated
with worse survival in CRC (24).
There are several studies which have begun to
elucidate the mechanism by which BCL-3 promotes colorectal
carcinogenesis; in our previous study, it was shown that BCL-3
expression promotes AKT mediated cell survival and drives
colorectal tumour growth in vivo (23), and BCL-3 has also been reported to
increase the stability of c-MYC via activation of ERK (25). In addition, BCL-3 has been
implicated in promoting tumorigenesis through inhibition of DNA
damage induced p53 activation by upregulating MDM2 expression
(47), and in inducing cyclin D
activity, and thus, cell cycle progression (48). More recently, it was shown that
BCL-3 promoted development of a cancer stem cell phenotype by
enhancing β-catenin signalling in colorectal tumour cells (24).
Given the importance of upregulated COX-2 expression
in colorectal tumorigenesis, the novel finding that BCL-3 regulates
PTGS2 gene expression and COX-2/PGE2 signalling,
highlights a new mechanism underlying the oncogenic actions of
BCL-3 in colorectal cancer. Although not required for COX-2
expression, it may be hypothesized that nuclear BCL-3 serves as a
transcriptional co-factor, as recently proposed for
β-catenin-dependent gene targets (24). It remains to be determined whether
BCL-3 enhances NF-κB-dependent COX-2 regulation or acts via
recruitment of other co-regulators, such as Pirin, Tip60, Jab1 and
Bard1 (49). The ability of BCL-3
to regulate PGE2 production is of particular importance;
not only does PGE2 promote expression of the intestinal
cancer stem cell marker LGR5 (41), but PGE2 production by
colorectal cancer cells, as well as by melanoma and breast cancer
cells, suppresses tumour immunity and drives tumour promoting
inflammation (50). COX-dependent
immune evasion has been shown to be critical for tumour growth in
colorectal cancer mouse models, as COX-2 has been reported to be
responsible for the majority of circulating PGE2
(31,50). A limitation of the present study is
that TNF-α and IL-1β are only two of the upstream regulators of
COX-2 activity, whether BCL-3 expression regulates the response to
other factors remains to be determined. Furthermore, the potential
effects of the identified mechanism on the numerous downstream
targets of COX-2/PGE2 signalling remains to be
determined (6).
In the present study, it was shown that targeting
BCL-3 expression may suppress COX-2/PGE2 signalling,
which not only impacts the growth of other tumour cells, but may
also potentially enhance the immune response to the cancer.
Taken together, and given the importance of these
pathways in tumour progression, targeting BCL-3 protein-protein
interactions (via disruption of the ankyrin repeat domain for
example) remains a priority. Inhibitors of the BCL-3 pathway
currently under development (Dr Richard Clarkson, Cardiff
University) may not only suppress tumour cell survival and block
tumour growth, but also increase anti-cancer immunity through
suppressing COX-2/PGE2 signalling. Hence, these findings
suggest that BCL-3 inhibitors may be used as an alternative or
complementary approach to using NSAIDs for prevention and/or
preventing recurrence in PGE2-driven tumorigenesis
(51), and may improve long-term
prognosis for patients with colorectal cancer.
Supplementary Data
Funding
This work was funded by a Cancer Research UK
programme (grant no. C19/A11975), a Saudi Government Studentship
(grant no. S6692), and support from the John James Bristol
Foundation.
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ACW, HF, TJC, CP and AG conceived and designed the
study. ACW, HF, TJC and AG developed the methodology. ACW, HF, TJC,
CP and AG acquired the data. ACW, HF, TJC, CP and AG analysed and
interpreted the data. TJC and ACW wrote, reviewed and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We would like to thank Dr Mark Jepson of the School
of Medical Sciences Wolfson Bioimaging Facility for support and
guidance.
Abbreviations:
|
BCL-3
|
B-cell chronic lymphocytic leukaemia
3
|
|
COX
|
cyclooxygenase
|
|
CRC
|
colorectal cancer
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
PGE2
|
prostaglandin E2
|
|
siRNA
|
small interfering RNA
|
|
TNF-α
|
tumour necrosis factor α
|
|
IL-1β
|
interleukin-1β
|
References
|
1
|
Stewart BW and Wild CP: World cancer
report 2014. International Agency for Research on Cancer; Lyon:
2014, https://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-2014.
|
|
2
|
Bowel cancer mortality statistics, 2014.
Cancer Research UK; Oxford; 2014, http://www.cancerresearchuk.org/health-professional/cancer-statistics/statisticsby-cancer-type/bowel-cancer/mortality
Accessed September 6, 2017.
|
|
3
|
Lucas C, Barnich N and Nguyen HTT:
Microbiota, inflammation and colorectal cancer. Int J Mol Sci.
18:pii: E1310. 2017.PubMed/NCBI
|
|
4
|
Long AG, Lundsmith ET and Hamilton KE:
Inflammation and colorectal cancer. Curr Colorectal Cancer Rep.
13:341–331. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Janakiram NB and Rao CV: The role of
inflammation in colon cancer. Adv Exp Med Biol. 816:25–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Greenhough A, Smartt HJ, Moore AE, Roberts
HR, Williams AC, Paraskeva C and Kaidi A: The COX-2/PGE2 pathway:
Key roles in the hallmarks of cancer and adaptation to the tumour
micro-environment. Carcinogenesis. 30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang S, Liu Z, Wang L and Zhang X:
NF-kappaB signaling pathway, inflammation and colorectal cancer.
Cell Mol Immunol. 6:327–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taniguchi K and Karin M: NF-κB,
inflammation, immunity and cancer: Coming of age. Nat Rev Immunol.
18:309–324. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nguyen LK, Cavadas MA, Kholodenko BN,
Frank TD and Cheong A: Species differential regulation of COX2 can
be described by an NFκB-dependent logic AND gate. Cell Mol Life
Sci. 72:2431–2443. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pereira SG and Oakley F: Nuclear
factor-kappaB1: Regulation and function. Int J Biochem Cell Biol.
40:1425–1430. 2008. View Article : Google Scholar
|
|
12
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar
|
|
13
|
McKeithan TW, Ohno H and Diaz MO:
Identification of a transcriptional unit adjacent to the breakpoint
in the 14;19 trans-location of chronic lymphocytic leukemia. Genes
Chromosomes Cancer. 1:247–255. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohno H, Takimoto G and McKeithan TW: The
candidate proto-oncogene bcl-3 is related to genes implicated in
cell lineage determination and cell cycle control. Cell.
60:991–997. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bours V, Franzoso G, Azarenko V, Park S,
Kanno T, Brown K and Siebenlist U: The oncoprotein Bcl-3 directly
transactivates through kappa B motifs via association with
DNA-binding p50B homodimers. Cell. 72:729–739. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nolan GP, Fujita T, Bhatia K, Huppi C,
Liou HC, Scott ML and Baltimore D: The bcl-3 proto-oncogene encodes
a nuclear I kappa B-like molecule that preferentially interacts
with NF-kappa B p50 and p52 in a phosphorylation-dependent manner.
Mol Cell Biol. 13:3557–3566. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Franzoso G, Bours V, Azarenko V, Park S,
Tomita-Yamaguchi M, Kanno T, Brown K and Siebenlist U: The
oncoprotein Bcl-3 can facilitate NF-kappa B-mediated
transactivation by removing inhibiting p50 homodimers from select
kappa B sites. EMBO J. 12:3893–3901. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Palmer S and Chen YH: Bcl-3, a
multifaceted modulator of NF-kappaB-mediated gene transcription.
Immunol Res. 42:210–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Legge DN, Chambers AC, Parker CT, Timms P,
Collard TJ and Williams AC: The role of B-Cell Lymphoma-3 (BCL-3)
in enabling the hallmarks of cancer: Implications for the treatment
of colorectal carcinogenesis. Carcinogenesi. Jan 13–2020.Epub ahead
of print. View Article : Google Scholar
|
|
20
|
Maldonado V and Melendez-Zajgla J: Role of
Bcl-3 in solid tumors. Mol Cancer. 10:1522011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cogswell PC, Guttridge DC, Funkhouser WK
and Baldwin AS Jr: Selective activation of NF-kappa B subunits in
human breast cancer: Potential roles for NF-kappa B2/p52 and for
Bcl-3. Oncogene. 19:1123–1131. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wakefield A, Soukupova J, Montagne A,
Ranger J, French R, Muller WJ and Clarkson RW: Bcl3 selectively
promotes metastasis of ERBB2-driven mammary tumors. Cancer Res.
73:745–755. 2013. View Article : Google Scholar
|
|
23
|
Urban BC, Collard TJ, Eagle CJ, Southern
SL, Greenhough A, Hamdollah-Zadeh M, Ghosh A, Poulsom R, Paraskeva
C, Silver A and Williams AC: BCL-3 expression promotes colorectal
tumorigenesis through activation of AKT signalling. Gut.
65:1151–1164. 2016. View Article : Google Scholar :
|
|
24
|
Legge DN, Shephard AP, Collard TJ,
Greenhough A, Chambers AC, Clarkson RW, Paraskeva C and Williams
AC: BCL-3 promotes a cancer stem cell phenotype by enhancing
beta-catenin signalling in colorectal tumour cells. Dis Model Mech.
12:pii: dmm037697. 2019. View Article : Google Scholar
|
|
25
|
Liu Z, Jiang Y, Hou Y, Hu Y, Cao X, Tao Y,
Xu C, Liu S, Wang S, Wang L, et al: The IκB family member Bcl-3
stabilizes c-Myc in colorectal cancer. J Mol Cell Biol. 5:280–282.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Puvvada SD, Funkhouser WK, Greene K, Deal
A, Chu H, Baldwin AS, Tepper JE and O'Neil BH: NF-kB and Bcl-3
activation are prognostic in metastatic colorectal cancer.
Oncology. 78:181–188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saamarthy K, Björner S, Johansson M,
Landberg G, Massoumi R, Jirström K and Masoumi KC: Early diagnostic
value of Bcl-3 localization in colorectal cancer. BMC Cancer.
15:3412015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Collins PE, Kiely PA and Carmody RJ:
Inhibition of transcription by B cell Leukemia 3 (Bcl-3) protein
requires interaction with nuclear factor κB (NF-κB) p50. J Biol
Chem. 289:7059–7067. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brocke-Heidrich K, Ge B, Cvijic H, Pfeifer
G, Löffler D, Henze C, McKeithan TW and Horn F: BCL3 is induced by
IL-6 via Stat3 binding to intronic enhancer HS4 and represses its
own transcription. Oncogene. 25:7297–7304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Y, Sun H, Hu M, Zhang Y, Chen S, Tighe
S and Zhu Y: The role of cyclooxygenase-2 in colorectal
carcinogenesis. Clin Colorectal Cancer. 16:165–112. 2017.
View Article : Google Scholar
|
|
31
|
Wang D and Dubois RN: The role of COX-2 in
intestinal inflammation and colorectal cancer. Oncogene.
29:781–788. 2010. View Article : Google Scholar
|
|
32
|
Skeen VR, Collard TJ, Southern SL,
Greenhough A, Hague A, Townsend PA, Paraskeva C and Williams AC:
BAG-1 suppresses expression of the key regulatory cytokine
transforming growth factor β (TGF-β1) in colorectal tumour cells.
Oncogene. 32:4490–4499. 2013. View Article : Google Scholar
|
|
33
|
Paraskeva C, Finerty S, Mountford RA and
Powell SC: Specific cytogenetic abnormalities in two new human
colorectal adenoma-derived epithelial cell lines. Cancer Res.
49:1282–1286. 1989.PubMed/NCBI
|
|
34
|
Williams AC, Harper SJ and Paraskeva C:
Neoplastic transformation of a human colonic epithelial cell line:
In vitro evidence for the adenoma to carcinoma sequence. Cancer
Res. 50:4724–4730. 1990.PubMed/NCBI
|
|
35
|
Elder DJ, Halton DE, Crew TE and Paraskeva
C: Apoptosis induction and cyclooxygenase-2 regulation in human
colorectal adenoma and carcinoma cell lines by the
cyclooxygenase-2-selective non-steroidal anti-inflammatory drug
NS-398. Int J Cancer. 86:553–560. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Southern SL, Collard TJ, Urban BC, Skeen
VR, Smartt HJ, Hague A, Oakley F, Townsend PA, Perkins ND,
Paraskeva C and Williams AC: BAG-1 interacts with the p50-p50
homodimeric NF-κB complex: Implications for colorectal
carcinogenesis. Oncogene. 31:2761–2772. 2012. View Article : Google Scholar
|
|
37
|
Hague A, Manning AM, Hanlon KA, Huschtscha
LI, Hart D and Paraskeva C: Sodium butyrate induces apoptosis in
human colonic tumour cell lines in a p53-independent pathway:
Implications for the possible role of dietary fibre in the
prevention of large-bowel cancer. Int J Cancer. 55:498–505. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
39
|
Williams AC, Browne SJ, Yeudal WA,
Paterson IC, Marshall CJ, Lane DP and Paraskeva C: Molecular events
including p53 and k-ras alterations in the in vitro progression of
a human colorectal adenoma cell line to an adenocarcinoma.
Oncogene. 8:3063–3072. 1993.PubMed/NCBI
|
|
40
|
Moore AE, Greenhough A, Roberts HR, Hicks
DJ, Patsos HA, Williams AC and Paraskeva C: HGF/Met signalling
promotes PGE(2) biogenesis via regulation of COX-2 and 15-PGDH
expression in colorectal cancer cells. Carcinogenesis.
30:1796–1804. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Al-Kharusi MR, Smartt HJ, Greenhough A,
Collard TJ, Emery ED, Williams AC and Paraskeva C: LGR5 promotes
survival in human colorectal adenoma cells and is upregulated by
PGE2: Implications for targeting adenoma stem cells with NSAIDs.
Carcinogenesis. 34:1150–1157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Popivanova BK, Kitamura K, Wu Y, Kondo T,
Kagaya T, Kaneko S, Oshima M, Fujii C and Mukaida N: Blocking
TNF-alpha in mice reduces colorectal carcinogenesis associated with
chronic colitis. J Clin Invest. 118:560–570. 2008.PubMed/NCBI
|
|
43
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ahlqvist K, Saamarthy K, Syed Khaja AS,
Bjartell A and Massoumi R: Expression of Id proteins is regulated
by the Bcl-3 proto-oncogene in prostate cancer. Oncogene.
32:1601–1608. 2013. View Article : Google Scholar
|
|
45
|
Pallares J, Martinez-Guitarte JL, Dolcet
X, Llobet D, Rue M, Palacios J, Prat J and Matias-Guiu X:
Abnormalities in the NF-kappaB family and related proteins in
endometrial carcinoma. J Pathol. 204:569–577. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thornburg NJ, Pathmanathan R and
Raab-Traub N: Activation of nuclear factor-kappaB p50
homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res.
63:8293–8301. 2003.PubMed/NCBI
|
|
47
|
Kashatus D, Cogswell P and Baldwin AS:
Expression of the Bcl-3 proto-oncogene suppresses p53 activation.
Genes Dev. 20:225–235. 2006. View Article : Google Scholar :
|
|
48
|
Westerheide SD, Mayo MW, Anest V, Hanson
JL and Baldwin AS Jr: The putative oncoprotein Bcl-3 induces cyclin
D1 to stimulate G(1) transition. Mol Cell Bio. 21:8428–8436. 2001.
View Article : Google Scholar
|
|
49
|
Dechend R, Hirano F, Lehmann K, Heissmeyer
V, Ansieau S, Wulczyn FG, Scheidereit C and Leutz A: The Bcl-3
oncoprotein acts as a bridging factor between NF-kappaB/Rel and
nuclear co-regulators. Oncogene. 18:3316–3323. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zelenay S, van der Veen AG, Böttcher JP,
Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais
R, Quezada SA, et al: Cyclooxygenase-dependent tumor growth through
evasion of immunity. Cell. 162:1257–1270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Süleyman H, Demircan B and Karagöz Y:
Anti-inflammatory and side effects of cyclooxygenase inhibitors.
Pharmacol Rep. 59:247–258. 2007.PubMed/NCBI
|