Introduction
Bladder cancer is the fourth most common type of
cancer, with ~25,000 estimated cases of mortality in the United
States in 2019 (1). Carcinoma in
the urinary bladder occurs in multiple forms, such as low-grade
superficial or high-grade muscle-invasive tumor (2). Urothelial cell carcinoma is the most
common form of bladder cancer that accounts >90% of all cases
(2). The superficial tumor can be
removed surgically even though the recurrence rate is higher, and
the disease can progress to high-grade stage eventually. Metastatic
muscle-invasive bladder cancer, on the other hand, is extreme and
accounts for fatality in 50% of the patients (3). The most reliable prognostic
evaluation for recurrence includes tumor stage and grade (4). Transurethral resection combined with
immunotherapy for superficial tumor, and cystectomy and
chemo/radiation therapy for invasive carcinoma are the most common
treatment strategies (5,6). Nevertheless, identification of novel
targets or molecules may provide insights into prognosis and
therapeutic options for bladder cancer, since the available
prognosis and treatment strategies fail to meet expectations.
Protein kinase C (PKC) is a family of
serine/threonine enzymes consisting of three subclasses of enzymes,
including conventional, novel and atypical. All PKCs function by
phosphorylating and activating different downstream proteins and
are involved in various transmembrane cross-talks and signal
transduction pathways (7).
Activation of conventional PKCs (α, β1, β2 and γ) depends on
diacylglycerol and Ca2+, whereas novel PKCs (θ, δ, η and
ε) are activated by diacylglycerol. However, atypical PKCs (ι and
ζ) require neither diacylglycerol, nor calcium, instead they
require protein-protein interaction (8). PKCs are involved in various cell
signaling events that stimulate cell growth, proliferation,
apoptosis, metastasis and regulation of gene expression (9).
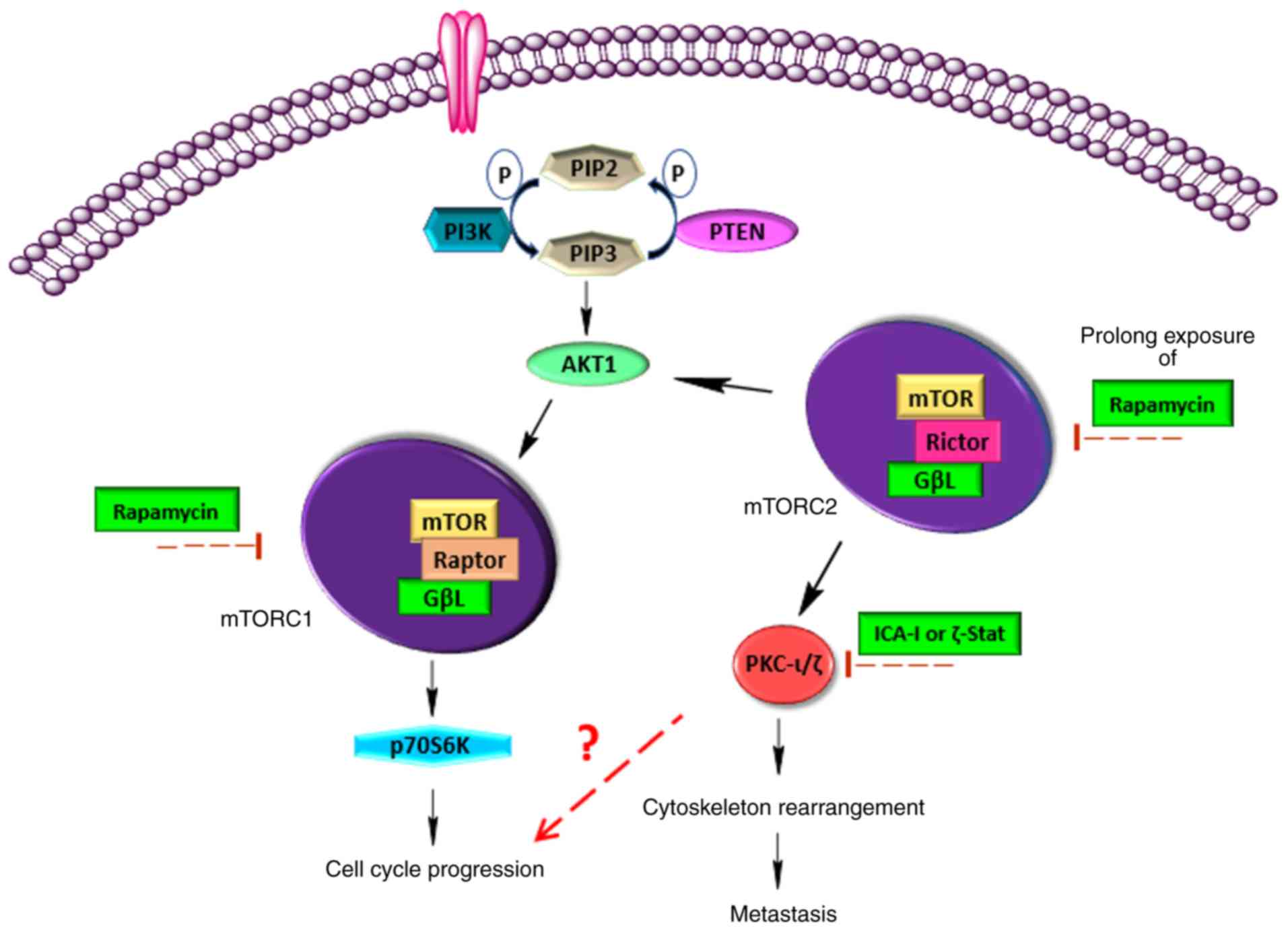
mTOR is considered to be an essential downstream
molecule of the PI3K/AKT1 signaling pathway, which triggers various
signaling cascades that mediate cellular growth, survival,
metastasis, metabolism and angiogenesis, and is often
hyperactivated in different types of cancer (10-12).
mTOR is a serine/threonine kinase which can exist in
two different complexes, namely mTOR complex 1 (mTORC1) and mTOR
complex 2 (mTORC2) (13). Raptor
and Rictor are two critical components of mTORC1 and mTORC2,
respectively. Furthermore, previous studies have demonstrated that
mTORC2 phosphorylates turn motif (TF) and hydrophobic motif (HM) of
conventional PKCs and AKT1, which ultimately leads to the
maturation and stabilization of these kinases (14-16).
Protein kinases, specifically atypical PKCs, have
been implicated in numerous types of cancer, such as breast cancer,
colon cancer, melanoma, ovarian cancer and glioblastoma (8,9,17-20).
Additionally, unlike other PKCs, atypical PKC has a glutamic acid
at the HM region, which makes the TM of the atypical PKC sensitive
to mTOR-mediated phosphorylation and stabilization of PKC-ι and
PKC-ζ (14). Therefore, in the
present study, bladder cancer TCCSUP cells were treated with a
combination of atypical PKC inhibitors (either ICA-I or ζ-Stat) and
rapamycin (mTOR inhibitor; Fig. 1)
to examine the effect of mTOR and atypical PKC, as well as the mTOR
complex, on atypical PKCs associated with bladder cancer cell
survival. The healthy bladder epithelial MC-SV-HUCT2 cell line, was
used in the present study as a healthy bladder cell line. The data
revealed that the combination of atypical PKC inhibitor and
rapamycin reduced metastatic bladder cancer cell progression by
inducing cell cycle arrest and eventual senescence.
Materials and methods
Antibodies and reagents
ICA-I [5-amino-1-(2, 3-dihydroxy-4- hydroxymethyl)
cyclopentyl)-1H-imidazole-4-carboxamide)] and ζ-Stat
(8-hydroxynaphthalene-1, 3, 6-trisulfonic acid) were obtained from
the National Cancer Institute, and rapamycin (cat. no. BP29631) was
procured from Thermo Fisher Scientific, Inc. Anti-phospho PKC-ι
(T555; cat. no. ab5813) and anti-phospho PKC-ζ (T560; cat. no.
ab59412) were purchased from Abcam, Inc.. Anti-PKC-ι (cat. no.
610176) and anti-E-cadherin (cat. no. 610181) antibodies were
purchased from BD Biosciences. The antibodies, anti-PKC-ζ (cat. no.
9372), anti-phospho Rictor (T1135; cat. no. 3806), anti-Rictor
(cat. no. 9476), anti-β-galactosidase (cat. no. 2372),
anti-β-catenin (cat. no. 25362), anti-P70S6K (cat. no. 2708),
anti-phospho P70S6K (T421/S424; cat. no. 9204), anti-AKT1 (cat. no.
4691), anti-phospho AKT1 (S473; cat. no. 4058), anti-phospho AKT1
(T308; cat. no. 4056), anti-phospho Src homology 2 domain cotaining
transforming protein (SHC) (S239/240) (cat. no. 2434) and anti-SHC
(cat. no. 2432), and the Senescence β-Galactosidase Staining kit
(cat. no. 9860) were procured from Cell Signaling Technology, Inc.
The antibodies purchased from Santa Cruz Biotechnology, Inc. were:
Anti-cyclin D1 (cat. no. sc70899), anti-CDK4 (cat. no. sc53636),
anti-p27 (cat. no. sc527), anti-phospho p53 (S315; sc101763),
anti-p53 (cat. no. sc126), anti-Lamin B1 (cat. no. sc374015),
anti-p21 (cat. no. sc817), anti-phospho mouse double minute 2
homolog (MDM2; S166; cat. no. sc53368) and anti-MDM2 (cat. no.
sc965). Anti-β-actin (cat. no. A3854) was obtained from
Sigma-Aldrich; Merck KGaA. The Annexin-V APC apoptosis assay kit
(cat. no. 601411) was procured from Cayman Chemical Company. Super
Signal West Pico
Chemiluminescent substrate (cat. no. 34580) was
purchased from Pierce; Thermo Fisher Scientific, Inc. Horseradish
peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) (H+L)
(cat. no. 1706516) and goat anti-rabbit IgG (H+L) (cat. no.
1706515) secondary antibodies were purchased from Bio-Rad
Laboratories, Inc., and water-soluble tetrazolium salts (WST-1;
cat. no. 11644807001) reagent was purchased from Sigma-Aldrich;
Merck KGaA. Eagle's minimum essential medium (EMEM, cat. no
ATCC® 30-2003) was procured from American Type Culture
Collection. F12K was obtained from Corning Inc. (cat. no. 10025CV),
and Trypsin-EDTA solution (cat. no. 25200056) was obtained from
Thermo Fisher Scientific, Inc.
Cell lines and subculture
The metastatic bladder cancer TCCSUP
(ATCC® HTB-5) and healthy bladder epithelial MC-SV-HUCT2
(ATCC® CRL-9519) cell lines were obtained from American
Type Culture Collection. The TCCSUP and MC-SV-HUCT2 cells were
sub-cultured and maintained in T75 flasks containing EMEM and F12K
media, respectively. Both flasks were supplemented with 10% FBS
(R&D Systems, Inc.) and 1% antibiotics (penicillin, 10 U/ml;
streptomycin, 10 mg/ml). Cells were incubated at 37°C with 5%
CO2. Cells were used for the experiments a few days
after subculture when cells were 70-80% confluent.
Viability of metastatic TCCSUP cells and
normal bladder MC-SV-HUCT2 cells following treatment with ICA-I,
ζ-Stat and rapamycin
Metastatic TCCSUP cells (3.0×103/well)
and healthy MC-SV-HUCT2 cells (3.0×103/well) were
subcultured in a 96-well plate. Following incubation for 24 h at
37°C with 5% CO2, the cells were treated with either
ICA-I (7.5 μM), ζ-Stat (7.5 μM) or rapamycin (100 nM)
separately, or combination of ICA-I and rapamycin or ζ-Stat for 72
h at 37°C with 5% CO2. Subsequently, cells were washed
with 200 μl 1X DPBS buffer and incubated with suitable media
(either EMEM or F12K) and WST-1 reagent (final dilution, 1:10) at
37°C with 5% CO2 for 3 h according to the manufacturer's
protocol. Finally, the cells were analyzed after 3 h at 450 nm
using a microplate reader (BioTek Instruments, Inc.) (21).
Flow cytometric analysis of apoptosis of
combination- treated bladder cancer cells
Annexin-V/APC and DAPI-based flow cytometry was used
to distinguish the apoptotic population from a healthy population.
Cells were plated in 100-mm cell culture plates, treated for 5
consecutive days as aforementioned for the cell viability test,
lifted, harvested and analyzed according to the manufacturer's
protocol. Briefly, the treated and untreated cells were then
resuspended in 100 μl Annexin-V/APC and DAPI solution.
Subsequently, the cells were incubated in the dark for 10 min at
room temperature. The samples were run at 633 nm excitation and 700
nm emission wavelengths for APC, and 350 nm excitation and 450 nm
emission wavelengths for DAPI using the BD FacsCanto II instrument
(Becton, Dickinson and Company), and analyzed using FacsDiva 6.3.1
software (BD Biosciences).
Cell lysate preparation and immunoblot
analysis
Cells (1.5×105) were cultured in 100-mm
cell culture flasks. After incubation for 24 h at 37°C with 5%
CO2 (to obtain ≥50% confluency), new medium was
supplied, and each experimental flask was treated with 7.5
μM ICA-I or ζ-Stat, 100 nM rapamycin or a combination of
ICA-I/ζ-Stat and rapamycin for 72 h at 37°C with 5% CO2.
An equal volume of DMSO (vehicle control), as the volume of the
drug, was added to the control. After continuing the treatment for
3 consecutive days, cells were placed on ice, washed with 1X DPBS,
scraped into a 1.5-ml centrifuge tube and resuspended in 500
μl cell 1× Cell Lysis buffer (cat. no. 9803; Cell Signaling
Technology, Inc.). The re-suspended cells were subsequently
sonicated and centrifuged at 16,128 x g at 4°C for 30 min, followed
by the determination of protein concentration using a Bradford
assay (22). An equal amount (30
μg/lane) of protein was loaded and separated by 10%
SDS-PAGE, followed by electroblotting onto a nitrocellulose
membrane (0.45-μm). The proteins were then blocked for 1 h
in 5% fat-free milk in TBS with 0.05% Tween-20 (TBST) at room
temperature. Subsequently, the membranes were incubated with
primary antibody solution in 5% BSA (cat. no. BP1600-100; Thermo
Fisher Scientific, Inc.) in TBST (dilution, 1:1,000) overnight at
4°C, followed by incubation with secondary antibody of either goat
anti-mouse IgG HRP conjugate or goat anti-rabbit IgG HRP conjugate
in 5% fat-free milk in TBST (dilution, 1:3,000) for 1.5 h at room
temperature. The immune reactive bands were finally visual-ized
using Supersignal West Pico Chemiluminescent substrate under
Amersham Imager 600 (GE Healthcare Life Sciences), as previously
described (23).
β-galactosidase staining of bladder
cancer cells for senescence
TCCSUP metastatic bladder cells (1×103
cells/well) were plated in 6-well plates, followed by treatment
with the combination of atypical PKC inhibitors and rapamycin for 7
consecutive days. Cells were then fixed and stained according to
the manufacturer's protocol using the senescence β-Gal staining
kit. Briefly, TCCSUP cells were fixed with 1× β-Gal fixative for 20
min at room temperature and stained with complete 1× β-Gal stain
solution in a dry incubator overnight at 37°C. Images were captured
using Jenoptik Gryphax® software (Jenoptik AG) with
differential interference contrast brightfield microscopy (Motic
AE31E; magnification, x20).
Scratch wound healing assay
TCCSUP malignant cells were plated into 6-well
plates. At ~90% confluency, old media were discarded, a line was
scratched across the cell monolayer using a 100-μl sterile
pipette tip and the cells were washed with 1X DPBS to remove
floating cells and debris. Subsequently, fresh complete media
(supplemented with 10% FBS) was supplied and the cells were treated
using ICA-I (7.5 μm), ζ-Stat (7.5 μm), rapamycin (100
nM), combination of ICA-I and rapamycin, or combination of ζ-Stat
and rapamycin for 72 h and incubated at 37°C with 5%
CO2. Finally, the cells that moved into the interspace
of the wound were observed after 72 h using a phase contrast
microscope (Motic Incorporation, Ltd.).
Immunoprecipitation (IP)
Protein (3 μg) was immuno-precipitated from
300 μg protein containing cell lysate suspension using
primary antibody of interest and separated by SDS-PAGE and finally
analyzed using the western blotting technique to determine the
associated proteins with the IP protein as described in our
previous study (8).
Densitometry
The intensity of each band was quantified using 1D
analysis software, Alpha View (v3.4.0.0; ProteinSimple). The
background intensity was subtracted from each band to quantify the
correct intensity.
Statistical analysis
To determine the statistical significance of the
data, the results were presented as the mean ± SEM of at least
three independent experiments. The data were compared by one-way
ANOVA (Dunnett's multiple comparison tests) or two-way ANOVA
(Tukey's multiple comparison test) using GraphPad Prism 8 software
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Atypical PKC inhibitors and rapamycin
reduce the survival of bladder cancer cells without affecting
healthy bladder cells
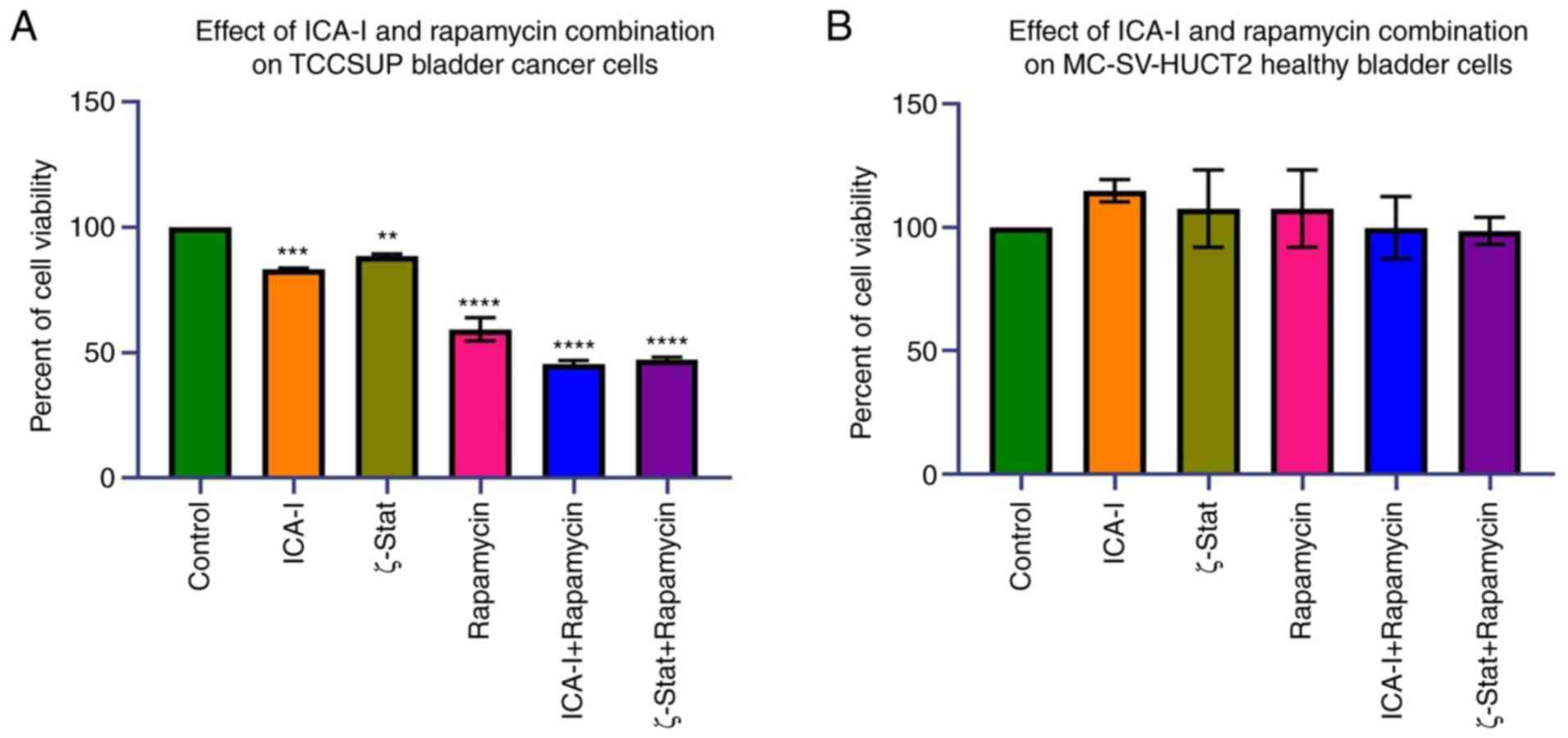
To examine the effects of the combination of
atypical PKC inhibitors and rapamycin on bladder cancer TCCSUP
cells, the present study first examined the viability of bladder
cancer following the combination treatment. The data indicated that
the combination of ICA-I with rapamycin and the combination of
ζ-Stat with rapamycin decreased the viability of bladder cancer
TCCSUP cells by >50% (P<0.0001) compared with the control
(Fig. 2A). Additionally, the
reduction in the viability of the cells treated with rapamycin or
atypical PKC inhibitors alone was less compared with
combination-treated cells (Fig.
2A). By contrast, the use of either combination or single drug
therapy did not produce any significant effect in healthy bladder
MC-SV-HUCT2 cells (Fig. 2B). These
discoveries suggested that the concomitant administration of mTOR
and atypical PKC inhibitors can be used to treat bladder cancer
cells without affecting healthy bladder cells.
Combination of atypical PKC inhibitors
and rapamycin induces cell cycle arrest by activating tumor
suppressors in bladder cancer cells
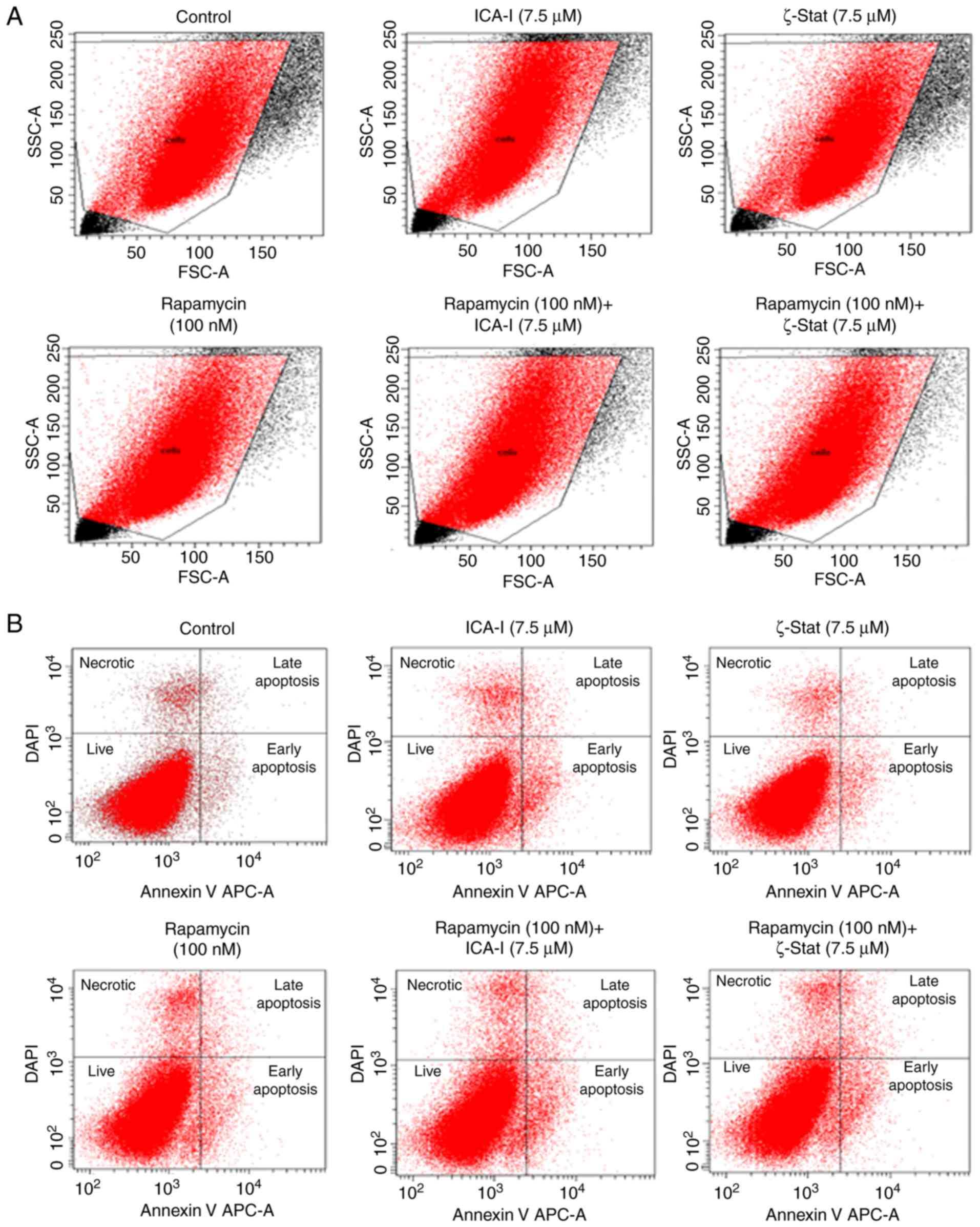
Since reduced viability of TCCSUP bladder cancer
cells was observed for the combination therapy of rapamycin and
atypical PKC inhibitor, the present study investigated the reason
for the reduced bladder cancer cell viability. The flow cytometric
analysis of apoptosis revealed that there was no substantial change
in the early and late apoptotic population of TCCSUP cells treated
with the combination even after 5 days (Fig. 3). Subsequently, the present study
investigated the status of cell cycle progression. During cell
cycle progression, cyclin E can work in conjunction with CDK2 to
transit the cells from G1 to S phase (24). The CDK interacting protein/kinase
inhibitory protein family members, such as p21 and p27, and
inhibitor of kinase 4/alternative reading frame family members,
such as p16, serve a critical role in inhibiting cell cycle
progression by binding and inactivating cyclin-CDK complexes
(Fig. 4A) (25). The findings indicated that there
was no significant change in the expression of cyclin D1 and CDK4
(Fig. 4B and C). However, the data
demonstrated that the simultaneous inhibition of mTOR and atypical
PKC by the combination of ICA-I and rapamycin significantly
increased the expression levels of p53 (P<0.0001),
p21(P<0.01), p27 (P<0.05; Fig.
4D and E) in treated cancerous urinary tract cells compared
with control cells.
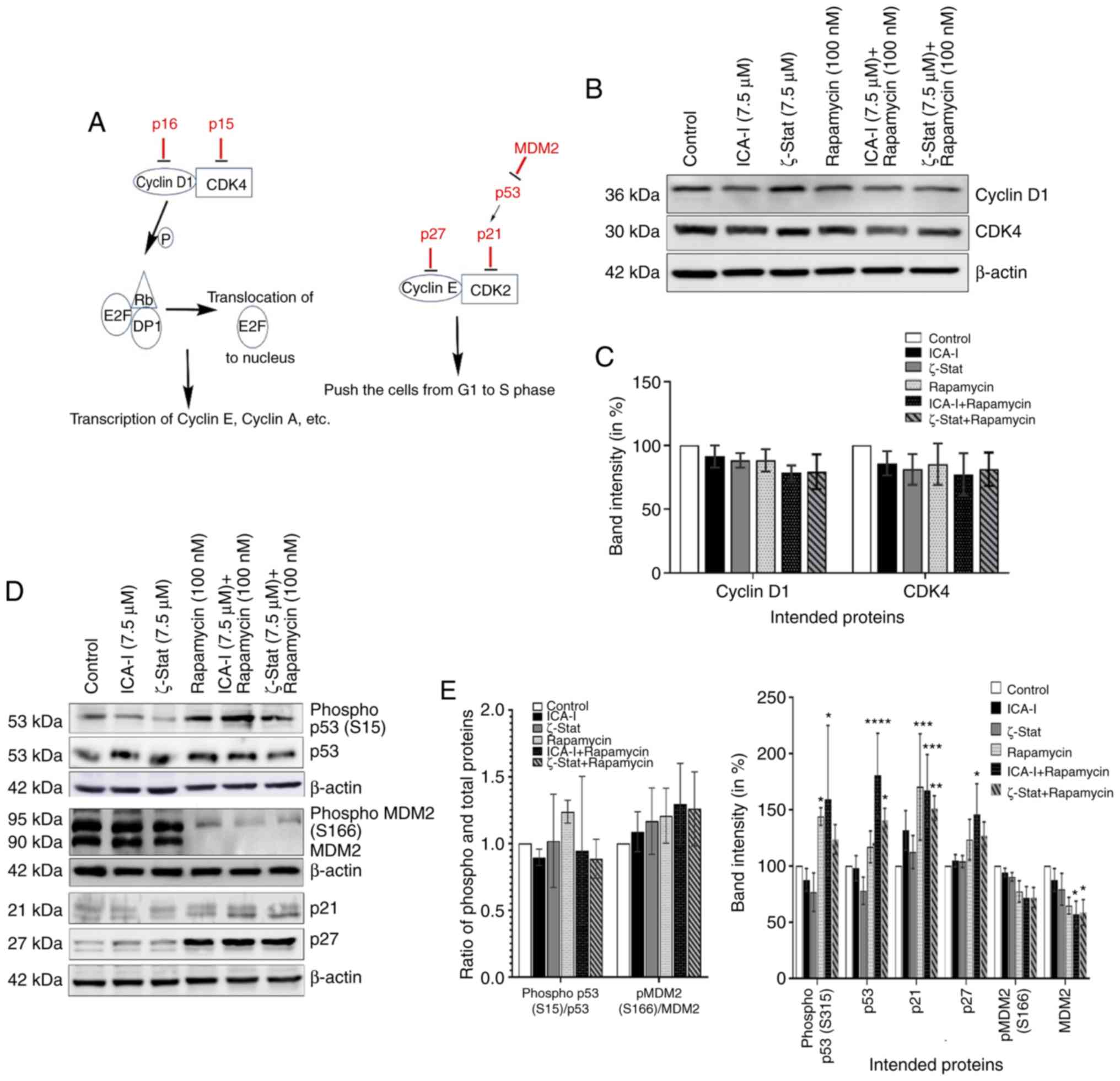 | Figure 4Combination of atypical PKC
inhibitors and rapamycin decreases bladder cancer cell progression.
(A) Proteins involved in cell cycle progression. Cells were grown
in 100-mm cell culture plates, followed by treatment with either
ICA-I (7.5 μM), ζ-Stat (7.5 μM), rapamycin (100 nM)
or a combination. The treated lysates were then subjected to
western blot analysis to observe the expression of cell cycle
regulatory proteins. (B) Western blotting bands of cyclin D1 and
CDK4 expression. (C) Expression levels of cyclin D1 and CDK4
presented as corresponding band intensity. (D) Western blotting
bands of phospho and total p53 and MDM2, as well as p21 and p27
expression in TCCSUP cells following 3 days of treatment of ICA-I,
ζ-Stat and rapamycin alone or in combination. (E) Densitometric
analysis of the phospho and total p53, and MDM2, p21 and p27. on
β-actin was used as a loading control (different β-actin loading
controls represent the protein bands from different blots). All
experiments were performed for at least three times. Data are
presented as the mean ± SEM. *P<0.05,
**P<0.02, ***P<0.01 and
****P<0.0001 vs. untreated control cells. MDM2, mouse
double min 2 homolog; p, phosphorylated; PKC, protein kinase C. |
Furthermore, in bladder cancer, p53 is known as the
guardian of the cell cycle and is the most important tumor
suppressor that serves a crucial role in regulating the cell cycle
via p21 (26). However, the
phosphorylation of p53, in turn, depends on the status of tumor
promoting MDM2 (26). The results
demonstrated that there was a notable change in the expression of
total MDM2 (P<0.05) in TCCSUP cells following treatment with
atypical PKC inhibitor and rapamycin combination (Fig. 4D and E). These findings suggested
that the combination of rapamycin with ICA-I or ζ-Stat reduced
malignant bladder cell growth by retarding cell cycle
progression.
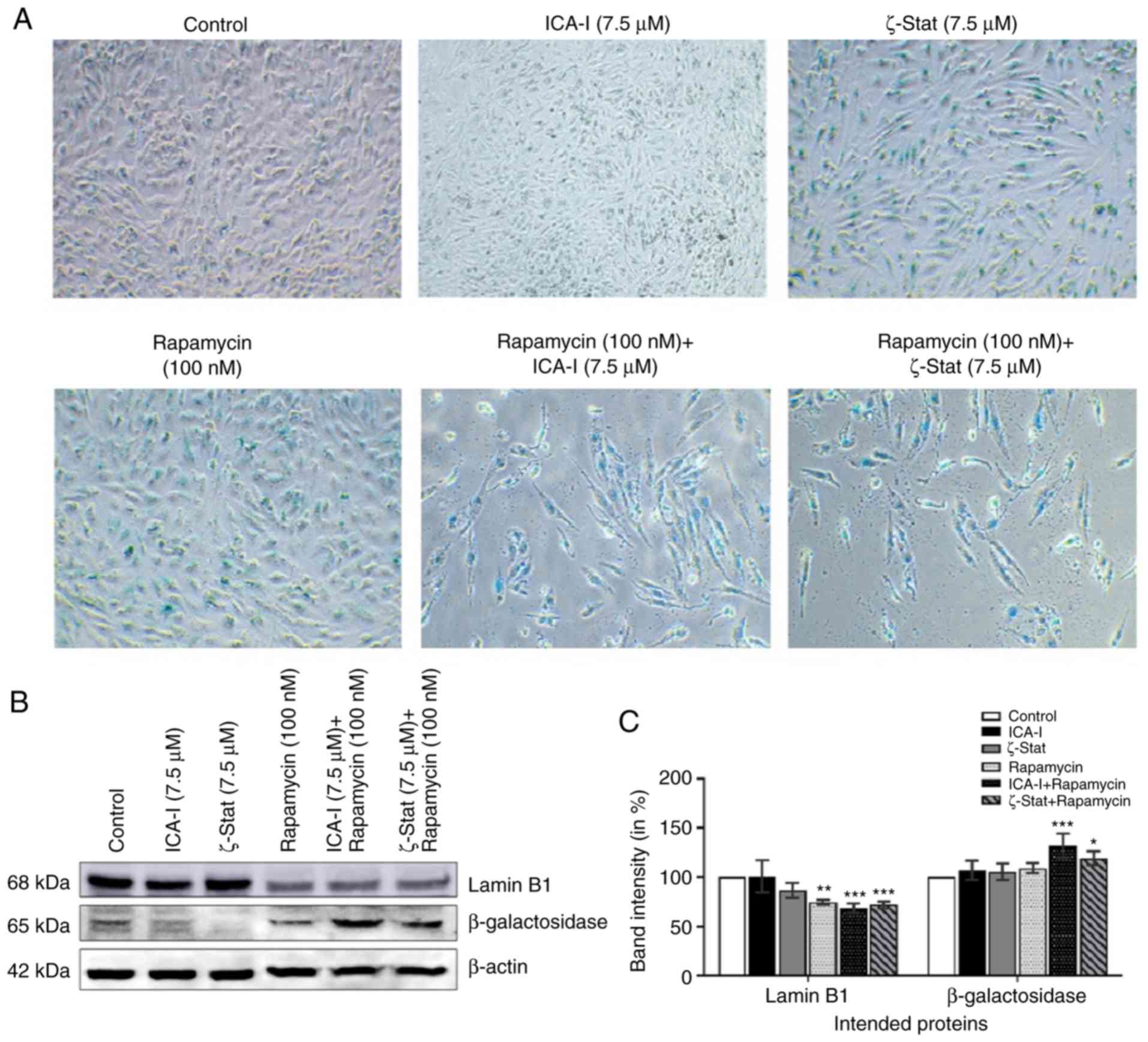
TCCSUP cells undergo senescence following
prolonged treatment with atypical PKC inhibitor and rapamycin
Cellular aging or senescence, gradual degradation of
functional characteristics, can be identified by the upregulation
of senescence associated β-Galactosidase (SA β-Gal) activity and
downregulation of a nuclear membrane protein, Lamin B1 (27,28).
To investigate whether SA β-Gal activity and Lamin B1 could be used
to quantify senescence in bladder cancer cells, metastatic cancer
cells were treated with atypical PKC inhibitors, rapamycin or the
combination for 7 days. The immunofluorescence analysis
demonstrated that with the combination treatment of rapamycin with
either ICA-I or ζ-Stat there was an increased SA β-Gal activity
(Fig. 5A), as well as, significant
overexpression of β-Galactosidase by >20% (P<0.05) in TCCSUP
cells (Fig. 5B and C).
Additionally, the combination of rapamycin and atypical PKC
inhibitor (either ICA-I or ζ-Stat) reduced the expression levels of
Lamin B1 by >30% (P<0.01; Fig.
5B and C). These results demonstrated that the prolonged
simultaneous inhibition of atypical PKC and mTOR induced senescence
in TCCSUP cells.
Concurrent inhibition of atypical PKCs
and mTOR reduces the migration of bladder cancer cells
To detect the anti-metastatic potential of the
combination, a preliminary scratch wound healing assay was
performed. The data depicted that the combination of ICA-I and
rapamycin treatment significantly prolonged the wound closure time
comapred with the control in TCCSUP bladder cancer cells (Fig. 6A and B). During metastasis, the
alterations of two critical proteins, namely E-cadherin and
β-catenin, in cells contribute to the acquisition of the motile
phenotype. E-cadherin, a cell adhesion protein, works in a reverse
relationship with β-catenin, a key molecule of the WNT/β-catenin
signaling pathway, to form an adherent junction which ultimately
gives the rigid structure of healthy cells (29,30).
The results revealed that the atypical PKC inhibitor and rapamycin
combination significantly increased the expression of E-cadherin
(P<0.0001; Fig. 6C and D).
Additionally, the combination of atypical PKC inhibitor and
rapamycin decreased the β-catenin levels in treated metastatic
bladder cancer cells; however, this was not statistically
significant (Fig. 6C and D).
Furthermore, SHC is a substrate for receptor tyrosine kinase that
has previously been identified to be involved in the dynamic
regulation of cell adhesion and motility of cancer cells (31,32).
To determine whether the atypical PKC and mTOR serve any role in
regulating the functionality of SHC protein in metastatic bladder
cancer cells, the level of SHC was examined. The data demonstrated
that the combination can also impeded the migration of bladder
cancer cells by reducing both phosphorylated and the total level of
SHC by >30% (P<0.0001) in treated cancerous cells compared
with control cells; however, the change in the ratio of
phosphorylated to total SHC levels was not significant (Fig. 6E and F). These results suggested
that the atypical PKC and mTOR mediated expression of E-cadherin,
β-catenin and SHC control the migration of TCCSUP cells.
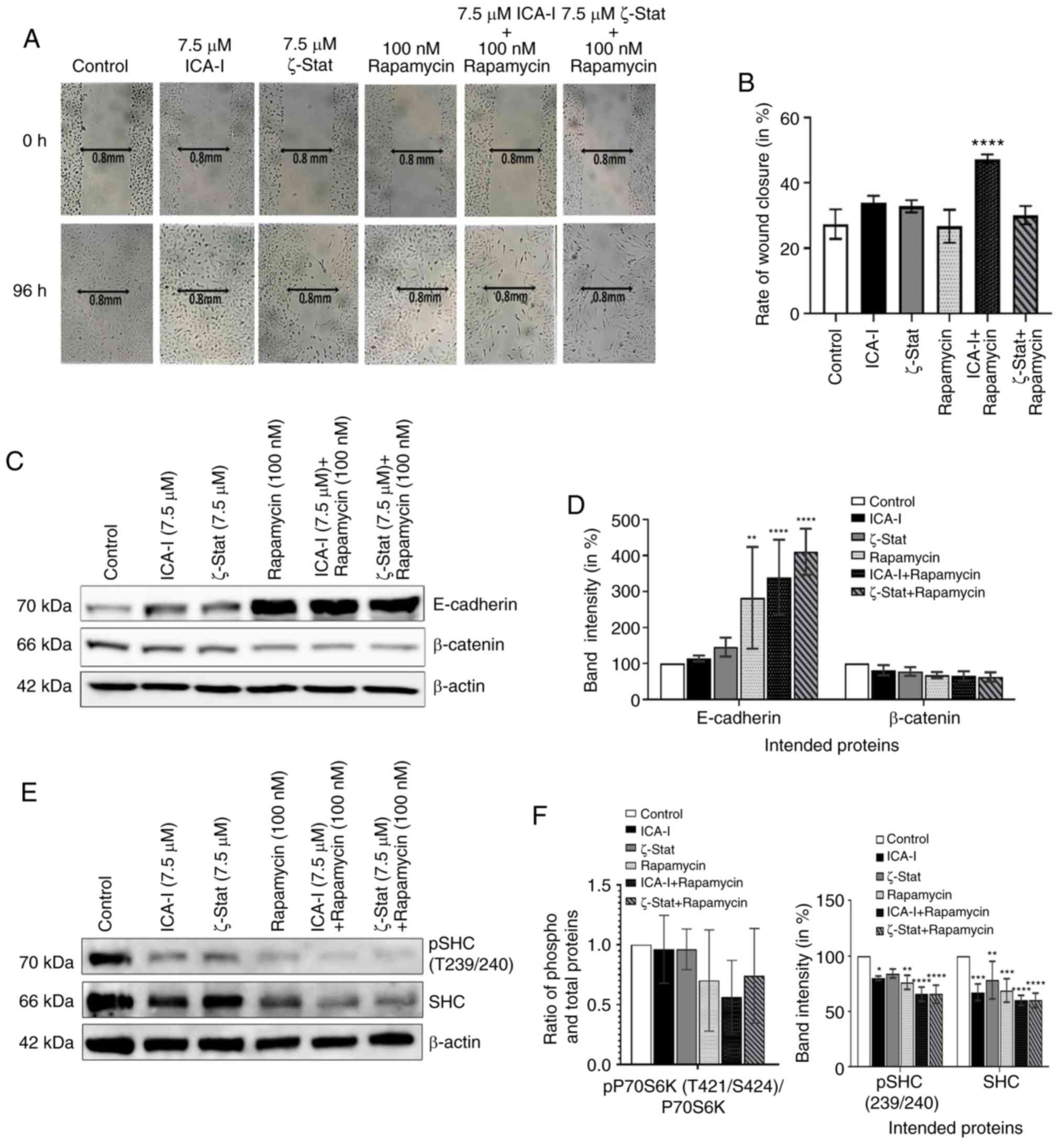 | Figure 6Combination of atypical PKC
inhibitors and rapamycin blocks migration of bladder cancer cells.
(A) Microscopic images of metastatic TCCSUP cells which were grown
to ~90% confluency and subjected to a wound healing assay by making
a scratch using a sterile 100-μl pipette tip, followed by
concurrent inhibition of atypical PKC and mTOR. Original
magnification, x10. (B) Bar charts presenting the rate of wound
closure after treating the cells for 3 consecutive days. In
addition, TCCSUP cells were grown in 100-mm cell culture plates
followed by treatment with either ICA-I (7.5 μM) or ζ-Stat
(7.5 μM) or rapamycin (100 nM) or combination for 3 days.
(C) Western blot analysis for the effect of ICA-I, ζ-Stat and
rapamycin alone or in combination on E-cadherin and β-catenin
expression. (D) Densitometric analysis of E-cadherin and β-catenin
levels. In addition, the levels of phospho and total SHC in TCCSUP
cells were assessed. (E) Bands and (F) their intensities (in
percent). β-actin was used as a loading control. All experiments
were performed at least three times. All data are presented as the
mean ± SEM. *P<0.05, **P<0.02,
***P<0.01 and ****P<0.0001 vs.
untreated control cells. p, phosphorylated; PKC, protein kinase C;
SHC, Src homology 2 domain cotaining transforming protein. |
Combination treatment of ICA-I and
rapamycin or ζ-Stat and rapamycin inhibits mTOR and atypical PKC
signaling pathways in bladder cancer cells
To examine whether the concurrent administration of
rapamycin and ICA-I or ζ-Stat could block the mTOR and atypical PKC
signaling pathways in bladder cancer cells, the present study
tested the changes in the functionality and expression of major
components of mTOR and atypical PKC signaling pathways in treated
TCCSUP cells. Our observed data indicated that the simultaneous
inhibition of mTOR and atypical PKC reduced the level of both
phosphorylated PKC-ι at T555 (by >50%; P<0.01) and
phosphorylated PKC-ζ at T560 (by >30%; P<0.02; Fig. 7A and B). The ratio of
phosphorylated to total PKC-ι was decreased, whereas, the ratio of
phosphorylated to total PKC-ζ was increased (Fig. 7A and B). Additionally, the
phosphorylation of the critical component of mTORC2, Rictor at
T1135 was also decreased by >50% (P<0.0001) in treated TCCSUP
cells. Furthermore, the total expression levels of PKC-ι, PKC-ζ and
Rictor (P<0.0001) were decreased following combination treatment
compared with the control (Fig. 7A and
B). The ratio of phosphorylated and total PKC-ι and Rictor
decreased (Fig. 7A and B);
however, this was not statistically significant. Consistent with
the aforementioned findings, the present study examined whether
atypical PKC and mTOR signaling pathways occur downstream and
upstream of each other by immunoprecipitating Rictor and checking
for PKC-ι and PKC-ζ. It was observed that Rictor was associated
with atypical PKC and the combination therapy of rapamycin and
atypical PKC inhibitors minimized the association of Rictor with
both PKC-ι by >30% (P<0.05) and PKC-ζ by >50%
(P<0.0001) in atypical PKC inhibitors and rapamycin
combination-treated cells compared with the control (Fig. 7C and D). Finally, the expression of
both total and phosphorylated P70S6K, a downstream molecule of mTOR
responsible for triggering cell cycle progression (33), also declined as a function of
combination treatment compared with control. Contrarily, the
reduction in the ratio of phospho P70S6K and P70S6K was found to be
statistically insignificant (Fig. 7E
and F). These findings suggest that the concomitant inhibition
of atypical PKC and mTOR can be used effectively to target both
atypical PKC and mTOR signaling to treat bladder cancer cells when
atypical PKC and mTOR are overexpressed.
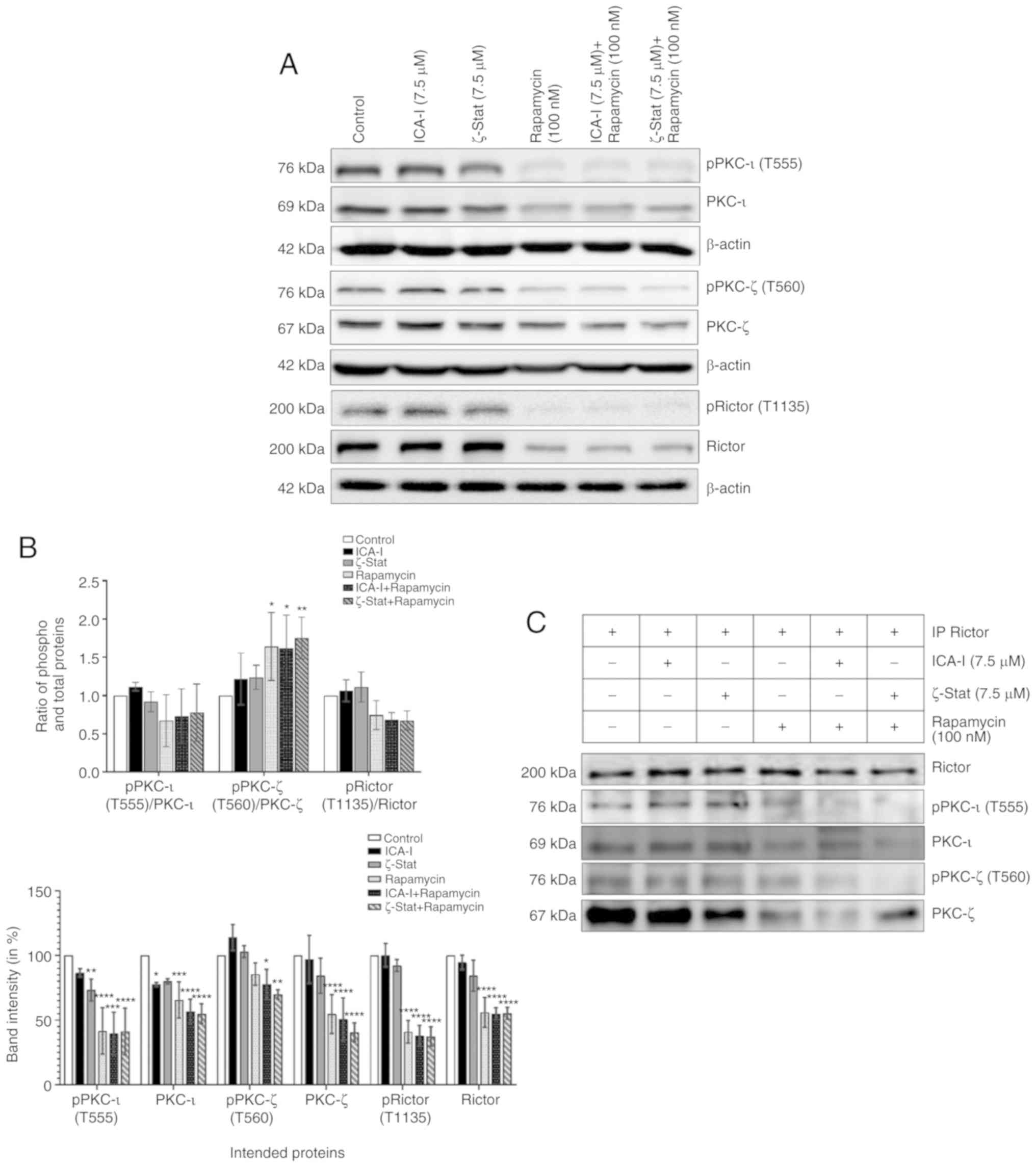 | Figure 7Combination of atypical PKC
inhibitors and rapamycin reduces the functionality of mTOR
complexes and atypical PKC in bladder cancer cells. (A) Western
blot analysis for the effect of ICA-I, ζ-Stat and rapamycin alone
or in combination on phospho and total PKC-ι, PKC-ζ and Rictor in
TCCSUP cells following 3 days of treatment. β-actin was used as a
loading control (different β-actin loading controls represent the
protein bands from different blots). (B) Densitometric analysis of
phospho and total PKC-ι, PKC-ζ and Rictor bands. (C) Rictor, key
component of mTORC2, was used in IP (3 μg) separately from
whole cell extracts (300 μg) of both treated (72 h) and
untreated metastatic TCCSUP cells using Rictor antibody, and
evaluated for atypical PKC molecules. The IP samples were separated
by SDS-PAGE and subjected to western blot analysis. Data are
presented as the mean ± SEM of three independent experiment.
*P<0.05, **P<0.02,
***P<0.01 and ****P<0.0001 vs.
untreated control cells. Combination of atypical PKC inhibitors and
rapamycin reduces the functionality of mTOR complexes and atypical
PKC in bladder cancer cells. (D) Effect of atypical PKC inhibitors
on total and phospho PKC-ι and PKC-ζ were quantified using
densitometry. (E) Effect of the combination on a mTORC1 downstream
protein, P70S6K and its phosphorylated form. (F) Densitometric
analysis of phospho and total P70S6K. Data are presented as the
mean ± SEM of three independent experiment. *P<0.05,
**P<0.02, ***P<0.01 and
****P<0.0001 vs. untreated control cells. IP,
immunoprecipitation; p, phosphorylated; PKC, protein kinase C. |
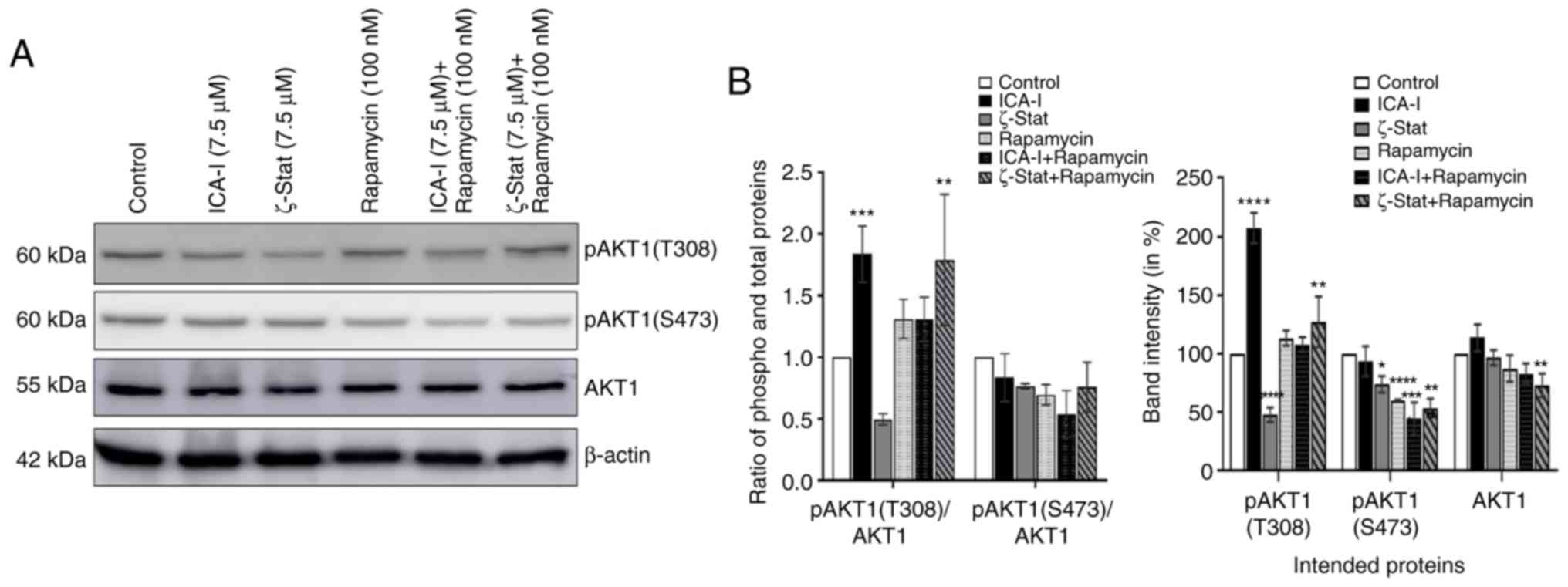
Combination of atypical PKC inhibitors
and rapamycin reduces the expression of AKT1 in metastatic TCCSUP
cells
AKT1 serves an interesting role in mediating mTOR
complexes driven pathways; it acts as an upstream regulator of
mTORC1 and downstream molecule of mTORC2 (34). The present study examined the
expression levels of AKT1 to observe whether the inhibition of
atypical PKCs and mTOR affects its functionality. The results
demonstrated that compared with the control, the combination
therapy significantly reduced the phosphorylation of AKT1 at S473
position by >45% (P<0.0001; Fig.
8) which is a phosphorylation site for mTORC2. However, there
was no significant change in the ratio of phosphorylated to total
AKT1 (Fig. 8). These findings
suggested that the combination of atypical PKC inhibitor and
rapamycin affects the activation of AKT1 in bladder cancer
cells.
Discussion
Recent advances in life sciences have shifted the
therapeutic approach of bladder cancer and are currently focusing
on segregating bladder cancer based on the genetic profile. The
activation of the PI3K/mTOR signaling pathway is crucial in bladder
cancer and is associated with resistance to chemotherapeutic agents
(35). The genetic profile of a
patient with bladder cancer seems to be promising in designing
targeted therapy, since the genomic analysis of patients with
bladder cancer who responded well to everolimus
(rapamycin-containing drug) indicated that they have high
expression levels of tuberous sclerosis 1, TSC1 (36). One of the main challenges in using
mTOR inhibitor is that it can cause reactivation of oncogenic AKT1
signaling resulting from the attenuation of the negative regulatory
loop from mTOR which can ultimately lead to disease recurrence
(36). Therefore, the present
study aimed to establish a novel therapeutic regimen that can cause
dual inhibition of cancer-causing pathways, namely atypical PKCs
and mTOR signaling in bladder cancer.
In our previous studies, it was observed that
atypical PKCs are overexpressed in prostate cancer, colon cancer,
melanoma, breast cancer, glioma and neuroblastoma cells (8,19,23,37,38).
Similarly, hyperactivation of mTOR is associated with different
types of cancer, such as bladder and breast cancer (11,39).
Notably, combination regimen, such as rapamycin and resveratrol,
have been identified to be effective in treating breast and bladder
cancer by inducing autophagy and preventing reactivation of AKT1
in vitro and in a mouse xenograft model (40,41).
Keeping the hypothesis in mind that both atypical PKC and mTOR
serve crucial carcinogenic roles in bladder cancer cells, the
present study aimed to inhibit both atypical PKC and mTOR in
bladder cancer cells. Another reason for trying this combination is
that in a recent study, a combination of atypical PKC inhibitor and
a widely used clinical agent, known as 5-flouorouracil, was trialed
in CRC cells, and it was observed that the combination can reduce
the growth and proliferation of CRC cells by blocking the DNA
repair mechanism of the cancer cells (42). First, the present study
investigated the effi-cacy of the inhibitors in bladder cancer
cells compared with healthy bladder cells. The cell viability
investigation revealed that the simultaneous inhibition of atypical
PKC and mTOR using the combination of either ICA-I or Stat and
rapamycin for 3 days reduced the viability of TCCSUP bladder cancer
cells markedly (>50%; P<0.0001) compared with control
untreated bladder cancer cells. However, the combination therapy
did not induce any significant changes in the MC-SV-HUCT2 healthy
bladder cell viability. It is interesting to note that the flow
cytometry based apoptosis assay did not detect any significant
apoptotic population even after treating the cells for 5 days. The
subsequent western blot analysis of cell cycle proteins following
treatment of TCCSUP cells with atypical PKC and mTOR inhibitors
revealed that there was an upregulation of p27 and p21, which are
two important tumor suppressors that work by inhibiting cyclin E
and CDK2, respectively, of the cyclin E-CDK2 cell cycle regulatory
complex (25,43). The activation of p21 depends on
another critical tumor suppressor protein known as p53, which in
turn, is negatively regulated by MDM2 (43). The further investigation revealed
that the combination of atypical PKC inhibitor and rapamycin
increased the functionality of tumor suppressing p53 while
retarding MDM2 expression. However, the combination treatment did
not induce any significant changes in other upstream cell cycle
regulatory molecules, such as cyclin D1and CDK4.
Interestingly, treatment was continued for 7
consecutive days to examine the fate of cells following cell cycle
arrest, and it was observed that prolonged treatment made the cells
undergo irreversible growth arrest or senescence. Two of the
crucial factors that are indicative of cellular senescence are: i)
Downregulation of Lamin B1, a nuclear membrane component important
in maintaining normal cellular function; and ii) increased SA β-Gal
activity (27). Based on this
observation, it was speculated that the prolonged inhibition of
atypical PKC and mTOR induced senescence as evident by reduced
Lamin B1 expression and increased SA β-Gal activity. Considering
the fact that mTOR and atypical PKCs may stimulate bladder cancer
cell progression, the present study also examined the metastatic
profile of bladder cancer cells as a function of combination
treatment. Similar to our previous study (20), combined inhibition of atypical PKC
and mTOR using ICA-I and rapamycin prolonged the rate of wound
closure in TCCSUP cells, as demonstrated by the scratch wound
healing assay. Although serum has a significant impact on the
proliferation of cells, the scratch wound healing assay was
performed using media containing 10% FBS to maintain consistency
across all experimental protocols, since changes in serum
concentration could affect the phosphorylation of proteins that
have a critical role in the cell signaling pathway (44). One of the first metastatic events
that takes place inside the cell is the loss of E-cadherin, a cell
adhesion molecule. Loss of E-cadherin causes loose junction between
cells and induces the cells to acquire a motile phenotype (29). Additionally, increased activity of
WNT/β-catenin signaling is another hallmark of cancer cell
metastasis (30). The present
study revealed that the combination treatment induced an increased
expression of E-cadherin and decreased expression of β-catenin.
Furthermore, the expression of SHC, another dynamic regulator of
adhesion and migration has been implicated in metastasis of
cancerous cells (31,32). Interestingly, the combination of
atypical PKC inhibitors and rapamycin notably reduced the
expression of SHC in bladder cancer cells.
One of the purposes of the present study was to
establish a link between atypical PKCs and mTOR complexes in
bladder cancer cells. It was considered that atypical PKCs may
serve a role as a downstream molecule of the mTOR signaling
pathway, since PKCs, in general, are found as a downstream protein
of mTORC2 that facilitates cancer cell migration and invasion by
cytoskeleton remodeling (11). The
results demonstrated that the combination of atypical PKC inhibitor
and rapamycin induced a pronounced inhibition in the expression of
phos-phorylated atypical PKC which may occur because of the role of
mTOR in mediating the phosphorylation of protein kinases at their
turn and hydrophobic region (14-16).
Rapamycin is the most commonly used extremely selective drug to
target mTOR, which is already in clinical use. It is well
established that mTOR complexes, specifically mTORC1, are sensitive
towards rapamycin but mTORC2 is somewhat insensitive to rapamycin
(45); hence, the goal of the
present study was to examine the effect of rapamycin and atypical
PKC inhibitor combination on mTORC2. It was demonstrated that the
combination induced an inactivation and downregulation of the most
crucial mTORC2 component, Rictor. Additionally, atypical PKCs were
associated with Rictor and the combination treatment reduced the
association in the treated bladder cancer cells. Furthermore, there
was a marked decrease in the expression of phosphorylated P70S6K, a
mTORC1 downstream protein responsible for the cell cycle
progression, following treatment with the combination (33). This finding was consistent with the
cell cycle protein analysis, which could result from the direct
inhibition of both mTORC1 and atypical PKC, or there may be some
links between these two proteins. Furthermore, AKT1 serves a role
in mTOR pathways; it acts as an upstream molecule of mTORC1 where
AKT1 acts by inhibiting tuberous sclerosis 2 (TSC2) required for
mTORC1 activation and is a downstream protein of mTORC2, whereas
mTORC2 phosphorylates AKT1 at Serine 473 (34). The results demonstrated that the
combination of both ICA-I and rapamycin, and ζ-Stat and rapamycin
therapy reduced the phosphorylation of AKT1 at Serine 473; which
may be due to the inactivation of upstream mTORC2 or indirect
inhibition of mTORC2 due to decreased activation of atypical PKCs.
However, more investigations need to be performed to validate this
statement.
The present study has some limitations; only in
vitro study is not adequate to justify the efficacy of the
proposed combination treatment in bladder cancer; hence, future
in vivo studies would increase the credibility of the
proposed statement. Furthermore, in this investigation, even though
a significant change in the phosphorylated and total levels of
intended proteins was observed following treatment with the
combination of atypical PKC inhibitors and rapamycin compared with
the control, the change in the ratio between the phosphorylated and
total protein levels appeared to be statistically insignificant in
most cases. One possible reason for not achieving statistical
significance is that in some cases the combination treatment
reduced the phosphorylated form of the protein without affecting
total protein levels, or there was fluctuating change between the
reduction of phosphorylated and total levels of protein, or in some
cases the phosphorylated and total proteins changed at the same
rate. Therefore, further studies need to be performed to understand
the change in effect between phosphorylated and total protein and
their ratios. Furthermore, the present study used only one
aggressive bladder cancer cell line that represents grade IV
transitional cell carcinoma; thus, the proposed combination should
be evaluated in another bladder cancer cell line.
In conclusion, atypical PKC and mTOR serve a crucial
role in the carcinogenesis of bladder cancer cells (Fig. 9). Additionally, mTOR can enhance
the activity of atypical PKC. Hence, the combination of atypical
PKC inhibitors and rapamycin can be used as a novel therapeutic
regimen for individualized therapy.
Funding
The present study was supported by the Leo and Anne
Albert Charitable Trust, Frederick H (grant no. 42-0142). Leonhardt
Foundation (grant no. 42-0044), Kyrias Foundation (grant no.
42-0044), Jin and Joo Lee Family Foundation (grant no. 42-0044),
and Miami Foundation for Cancer Research, Inc. (grant no.
42-0044).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RP and SMAI designed the experiment. SMAI wrote the
draft of the manuscript. RP performed most of the experiments and
analyzed the data. TS and RRB assisted RP in western blotting and
immunoprecipitation analyses. MAD was involved in the conception,
design and mentoring, as well as critical interpretation and
evaluation of the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mr. Gene Prenzo,
Trustee of the Leo and Anne Albert Charitable Trust for his
generous financial support of this work. The abstract of the
present study was presented at the AACR Annual Meeting 2019; March
29-April 3, 2019; Atlanta, GA, USA, and was published as Abstract
no. 4290.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Montironi R, Cheng L, Scarpelli M and
Lopez-Beltran A: Pathology and genetics: Tumours of the urinary
system and male genital system: Clinical implications of the 4th
edition of the WHO classification and beyond. Eur Urol. 70:120–123.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abraham S, Knapp DW, Cheng L, Snyder PW,
Mittal SK, Bangari DS, Kinch M, Wu L, Dhariwal J and Mohammed SI:
Expression of EphA2 and Ephrin A-1 in carcinoma of the urinary
bladder. Clin Cancer Res. 12:353–360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reuter VE: Pathology of bladder cancer:
Assessment of prognostic variables and response to therapy. Semin
Oncol. 17:524–532. 1990.PubMed/NCBI
|
|
5
|
Sylvester RJ, van der MEIJDEN AP and Lamm
DL: Intravesical bacillus Calmette-Guerin reduces the risk of
progression in patients with superficial bladder cancer: A
meta-analysis of the published results of randomized clinical
trials. J Urol. 168:1964–1970. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gschwend JE, Dahm P and Fair WR: Disease
specific survival as endpoint of outcome for bladder cancer
patients following radical cystectomy. Eur Urol. 41:440–448. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nelson DL and Cox MM: Lehninger principles
of biochemistry. 5th ed. Book; pp. 1–1294. 2008
|
|
8
|
Islam SMA, Patel R and Acevedo-Duncan M:
Protein Kinase C-ζ stimulates colorectal cancer cell carcinogenesis
via PKC-ζ/Rac1/Pak1/β-Catenin signaling cascade. Biochim Biophys
Acta Mol cell Res. 1865:650–664. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Islam SMA, Patel R, Bommareddy RR, Khalid
KM and Acevedo-Duncan M: The modulation of actin dynamics via
atypical Protein Kinase-C activated Cofilin regulates metastasis of
colorectal cancer cells. Cell Adh Migr. 13:106–120. 2019.
View Article : Google Scholar :
|
|
10
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phos-phoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sengupta S, Peterson TR and Sabatini DM:
Regulation of the mTOR complex 1 pathway by nutrients, growth
factors, and stress. Mol Cell. 40:310–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Facchinetti V, Ouyang W, Wei H, Soto N,
Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al:
The mammalian target of rapamycin complex 2 controls folding and
stability of Akt and protein kinase C. EMBO J. 27:1932–1943. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikenoue T, Inoki K, Yang Q, Zhou X and
Guan KL: Essential function of TORC2 in PKC and Akt turn motif
phosphorylation, maturation and signalling. EMBO J. 27:1919–1931.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eder AM, Sui X, Rosen DG, Nolden LK, Cheng
KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, et al:
Atypical PKC contributes to poor prognosis through loss of
apical-basal polarity and Cyclin E overexpression in ovarian
cancer. Proc Natl Acad Sci. 102:12519–12524. 2005. View Article : Google Scholar
|
|
18
|
Guo H, Ma Y, Zhang B, Sun B, Niu R, Ying G
and Zhang N: Pivotal Advance: PKC is required for migration of
macrophages. J Leukoc Biol. 85:911–918. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smalley T, Islam SMA, Apostolatos C,
Apostolatos A and Acevedo-Duncan M: Analysis of PKC-ζ protein
levels in normal and malignant breast tissue subtypes. Oncol Lett.
17:1537–1546. 2019.PubMed/NCBI
|
|
20
|
Islam SMA, Dey A and Acevedo-Duncan M:
Abstract 719: Combination of atypical protein kinase-C inhibitor
and 5-fluo-rouracil retards the proliferation of colorectal cancer
cells. Mol Cell Biol. 719:2019.
|
|
21
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Patel R, Win H, Desai S, Patel K, Matthews
JA and Acevedo-Duncan M: Involvement of PKC-ι in glioma
proliferation. Cell Prolif. 41:122–135. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sherr CJ and Roberts JM: Living with or
without cyclins and cyclin-dependent kinases. Genes Dev.
18:2699–2711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hardie DG and Hanks S: The protein kinase
factsbook. Academic Press; 1995
|
|
26
|
Kriegmair MC, Balk M, Wirtz R, Steidler A,
Weis CA, Breyer J, Hartmann A, Bolenz C and Erben P: Expression of
the p53 inhibitors mdm2 and mdm4 as outcome predictor in
muscle-invasive bladder cancer. Anticancer Res. 36:5205–5213. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang AS, Ong PF, Chojn owski A, Clavel C
and Dreesen O: Loss of lamin B1 is a biomarker to quantify cellular
senescence in photoaged skin. Sci Rep. 7:156782017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goldstein S: Replicative senescence: The
human fibroblast comes of age. Science. 249:1129–1133. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wheelock MJ and Johnson KR: Cadherins as
Modulators of Cellular Phenotype. Annu Rev Cell Dev Biol.
19:207–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
De P, Carlson JH, Wu H, Marcus A,
Leyland-Jones B and Dey N: Wnt-beta-catenin pathway signals
metastasis-associated tumor cell phenotypes in triple negative
breast cancers. Oncotarget. 7:43124–43149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mauro L, Sisci D, Bartucci M, Salerno M,
Kim J, Tam T, Guvakova MA, Ando S and Surmacz E: SHC-α5β1 integrin
interactions regulate breast cancer cell adhesion and motility. Exp
Cell Res. 252:439–448. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jackson JG, Yoneda T, Clark GM and Yee D:
Elevated levels of p66 Shc are found in breast cancer cell lines
and primary tumors with high metastatic potential. Clin Cancer Res.
6:1135–1139. 2000.PubMed/NCBI
|
|
33
|
Xiao L, Wang YC, Li WS and Du Y: The role
of mTOR and phospho-p70S6K in pathogenesis and progression of
gastric carcinomas: An immunohistochemical study on tissue
micro-array. J Exp Clin Cancer Res. 28:1522009. View Article : Google Scholar
|
|
34
|
Malley CO and Pidgeon GP: The mTOR pathway
in obesity driven gastrointestinal cancers: Potential targets and
clinical trials. BBA Clin. 5:29–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ching CB and Hansel DE: Expanding
therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway.
Lab Invest. 90:1406–1414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carneiro BA, Meeks JJ, Kuzel TM, Scaranti
M, Abdulkadir SA and Giles FJ: Emerging therapeutic targets in
bladder cancer. Cancer Treat Rev. 41:170–178. 2015. View Article : Google Scholar
|
|
37
|
Apostolatos AH, Ratnayake WS, Win-Piazza
H, Apostolatos CA, Smalley T, Kang L, Salup R, Hill R and
Acevedo-Duncan M: Inhibition of atypical protein kinase C-ι
effectively reduces the malignancy of prostate cancer cells by
downregulating the NF-κB signaling cascade. Int J Oncol.
53:1836–1846. 2018.PubMed/NCBI
|
|
38
|
Ratnayake WS, Apostolatos AH, Ostrov DA
and Acevedo-Duncan M: Two novel atypical PKC inhibitors; ACPD and
DNDA effectively mitigate cell proliferation and epithelial to
mesenchymal transition of metastatic melanoma while inducing
apoptosis. Int J Oncol. 51:1370–1382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pinto-Leite R, Arantes-Rodrigues R, Sousa
N, Oliveira PA and Santos L: mTOR inhibitors in urinary bladder
cancer. Tumor Biol. 37:11541–11551. 2016. View Article : Google Scholar
|
|
40
|
Alayev A, Salamon RS, Schwartz NS, Berman
AY, Wiener SL and Holz MK: Combination of rapamycin and resveratrol
for treatment of bladder cancer. J Cell Physiol. 232:436–446. 2017.
View Article : Google Scholar
|
|
41
|
Alayev A, Berger SM, Kramer MY, Schwartz
NS and Holz MK: The combination of rapamycin and resveratrol blocks
autophagy and induces apoptosis in breast cancer cells. J Cell
Biochem. 116:450–547. 2015. View Article : Google Scholar :
|
|
42
|
Islam SMA, Dey A, Patel R, Smalley T and
Acevedo-Duncan M: Atypical Protein Kinase-C inhibitors exhibit a
synergistic effect in facilitating DNA damaging effect of
5-fluorouracil in colorectal cancer cells. Biomed Pharmacother.
120:1096652020. View Article : Google Scholar
|
|
43
|
Orlando DA, Lin CY, Bernard A, Wang JY,
Socolar JE, Iversen ES, Hartemink AJ and Haase SB: Global control
of cell-cycle transcription by coupled CDK and network oscillators.
Nature. 453:944–947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Levin VA, Panchabhai SC, Shen L, Kornblau
SM, Qiu Y and Baggerly KA: Different changes in protein and
phosphoprotein levels result from serum starvation of high-grade
glioma and adenocarcinoma cell lines. J Proteome Res. 9:179–191.
2010. View Article : Google Scholar
|
|
45
|
Ballou LM and Lin RZ: Rapamycin and mTOR
kinase inhibitors. J Chem Biol. 1:27–36. 2008. View Article : Google Scholar
|