Introduction
It is estimated that ~50% of patients with melanoma
harbour B-Raf (BRAF)V600 driver mutations, the most common of which
being BRAFV600E; this mutation leads to the activation of
mitogen-activated protein kinase (MAPK) proliferative and survival
pathways (1). BRAF inhibitors,
alone or in combination with MAPK kinase (MEK) inhibitors, are
extensively used to treat BRAF-mutated metastatic melanoma;
however, acquired resistance unfortunately occurs in the majority
of patients (2). Resistance
mechanisms involve mutations or changes in gene expression that
result in the reactivation of MAPK signalling, or the activation of
other proliferative and survival pathways, such as PI3K signaling
(3-7). Increasing evidence suggests that
these resistant states exist prior to treatment and may be selected
by treatment with BRAF inhibitors (8,9).
These subpopulations can contribute to tumour progression by
presenting a slow-cycling behaviour or phenotype associated with
epithelial-to-mesenchymal transition. This plasticity may allow a
rapid and dynamic response, generating a phenotype that could more
effectively tolerate drug treatment, underlying the limited cell
death observed in vitro and in vivo during targeted
therapy (8).
Acquired drug resistance could also be driven by
epigenetic events; it has been shown that epigenetic alterations
contribute to chemotherapy resistance in different types of
tumours, including breast, colorectal and ovarian cancers (10-13).
Recent evidence suggests that chromatin architecture reprogramming
could be also implicated in drug resistance to MAPK inhibitors in
melanoma cells (14,15). Several groups have reported that
treatment with different epigenetic inhibitors, such as histone
deacetylate (HDAC) inhibitors (16,17),
bromodomain and extra-terminal motif (BET) inhibitors (18) and DNA methyltransferase (DNMT)
inhibitors (19), in combination
with BRAF inhibitors, could overcome resistance.
Aside from the resistant cells that are able to
proliferate in the presence of MAPK inhibitors, our and other
previous studies have shown that BRAF and MEK inhibitors can lead
to the enrichment of a drug-tolerant tumour cell population that
persists in a slow-cycling or quiescent state (9,20-22).
This evidence now may be of greater clinical relevance, as combined
BRAF and MEK inhibitor treatment has been recently approved in the
adjuvant setting for patients with stage III recurrent
BRAFV600-mutated melanoma, as it was reported to result in a
significantly reduced risk of recurrence (23). If a population of persisting
melanoma cells is present, once the treatment is discontinued, they
could give rise to relapses.
Our previous study described a persistent melanoma
cell population [surviving (SUR) cells], obtained following
long-term PLX4032 treatment, of sensitive BRAFV600E-mutated
melanoma cell lines (20). SUR
cells express the cancer stem cell markers CD271 and ATP-binding
cassette B5, and present senescence-associated characteristics,
such as senescence-associated (SA) β-galactosidase activity.
Discontinuing MAPK inhibitor treatment of SUR cells permits their
regrowth, and they eventually regain drug sensitivity equal to
parental cells, demonstrating the plasticity of the SUR phenotype.
SUR cells exhibit an increased tumorigenicity compared with
parental cells when injected subcutaneously in NOD/SCID-γ (NSG)
mice, yet retain melanoma differentiation antigens (Ags) and human
leukocyte Ag class I expression, and are therefore susceptible to
Ag-specific cytotoxic T lymphocytes lysis (20).
It was hypothesized that the SUR phenotype may be
determined by epigenetic changes. Thus, the aim of the present
study was to determine if treatment with epigenetic inhibitors
could efficiently eliminate the SUR population. SUR cell
sensitivity to different epigenetic inhibitors was analysed, and it
was found that both parental and SUR cells were sensitive to HDAC
inhibitors. It is proposed that the combination of PLX4032 with
epigenetic inhibitors could be efficacious to achieve complete
elimination of SUR cells that persist after long-term BRAF
inhibitor treatment.
Materials and methods
Cell lines and drugs
The MEL-XY3 and MEL-XY13 cell lines have already
been described (20). MEL-XX12 and
MEL-XX15 were obtained in-house from metastatic melanoma biopsies.
Both cell lines present the BRAFV600E mutation. MEL-XX12 was
established from a 58-year-old female diagnosed with cutaneous
melanoma in the right side of the back. The patient developed a
local recurrence in the back which was excised at the Hospital
Naval Dr Pedro Mallo (Buenos Aires, Argentina). The patient
provided oral consent (as the patient was hospitalised in a
different institution at the time of collection, oral rather than
written consent was obtained) for the collection of part of the
fresh tumour to be used for the establishment of experimental cell
lines for cancer research purposes. The MEL-XX15 cell line was
established from a 38-year-old female diagnosed with cutaneous
melanoma in the right forearm. The cell line was derived from a
metastatic subcutaneous nodule excised from the breastbone site at
4 years after diagnosis. Surgery was conducted at the Instituto
Alexander Fleming (Buenos Aires, Argentina), and the patient
provided signed informed consent for surgery and the use of part of
the tumour sample for scientific research. Ethical approval of the
informed consent for surgery was granted by the Comité de Etica en
Investigación del Instituto Alexander Fleming prior to the
isolation and establishment of these two cell lines.
Melanoma cell lines were cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.; MEL-XY3 cells only) or melanoma
medium [DMEM:F-12 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 2 mM glutamine, 20 nM sodium selenite, 100
µM ascorbic acid, 0.3 mg/ml galactose, 0.15 mg/ml sodium
pyruvate, 5 µg/ml insulin; all cell lines] plus 10% fetal
calf serum (AusGeneX), 100 IU/ml penicillin and 10 µg/ml
streptomycin. All cells were maintained at 37°C in a 5%
CO2 humidified atmosphere. Mycoplasma detection was
regularly assayed by PCR. PLX4032, GDC-0973, MGCD0103, SAHA,
S63845, panobinostat and tubacin were purchased from MedChemExpress
LLC. IBET151, JQ-1, OTX-015, decitabine, EPZ-6438 and GSK126 were
purchased from Selleck Chemicals. MC1568, sodium butyrate and
trichostatin A (TSA) were purchased from Sigma-Aldrich (Merck
KGaA). CDKI-73 was a gift from Dr Shudong Wang (University of South
Australia).
Generation of SUR cells
SUR cells were generated by exposing parental cells
to 10 µM PLX4032 or 10 µM PLX4032 + 1 µM
GDC-0973 combined treatment for 5 weeks. Media was changed twice
weekly.
RNA sequencing (RNA-seq)
RNA-seq was performed for MEL-XY3 and SUR MEL-XY3
cells following PLX4032 exposure using the Illumina Hiseq 4000
sequencing platform (Illumina, Inc.) with >20 million
high-quality single-end reads per sample (sequencing was performed
by BGI Americas). Quality control of reads was performed with
FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and FastQC
(http://www.bioinformatics.babraham.ac.uk/proj-ects/fastqc/).
Reads were aligned to the latest human Hg38 reference genome using
the STAR spliced read aligner (24). Fragment counts were derived using
the HTSeq package (25).
Differentially expressed genes were identified by a ranking based
on adjusted P-values ≤0.005 and a false discovery rate (FDR) ≤0.1
using the R package edgeR (R version 3.2) (26,27).
Gene set enrichment analysis (GSEA) (28) was performed using GSEA software
from Broad Institute (https://www.gsea-msigdb.org/gsea/index.jsp). The
present RNA-seq dataset was compared against a Molecular Signature
Database gene set composed of a Gene Ontology (GO; http://geneontology.org/) gene set library (C5
collection), with an FDR <0.25 and P<0.01. The data discussed
in this publication have been deposited in Gene Expression Omnibus
(GEO) (29) and are accessible via
GEO accession number GSE126960 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126960).
Normalized RNA-seq data were retrieved from GSE50509
dataset of the GEO database (29,30).
Graphs were plotted using GraphPad Prism software (GraphPad
Software, Inc.). A two-tailed paired t-test was performed to
analyse the statistical significance of the given samples.
Reverse phase protein array (RPPA)
RPPA experiments were performed to analyze protein
expression in MEL-XY3 cells, in MEL-XY3 cells treated for 7 days
with PLX4032, in MEL-XY3 SUR cells and in MEL-XY3 SUR cells 7 days
after drug removal, at which point the cells had resumed
proliferating. RPPA was performed at the Victorian Centre for
Functional Genomics RPPA platform at the Peter MacCallum Cancer
Centre (Melbourne, Australia). Protein lysates were prepared in
biological duplicates from cell lines freshly lysed in Zeptosens
Cell Lysis Buffer 1 (CLB1; Bayer AG) and quantified using a Pierce™
Coomassie Plus Bradford Protein Assay Kit (Thermo Fisher
Scientific, Inc.); triplicate measurements were performed for each
sample. Employing a Sciclone/Caliper ALH3000 liquid handling robot
(PerkinElmer, Inc.), samples were serially diluted in 10% CLB1:90%
CSBL1 spotting buffer (Zeptosens AG; Bayer AG) and spotted onto
ZeptoChips (Zeptosens AG; Bayer AG) in duplicate using a
Nano-plotter-NP2.1 noncontact microarray system (GeSiM mbH). Chips
were blocked under non-contact conditions for 1 h with BB1 buffer
(Zeptosens AG; Bayer AG) and incubated with prevalidated primary
antibodies (1:500) for 20 h (details of the antibodies used by the
RPPA platform can be found at https://research.unimelb.edu.au/infrastructure/acrf-translational-reverse-phase-protein-array-platform),
and Zenon™ Alexa Fluor® 647 anti-rabbit secondary
antibody (1:1,000; cat. no. Z-25308; Thermo Fisher Scientific) for
4 h. Chips were read on a Zeptosens instrument (Zeptosens AG; Bayer
AG), and ZeptoView software version 3.1 was utilized to calculate
the relative fluorescence intensity. All samples were normalized to
the background values reported in the negative control containing
secondary antibody only. Duplicates were aver-aged, samples
normalised by global rank invariant method and the data
log2-normalised; log2 fold changes compared to the parental MEL-XY3
cell line were presented. The RPPA heat map was generated with
Plotly software (https://chart-studio.plot.ly/create/#/).
Western blotting
Cell pellets were lysed with RIPA buffer (150 mM
sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50
mM Tris pH 8.0) plus protease inhibitor (cat. no. P8340) and
phosphatase inhibitor (cat. no. P5726; both Sigma-Aldrich; Merck
KGaA) at recommended rates. Western blotting was conducted as
described previously (20). Total
protein was determined using a BCA assay (Bio-Rad Laboratories,
Inc.). Blocking of membranes was performed at room temperature for
1 h; primary antibody incubations were performed overnight at 4°C,
with secondary antibody incubations performed at room temperature
for 1 h. Labelled bands were detected by Clarity ECL kit (Bio-Rad
Laboratories, Inc.) and images were captured by the ChemiDoc MP
Bio-Rad image system. Antibodies directed against HDAC6 (1:1,000;
cat. no. 7558), DNMT3a (1:1,000; cat. no. 3598), DNMT3b (1:1,000;
cat. no. 67259) and enhancer of zeste homolog 2 (1:2,000; EZH2;
cat. no. 5246) were purchased from Cell Signaling Technology, Inc.
Anti-β-actin antibody (1:3,000; cat. no. AC-74) was acquired from
Sigma-Aldrich (Merck KGaA). The secondary antibodies used were
horseradish peroxidase-conjugated goat anti-mouse IgG (H+L) and
goat-anti-rabbit IgG (H+L) antibodies (1:3,000; cat. nos. 1706516
and 1706515; Bio-Rad Laboratories, Inc.).
Transmission electron microscopy
(TEM)
MEL-XY3 cells, MEL-XY3 SUR cells, and MEL-XY3 SUR
cells at 2 and 7 days after PLX4032 removal were analyzed by TEM.
Cells were harvested, fixed for 4 h at 4°C in 2.5% glutaraldehyde,
post-fixed in 1% (v/v) osmium tetroxide. Subsequently, samples were
embedded in Durcupan Epoxy resin (Sigma-Aldrich; Merck KGaA), and
ultrathin sections (70-90 nm) were mounted on copper grids and
stained with uranyl acetate. Stained samples on grids were
visualized using a Zeiss EM 109T TEM (Carl Zeiss AG), and digital
micrographs were captured with a Gatan ES1000W digital camera
(Gatan, Inc.) in the LANAIS-MIE facility (Buenos Aires,
Argentina).
Cell viability assay
Cells were seeded at a density of 3,000 cells/well
into 96-well plates in complete medium. After 24 h, different
inhibitors were added, and cells were maintained at 37°C in a 5%
CO2 humidified atmosphere. For each inhibitor, 7
different concentrations were assayed, starting from 10 µM
(decitabine, JQ-1), 20 µM (IBET-151, OTX-015, S63845,
CDKI-73, EPZ-6438 and GSK126), 500 nM (pano-binostat), 1.581
µM (TSA), 63.245 µM (SAHA, MGCD0103, sodium
butyrate), 94.868 µM (MC1568) and 316.22 µM
(tubacin), with subsequent 3.16-fold serial dilutions. After 72 h,
cell viability was assessed using CellTiter-Glo® reagent
(Promega Corporation). Luminescence was recorded with a POLARstar
Omega microplate reader (BMG Labtech). Data generated using this
assay are presented in Fig. 2.
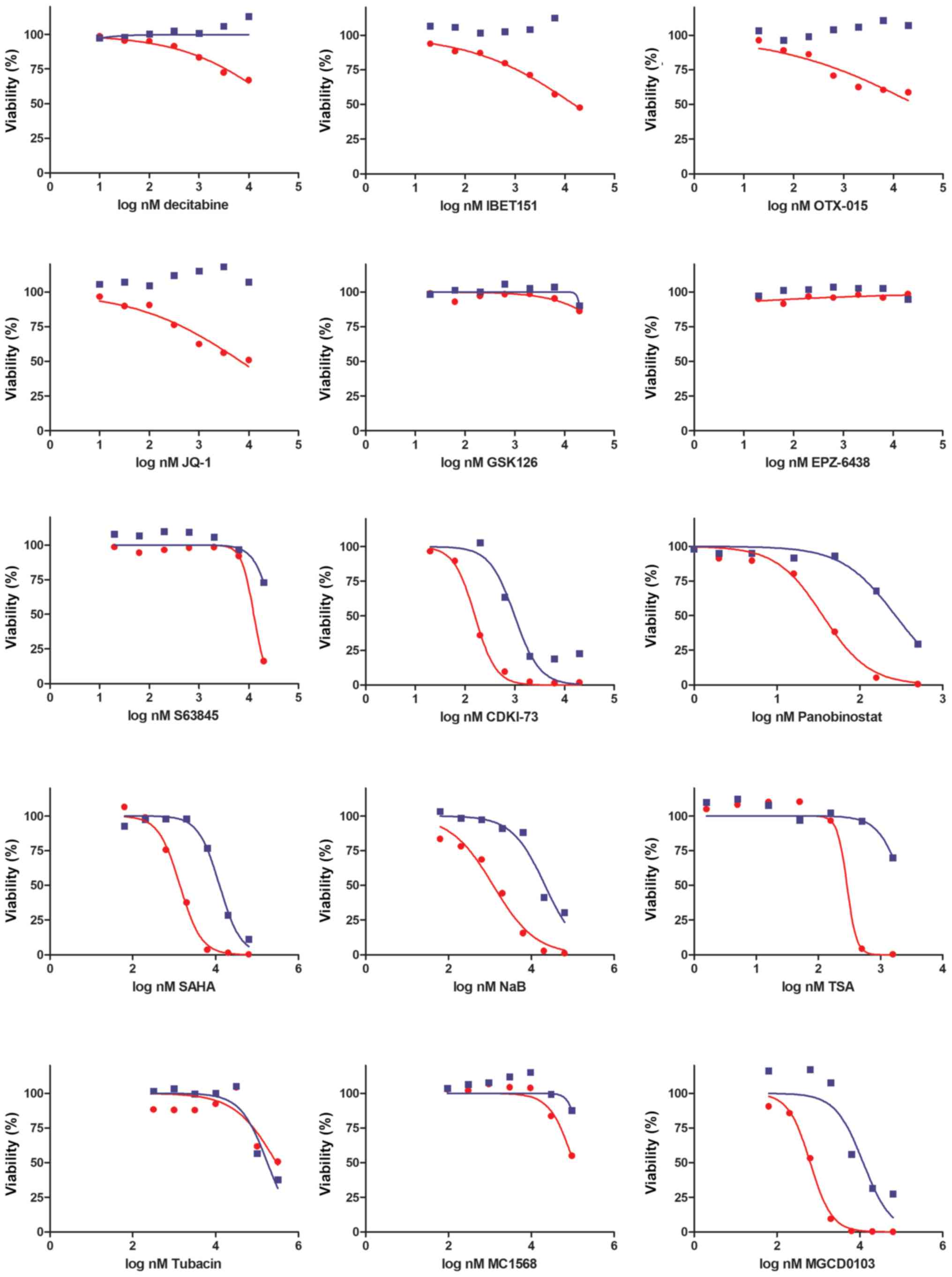 | Figure 2Sensitivity of MEL-XY3 and MEL-XY3
SUR cells to epigenetic inhibitors. Viability was determined with a
CellTiter-Glo assay after 72 h of drug treatment. DNA
methyltransferase (decitabine), bromodomain and extra-terminal
motif inhibitors (IBET151, OTX-015, JQ-1), enhancer of zeste
homolog 2 (GSK126, EPZ-6438), myeloid chronic leukaemia-1 (S63845),
cyclin-dependent kinase 9 (CDKI-73) and histone deacetylase
inhibitors (panobinostat, SAHA, NaB, TSA, tubacin, MGCD0103,
MC1568) were assayed. Representative inhibition curves of 2
independent experiments are shown. Lines represent the nonlinear
regression curve fit. Red, parental cells; blue, SUR cells. NaB,
sodium butyrate; SUR, surviving cells following long-term PLX4032
treatment; TSA, trichostatin A. |
Alternatively, after 72 h, cells were stained with 2
µg/ml Hoechst for 15 min at room temperature to visualize
the nuclei and plates were scanned (six randomly selected
fields/well) using an automated In Cell Analyzer 2200 microscope
(GE Healthcare Life Sciences). Quantitative analysis of the
acquired images was performed using In Cell Analyzer 1000
Workstation 3.7.3 (GE Healthcare Life Sciences). Data generated
using this assay are presented in Fig.
4. Non-linear regression analysis and IC50
determination were performed using GraphPad Prism 5 software.
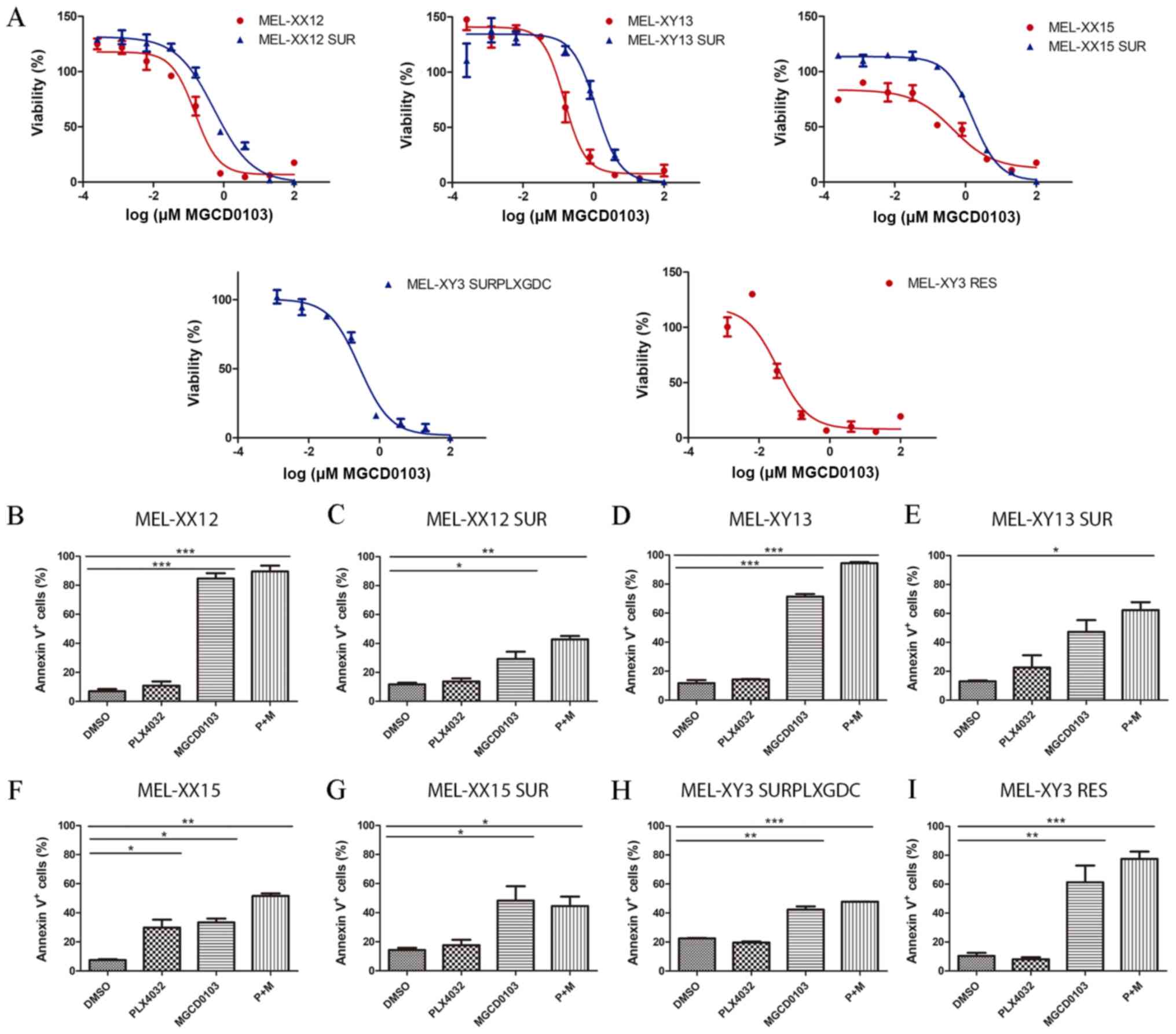 | Figure 4Sensitivity of SUR melanoma cell
lines to MGCD0103. (A) Sensitivity to MGCD0103 of parental (red)
and SUR (blue) cells obtained from BRAF-mutated melanoma cell
lines. Viability was determined after 72 h of drug treatment. Lines
represent the nonlinear regression curve fit. Percentage of Annexin
V+ apoptotic cells after 72 h treatment with 10
µM PLX4032, 2 µM MGCD0103 or combined treatment is
shown for (B) MEL-XX12, (C) MEL-XX12 SUR, (D) MEL-XY13, (E)
MEL-XY13 SUR, (F) MEL-XX15, (G) MEL-XX15 SUR, (H) MEL-XY3 SURPLXGDC
and (I) MEL-XY3 RES cells. *P<0.05,
**P<0.01, ***P<0.001. RES, resistant to
PLX4032; SUR, surviving cells following long-term PLX4032
treatment; SURPLXGDC, cells that survive long-term PLX4032 and
GDC-0973 treatment. |
In order to study the effect of inhibitors for
longer periods of time, 50,000 cells were seeded in MW24 plates in
complete medium. After 24 h, DMSO, 10 µM PLX4032, 2
µM MGCD0103 or 10 µM PLX4032 + 2 µM MGCD0103
were added, and cells were maintained at 37°C for 10 days, changing
media every 3 days. At day 10, cells were fixed with 4%
paraformaldehyde at room temperature for 15 min and stained with
0.05% cristal violet solution at room temperature for 20 min.
Analysis of apoptosis and cell cycle
Cell cycle and apoptosis were analysed using BD FACS
Canto II or BD FACScalibur flow cytometers (both BD Biosciences).
Cells were seeded in a MW12 plate (50,000 cells/well) and 24 h
later, the tested drugs were added to the media. Apoptotic cells
were quantified using Annexin V/propidium iodide (PI) staining
according to the manufacturer's protocol (BD Biosciences)
incubating cells for 15 min at room temperature. For cell cycle
analysis, cells were stained with PI staining solution (50
µg/ml PI, RNAse 100 µg/ml, 0.1% Triton-X, 1 mM
Na3Citrate) for 30 min on ice. The cell cycle was
analysed using PI-stained cells, and the cell cycle was fitted to
viable cells using FlowJo v10.2 software (FlowJo LLC) to exclude
dead cells from analysis. Experiments were performed in triplicate
and 20,000 events were acquired for each point.
NRAS status of MEL-XY3 BRAF inhibitor
resistant (RES) cells
MEL-XY3 RES cells were obtained from resistant
colonies generated in one experiment when MEL-XY3 were treated with
PLX4032 for several weeks to obtain SUR cells. The NRAS status of
MEL-XY3 RES cells was determined after DNA extraction with DNAzol™
(Invitrogen; Thermo Fisher Scientific, Inc.), PCR amplification and
Sanger sequencing (performed by Macrogen, Inc.). For PCR
amplification, GoTaq polymerase (Promega coproration) was used and
the thermocycling conditions used were: Initial denaturalization
for 2 min at 95°C, then 35 cycles of denaturation at 95°C for 30
sec, annealing at 60°C for 30 sec and extension at 72°C for 30 sec,
with a final extension at 72°C for 5 min. The primers used were:
Forward, 5′-CCC CTT ACC CTC CAC ACC-3′ and reverse, 5′-CAC AAA GAT
CAT CCT TTC AGA GAA-3′.
Three-dimensional (3D) spheroid
cultures
A total of 3,000 cells were seeded in 96-well
ultra-low attachment plates to induce anchorage-independent
spheroid formation. After 72 h, multiple spheroids were harvested
and embedded in 0.5 ml of collagen solution (Cultrex bovine
collagen 1; Trevigen, Inc.) in a 24-well plate, and left to
solidify at 37°C. After 15 min, 1 ml of media was added on top of
the collagen layer in order to obtain a final concentration of 10
µM PLX4032, 2 µM SAHA, 2 µM MGCD0103 and 2
µM CDKI-73. Spheroids were main-tained at 37°C in a 5%
CO2 humidified atmosphere. Following 5 days of
treatment, spheroids were photographed using a Nikon Ti inverted
microscope (magnification, 40×) and Image J software v3.5.0
(National Institutes of Health) was used to establish the spheroid
diameter.
To assess the viability of cells grown in a 3D
culture, spheroids were prepared. After 72 h, spheroids were grown
for an additional 72 h in the presence of drugs without collagen.
For each condition, 24 spheroids were disaggregated with
trypsin-EDTA, stained with Annexin V/PI and analysed by flow
cytometry as aforementioned. Three independent experiments were
performed. Percentage of viability was determined normalizing the
percentage of viable cells (Annexin V−/PI−)
to DMSO treated cells.
Statistical analysis
Experiments were performed with at least two
independent repeats; representative experiments were selected for
inclusion in figures. GraphPad Prism version 5.0 software was used
to graph data and perform statistical analysis. Data are presented
as the mean ± SD. The significance of differences between
experimental data was determined via ANOVA with Tukey's multiple
comparison tests. The significance level was set as P<0.05.
Results
Molecular characterization of SUR
cells
Preliminary characterization of SUR cells obtained
after 5 weeks of PLX4032 treatment has been previously described
(20). In order to obtain a more
complete picture of the changes that accompany the transition from
the actively dividing MEL-XY3 tumour cells to the resting SUR
cells, TEM was performed to characterize SUR cell morphology and
intracellular structure. Parental MEL-XY3 cells presented round
nuclei and smooth plasma membranes (Fig. S1A). In contrast, SUR cells
presented irregular nuclei, a large degree of membrane blebbing and
an increased number of probably fragmented mitochondria (Fig. S1B). After drug removal, SUR cells
started to resemble parental cells, with less distorted nuclei and
a smoothed plasma membrane, even though a large number of
mitochondria were still observed (Fig. S1C and D).
Next, SUR cells were subjected to RNA-seq analysis
and their expression profiles were compared to the parental
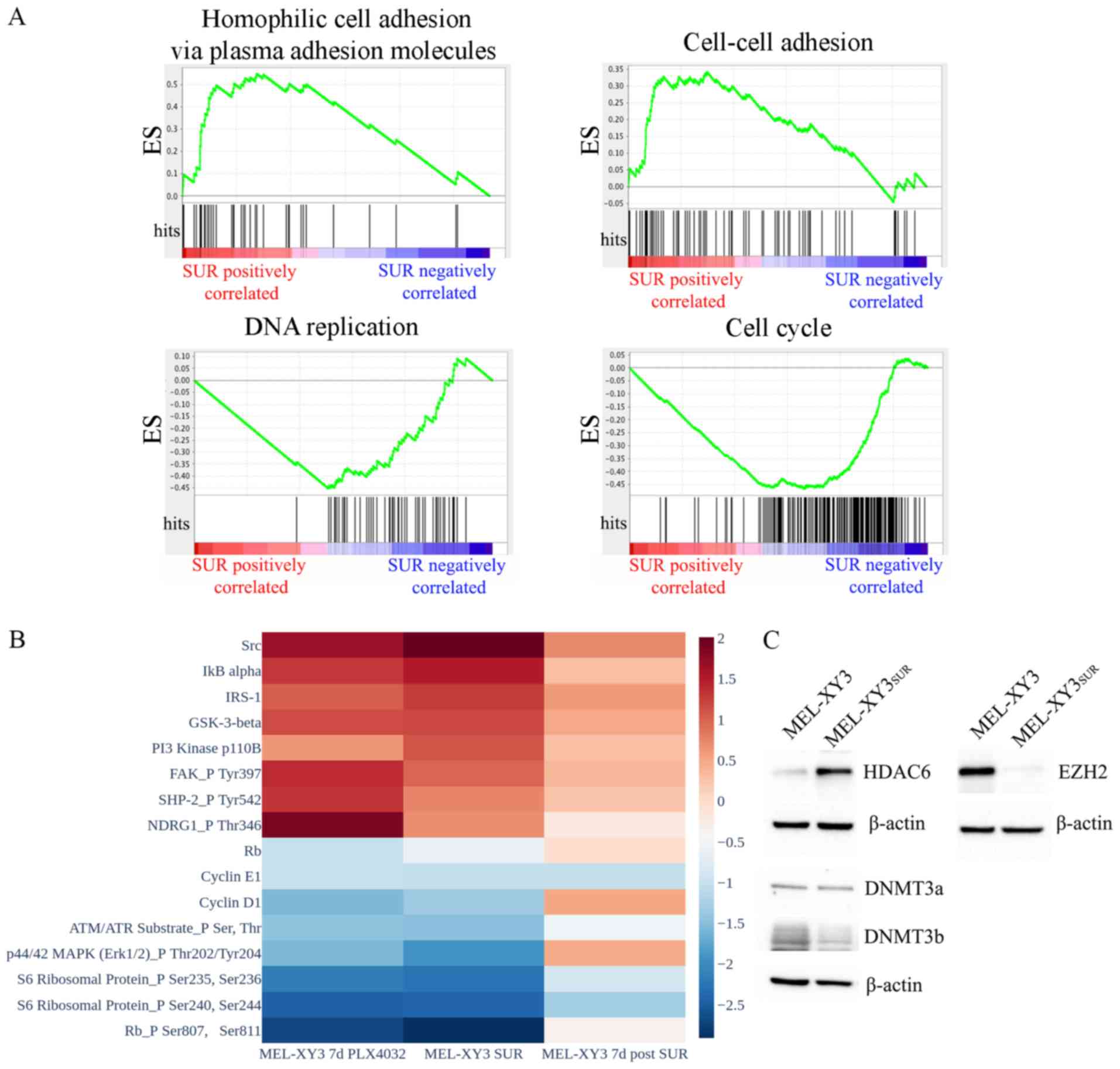
population (Fig. 1A). Considerable
differences in gene expression were observed as 1,415 genes had
significantly modified expression levels; 784 genes were
downregulated and 631 genes were upregulated in SUR cells
(P<0.05). GSEA revealed that SUR cells exhibited significantly
enriched GO molecular signatures associated with cell adhesion
(cell-cell adhesion, homophilic cell adhesion via plasma membrane
adhesion molecules, cell-cell adhesion via plasma membrane adhesion
molecules and calcium-dependent cell-cell adhesion via plasma
membrane cell-adhesion molecules). In contrast, gene sets
associated with proliferation and the cell cycle were downregulated
(cell cycle, cell division and DNA replication; FDR <0.25,
P<0.01), as predicted based on their phenotype.
RPPA experiments validated the downregulation of
proteins related to cell cycle progression (cyclin D1, cyclin E1,
retinoblastoma protein total and phosphorylation levels, S6
ribosomal protein phosphorylation) in SUR cells. At 7 days after
drug removal, the levels of cell cycle proteins started to return
towards those of the parental MEL-XY3 cells (Figs. 1B and S2), confirming the reversibility of the
SUR phenotype. Phosphorylated ERK1 levels decreased in SUR cells
and returned to higher levels once the BRAF inhibitor was removed
from culture media, indicative of continued but reversible
suppression of MAPK signalling in SUR cells. Other proteins were
upregulated in SUR cells, including proteins associated with
proliferation pathways and drug resistance (Fig. 1B). These included Src and
phosphorylated NDRG1, which have been linked to resistance to MAPK
inhibitors and chemotherapy, respectively (31,32).
PI3K protein was increased in SUR cells, possibly indicating a
survival mechanism compensating for the loss of MAPK signalling.
Even though there was an increase in the expression levels of these
proteins or phosphorylated proteins, SUR cells remained in a
quiescent state, suggesting that these changes were insufficient to
promote proliferation and complete drug resistance. Once the drug
was removed from culture media, the expression levels of these
upregulated proteins decreased. Expression levels were similar in
cells maintained with PLX4032 for 7 days and SUR cells, but a more
pronounced change was observed in SUR cells.
In order to look for epigenetic changes in SUR cells
with respect to the parental line XY3, the expression of different
epigenetic enzymes were evaluated via western blotting. SUR cells
presented a strong decrease in the expression of EZH2 (Fig. 1C). DNMT3b expression was slightly
decreased in SUR cells, whereas DNMT3a expression remained stable.
In contrast, HDAC6 was markedly increased in SUR cells compared
with parental cells. Consistently, RNA-seq data from GSE50509 from
patients with metastatic melanoma resistant to BRAF inhibitors
revealed significant upregulation of several HDACs after BRAF
inhibitor treatment, including HDAC6 (P<0.05; Fig. S3). These results demonstrated that
SUR cells presented altered expression of epigenetic modifiers and,
together with the apparently reversible changes observed in gene
and protein expression, these data suggested that the SUR phenotype
emerging after MAPK inhibition could be associated with epigenetic
regulation.
Sensitivity of SUR cells to epigenetic
inhibitors
To investigate whether SUR cells could be
eliminated, the sensitivity of MEL-XY3 parental and SUR cells to
different epigenetic inhibitors were analysed in vitro. Both
cell types shared similar sensitivity to the assayed drugs. SUR
cells were resistant to EZH2 histone methylase inhibitors (EPZ-6438
and GSK126), BET inhibitors (IBET151, JQ-1 and OTX-015), DNMT
inhibi-tors (decitabine), HDAC class II inhibitors (MC1568) and
HDAC6 inhibitors (tubacin; Fig.
2). Moreover, SUR cells were resistant to inhibition of the
antiapoptotic protein member of Bcl-2 family myeloid cell
leukaemia-1 (S63845).
However, parental and SUR MEL-XY3 cells were
sensitive to certain pan-HDAC inhibitors, including SAHA
(IC50: MEL-XY3 1.3 µM, SUR 6.98 µM),
panobinostat (IC50: MEL-XY3 35 nM, SUR 269 nM) and
sodium butyrate (IC50: MEL-XY3 1.2 µM, SUR 21
µM), whilst not exhibiting sensitivity to others, such as
TSA. In addition, they were sensitive to MGCD0103 (IC50:
MEL-XY3 0.63 µM, SUR 1.20 µM), an HDAC inhibitor
which preferentially inhibits class I enzymes, with the exception
of HDAC8 (33). Further, both
parental and SUR cells were sensitive to the CDK9 inhibitor CDKI-73
(IC50: MEL-XY3 0.15 µM, SUR 0.99 µM;
Fig. 2).
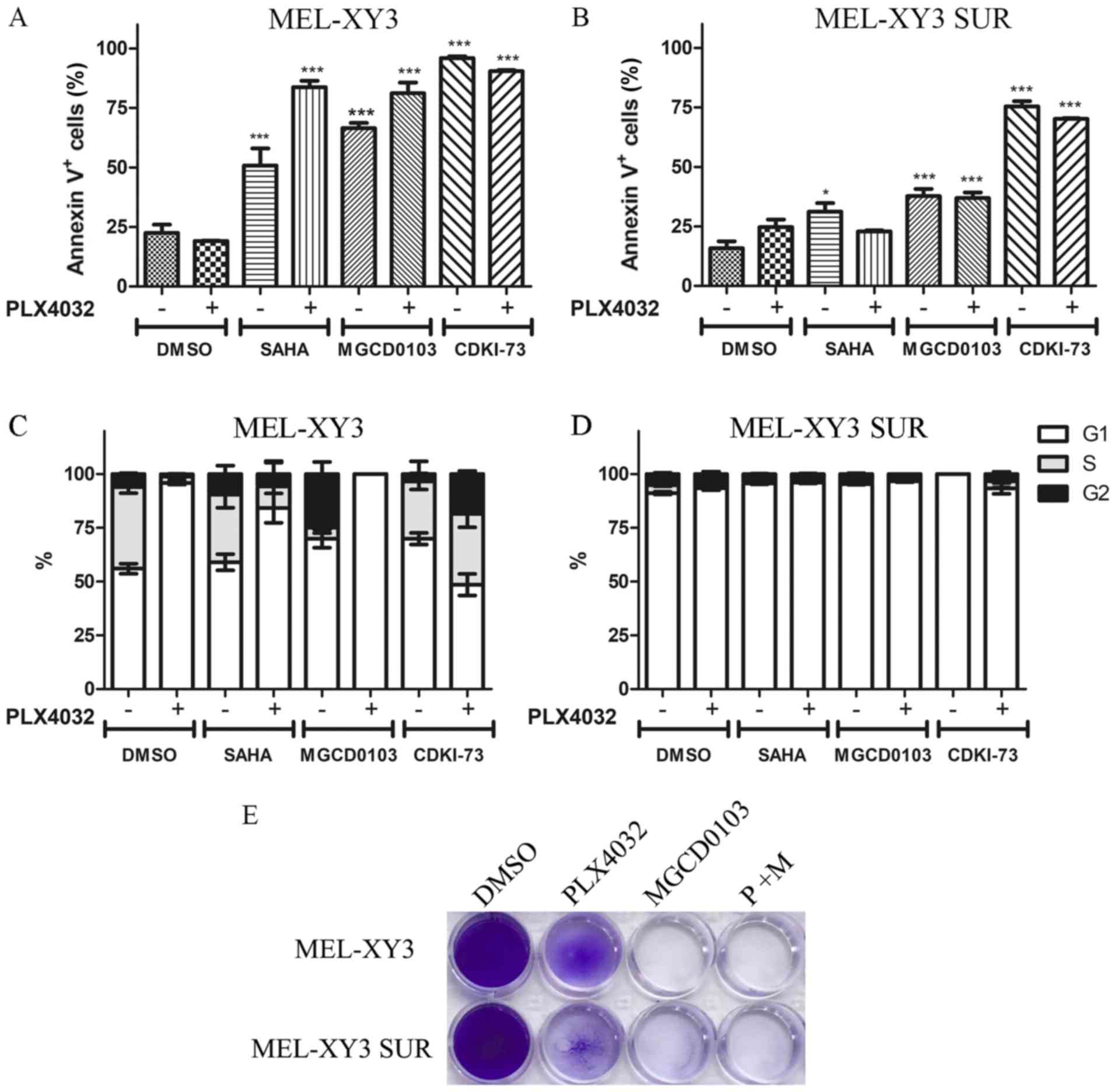
Even though PLX4032 did not induce apoptosis in
MEL-XY3 parental cells or SUR cells, treatment with HDAC inhibitors
and CDK9 inhibitors induced apoptosis both in parental and (to a
lesser degree) SUR cells (Figs. 3A and
B, and S4A). No significant
differences were observed in the proportion of necrotic (Annexin
V+/PI+) and apoptotic (Annexin
V+/PI-) cells between the different
treatments (data not shown).
PLX4032 induced G1 cell cycle arrest in MEL-XY3 and
SUR cells maintained in the presence of the different inhibitors
tested (Figs. 3D and S4B). MGCD0103 alone markedly increased
the proportion of G2 cells, whereas SAHA and CDKI-73 alone did not
notably alter cell cycle distribution (Fig. 3C). Although some melanoma cells
were still viable after 72 h of treatment, after 10 days of
treatment with MGCD0103, both parental MEL-XY3 cells and SUR cells
were completely eliminated (Fig.
3E). Additionally, the presence of a sub-G0/G1 population was
observed in cell cycle analysis, indicating the presence of dead
cells, consistent with Annexin V/PI staining and the results
obtained after 10 days of treatment (data not shown).
To determine whether the findings from the MEL-XY3
and MEL-XY3 SUR cells could be extended to other V600BRAF-mutated
cell lines, the effects of MGCD0103 against three other BRAFV600E
melanoma cells, and their corresponding SUR populations obtained
after treatment with PLX4032 for 5 weeks, were evaluated. SUR cells
obtained from MEL-XX12, MEL-XY13 and MEL-XX15 cells were sensitive
to MGCD0103 (IC50: MEL-XX12 SUR 0.25 mM, MEL-XY13 SUR
0.82 mM and Mel-XX15 SUR 1.04 mM; Fig.
4A) as well as their parental cells. Apoptotic cell death was
induced both in parental and SUR cells obtained from the different
cell lines (Fig. 4B-G) by
MGCD0103, alone or in combination with PLX4032. Additionally,
MEL-XY3 SUR cells obtained after combined BRAF and MEK inhibitor
treatment were sensitive to MGCD0103 (IC50: 0.27 mM;
Fig. 4A), with apoptosis induced
(Fig. 4H).
Although MEL-XY3 cells predominantly turned into SUR
cells following PLX4032 treatment, a permanent BRAF
inhibitor-resistant MEL-XY3 RES cell population capable of
proliferating in the presence of PLX4032 was obtained in one of the
experiments, when MEL-XY3 cells were cultured for several weeks in
the presence of PLX4032. These cells presented a Q61K mutation in
NRAS, a mutation already reported to confer resistance to BRAF
inhibitors (4). These cells
underwent apoptosis when treated with MGCD0103 (Figs. 4A and I, and S4C).
To determine the efficacy of the epigenetic
inhibitors in a 3D growth model, the effects of HDAC and CDK9
inhibitors in spheroids that mimic tumour architecture were
evaluated. Spheroids were incubated in a collagen matrix with the
different drugs in order to analyse their impact on cell
proliferation and migration. Treatment with PLX4032 significantly
reduced spheroid diameter both in parental and SUR cells,
consistent with cell cycle arrest observed after PLX4032 treatment
(Fig. 5A-C). Moreover, the results
suggested that BRAF inhibitor treatment also inhibited the
migration of cells into the surrounding collagen matrix, as the
spheroids appeared more compact. HDAC and CDK9 inhibitors both
significantly reduced the diameter of MEL-XY3 spheroids (Fig. 5A and B); combined treatment with
PLX4032 did not alter the effects observed with monotherapy. SUR
cell spheroid diameter (Fig. 5A and
C) was decreased by CDK9 inhibitor treatment; however, HDAC
inhibitors did not significantly alter SUR cell spheroid diameter,
except when they were combined with PLX4032. SUR cell spheroids
treated with HDAC inhibitors may exhibit invasive behaviour, with
morphology suggestive of an invasive phenotype. It has been
previously demonstrated that several HDAC inhibitors, such as SAHA,
TSA, valproic acid and sodium butyrate, promote melanoma cell
invasion in vitro via the upregulation of N-cadherin and
inhibition of Ras homolog A activity (34). Using flow cytometry, it was found
that parental and SUR cell spheroids treated with MGCD0103 alone or
in combination with PLX4032 for 72 h had significantly reduced
viability, whereas PLX4032 treatment alone did not lead to
significant loss of viability (Fig
S4D; Fig. 5D and E;).
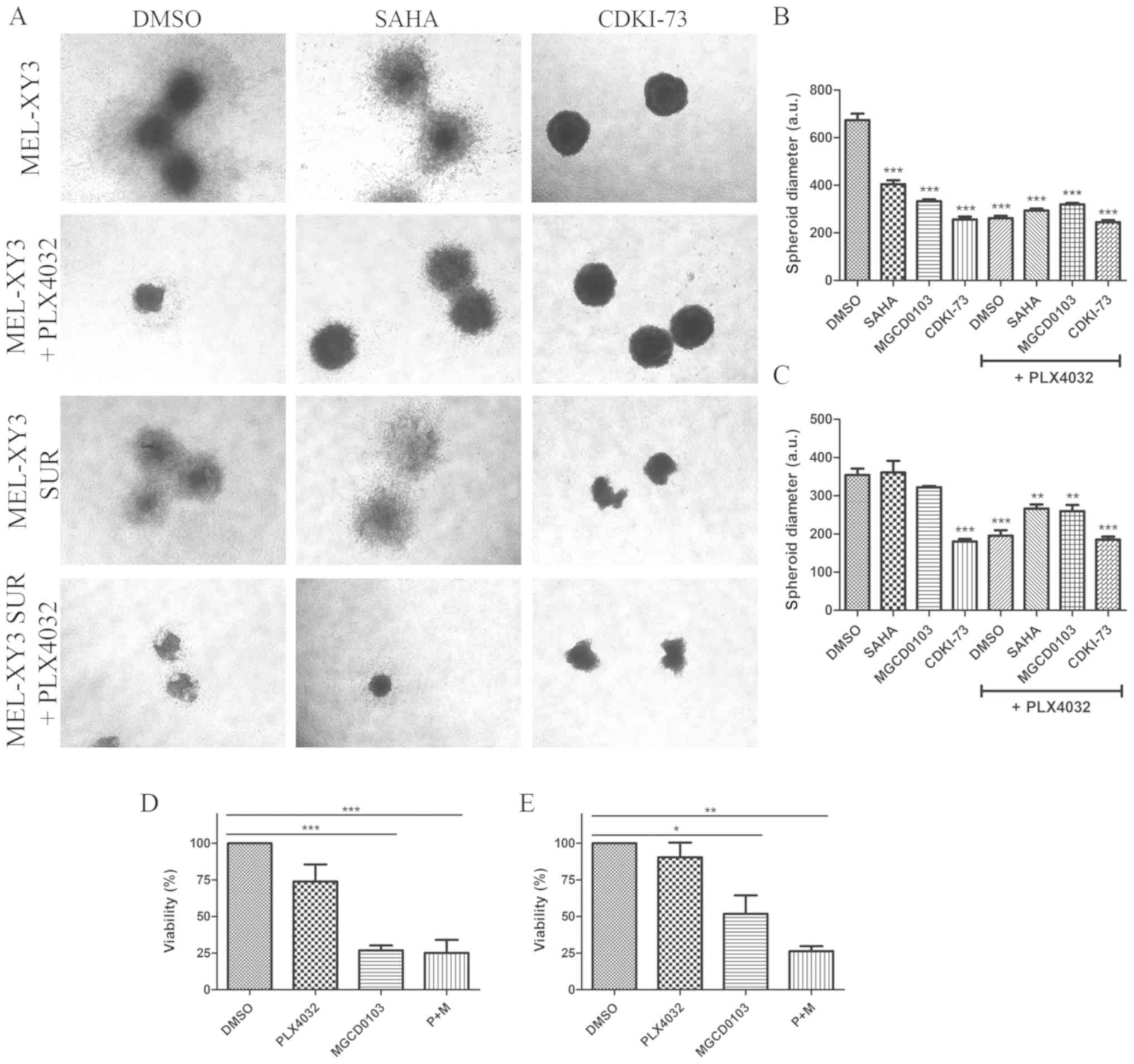 | Figure 5Effect of treatment with epigenetic
inhibitors on melanoma cell spheroids. Collagen-embedded spheroids
obtained from MEL-XY3 and MEL-XY3 SUR cells were treated for 5 days
with DMSO, 2 µM SAHA, 2 µM MGCD0103 or 2 µM
CDKI-73 in the presence or absence of 10 µM PLX4032. (A)
Phase contrast images of collagen-embedded spheroids after 5 days
of treatment. Magnification, ×40. Diameters of (B) MEL-XY3 and (C)
MEL-XY3 SUR spheroids. Data are presented as the mean ± SD.
**P<0.01, ***P<0.001 vs. DMSO.
Percentage of viable cells obtained from (D) MEL-XY3 and (E)
MEL-XY3 SUR spheroids after 72 h of treatment relative to
DMSO-treated cells. Following treatment, spheroids within each
treatment were pooled, and the viability of cells was determined by
Annexin V/PI staining and flow cytometry. *P<0.05,
**P<0.01, ***P<0.001. a.u., arbitrary
units; SUR, surviving cells following long-term PLX4032
treatment. |
Discussion
Our previous study described SUR cells, a population
of BRAF-mutated melanoma cells that persists after MAPK inhibitor
treatment, and which represent a reversible state characterized by
changes in gene expression, cell cycle arrest and SA
β-galactosidase activity (20). To
expand upon this previous study, the present study involved a
molecular characterization of SUR cells by RNA-seq and RPPA
experiments. RNA-seq showed that SUR cells exhibited decreased
expression of proliferation-associated genes and increased
expression of cell adhesion molecules. This is consistent with
their phenotype; SUR cells do not proliferate and are strongly
attached to culture dishes. At 7 days after removal of MAPK
inhibitor, SUR cells revert to a proliferative state (20). RPPA data showed that the expression
levels of proteins that are up- or downregulated in SUR cells
compared with parental cells return to their basal expression
levels after drug removal, suggesting phenotypic plasticity
(21,35). Previous studies showed that
aberrant expression of epigenetic modifiers such as EZH2, SET
domain bifurcated histone lysine methyltransferase 1, G9a histone
methyltransferase and HDACs led to adaptive resistance in cancer
cells, including melanoma (21,35,36).
In the present study, SUR cells presented changes in the protein
expression levels of different epigenetic enzymes, such as EZH2 and
HDAC6. These findings suggested that the SUR phenotype could be
determined by epigenetic changes. It has been suggested that
changes in nuclear morphology and structure could cause global
epigenetic changes (37). The
alterations observed by TEM in the nuclear morphology of SUR cells
may be consistent with this hypothesis.
Previously, our study showed that SUR cells present
SA characteristics, such as increased SA β-galactosidase activity
and changes in the expression levels of several proteins associated
with senescence, such as p16ink4 and p21 (20). Induction of senescence by BRAF
inhibition with PLX4032 has also been reported by other groups
(38,39). The acquisition of a senescent
phenotype could contribute to resistance to apoptosis; cells that
are resistant to apoptosis often exhibit an increase in senescence
features (40). The induction of a
senescent state could explain the limited proapoptotic effect of
BRAF inhibition, which was observed in the different SUR cell
lines. Furthermore, it has been shown that melanoma cells treated
with PLX4032 present an SA secretory phenotype, which leads to the
secretion of factors that stimulate naïve melanoma cells and
fibroblasts, thereby limiting the antitumorigenic effect of BRAF
inhibition (41). On the other
hand, epigenetic control is a key factor in the induction of
senescence. Recently, it has been proposed that oncogene-induced
senescent melanocytes can bypass the senescent state through the
activation of epigenetic modifying agents (including histone
demethylases such as lysine-specific histone demethylase 1 and
Jumonji domain-containing protein 2C), re-entering into the cell
cycle as malignant melanoma cells (42). Moreover, the inhibition of these
enzymes in melanoma cells restored senescence.
In the present study, in vitro SUR cell
sensitivity to different epigenetic inhibitors was analysed,
including DNMT, BET, HDAC, EZH2 and CDK9 inhibitors. It was found
that both HDAC inhibitors (SAHA and MGCD0103) and a CDK9 inhibitor
(CDKI-73) were able to induce apoptosis in SUR cells. Although
parental cells were more sensitive than SUR cells to HDAC and CDK9
inhibitors, following 10 days of treatment with MGCD0103, complete
elimination of SUR cells was observed in vitro. It should be
stressed that parental non-treated cells are actively
proliferating, whereas SUR cells are in a resting state; therefore
the observed differences in the sensitivity to the assayed drugs
may be based on these conditions. The main aim was to find drugs or
combinations that resulted in complete elimination of SUR cells.
The fact that the same inhibitors also effectively eliminated the
parental cells is a therapeutic advantage that may increase the
efficacy of first-line treatment. Regarding the spheroid growth
assay results, PLX4032 efficiently reduced spheroid diameter in
parental and SUR cells, and this effect was not reverted by the
presence of the other inhibitors.
Different types of epigenetic inhibitors are
currently being applied for cancer treatment. Decitabine inhibits
DNMTs, producing DNA hypomethylation (43). It is approved for treatment of
haematological malignancies, such as myelo-dysplastic syndrome,
chronic myelomonocitic leukaemia and acute myeloid leukaemia. An
EZH2 inhibitor, tazemetostat, has exhibited promising results in a
phase I clinical trial, both in lymphoma and solid tumours such as
sarcoma (44). Different BET
inhibitors have been tested for the treatment of haematological
malignancies and solid tumours, presenting limited efficacy as
single agents; however, recent studies have proposed combination
treatments to obtain improved clinical results (45,46).
To date, monotherapy with HDAC inhibitors has had
limited success in the treatment of solid tumours; however,
combination therapy has shown more encouraging results (47). It has been previously shown in
different tumour models that drug-tolerant persister cells can be
eliminated by HDAC inhibitors such as TSA (36,48).
SAHA, also known as vorinostat, is a pan-HDAC inhibitor. Even
though treatment of patients with advanced melanoma with SAHA as a
monotherapy did not produce encouraging results (49), combination or sequential treatment
with BRAF inhibitors could be a promising alternative. Wang et
al (16) demonstrated that
BRAF inhibitor-resistant melanoma cells present increased reactive
oxygen species (ROS) levels, and that this characteristic produces
vulnerability to HDAC inhibitors, as HDAC inhibitors yield a
further increase in ROS levels, thereby leading to DNA damage and
apoptosis. They proposed a sequential treatment (BRAF + MEK
inhibitors followed by SAHA) to reach lethal ROS levels, with the
benefit of avoiding toxicity due to combined treatment. A phase I
clinical trial analysing SAHA in resistant BRAF-mutated advanced
melanoma is currently ongoing (clinical trial no. NCT02836548). In
the present study, it was observed that SAHA alone induced greater
apoptosis than the combination of SAHA + PLX4032 in SUR cells,
consistent with Wang et al (16); however, this effect was not
significant.
Epigenetic alterations have been described in the
pathogenesis of autoimmunity (50). Evidence suggests that HDAC
inhibitors, including SAHA, may also be applicable to the treatment
of autoimmune diseases (51). HDAC
inhibitors have been shown to inhibit inflammatory mediator
production (including cytokines, chemokines and nitric oxide) and
lead to M2 polarization of macrophages (52). Moreover, experiments conducted in
murine models suggest that DNMT inhibitors could be deployed in the
treatment of autoimmune diseases (53,54).
MGCD0103, also known as mocetinostat, is a class I
and IV (isoforms 1, 2, 3 and 11) HDAC-selective inhibitor (33). Preclinical studies have shown
potent antitumor activity of MGCD0103 against various human cancer
cell lines and human tumour xenografts in mice (33). A clinical trial demonstrated
MGCD0103 clinical activity and acceptable safety profiles in
patients with haematological malignancies (55). MGCD0103 increases antigen
presentation and decreases immune-suppressive cell types (56). Currently, mocetinostat is being
tested in combination with durvalumab, an anti-programmed cell
death (PD)-ligand 1 antibody, in patients with non-small cell lung
cancer (clinical trial no. NCT02805660). Further, a phase 1b
clinical study of MGCD0103 in combination with ipilimumab
(anti-cytotoxic T-lymphocyte-associated protein 4) and nivolumab
(anti-PD-1) in patients with stage IV unresectable melanoma is
being conducted (cat. no. NCT03565406). The present results showed
that SUR cells present a stronger response to MGCD0103 than SAHA,
with the advantage that MGCD0103 is a more selective inhibitor.
MGCD0103 induced cytotoxicity in populations of SUR cells obtained
from different melanoma cell lines, as well as in a
PLX4032-resistant cell line harbouring an acquired NRAS
mutation.
Although it was observed that HDAC6 was upregulated
in SUR cells, these cells were not highly sensitive to HDAC6
inhibition. HDAC6 may not be the primary driver of cell survival in
the SUR cells. Other alterations were observed, including EZH2
downregulation. SAHA and MGCD0103 are pan and class I/IV HDAC
inhibitors, respectively, and they were effective in the
elimination of SUR cells. These drugs can not only directly inhibit
HDAC, but could have additional effects on HDAC-associated
molecules.
CDK9, the catalytic subunit of positive
transcription elongation factor b, is a key regulatory kinase of
RNA polymerase II (57). Recently,
it was reported that CDK9 is essential for maintaining gene
silencing at heterochromatic loci, validating its role as an
epigenetic target. CDK9 targeting reactivates tumour-suppressor
genes and induces a cellular immune response that may sensitize to
checkpoint inhibitors (57).
CDKI-73 is a very potent CDK9 inhibitor that showed high
cytotoxicity against different human cancer cell lines (58), and the present findings indicated
that it effectively eliminates SUR cells.
The main limitation of the present study is the lack
of in vivo experiments. It would be of major interest to
perform in vivo experiments to validate the observed in
vitro results. One potential experiment would be to grow
xenograft tumours derived from human melanoma cells in the skin of
immuno-compromised NSG mice. After the tumours have established,
the mice should be treated with the BRAF inhibitor PLX4032 to
reduce the size of the tumours and allow generation of SUR cells
in vivo. Afterwards, mice should be treated with DMSO,
PLX4032, MGCD0103 or PLX4032 + MGCD0103. It is predicted that
tumours from mice treated with either MGCD0103 alone or in
combination with BRAF inhibitor will not regrow, as these
treatments should effectively kill the SUR cell population based on
the present in vitro results.
In conclusion, in the present study a molecular
characterization of SUR cells was performed, and evidence
indicating that the SUR phenotype may be determined by epigenetic
changes was found. Subsequently, it was investigated as to whether
SUR cells were sensitive to epigenetic inhibitors, with the results
suggesting that co-targeting of BRAF and HDAC/CDK9 can efficiently
eliminate BRAF-mutated melanoma cells, whilst also eliminating the
SUR population that remains viable after prolonged MAPK inhibitor
treatment. The present study is limited to in vitro and 3D
spheroid models; future preclinical studies will aid in designing
therapeutic modalities that may provide a solid base for clinical
trials aiming to overcome the persistence of melanoma cells after
MAPK inhibition.
Supplementary Data
Acknowledgments
CDKI-73 was kindly provided by Dr Shudong Wang
(University of South Australia). We acknowledge Dr Patricio
Yankilevich (IBioBA-CONICET) for RNA-seq data analysis, and
Professor Kaylene J. Simpson, Ms. Arthi Macpherson and Ms. Lucy Liu
(Victorian Centre for Functional Genomics RPPA platform) for
assistance with the RPPA experiments in this study.
Funding
This work was supported by grants from CONICET,
Agencia Nacional de Promoción Científica y Tecnológica (grant nos.
PICT 2014-1932 and PICT 2017-1141) and Fundación Sales, as well as
by an Australian National Health and Medical Research Council
program (grant no. 633004). The Victorian Centre for Functional
Genomics RPPA platform is funded by the Australian Cancer Research
Foundation, the Australian Phenomics Network through funding from
the Australian Government's National Collaborative Research
Infrastructure Strategy program, the Peter MacCallum Cancer
Foundation and the University of Melbourne Collaborative Research
Infrastructure Program.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Gene Expression Omnibus
repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126960).
Author contributions
FPMR participated in the conception and design of
the study, collection and assembly of data, data analysis and
interpre-tation, and manuscript writing. AB and EMVE participated
in sample collection, and the assembly, analysis and interpretation
of data. SJG, AAE and PH provided reagents, and contributed to the
analysis and interpretation of data. MMB and JM conceived and
designed the study, and contributed to data analysis and
interpretation, and manuscript writing. All authors contributed to
manuscript revision, read and approved the submitted version.
Ethics approval and consent to
participate
Ethical approval was granted prior to the
establishment of the MEL-XX12 and MEL-XX15 cell lines by the Comite
de Etica en Investigacion of the Instituto Alexander Fleming, and
informed consent was obtained from patients prior to the collection
of samples.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Villanueva J, Vultur A and Herlyn M:
Resistance to BRAF inhibitors: Unraveling mechanisms and future
treatment options. Cancer Res. 71:7137–7140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Allen EM, Wagle N, Sucker A, Treacy
DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker
S, Kryukov GV, et al: The genetic landscape of clinical resistance
to RAF inhibition in metastatic melanoma. Cancer Discov. 4:94–109.
2014. View Article : Google Scholar :
|
|
4
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shi H, Hugo W, Kong X, Hong A, Koya RC,
Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al:
Acquired resistance and clonal evolution in melanoma during BRAF
inhibitor therapy. Cancer Discov. 4:80–93. 2014. View Article : Google Scholar :
|
|
6
|
Shi H, Moriceau G, Kong X, Lee MK, Lee H,
Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al: Melanoma
whole-exome sequencing identifies (V600E)B-RAF
amplification-mediated acquired B-RAF inhibitor resistance. Nat
Commun. 3:7242012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arozarena I and Wellbrock C: Overcoming
resistance to BRAF inhibitors. Ann Transl Med. 5:3872017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levesque MP, Cheng PF, Raaijmakers MI,
Saltari A and Dummer R: Metastatic melanoma moves on: Translational
science in the era of personalized medicine. Cancer Metastasis Rev.
36:7–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rambow F, Rogiers A, Marin-Bejar O, Aibar
S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter
M, et al: Toward minimal residual disease-directed therapy in
melanoma. Cell. 174:843–855 e819. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Strauss J and Figg WD: Epigenetic
approaches to overcoming chemotherapy resistance. Lancet Oncol.
16:1013–1015. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeller C, Dai W, Steele NL, Siddiq A,
Walley AJ, Wilhelm-Benartzi CS, Rizzo S, van der Zee A, Plumb JA
and Brown R: Candidate DNA methylation drivers of acquired
cisplatin resistance in ovarian cancer identified by methylome and
expression profiling. Oncogene. 31:4567–4576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giessrigl B, Schmidt WM, Kalipciyan M,
Jeitler M, Bilban M, Gollinger M, Krieger S, Jäger W, Mader RM and
Krupitza G: Fulvestrant induces resistance by modulating GPER and
CDK6 expression: Implication of methyltransferases, deacetylases
and the hSWI/SNF chromatin remodelling complex. Br J Cancer.
109:2751–2762. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen Y, Tong M, Liang Q, Guo Y, Sun HQ,
Zheng W, Ao L, Guo Z and She F: Epigenomics alternations and
dynamic transcriptional changes in responses to 5-fluorouracil
stimulation reveal mechanisms of acquired drug resistance of
colorectal cancer cells. Pharmacogenomics J. 18:23–28. 2018.
View Article : Google Scholar :
|
|
14
|
Song C, Piva M, Sun L, Hong A, Moriceau G,
Kong X, Zhang H, Lomeli S, Qian J, Yu CC, et al: Recurrent tumor
Cell-intrinsic and -extrinsic alterations during MAPKi-induced
melanoma regression and early adaptation. Cancer Discov.
7:1248–1265. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Strub T, Ghiraldini FG, Carcamo S, Li M,
Wroblewska A, Singh R, Goldberg MS, Hasson D, Wang Z, Gallagher SJ,
et al: SIRT6 haploinsufficiency induces BRAFV600E
melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat
Commun. 9:34402018. View Article : Google Scholar
|
|
16
|
Wang L, Leite de Oliveira R, Huijberts S,
Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song JY, Zevenhoven J,
et al: An acquired vulnerability of Drug-resistant melanoma with
therapeutic potential. Cell. 173:1413–1425.e14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gallagher SJ, Gunatilake D, Beaumont KA,
Sharp DM, Tiffen JC, Heinemann A, Weninger W, Haass NK, Wilmott JS,
Madore J, et al: HDAC inhibitors restore BRAF-inhibitor sensitivity
by altering PI3K and survival signalling in a subset of melanoma.
Int J Cancer. 142:1926–1937. 2018. View Article : Google Scholar
|
|
18
|
Zhao B, Cheng X and Zhou X: The
BET-bromodomain inhibitor JQ1 mitigates vemurafenib drug resistance
in melanoma. Melanoma Res. 28:521–526. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zakharia Y, Monga V, Swami U, Bossler AD,
Freesmeier M, Frees M, Khan M, Frydenlund N, Srikantha R, Vanneste
M, et al: Targeting epigenetics for treatment of BRAF mutated
metastatic melanoma with decitabine in combination with
vemurafenib: A phase lb study. Oncotarget. 8:89182–89193. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Madorsky Rowdo FP, Barón A, von Euw EM and
Mordoh J: In vitro long-term treatment with MAPK inhibitors induces
melanoma cells with resistance plasticity to inhibitors while
retaining sensitivity to CD8 T cells. Oncol Rep. 37:1367–1378.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ravindran Menon D, Das S, Krepler C,
Vultur A, Rinner B, Schauer S, Kashofer K, Wagner K, Zhang G,
Bonyadi Rad E, et al: A stress-induced early innate response causes
multidrug tolerance in melanoma. Oncogene. 34:4448–4459. 2015.
View Article : Google Scholar :
|
|
22
|
Gallagher SJ, Tiffen JC and Hersey P:
Histone modifications, modifiers and readers in melanoma resistance
to targeted and immune therapy. Cancers (Basel). 7:1959–1982. 2015.
View Article : Google Scholar
|
|
23
|
Long GV, Hauschild A, Santinami M,
Atkinson V, Mandalà M, Chiarion-Sileni V, Larkin J, Nyakas M,
Dutriaux C, Haydon A, et al: Adjuvant dabrafenib plus trametinib in
stage III BRAF-mutated melanoma. N Engl J Med. 377:1813–1823. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar
|
|
25
|
Anders S, Pyl PT and Huber W: HTSeq-a
Python framework to work with high-throughput sequencing data.
Bioinformatics. 31:166–169. 2015. View Article : Google Scholar
|
|
26
|
R Core Team: R: a language and environment
for statistical computing. R Foundation for Statistical Computing;
Vienna: 2015, https://www.R-project.org/.
|
|
27
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar
|
|
28
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Edgar R, Domrachev M and Lash EM: Gene
expression omnibus: NCBI Gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar :
|
|
30
|
Rizos H, Menzies AM, Pupo GM, Carlino MS,
Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al:
BRAF inhibitor resistance mechanisms in metastatic melanoma:
Spectrum and clinical impact. Clin Cancer Res. 20:1965–1977. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Girotti MR, Lopes F, Preece N,
Niculescu-Duvaz D, Zambon A, Davies L, Whittaker S, Saturno G,
Viros A, Pedersen M, et al: Paradox-breaking RAF inhibitors that
also target SRC are effective in drug-resistant BRAF mutant
melanoma. Cancer Cell. 27:85–96. 2015. View Article : Google Scholar :
|
|
32
|
Weiler M, Blaes J, Pusch S, Sahm F,
Czabanka M, Luger S, Bunse L, Solecki G, Eichwald V, Jugold M, et
al: mTOR target NDRG1 confers MGMT-dependent resistance to
alkylating chemotherapy. Proc Natl Acad Sci USA. 111:409–414. 2014.
View Article : Google Scholar
|
|
33
|
Fournel M, Bonfils C, Hou Y, Yan PT,
Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, et
al: MGCD0103, a novel isotype-selective histone deacetylase
inhibitor, has broad spectrum antitumor activity in vitro and in
vivo. Mol Cancer Ther. 7:759–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Diaz-Nunez M, Diez-Torre A, De Wever O,
Andrade R, Arluzea J, Silió M and Aréchaga J: Histone deacetylase
inhibitors induce invasion of human melanoma cells in vitro via
differential regulation of N-cadherin expression and RhoA activity.
BMC Cancer. 16:6672016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Emran Al A, Marzese DM, Menon DR, Stark
MS, Torrano J, Hammerlindl H, Zhang G, Brafford P, Salomon MP,
Nelson N, et al: Distinct histone modifications denote early
stress-induced drug tolerance in cancer. Oncotarget. 9:8206–8222.
2017.
|
|
36
|
Guler GD, Tindell CA, Pitti R, Wilson C,
Nichols K, KaiWai Cheung T, Kim HJ, Wongchenko M, Yan Y, Haley B,
et al: Repression of Stress-induced LINE-1 expression protects
cancer cell subpopulations from lethal drug exposure. Cancer Cell.
32:221–237 e213. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Easwaran HP and Baylin SB: Role of nuclear
architecture in epigenetic alterations in cancer. Cold Spring Harb
Symp Quant Biol. 75:507–515. 2010. View Article : Google Scholar
|
|
38
|
Haferkamp S, Borst A, Adam C, Becker TM,
Motschenbacher S, Windhövel S, Hufnagel AL, Houben R and
Meierjohann S: Vemurafenib induces senescence features in melanoma
cells. J Invest Dermatol. 133:1601–1609. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Krayem M, Najem A, Journe F, Morandini R,
Sales F, Awada A and Ghanem GE: Acquired resistance to BRAFi
reverses senescence-like phenotype in mutant BRAF melanoma.
Oncotarget. 9:31888–31903. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Childs BG, Baker DJ, Kirkland JL, Campisi
J and van Deursen JM: Senescence and apoptosis: Dueling or
complementary cell fates? EMBO Rep. 15:1139–1153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grimm J, Hufnagel A, Wobser M, Borst A,
Haferkamp S, Houben R and Meierjohann S: BRAF inhibition causes
resilience of melanoma cell lines by inducing the secretion of
FGF1. Oncogenesis. 7:712018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu Y, Schleich K, Yue B, Ji S, Lohneis P,
Kemper K, Silvis MR, Qutob N, van Rooijen E, Werner-Klein M, et al:
Targeting the Senescence-overriding cooperative activity of
structurally unrelated H3K9 demethylases in melanoma. Cancer Cell.
33:322–336.e8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Duchmann M and Itzykson R: Clinical update
on hypomethylating agents. Int J Hematol. 110:161–169. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Italiano A, Soria JC, Toulmonde M, Michot
JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A,
Suttle B, et al: Tazemetostat, an EZH2 inhibitor, in relapsed or
refractory B-cell non-Hodgkin lymphoma and advanced solid tumours:
A first-in-human, open-label, phase 1 study. Lancet Oncol.
19:649–659. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Doroshow DB, Eder JP and LoRusso PM: BET
inhibitors: A novel epigenetic approach. Ann Oncol. 28:1776–1787.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pawar A, Gollavilli PN, Wang S and
Asangani IA: Resistance to BET inhibitor leads to alternative
therapeutic vulnerabilities in castration-resistant prostate
cancer. Cell Rep. 29:13972019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Suraweera A, O'Byrne KJ and Richard DJ:
Combination therapy with histone deacetylase inhibitors (HDACi) for
the treatment of cancer: Achieving the full therapeutic potential
of HDACi. Front Oncol. 8:922018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sharma SV, Lee DY, Li B, Quinlan MP,
Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach
MA, et al: A chromatin-mediated reversible drug-tolerant state in
cancer cell subpopulations. Cell. 141:69–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Haas NB, Quirt I, Hotte S, McWhirter E,
Polintan R, Litwin S, Adams PD, McBryan T, Wang L, Martin LP, et
al: Phase II trial of vorinostat in advanced melanoma. Invest New
Drugs. 32:526–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jeffries MA and Sawalha AH: Autoimmune
disease in the epigenetic era: How has epigenetics changed our
understanding of disease and how can we expect the field to evolve?
Expert Rev Clin Immunol. 11:45–58. 2015. View Article : Google Scholar :
|
|
51
|
Mohammadi A, Sharifi A, Pourpaknia R,
Mohammadian S and Sahebkar A: Manipulating macrophage polarization
and function using classical HDAC inhibitors: Implications for
autoimmunity and inflammation. Crit Rev Oncol Hematol. 128:1–18.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Patel U, Rajasingh S, Samanta S, Cao T,
Dawn B and Rajasingh J: Macrophage polarization in response to
epigenetic modifiers during infection and inflammation. Drug Discov
Today. 22:186–193. 2017. View Article : Google Scholar :
|
|
53
|
Mangano K, Fagone P, Bendtzen K, Meroni
PL, Quattrocchi C, Mammana S, Di Rosa M, Malaguarnera L, Coco M and
Magro G: Hypomethylating agent 5-aza-2′-deoxycytidine (DAC)
ameliorates multiple sclerosis in mouse models. J Cell Physiol.
229:1918–1925. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fagone P, Mazzon E, Chikovani T, Saraceno
A, Mammana S, Colletti G, Mangano K, Bramanti P and Nicoletti F:
Decitabine induces regulatory T cells, inhibits the production of
IFN-gamma and IL-17 and exerts preventive and therapeutic efficacy
in rodent experimental autoimmune neuritis. J Neuroimmunol.
321:41–48. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Younes A, Oki Y, Bociek RG, Kuruvilla J,
Fanale M, Neelapu S, Copeland A, Buglio D, Galal A, Besterman J, et
al: Mocetinostat for relapsed classical Hodgkin's lymphoma: An
open-label, single-arm, phase 2 trial. Lancet Oncol. 12:1222–1228.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Briere D, Sudhakar N, Woods DM, Hallin J,
Engstrom LD, Aranda R, Chiang H, Sodré AL, Olson P, Weber JS and
Christensen JG: The class I/IV HDAC inhibitor mocetinostat
increases tumor antigen presentation, decreases immune suppressive
cell types and augments checkpoint inhibitor therapy. Cancer
Immunol Immunother. 67:381–392. 2018. View Article : Google Scholar
|
|
57
|
Zhang H, Pandey S, Travers M, Sun H,
Morton G, Madzo J, Chung W, Khowsathit J, Perez-Leal O, Barrero CA,
et al: Targeting CDK9 reactivates epigenetically silenced genes in
cancer. Cell. 175:1244–1258.e1226. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lam F, Abbas AY, Shao H, Teo T, Adams J,
Li P, Bradshaw TD, Fischer PM, Walsby E, Pepper C, et al: Targeting
RNA transcription and translation in ovarian cancer cells with
phar-macological inhibitor CDKI-73. Oncotarget. 5:7691–7704. 2014.
View Article : Google Scholar : PubMed/NCBI
|