Introduction
Hepatocellular carcinoma (HCC) accounts for ~90% of
all liver cancer cases, and HCC has one of the highest mortality
rates of all cancer types (1). The
prognosis of patients with HCC has improved in recent years due to
therapeutic advancements (2).
However, the bone metastases of HCC is becoming a more frequent
occurrence (3). The bone is one of
the most common sites of HCC metastasis, and the most common site
of bone metastases is the spine. Spinal metastases are estimated to
account for 50-75% of HCC bone metastases (4,5).
When spinal metastasis occurs, it may result in bone destruction
and tumor invasion into the canal space, progressing to axial pain
and neurological deficits. Although the prevention and early
treatment of HCC with spinal metastasis is an urgent public health
concern, there are limited studies available to date on the
molecular mechanisms underlying HCC-derived spinal metastasis
(6).
Several types of cancer metastasize in an
organ-specific manner and organ-specific metastasis is dependent on
the interaction of cancer cells with the host microenvironment
(7). Tumor cells do not act
independently during the establishment of a metastatic
microenvironment. The metastatic microenvironment itself provides a
suitable environment for the colonization and growth of metastatic
cells by expressing several pro-tumorigenic factors. The metastatic
cells then regulate the microenvironment by releasing cytokines and
interacting with other cells (8).
Recently, it was reported that organ-specific endothelial cells
regulate organ-specific metastasis (9). Dysregulated endothelial cell behavior
is vital in establishing the tumor microenvironment by producing
several tumor-regulated factors, such as chemokines, that promote
angiogenesis, tumor cell survival and metastasis (10). In bone metastasis, communication
between endothelial cells and metastatic cells constitutes a
vicious cycle, leading to the substantial disruption in the
physiological homeostasis of the bone microenvironment. Endothelial
cells have been suggested to participate in the tumor metastatic
microenvironment, as they constitute a significant part of the
spinal vascular system (11,12).
However, the focus of studies on bone metastasis has almost always
been on osteoblasts and osteoclasts, not endothelial cells. Bone
marrow endothelial cells (BMECs) are an important component of the
hematopoietic microenvironment that regulate the homing,
differentiation, self-renewal and migration of hematopoietic stem
cells by releasing several cytokines, including chemokines
(13,14). BMECs also regulate immune cells in
the bone marrow (15). BMECs,
which exert their specific characteristics in the bone marrow
microenvironment, have been widely studied in multiple myeloma
(16,17). However, the role of BMECs in spinal
metastasis has not been extensively studied to date.
Chemokines are a family of small structurally
related secreted cytokines, which play essential roles in
inflammation and immunity (18).
In addition to the involvement of chemokines in immunology and
pathogenesis, recent studies have focused on the role of chemokines
in modulating several aspects of cancer metastasis. Among the
studied chemokines, C-X3-C motif chemokine ligand 1 (CX3CL1) acts
as key regulator in spinal metastasis. CX3CL1 is the only member of
the CX3C chemokine subfamily, and is involved in regulating the
migration, invasion and survival of cancer cells (19). More specifically, the mucin-like
domain of CX3CL1 contains a cleavage site that allows
metalloproteases to cleave and release the protein in a soluble
form (20). It has been reported
that CX3CL1 is closely associated with cancer cell metastasis in
prostate (21), gastric (22) and breast (23) cancer, as well as in renal (24) and colon carcinoma (25). According to previous studies by the
authors, the quantity of CX3CL1 in the cancellous bone of the spine
is higher compared with the limbs, and the inhibition of CX3CL1 has
been confirmed as an effective strategy to prevent spinal
metastasis from breast and prostate cancer in an in vivo
model (26,27). However, the role of CX3CL1 in
spinal metastasis from HCC has not yet been investigated, at least
to the best of our knowledge.
Considering that BMECs are specialized cells with
the capacity to release large quantities of cytokines in the spine,
and CX3CL1 found in the spine is released from BMECs and leads to
an increase in their associated functions, CX3CL1 may promote the
invasion and migration of HCC cells and activate the Src/PTK2
signaling pathway in BMECs. Protein tyrosine kinase 2 (PTK2) has
been widely studied and enhances in vivo tumorigenesis and
metastasis in HCC, as well as cell invasion and migration in
vitro (28,29). The occurrence of these phenotypic
changes has been determined to be driven by the activation of
downstream pathways, such as the RHOA/ROCK2 and PIK3CA/AKT1
signaling pathways (30,31) In the present study, it was
demonstrated that CX3CL1 may promote the activation of the
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha (PIK3CA)/AKT serine/threonine kinase 1 (AKT1) and Ras homolog
family member A (RHOA)/Rho associated coiled-coil containing
protein kinase 2 (ROCK2) signaling pathways via the Src/PTK2
signaling pathway. The specific mechanism used by BMECs to secrete
CX3CL1 was determined. A disintegrin and metalloproteinase 17
(ADAM17), which is expressed by BMECs, was activated by
mitogen-activated protein kinase (MAPK) and was essential for
CX3CL1 secretion. The results of an in vivo experiment
revealed that CX3CR1-expressing HCC cells were attracted to the
spine by CX3CL1, which was expressed in spinal cancellous bone. To
determine the significance of this observation, the malignant
capacities of HCC cells mixed with BMECs were determined. Taken
together, the results of the present study demonstrate that CX3CL1
is expressed in BMECs and acts as a driving force of HCC in the
spinal metastatic microenvironment.
Materials and methods
Patients and cell isolation
There were 25 clinical specimens (healthy vertebral
bone from 5 patients with fracture surgery, tumor bones and spinal
metastases from 15 HCC patients with spinal metastasis, and primary
tumors from 5 HCC patients) used in the present study which were
obtained from the Department of Orthopedic Surgery, Zhongshan
Hospital, Fudan University (Shanghai, China) between July, 2015 and
July, 2019. There were 5 cases of spinal fracture (51.21±18.57), 5
cases of HCC (55.29±13.44 years) and 15 cases of HCC with spinal
metastasis (62.12±9.69 years), and all participants were male. All
patients provided informed consent and agreed to participate in the
study. The present study was approved by the Ethics Committee of
Zhongshan Hospital, Fudan University (approval nos. Y2014-185 and
Y2019-085). BMECs were isolated from fresh, healthy human bone
marrow collected during surgery from 2 patients, a 57-year-old male
patient and a 64-year-old male patient. As BMECs exhibit a
different sensitivity to trypsin digestion and adaptability to
extracellular matrix (ECM), BMECs were purified from other cells
after 3 to 4 passages using trypsin digestion. Morphological
observation and immunofluorescence staining were performed using
p-selectin (cat no. ab6632; Abcam; 1:400) and CD106 (cat. no.
ab215380; Abcam; 1:400) to identify BMECs. These cells also tested
negative for the mesenchymal stromal cell markers CD117 (cat. no.
ab25022; Abcam; 1:400) and STRO-1 (cat. no. ab214086; Abcam;
1:400). The BMECs were maintained in endothelial cell medium
containing 10% fetal bovine serum (FBS) (cat. no. 10099; Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2.
Reagents
Matrigel was obtained from BD Biosciences (cat. no.
3433-005-01). The MAPK14 inhibitor, SB203580, was purchased from
Selleck Chemicals. Lipofectamine® 2000 was purchased
from Invitrogen (cat. no. 11668019; Thermo Fisher Scientific,
Inc.). The Src protein inhibitor, bosutinib (0.5 nmol/l for 3 h),
PTK2 inhibitor, PF562271 (3.3 μmol/l for 24 h), and PIK3CA
inhibitor, LY294002 (10 μmol/l for 24 h), were obtained from
Selleck Chemicals. Phorbol 12-myristate 13-acetate (PMA) (10 ng/ml
for 24 h) was purchased from Abcam. Other reagents were purchased
from EMD Millipore or Sigma-Aldrich (Merck KGaA).
Cell lines and cell culture
THP-1 (SCSP-567), MHCC97H (SCSP-528) and Hep3B
(SCSP-5045) cells were purchased from The Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences. Human umbilical vein
endothelial cells (HUVECs) (ATCC® CRL-1730™) were
purchased from the American Type Culture Collection. All cells were
maintained in a humidified atmosphere with 5% CO2 at
37°C. MHCC97H and Hep3B cells were maintained in high glucose DMEM
containing 10% FBS. Cell culture medium supplements were obtained
from Sigma-Aldrich (Merck KGaA). HUVECs were cultured in
endothelial cell medium (cat. no. 1001; ScienCell Research
Laboratories, Inc.).
Western blot analysis
Total protein was extracted from cells using a lysis
buffer with phenylmethanesulfonylfluoride (cat. no. ST505; Beyotime
Institute of Biotechnology) and a phosphorylase inhibitor (cat. no.
78445, Thermo Fisher Scientific, Inc.). The protein concentration
was determined by the Coomassie Brilliant Blue method. A total of
40 μg proteins were loaded on an SDS gel (12% lower gel and
5% upper gel), resolved using SDS-PAGE and transferred to
nitrocellulose membranes (EMD Millipore). The membranes were
blocked with 5% non-fat milk in TBS containing 0.05% Tween-20. The
membranes were incubated with the primary antibodies overnight at
4°C and then incubated with the secondary antibodies (cat. nos.
ab97040, ab6940 and ab6566; 1:2,000; Abcam) for 2 h at room
temperature. The anti-matrix metalloproteinase (MMP)13 antibody
(cat. no. sc-101564; 1:1,000) was obtained from Santa Cruz
Biotechnology, Inc. The anti-p-PTK2 (Tyr397) (cat. no. 8556;
1:1,000), anti-p-PTK2 (Tyr576) (cat. no. 3281; 1:1,000),
anti-p-PTK2 (Tyr925) (cat. no. 3284; 1:1,000), anti-total PTK2
(cat. no. 3285; 1:1,000), anti-p-Src (Tyr416) (cat. no. 6943;
1:1,000), anti-Src antibody (cat. no. 2108; 1:1,000) was purchased
from Cell Signaling Technology, Inc. The anti-CX3CL1 (cat. no.
ab89229; 1:1,000), anti-CX3CR1 (cat. no. ab95620; 1:1,000),
anti-ADAM17 (cat. no. ab2051; 1:1,000), anti-p-ADAM17 (Tyr735)
(cat. no. ab182630; 1:1,000), anti-p-MAPK14 (Tyr180) (cat. no.
ab4822; 1:1,000), anti-MAPK14 (cat. no. ab31828; 1:1,000),
anti-RHOA (cat. no. ab187027; 1:1,000), anti-ROCK2 (cat. no.
ab125025), anti-NIMA related kinase 2 (NEK2; cat. no. ab227958;
1:1,000), anti-AKT1 (cat. no. ab81283; 1:1,000), anti-p65-nuclear
factor (NF)-κB (cat. no. ab16502; 1:1,000), anti-CD163 (cat. no.
ab87099; 1:1,000), anti-MRC1 (cat. no. ab188269; 1:1,000),
anti-GAPDH (cat. no. ab8245; 1:1,000) and anti-β-actin antibodies
(cat. no. ab8226; 1:1,000) were obtained from Abcam. The enhanced
chemiluminescent (ECL) kit was purchased from Abcam (cat. no.
ab133406). The experiments were performed 3 times independently. To
statistically compare the differences between groups,
log2 transformation was applied to normalize the results
of densitometry using ImageJ 1.8.0 software (National Institutes of
Health).
Immunohistochemistry
Tissues were collected, sectioned into a thickness
of 8 μm, dewaxed in xylene and rehydrated in graded ethanol.
Subsequently, tissues were soaked in 3% H2O2
for 20 min to block endogenous peroxidase. Tissues were incubated
with anti-CX3CR1 (cat. no. ab95620; 1:2,000) or anti-CX3CL1
antibody (cat. no. ab89229; 1:2,000) (from Abcam) at 4°C overnight.
Following secondary antibodies (cat. nos. ab150077 and ab150115;
1:2,000; Abcam) incubation, immunoperoxidase staining was performed
using the VECTASTAIN Elite ABC kit (cat. no. AK-5001; Vector
Laboratories, Inc.) and 3,3′-diaminobidine-tetrachloric acid as a
chromogenic agent. The sections were observed using an optical
microscope (Olympus-IX51; Olympus Corporation).
Reverse transcription-quantitative PCR
(RT-qPCR)
TRIzol® reagent was used to extract total
RNA and a Thermo Script RT-PCR system was used to reverse
transcribe the RNA into cDNA (cat. no. 12594100; Invitrogen; Thermo
Fisher Scientific, Inc.). SYBR Premix Ex Taq™ II (Takara Bio, Inc.)
was used to measure mRNA expression, and GAPDH was used as
the endogenous reference gene. Primers were purchased from Sangon
Biotech, Co., Ltd. and the sequences of the primers are presented
in Table I. The thermocycling
conditions for PCR amplification were as follows: Denaturation for
2 min at 95°C; followed 30 cycles of 94°C for 20 sec, 58°C for 20
sec, and 72°C for 30 sec. The expression ratio was calculated using
the 2−ΔΔCq method normalized to GAPDH (32). To statistically compare the
differences between groups, log2 transformation was used
to normally distribute the data.
 | Table IPrimers used for RT-qPCR. |
Table I
Primers used for RT-qPCR.
| Gene | Primer
sequences |
|---|
| CX3CR1 | F:
5′-AGTGTCACCGACATTTACCTCC-3′ |
| R:
5′-AAGGCGGTAGTGAATTTGCAC-3′ |
| CX3CL1 | F:
5′-GAATTCCTGGCGGGTCAGCACCTCGGCATA-3′ |
| R:
5′-AAGCTTTTACAGGGCAGCGGTCTGGTGGT-3′ |
| ADAM17 | F: 5′-GTGAGCAGTTTCT
CGAACGC-3′ |
| R:
5′-AGCTTCTCAAGTCGCAGGTG-3′ |
| GAPDH | F:
5′-GTCGGTGTGAACGGATTTG-3′ |
| R:
5′-TCCCATTCTCAGCCTTGAC-3′ |
| IL6 | F:
5′-ACATCGTCGACAAAATCTCTGCA-3′ |
| R:
5′-AGCCAGTGTCTCCTTGCTGTTT-3′ |
| CXCL8 | F:
5′-CAGTGCATAAAGACATACTCC-3′ |
| R:
5′-TTTATGAATTCTCAGCCCTC-3′ |
| CCL3 | F:
5′-GCTGTGATCTTCAAGACC-3′ |
| R:
5′-AAGTCTTCGGAGTTTGGG-3′ |
| MMP2 | F:
5′-GTTGGCAGTGCAATACCTGA-3′ |
| R:
5′-GAGCAAAGGCATCATCCACT-3′ |
| MMP8 | F:
5′-CCAAAGAGATCACGGTGACA-3′ |
| R:
5′-GTTGCTGGTTTCCCTGAAAG-3′ |
| PTHLH | F:
5′-CAGTGGAGTGTCCTGGTATT-3′ |
| R:
5′-GATCTCCGCGATCAGATGGT-3′ |
Cell co-culture and invasion
Transwell chambers with 8 μm pores (cat. no.
3402; Corning, Inc.) were used for assessing cell invasion. The
upper chambers were pre-coated with Matrigel (cat. no. E6909;
Sigma-Aldrich; Merck KGaA). For co-culture of the HCC cells and
BMECs, the 24-well Transwell co-culture system was used. HCC cells
at a density of 5×104 cells and were plated in the upper
chamber, while BMECs at a density of 2×105 cells were
seeded in the lower chamber. Both chambers were supplemented with
serum-free medium. Subsequently, 100 nM CX3CL1 (R&D Systems,
Inc.) or 50 ng/ml neutralizing antibody of CX3CL1 (cat. no.
ab89229; Abcam) were added to the lower chamber. The cells were
incubated at 37°C for 48 h, and the upper surfaces were gently
swabbed to remove cells which had not migrated. The cells which had
migrated to the lower membrane were fixed with 1% glutaraldehyde
and stained with 0.1% crystal violet (cat. no. C0775;
Sigma-Aldrich) for 15 min at room temperature. The number of cells
which had invaded were counted in 5 randomly selected fields per
well. Invasion experiments were performed 3 times
independently.
Cell migration
Transwell chambers with 8 μm pores (cat. no.
3402; Corning, Inc.) were used for assessing cell migration. For
co-culture of the HCC cells and BMECs, the 24-well Transwell
co-culture system was used. HCC cells at a density of
2×105 cells and were placed in the lower chamber. While
BMECs at a density of 1×105 cells were seeded in the
upper chamber. Both chambers were supplemented with serum-free
medium. Subsequently, 100 nM CX3CL1 (R&D Systems) or 50 ng/ml
neutralizing antibody of CX3CL1 (cat. no. ab89229; Abcam) were
added to the lower chamber. Cells were grown to over the entire
bottom of each well in a monolayer, a 100-μl pipette tip was
used to scratch the monolayer, and cells were cultured in 2 ml
serum-free DMEM. Wound-healing was observed using an optical
microscope after 48 h. The experiments were repeated 3 times
independently.
Cell counting kit-8 (CCK-8) assays
A total of 1×104 cells/ml BMECs were
seeded into a 96-well plate. Cells were incubated at 37°C for 24 h,
after which the media was removed and 10 μl CCK-8 solution
was added to each well, and further incubated at room temperature
for 2 h. The absorbance at 450 nm was measured using a microplate
reader (Thermo Fisher Scientific, Inc.). The experiments were
repeated 3 times independently.
Hematoxylin and eosin (H&E)
staining
The tissues were fixed in 10% formaldehyde,
dehydrated in graded ethanol, paraffin-embedded, and sliced. The
tissue sections were then dewaxed in xylene, rehydrated in graded
ethanol, and stained with hematoxylin for 1 min and eosin for 1
min. Following dehydration in graded ethanol and vitrification in
dimethylbenzene, the tissue sections were observed under an optical
microscope (Olympus-IX51; Olympus Corporation). All experimental
procedures were carried out at room temperature.
ELISA
Cell medium was collected and used for detecting the
concentrations of CX3CL1. The levels of CX3CL1 were detected using
ELISA kits (Boster Bio) in accordance with the manufacturer's
instructions. The optical density (OD) at 450 nm was measured using
a microplate reader (Bio-Rad Laboratories, Inc.).
Small interfering (si)RNA and
transfection
The overexpression plasmids of ADAM17 (bank ID:
NM_003183) were purchased from GeneChem, Inc. The siRNA sequences
used were as follows: siCX3CL1, 5′-GGACAAGCCACATAGGAAA-3′ and
siADAM17, 5′-GCUUGUUCAUCGAGUGAAAdTdT-3′. Control cells were
subjected to mock transfection with scrambled sequences (GeneChem,
Inc.). CX3CR1 overexpression lentivirus (bank ID: NM_001337) and
CX3CL1 siRNA lentivirus (bank ID: NM_002996) were purchased from
GeneChem, Inc. Cells were first seeded into 6-well plates and
cultured to a cell density of 70%. Subsequently, lentiviral
transfection as performed according to the manufacturer's
instructions. ADAM17 overexpression, siADAM17, CX3CL1
overexpression and siCX3CL1 plasmids were transfected into BMECs,
respectively. CX3CR1 overexpression lentivirus was transfected into
HCC cell lines. CX3CL1 siRNA lentivirus was transfected into BMECs.
At 1 day prior to transfection with 100 nM siRNA or 1 μg/ml
plasmids, 1×105 cells/well were plated into 24-well
plates in DMEM and 10% FBS. Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for
transfection for 6 h in serum-free medium at 37°C according to the
manufacturer's protocol. Subsequently, the medium was replaced with
DMEM with 10% FBS.
Animal model
A total of 40 4-6-week-old male NOD/SCID mice were
obtained from Vital River Laboratory Animal Technology Co., Ltd.
All animal experiments were performed in compliance with the
Guidelines for the Care and Use of Research Animals established by
the Animal Ethics Committee of Zhongshan Hospital, Fudan
University. The animal studies were approved by the Animal Ethics
Committee of Zhongshan Hospital, Fudan University. Experiments met
the ethical requirements for animal experiments. MHCC97H cells were
infected with Luc-CX3CR1 lentivirus to establish CX3CR1-MHCC97H, a
stable HCC cell line overexpressing CX3CR1. A total of 30 mice were
randomly divided into 2 groups. Subsequently, 1×106
tumor cells were suspended in 200 μl serum-free medium, and
the cells were injected into the left ventricle of the anesthetized
mice. Mice were anesthetized by an intraperitoneal injection of
sodium pentobarbital (80 mg/kg). After 6-8 weeks, the mice were
analyzed using in vivo imaging, micro-CT scans. The mice
were sacrificed at 8 weeks by an intraperitoneal injection of
sodium pentobarbital (200 mg/kg) and histological analysis was then
performed. To determine whether locally administered BMECs mixed
with tumor cells promoted tumor growth, 5×106 MHCC97H
cells were mixed with 1×105 BMECs infected with
CX3CL1-silencing lentivirus or control lentivirus and injected into
the spine of the mice for primary tumor assessment. After 4 weeks,
the tissues were analyzed using immunohistochemistry. The tumor
long diameter (L) and short diameter (W) were measured to calculate
the tumor volume. Tumor volume (V) = (L x W2)/2
(33).
Flow cytometry
To stain for the cell surface markers, cells were
incubated with following fluorescence-labeled monoclonal antibodies
for 30 min on ice respectively: PE-conjugated anti-CD11b (cat. no.
333142; BD Biosciences; 1:300), FITC-conjugated anti-F4/80 (cat.
no. orb223773; Biorbyt; 1:300), PE-Cy7-conjugated-CD45 (cat. no.
147703; BioLegend; 1:300), APC-conjugated CX3CR1 (cat. no. 341609;
BioLegend; 1:300), PE-Cy3-conjugated MRC1 (cat. no. sc-376232;
Santa Cruz Biotechnology, Inc.; 1:300) and PE-Cy5-CD163 (cat. no.
sc-33715; Santa Cruz Biotechnology, Inc.; 1:300) antibodies.
Macrophages population in peripheral blood of mice was determined
based on the co-expression of CD45, CD11b and F4/80. Cells were
selected based on the higher expression of CD45 for macrophages.
CD11b and F4/80 expression were used for the further
characterization of the macrophages. Data were acquired using a
fluorescence activated cell sorting (FACS)Calibur (BD Biosciences),
and analyzed using CellQuest software (BD Biosciences).
Statistical analysis
The staining intensity of immunohistochemical and
fluorescence the intensity of immunofluorescent were measured
through integrated optical density/area in Image-Pro Plus 6.0. SPSS
version 16.0 (SPSS, Inc.) was used to analyze all data. Data are
presented as the means ± standard deviation of 3 experimental
repeats. The χ2 test was used to analyze the in
vivo experiments (spinal metastasis rate). Otherwise, ANOVA
with a post hoc Tukey's test, a Student's t-test, Mann-Whitney U or
Kruskal Wallis test followed by Dunn's non-parametric post hoc test
were used to compare differences between groups. P<0.05 was
considered to indicate a statistically significant difference. The
Mann-Whitney U test is a non-parametric test of the null hypothesis
that two samples are derived from the same population against an
alternative hypothesis, prinicipally that a particular population
tends to have larger values than the other. Unlike the t-test, it
does not require the assumption of normal distributions. It is
almost as efficient as the t-test on normal distributions (34).
Results
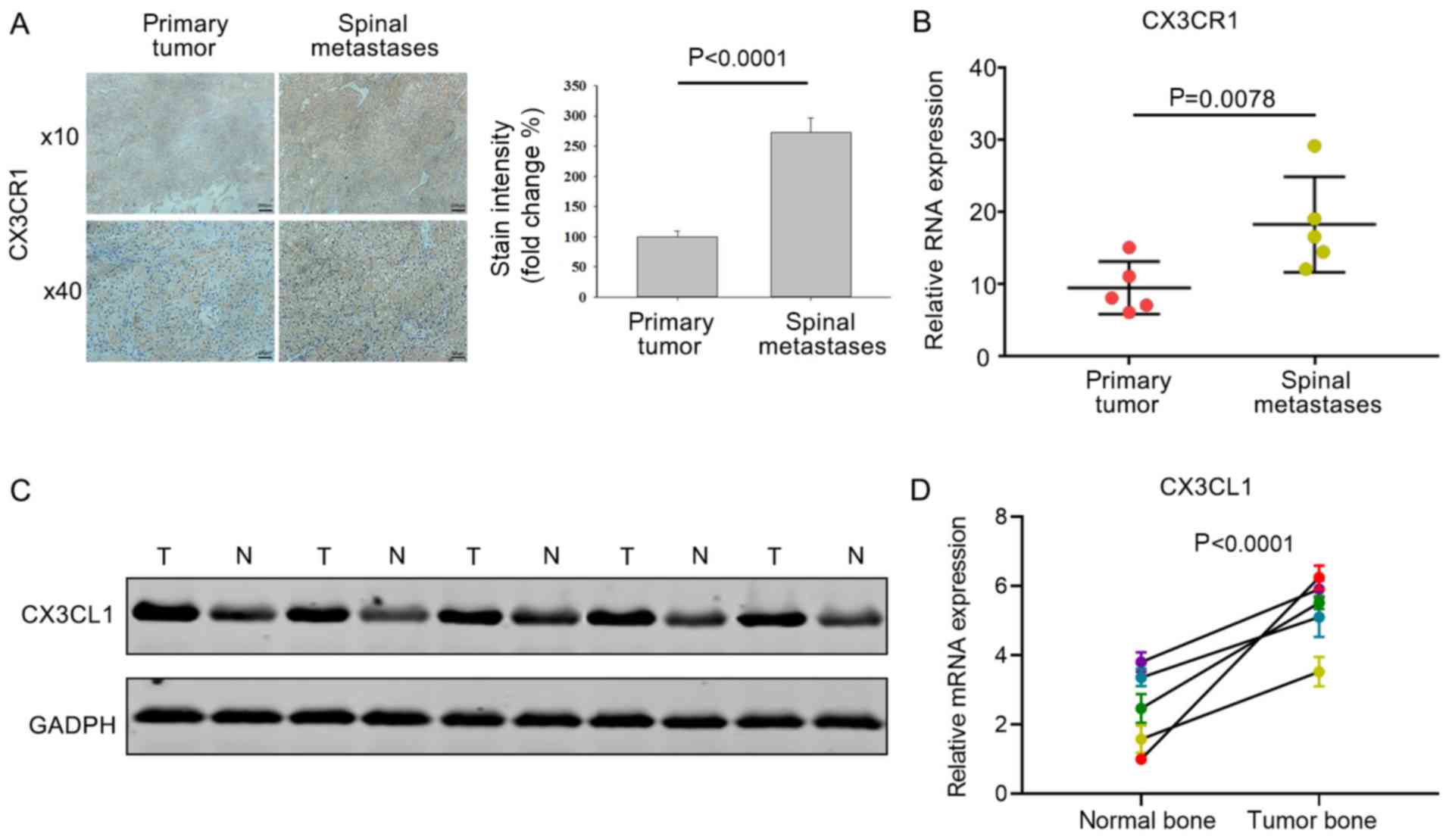
CX3CL1 and CX3CR1 expression are
upregulated in spinal metastases
To investigate the expression of CX3CR1 and CX3CL1
in primary and metastatic tumors, tissue sections and total protein
samples were isolated during surgery from patients with or without
HCC spinal metastases. Immunohistochemistry revealed that the
CX3CR1 levels in HCC spinal metastases were upregulated compared
with the primary tumors (Fig. 1A).
The mRNA expression levels of CX3CR1 were also higher in HCC
spinal metastases compared with primary tumors (Fig. 1B). CX3CL1 expression in healthy
vertebral bone was compared with that of bone tumors from patients
with HCC. Western blot analysis revealed that the protein
expression levels of CX3CL1 were higher in tumor bone compared with
healthy vertebral bone (Fig. 1C).
Consistent with the increase in protein levels, the mRNA expression
levels of CX3CL1 was also increased in tumor bone compared
with healthy vertebral bone (Fig.
1D). These differences in expression support the hypothesis
that CX3CR1 and CX3CL1 serve a critical role in spinal
metastases.
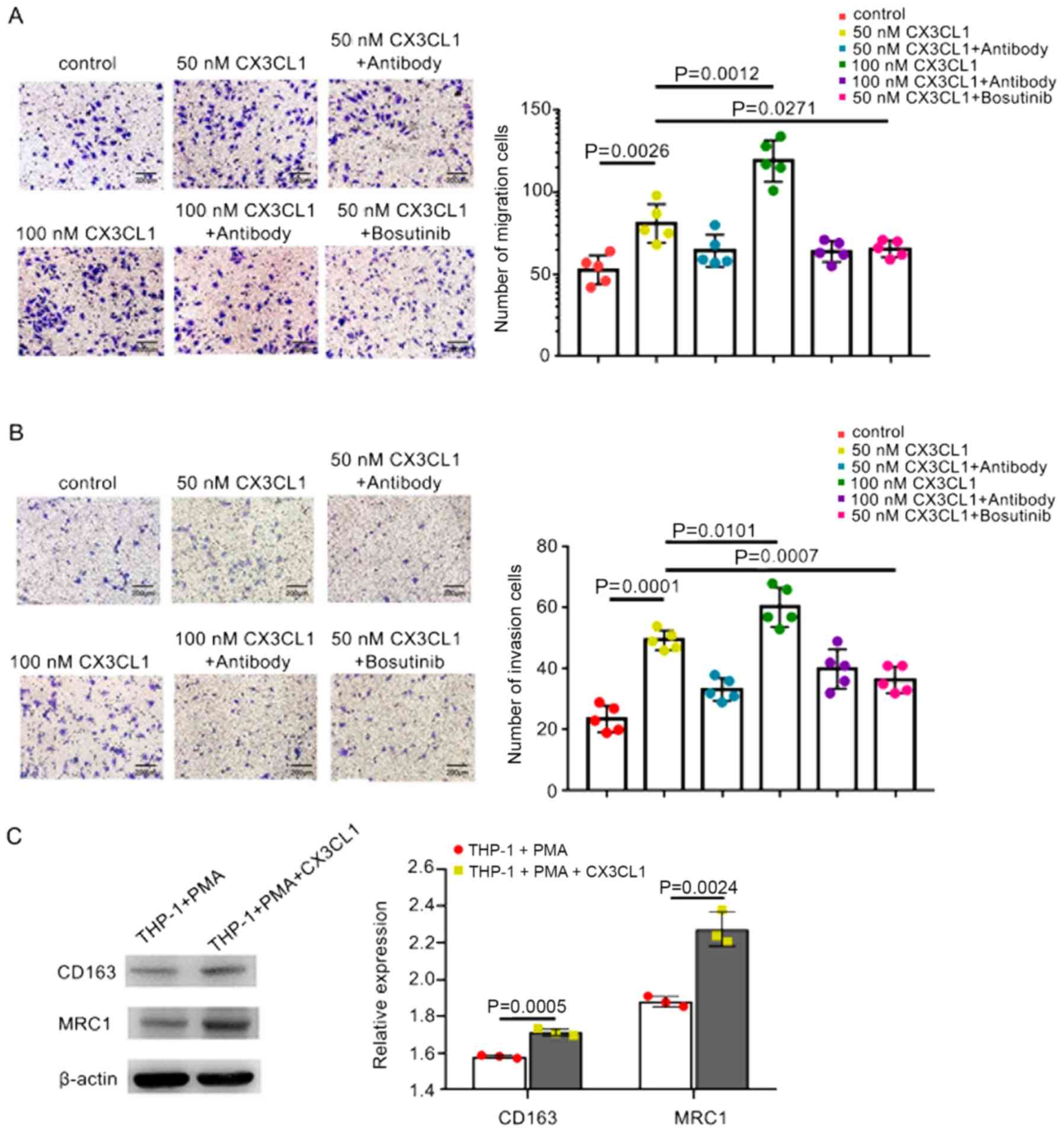
CX3CL1 promotes HCC cell invasion and
migration through the Src signaling pathway
A previous study demonstrated that CX3CL1 was not
only significantly expressed in spinal cancellous bone, but also in
the metastatic bone of patients with HCC (26). As a chemokine, CX3CL1 has been
reported to enhance increased cancer cell invasion and migration
(35). In order to investigate
whether CX3CL1 binds to its receptor, CX3CR1, to enhance the
migration of HCC cells, migration assays were performed using
Transwell chambers. CX3CL1 significantly increased the migration of
HCC cells compared with the control, and higher concentrations of
CX3CL1 further enhanced migration (Fig. 2A). Subsequently, invasion assays
were performed. The results confirmed that CX3CL1 significantly
increased the invasion of MHCC97H cells compared with the control
(Fig. 2B).
Through high-throughput analysis in our previous
studies, it was shown that the Src/PTK2 pathway may participate in
regulating CX3CL1-enhanced cell migration and invasion (27). Initially, the role of Src in cell
invasion and migration was determined. The Src protein inhibitor,
bosutinib, suppressed the migration and invasion of HCC cells
(Fig. 2A and B). These results
suggest CX3CL1 may enhance the migration and invasion of HCC via
Src.
CX3CL1/CX3CR1 has also been shown to promote
macrophages to exhibit M2 phenotypes (36). In addition, tumor-associated
macrophages (TAMs) also exhibit an M2 phenotype during tumor
development. Therefore, in the present study, the effect of CX3CL1
on macrophages was determined. THP-1 cells were induced to
differentiate into resting macrophages using PMA. CX3CL1 was
subsequently added to the PMA-induced THP-1 cells for 48 h. The
expression levels of the M2 markers, MRC1 and CD163, were examined
by western blot analysis and was found to be significantly
increased compared with the controls (Fig. 2D). These results suggest that TAMs
differentiate into the M2 morphological phenotype following CX3CL1
stimulation.
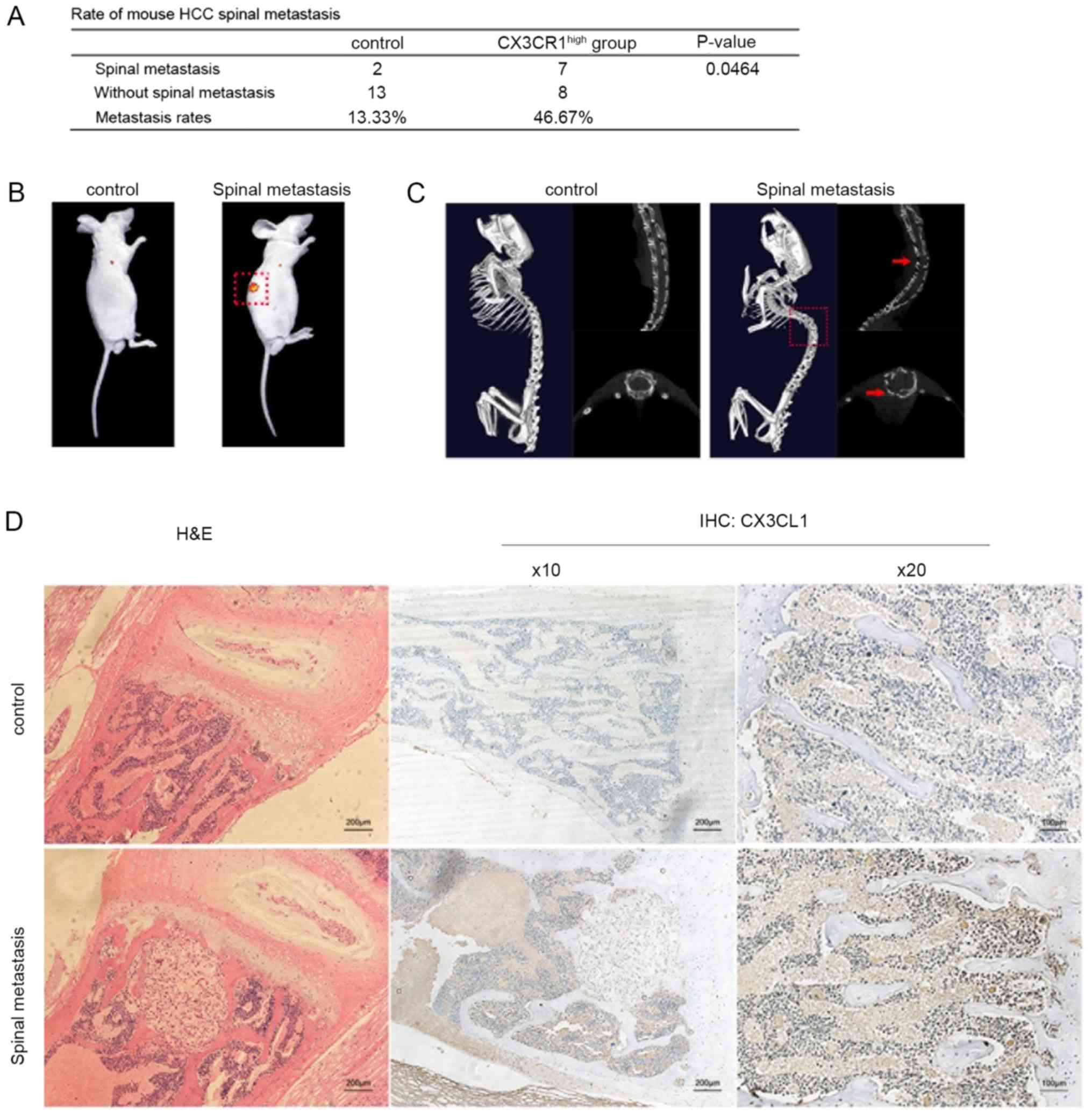
CX3CL1 facilitates HCC spinal metastasis
in a mouse model
To examine the effects of CX3CL1 in HCC spinal
metastases, a nude mouse model of HCC metastasis was established by
injecting HCC cells directly into the left ventricle of nude mice.
Lentiviral transfection was used to overexpress CX3CR1 in MHCC97H
cells and establish a CX3CR1-MHCC97H cell line (Fig. S1A). After 6 weeks, the mice were
examined by performing in vivo imaging to investigate
whether spinal metastasis had occurred. In total, 2 spinal
metastases were visualized in the control group; however, 7 spinal
metastases were visualized in the CX3CR1-MHCC97H group (Fig. 3A and B). In addition, Micro-CT was
also used to further analyze the spinal metastases by inspecting
for bone damage (Fig. 3C). The
results indicated that spinal metastasis was enhanced in nude mice
injected with CX3CR1-MHCC97H cells compared with the controls
(Fig. 3A). Subsequently, CX3CL1
levels in spinal cancellous bone from the control and
CX3CR1-MHCC97H groups were compared. The cancellous bone collected
from the mice with CX3CR1-expressing tumor cells exhibited higher
CX3CL1 levels compared with the controls (Fig. 3D). These results suggest that HCC
spinal metastasis is increased in spinal cancellous bone expressing
higher levels of CX3CL1.
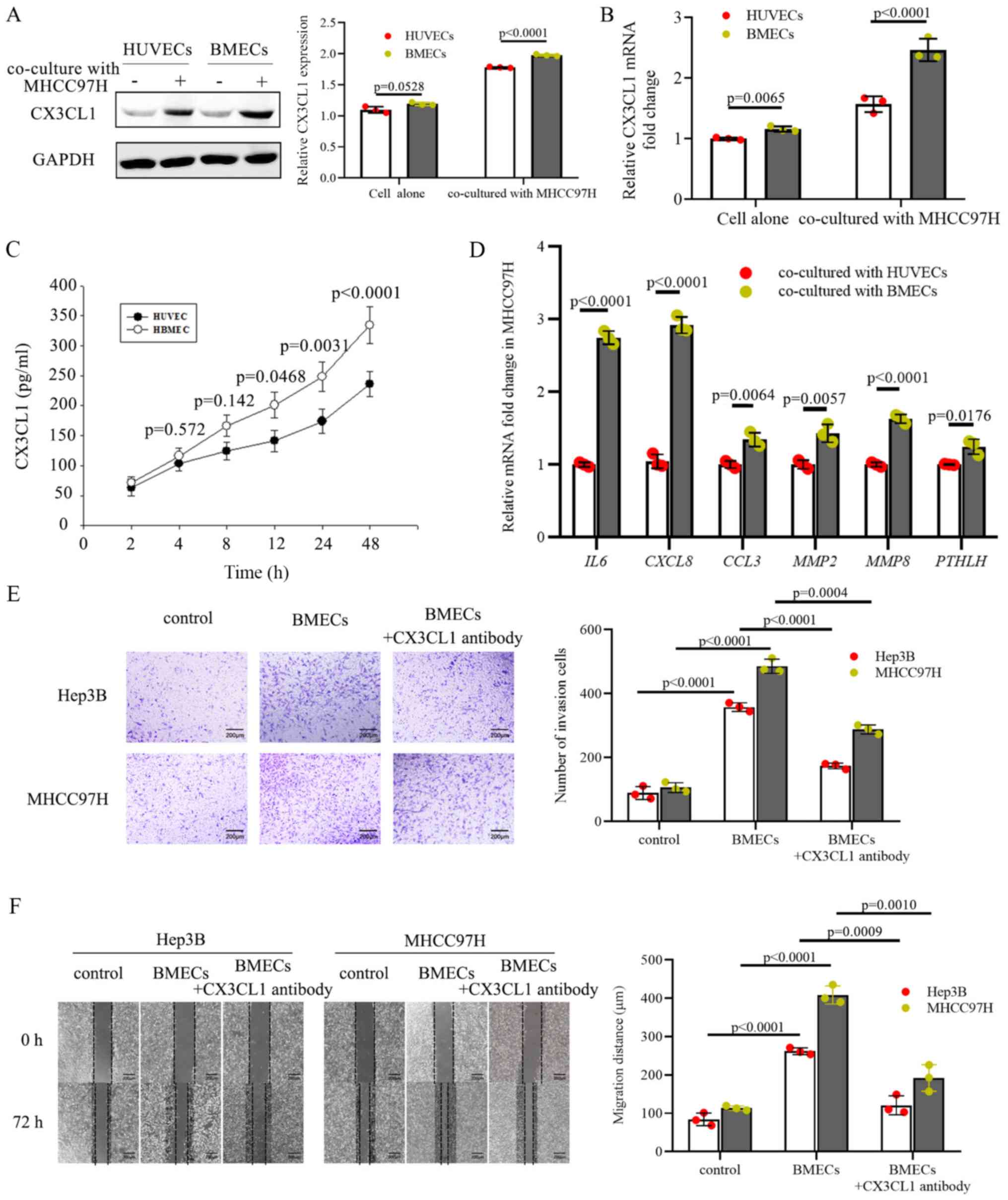
BMECs promote the invasion and migration
of HCC cells through CX3CL1
BMECs are responsible for forming the hematopoietic
microenvironment and modulating hematopoietic cells; thus, they
exhibit a relatively high ability to secrete cytokines. BMECs are a
major source of cytokines in the bone marrow microenvironment, and
a previous study reported that BMECs produce CX3CL1 (37). To elucidate the specific regulatory
mechanisms used by BMECs on CX3CL1, the CX3CL1 levels in BMECs were
compared with HUVECs. Primary BMECs were isolated from human bone
marrow samples and it was shown that the CX3CL1 protein and mRNA
levels were upregulated in BMECs co-cultured with MHCC97H cells and
were higher compared with HUVECs (Fig.
4A and B). Given that CX3CL1 is a secreted chemokine and can
promote chemotaxis, the CX3CL1 protein levels in media conditioned
by BMECs were measured using an ELISA kit. The results indicated
that BMECs released increased quantities of CX3CL1 into the
extracellular environment compared with HUVECs (Fig. 4C). Based on the above results, it
was hypothesized that BMECs serve as a crucial source of CX3CL1.
Bone destruction is one of the crucial steps required for tumor
metastasis. MHCC97H cells co-cultured with BMECs displayed higher
mRNA expression levels of CXCL8, interleukin
(IL)6, CCL3, MMP2, MMP8 and
parathyroid hormone like hormone (PTHLH) (Fig. 4D), all of which are strictly
associated with bone destruction in cancer (38). Taken together, these results
suggest that BMECs express and secrete CX3CL1 into the bone marrow
to attract metastatic cells into the vertebral body and to regulate
the surrounding tumor microenvironment.
To determine whether BMEC-derived CX3CL1 directly
regulates the function of tumor cells, cell invasion was measured
using Transwell assays and migration was measured using
wound-healing assays. The presence of BMECs increased the invasion
and migration of MHCC97H and Hep3B cells (Fig. 4E and F). When CX3CL1 in
BMEC-conditioned media was neutralized using an CX3CL1 antibody,
the invasion and migration activity of the MHCC97H and Hep3B cells
was significantly decreased (Fig. 4E
and F), suggesting that BMECs release soluble CX3CL1 to
facilitate the invasion and migration of HCC cells.
CX3CL1 increases the activation of the
PIK3CA/AKT1 and RHOA/ROCK2 signaling pathways
BMECs promoted the activation of p-PTK2
(Tyr397/Tyr576/Tyr925), as shown by the increase in p-Src (Tyr416)
protein expression in MHCC97H cells (Fig. 5A). Additionally, a reduction in
p-Src (Tyr416) and p-PTK2 (Tyr397/Tyr576/Tyr925) protein levels was
also observed when CX3CL1 was neutralized (Fig. 5A). These results suggest that BMECs
release CX3CL1 which upregulates the activation of Src/PTK2
signaling in MHCC97H cells.
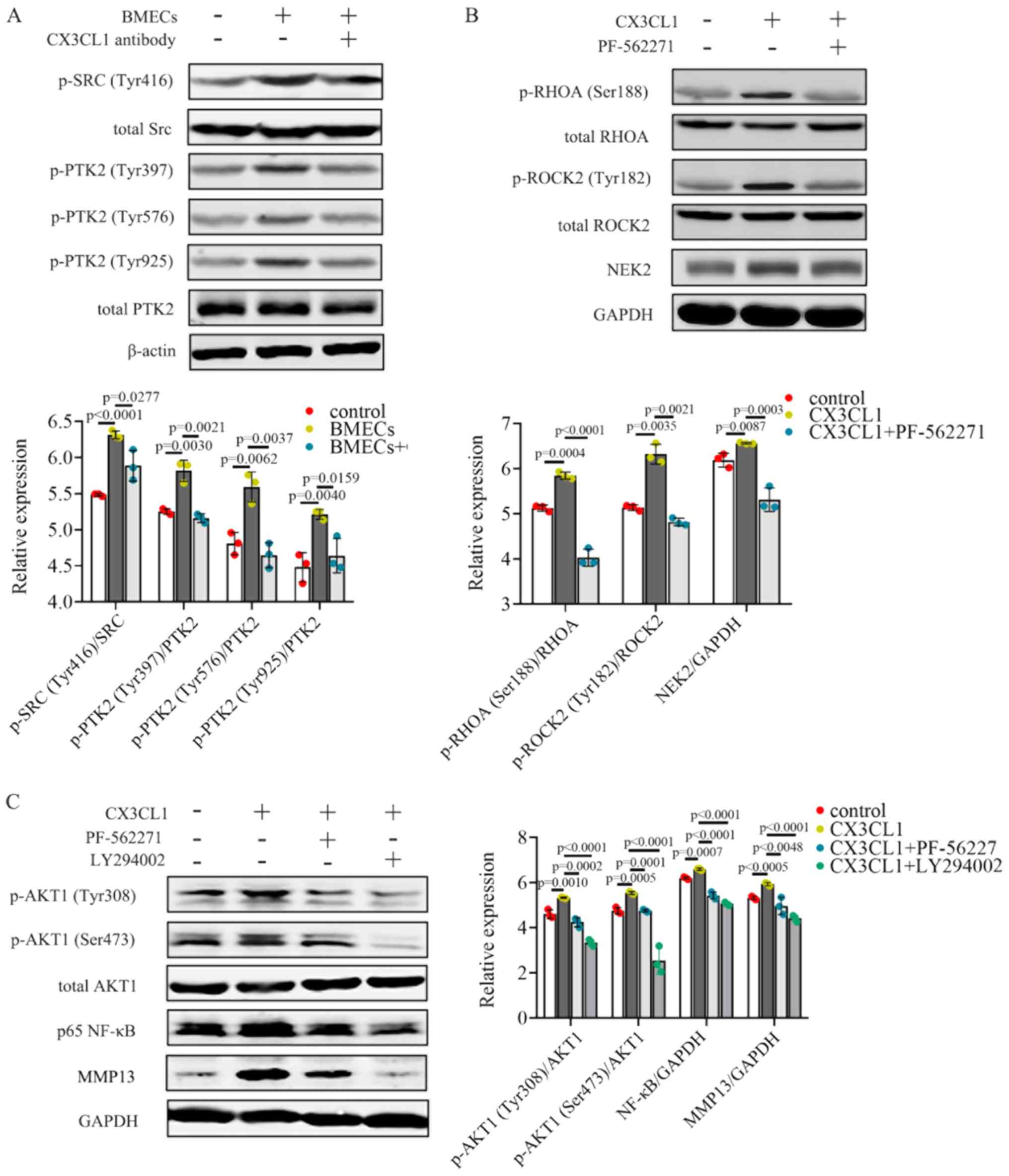 | Figure 5CX3CL1 increases the activation of
the PIK3CA/AKT1 and RHOA/ROCK2 signaling pathways. (A) Protein
expression levels of p-Src (Tyr416), total Src, p-PTK2
(Tyr397/Tyr576/Tyr925), total PTK2 and β-actin were assessed using
western blot analysis;. n=3. (B) p-RHOA (Ser188), total RHOA,
p-ROCK2 (Tyr182), total ROCK2, NEK2 and GAPDH were measured by
western blot analysis; n=3. (C) p-AKT1 (Tyr308/Ser473), total AKT1,
p65 NF-κB and MMP13 were detected by western blot analysis; n=3.
western blot data were analyzed using ANOVA and Tukey's test. p-,
phosphor; HCC, hepatocellular carcinoma; CX3CL1, C-X3-C motif
chemokine ligand 1. |
PIK3CA/AKT1 and RHOA/ROCK2 are both crucial for cell
invasion and migration. The PI3K/AKT signaling pathway plays an
important role in cancer for the regulation of cell growth and
survival, which have long been well described (39). RHOA/ROCK2 has been shown to be
linked to the contraction and elongation of the cytoskeleton which
is closely associated with cancer metastasis (40). These two pathways have been
confirmed to be involved in HCC progression (41,42).
To verify the effects of PTK2 on CX3CL1-induced MHCC97H cells,
cells were treated with the PTK2 inhibitor, PF562271. Exposure to
CX3CL1 resulted in the induction of p-RHOA (Ser188)/p-ROCK2
(Tyr182) and increased NEK2 expression levels (Fig. 5B). PF562271 treatment reversed
activation of the RHOA/ROCK2/NEK2 signaling pathway (Fig. 5B). NEK2 facilitates the migration
and invasion of HCC, and higher NEK2 levels are associated with a
less favorable prognosis of patients with HCC (43,44).
Both the PTK2 inhibitor, PF562271, and the PIK3CA inhibitor,
LY294002, downregulated the CX3CL1-induced activation of p-AKT1
(Tyr308/Ser473) (Fig. 5C). The
expression levels of both NF-κB and its downstream effector, MMP13,
were decreased when the cells were treated with PF562271 (Fig. 5C). Taken together, these data
illustrate that CX3CL1 induces the activation of the RHOA/ROCK2 and
PIK3CA/AKT1 signaling pathways in HCC cells.
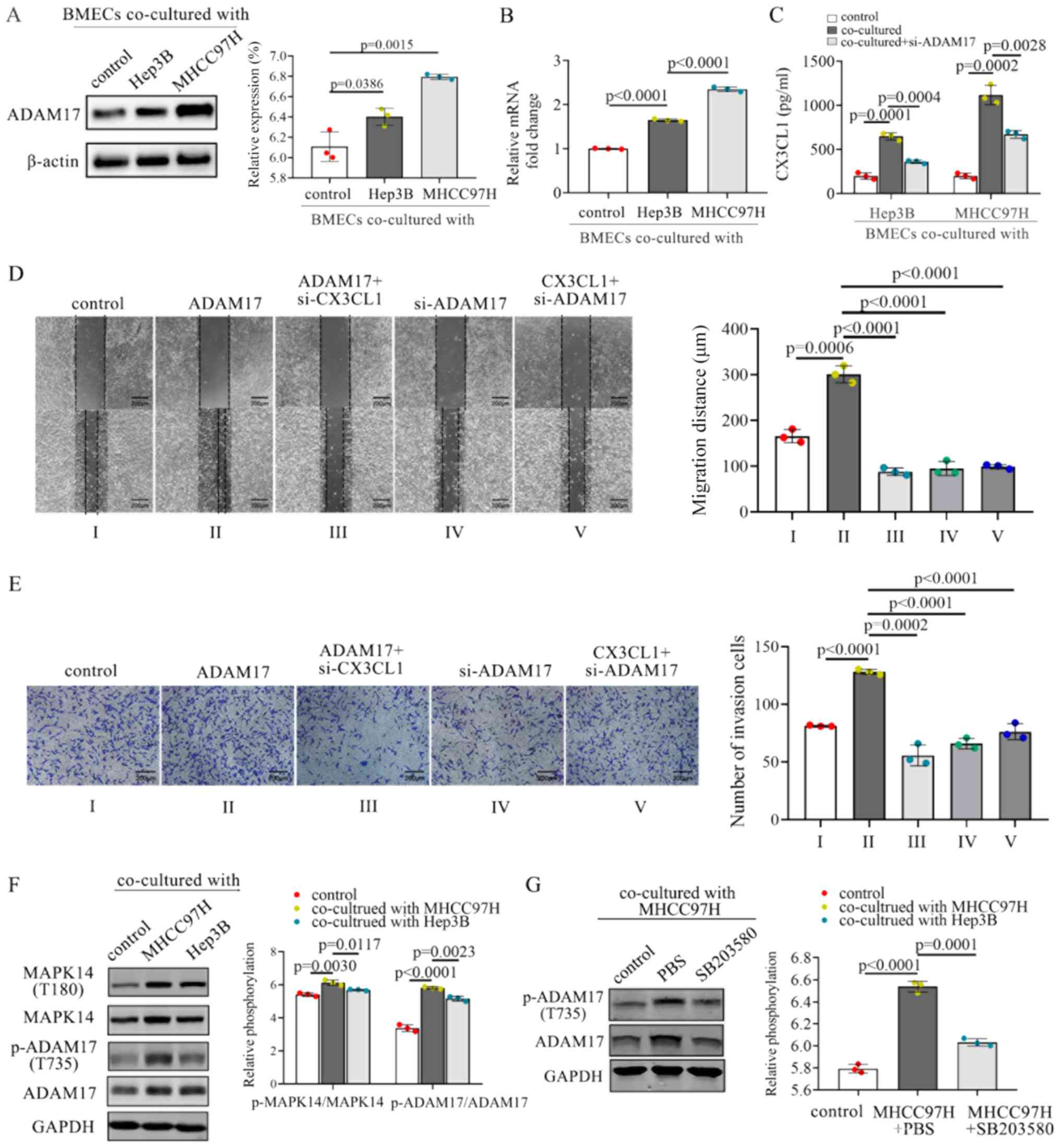
MAPK14 activation of ADAM17 in BMECs
promotes the invasion and migration of HCC cells by regulating
CX3CL1
Upon arriving at the metastatic site, tumor cells
can alter the expression of genes in endothelial cells, thereby
promoting tumor progression (45).
Thus, whether HCC cells can influence BMECs to alter related genes
in the process of CX3CL1 production was assessed in the present
study. It was hypothesized that adjacent HCC cells may impact the
regulation of normal BMECs in the bone marrow microenvironment.
CX3CL1 has a mucin-like domain which contains a cleavage site that
allows metalloproteases, such as ADAM17, to cleave and release a
soluble form of CX3CL1 (46). Both
MHCC97H and Hep3B cells promoted the expression of ADAM17 in BMECs
(Fig. 6A). The mRNA levels of
ADAM17 were also upregulated when the BMECs were co-cultured
with MHCC97H or Hep3B cells (Fig.
6B). Furthermore, the co-culture of BMECs with MHCC97H and
Hep3B cells resulted in increased levels of soluble CX3CL1 compared
with the controls (Fig. 6C).
Inhibition of ADAM17 in BMECs decreased the CX3CL1 secretion
(Fig. 6C). Above-mentioned results
suggest that ADAM17 may effectively promote the secretion of
soluble CX3CL1 from BMECs.
The expression levels of CX3CL1 were inhibited by
transfection with CX3CL1 siRNA, or were promoted by transfection
with CX3CL1 overexpression plasmid (Fig. S1B). ADAM17 overexpression or
si-ADAM17 was also transfected into BMECs to regulate the
overexpression of ADAM17 (Fig.
S1C). The knockdown of ADAM17 inhibited CX3CL1 secretion from
BMECs. Thus, whether the knockdown of ADAM17 reversed the
CX3CL1-induced promotion of cell invasion and migration was
examined. The knockdown of ADAM17 in BMECs resulted in a
downregulation of the invasion and migration of MHCC97H cells,
whereas ADAM17 overexpression increased cell proliferation and
migration (Fig. 6D and E).
Furthermore, treatment with recombinant CX3CL1 enhanced the
invasion and migration of HMCC97H, whereas inhibition of ADAM17
attenuated this enhancement (Fig. 6D
and E). These results suggest that the knockdown of ADAM17
significantly blocked the CX3CL1-induced invasion and migration of
HCC cells.
It has been reported that the activation of ADAM17
is dependent on phosphokinases, such as MAPK14 (47). Therefore, the role of MAPK14 in the
tumor cell-mediated activation of ADAM17 was assessed. The
co-culture of BMECs with MHCC97H or Hep3B cells induced the
activation of MAPK14 and the phosphorylation of ADAM17 (Fig. 6F). The MAPK14 inhibitor, SB203580,
inhibited the phosphorylation of ADAM17 at residue T735 (Fig. 6G). Thus, MAPK14 activation is
crucial for the activation of ADAM17 in BMECs co-cultured with HCC
cells.
Interaction between BMECs and HCC cells
enhances tumor growth in the spine
As both endothelial cells and cancer cells can
produce several cytokines, it was hypothesized that they may
mutually influence each other. Tumor cell growth and tumor
angiogenesis are increased by the recruitment of endothelial cells,
contributing to the formation of the metastatic niche (8). Therefore, the modulatory effect of
CX3CL1 of HCC on BMECs. CX3CL1 expression was knocked down by
lentiviral transfection into BMECs to construct
CX3CL1low BMECs; CX3CL1nor BMECs were used as
a control. Transwell assays were to assess cell migration, and
CCK-8 assays were used to assess proliferation. HCC cells enhanced
the recruitment and proliferation of CX3CL1nor BMECs,
but this enhancement was attenuated in CX3CL1low BMECs
(Fig. 7A and B). It was
hypothesized that soluble CX3CL1 from BMECs could stimulate HCC
cells, which in turn would act on BMECs to create a vicious
feedback cycle. Previous data have revealed that HCC cells induce
BMECs to exhibit an aggressive phenotype, which may contribute to
the formation of a bone metastatic niche (48). These data demonstrate the important
regulatory role that CX3CL1 plays in the communication between HCC
cells and BMECs.
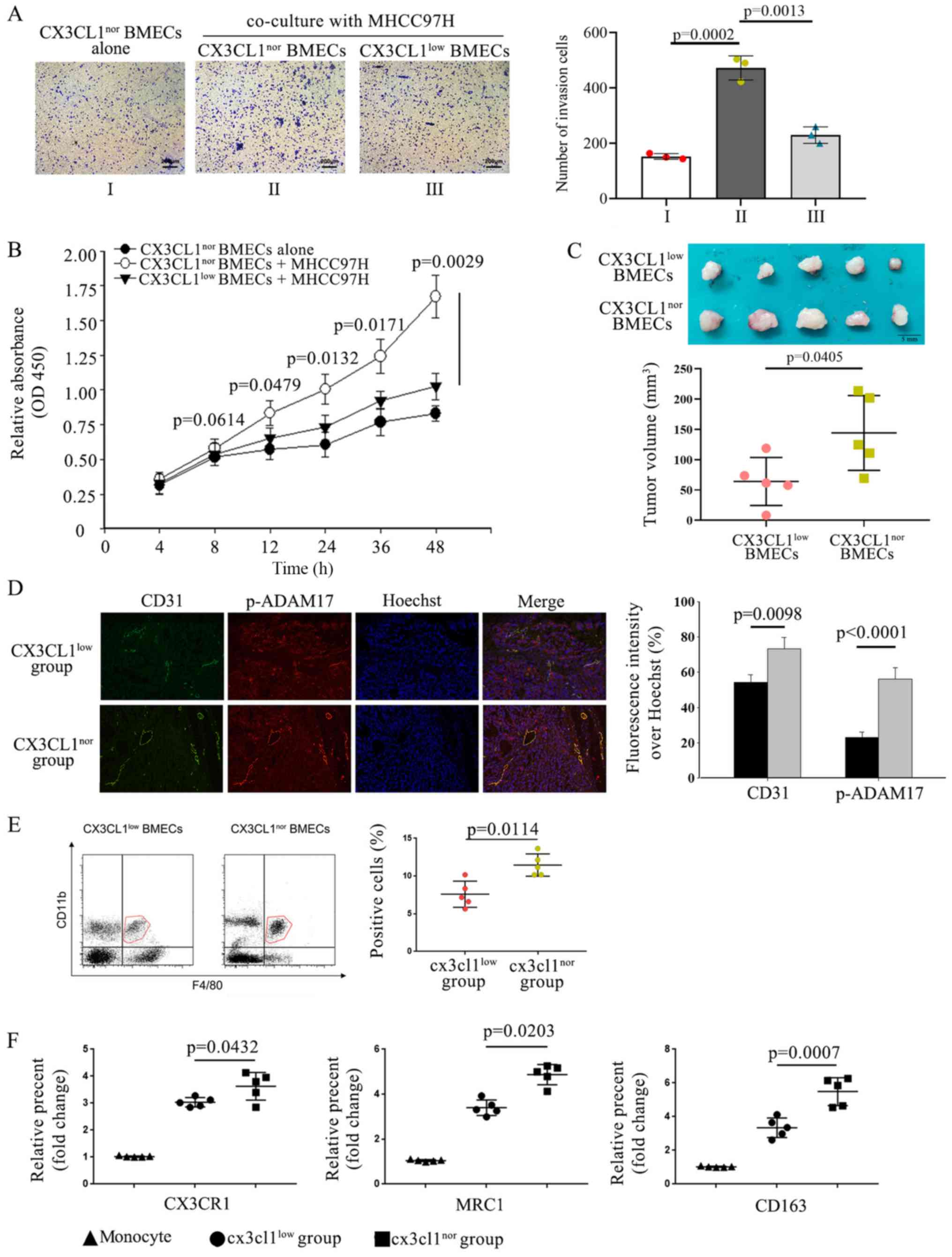 | Figure 7Interaction between BMECs and HCC
cells enhances tumor growth in the spine. (A) si-CX3CL1 was
transfected into BMECs to construct CX3CL1low BMECs.
Transwell assays were used to examine recruitment of BMECs
co-cultured with or without HMCC97H cells. The number of the
recruited cells was quantified. n=3. Cell recruitment were analyzed
using ANOVA test. (B) Cell vitality of BMECs was assessed using
CCK-8 assay; n=3. Data were analyzed using ANOVA followed by
Tukey's test. (C) Tumor size was measured based on the volume. n=5.
Scale bar, 5 mm. Data were analyzed using an unpaired t-test. (D)
Samples from 2 groups were examined using immunofluorescence.
Magnification, ×20. The result was analyzed using an unpaired
t-test. Gray bars represent the CX3CL1nor BMECs group
and black bars represent the CX3CL1low BMECs group (E)
Flow cytometry revealed the percentage and number of
F4/80+CD11b+ cells; n=5. (F) Flow cytometry
was used for detecting expression of M2 macrophage surface markers,
CX3CR1, MRC1 and CD163; n=5. Cells were selected based on higher
expression of CD45 for macrophages. Subsequently, CD11b and F4/80
expression were used for further characterization of macrophages.
FACS (fluorescent-activated cell sorting) data were analyzed using
a Kruskal Wallis test followed by Dunn's non-parametric post hoc
test. HCC, hepatocellular carcinoma; BMECs, bone marrow endothelial
cells; si, small interfering; CCK-8, Cell Counting kit-8; CX3CL1,
C-X3-C motif chemokine ligand 1. |
To verify the effects of BMECs on the metastasis and
growth of HCC cells in the spine, HCC cells were injected into the
spines of mice in situ. BMECs infected with CX3CL1-silencing
lentivirus were CX3CL1nor BMEC group while BMECs with
control lentivirus were CX3CL1nor BMEC group (Fig. S1D). In the in vivo
experiments, tumor cells mixed with CX3CL1nor BMECs
exhibited significantly larger tumors compared with those of mice
injected with tumor cells mixed with CX3CL1low BMECs
(Fig. 7C). Immunofluorescence
revealed that expression of the endothelial cell marker CD31 was
higher in the CX3CL1nor BMEC group compared with the
CX3CL1low BMEC group (Fig.
7D). The levels of activated p-ADAM17 in endothelial cells
mixed with CX3CL1low BMECs were lower compared with
those in the control group (Fig.
7D). Flow cytometry was applied to detect the macrophage
population. Additionally, it was also observed that the percentage
and number of F4/80+CD11b+ macrophages were
upregulated in the CX3CL1nor BMEC group compared with
the CX3CL1low BMECs group (Fig. 7E). Flow cytometry showed that the
M2 macrophage marker MRC in F4/80+CD11b+
cells were increased in the CX3CL1nor BMEC group
compared with the CX3CL1low BMEC group (Fig. 7F). Taken together, the results
demonstrate that BMECs are involved in the formation of a bone
metastatic microenvironment, which then supports and promotes the
growth of HCC cells.
Discussion
Although the communication between tumor cells and
endothelial cells has been widely studied during the metastatic
process of a variety of cancer types, few studies have focused on
the effect of BMECs in spinal metastasis. The present study
demonstrated that BMECs secrete CX3CL1 to enhance HCC metastasis.
CX3CL1 was regulated by ADAM17 in BMECs and promoted HCC spinal
metastasis via Src/PTK2 activation. The MHCC97H cell line was used
to establish an in vitro using a nude mice model of high
metastasis of HCC. Compared with MHCC97H cells, the metastasis of
Hep3B cells is lower (49). These
two types of cells can be used to compare the differential effects
of CX3CL1 on cells with different metastatic ability.
Increasing evidence has indicated that endothelial
cells play a crucial role in regulating metastatic tumor cells
(8). Endothelial cells display
numerous aggressive invasive behaviors, such as vessel formation,
the secretion of tumor-promoting cytokines and the modulation of
the metastatic niche (50). BMECs
play a critical role in maintaining and driving the homeostasis of
hematopoiesis under normal physiological conditions (13), and alter their cytoskeleton in
order to regulate and promote the adhesion of hematopoietic stem
cells and to construct the hematopoietic microenvironment.
Additionally, BMECs are a major source of secreted cytokines in the
bone marrow. However, the functions and mechanisms of BMECs in
spinal metastasis have not been thoroughly investigated.
Recently, increasing evidence has indicated that
CX3CL1 plays an important role in regulating cell invasion,
proliferation and migration of inflammatory cells and various
cancer cell lines (51). For
example, the expression of CX3CL1 is associated with the metastatic
rate and a poor prognosis of patients with prostate cancer
(19). A previous study by the
authors demonstrated that CX3CL1 expression was upregulated in
spinal cancellous bone (27).
Therefore, the present study aimed to examine the effects and
regulatory mechanisms of CX3CL1 in spinal metastasis. The
expression levels of CX3CL1 in HCC spinal metastases were
upregulated, and the in vitro experiments confirmed these
results. Cell invasion and migration experiments demonstrated that
the aggressive nature of cancer cells was decreased by the
administration of a CX3CL1 neutralizing antibody. The CX3CL1
neutralizing antibody also downregulated the activation of Src/PTK2
in HCC cells. Src promoted tumor progression, the activation of
which is increased in metastatic cells compared with non-metastatic
cells (52). Therefore, Src
activation is considered a biological marker of tumor progression
(53). PP1, a Src family
inhibitor, can inhibit Src activity and block downstream PTK2
signaling (54). PTK2 is a
tyrosine-phosphorylated protein that influences cell invasion and
migration (55). The
phosphorylation of PTK2 regulates its localization at focal
adhesion sites located on the plasma membrane (56). Recent studies have suggested that
PTK2 is closely associated with cancer survival, metastasis and
growth (57,58). In several cell lines, PTK2 is often
associated with integrin-related signaling at focal adhesion sites
which mediate cellular migration and adhesion (59). Rho family GTPases are known
regulators of cell adhesion and the inhibition of PTK2 destabilizes
RHOA activity (60,61). PTK2 is also a core modulator of the
PIK3CA/AKT1 signaling pathway, primarily through the formation of
complexes with PIK3CA (62).
PIK3CA/AKT1 signaling is increased in several types of cancer,
including HCC, and promotes cancer survival and metastasis
(63). In the present study, NEK2
expression was shown to be increased in cells treated with CX3CL1.
NEK2 expression has been reported to be correlated with that of
β-catenin in cancer (64). Taken
together, these results suggest that Src/PTK2 signaling may form an
essential component of the mechanisms underlying spinal metastasis
in HCC.
Once metastatic cells reach the distant metastasis
site, they alter the proliferation, migration and gene expression
profiles of normal endothelial cells (65). Thus, whether there were any
alterations to gene expression in BMECs, was assessed and it was
shown that CX3CL1 was highly expressed in the cancellous bone of
the spine. CX3CL1 differs from other chemokines as CX3CL1 has two
functional types; a membrane-bound form and a soluble form. This
dual identity of CX3CL1, both a chemokine and a receptor, allows it
to function as both a chemoattractant and an adhesion molecule. The
shedding of the CX3CL1 ectodomain is the process in which it is
cleaved at the juxtamembrane region by metalloproteases, leading to
the detachment of the extracellular region (66). This shedding process releases
soluble CX3CL1 from its membrane-bound form (67). One of the primary enzymes
responsible for converting CX3CL1 from a membrane-bound form to a
soluble form is the proteolytically active ADAM17 (68). In the present study, ELISA analyses
revealed the content of soluble form CX3CL1 in the medium was
significantly reduced in BMECs transfected with ADAM17-targeting
siRNA. Therefore, BMECs and HCC cells interact to enhance tumor
formation in the spinal metastatic environment.
Finally, it was found that CX3CL1 promoted HCC
spinal metastasis in vivo as well. Alternatively, studies
have indicated that CX3CL1 treatment may improve HCC outcomes in
mice by inducing antitumor immunity (69). Thus, it was hypothesized that the
secreted form and membrane-bound form of CX3CL1 may exert different
effects in vivo. Furthermore, the effect of CX3CL1, varying
from tumor suppression to tumor enhancement, is dependent on the
target tissue and the experimental model. TAMs secrete several
types of inflammatory cytokines in the tumor environment (70). Activated macrophages release
several molecules that may directly or indirectly affect the tumor
cell behavior. Certain studies have shown that TAMs are important
in the development of HCC (71).
TAMs predominantly differentiate into the M2 phenotype, which is
beneficial to both tumor formation and growth (72). In the present study, it was found
that TAMs that were stimulated by CX3CL1 became polarized towards
an M2 phenotype, thereby enhancing the invasive nature of HCC.
In conclusion, the present study demonstrated that
CX3CL1 secreted from BMECs played an important role in enhancing
spinal metastasis from HCC. BMECs promoted HCC invasion and
migration by producing CX3CL1. ADAM17 was activated by MAPK and
regulated the secretion of CX3CL1. These findings may partly
explain the mechanisms underlying HCC spinal metastasis, and
highlight potential novel targets for the treatment of HCC spinal
metastasis.
Supplementary Data
Funding
The present study was supported by the Natural
Science Foundation of China (grant nos. 81772855 and 81572629) and
the Science and Technology Commission of Shanghai Municipality
(17411950302).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
JD was involved in the conception and design of the
study, and in the preparation of the manuscript. CS, AH and SW were
involved in conducting the experiments, and in the collection and
supervision of the data. YL and HW analyzed and examined the data.
BT and LJ were involved in the drafting of the manuscript and in
collecting and pre-treating the clinical specimens. All authors
have read and approved the manuscript.
Ethics approval and consent to
participate
All patients provided informed consent and agreed to
participate in the study. The present study was approved by the
Ethics Committee of Zhongshan Hospital, Fudan University (approval
no. Y2014-185 and Y2019-085). All animal experiments were performed
in compliance with the Guidelines for the Care and Use of Research
Animals established by the Animal Ethics Committee of Zhongshan
Hospital, Fudan University. The animal studies were approved by the
Animal Ethics Committee of Zhongshan Hospital, Fudan
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Cabibbo G, Enea M, Attanasio M, Bruix J,
Craxì A and Cammà C: A meta-analysis of survival rates of untreated
patients in randomized clinical trials of hepatocellular carcinoma.
Hepatology. 51:1274–1283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Makuuchi M and Sano K: Surgical treatment
of HCC: Update topics. Updates Surg. 63:67–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Santini D, Pantano F, Riccardi F, Di
Costanzo GG, Addeo R, Guida FM, Ceruso MS, Barni S, Bertocchi P,
Marinelli S, et al: Natural history of malignant bone disease in
hepatocellular carcinoma: Final results of a multicenter bone
metastasis survey. PLoS One. 9:e1052682014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi C and Seong J: Predictive factors of
palliative radiotherapy response and survival in patients with
spinal metastases from hepatocellular carcinoma. Gut Liver.
9:94–102. 2015. View Article : Google Scholar :
|
|
5
|
Kim SU, Kim DY, Park JY, Ahn SH, Nah HJ,
Chon CY and Han KH: Hepatocellular carcinoma presenting with bone
metastasis: Clinical characteristics and prognostic factors. J
Cancer Res Clin Oncol. 134:1377–1384. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hang LH, Li SN, Luo H, Shu WW, Mao ZM,
Chen YF, Shi LL and Shao DH: Connexin 43 mediates CXCL12 production
from spinal dorsal horn to maintain bone cancer pain in rats.
Neurochem Res. 41:1200–1208. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Obenauf AC and Massagué J: Surviving at a
distance: Organ-specific metastasis. Trends Cancer. 1:76–91. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esposito M and Kang Y: Targeting
tumor-stromal interactions in bone metastasis. Pharmacol Ther.
141:222–233. 2014. View Article : Google Scholar :
|
|
9
|
Yousefi M, Nosrati R, Salmaninejad A,
Dehghani S, Shahryari A and Saberi A: Organ-specific metastasis of
breast cancer: Molecular and cellular mechanisms underlying lung
metastasis. Cell Oncol (Dordr). 41:123–140. 2018. View Article : Google Scholar
|
|
10
|
Hida K, Maishi N, Sakurai Y, Hida Y and
Harashima H: Heterogeneity of tumor endothelial cells and drug
delivery. Adv Drug Deliv Rev. 99:140–147. 2016. View Article : Google Scholar
|
|
11
|
Colmone A and Sipkins DA: Beyond
angiogenesis: The role of endothelium in the bone marrow vascular
niche. Transl Res. 151:1–9. 2008. View Article : Google Scholar
|
|
12
|
Singh S, Singh R, Sharma PK, Singh UP, Rai
SN, Chung LW, Cooper CR, Novakovic KR, Grizzle WE and Lillard JW
Jr: Serum CXCL13 positively correlates with prostatic disease,
prostate-specific antigen and mediates prostate cancer cell
invasion, integrin clustering and cell adhesion. Cancer Lett.
283:29–35. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kopp HG, Avecilla ST, Hooper AT and Rafii
S: The bone marrow vascular niche: Home of HSC differentiation and
mobilization. Physiology (Bethesda). 20:349–356. 2005.
|
|
14
|
Li WM, Huang WQ, Huang YH, Jiang DZ and
Wang QR: Positive and negative hematopoietic cytokines produced by
bone marrow endothelial cells. Cytokine. 12:1017–1023. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bade-Döding C, Göttmann W, Baigger A,
Farren M, Lee KP, Blasczyk R and Huyton T: Autocrine GM-CSF
transcription in the leukemic progenitor cell line KG1a is mediated
by the transcription factor ETS1 and is negatively regulated
through SECTM1 mediated ligation of CD7. Biochim Biophys Acta.
1840:1004–1013. 2014. View Article : Google Scholar
|
|
16
|
Lavi N, Kessler O, Ziv K, Nir-Zvi I,
Mumblat Y, Eiza N, Paran Y, Brenner B, Vadasz Z and Neufeld G:
Semaphorin-3A inhibits multiple myeloma progression in a mouse
model. Carcinogenesis. 39:1283–1291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lamanuzzi A, Saltarella I, Ferrucci A, Ria
R, Ruggieri S, Racanelli V, Rao L, Annese T, Nico B, Vacca A, et
al: Role of erythropoietin in the angiogenic activity of bone
marrow endothelial cells of MGUS and multiple myeloma patients.
Oncotarget. 7:14510–14521. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Griffith JW, Sokol CL and Luster AD:
Chemokines and chemokine receptors: Positioning cells for host
defense and immunity. Annu Rev Immunol. 32:659–702. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shulby SA, Dolloff NG, Stearns ME, Meucci
O and Fatatis A: CX3CR1-fractalkine expression regulates cellular
mechanisms involved in adhesion, migration, and survival of human
prostate cancer cells. Cancer Res. 64:4693–4698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wong HS, Jaumouillé V, Heit B, Doodnauth
SA, Patel S, Huang YW, Grinstein S and Robinson LA: Cytoskeletal
confinement of CX3CL1 limits its susceptibility to proteolytic
cleavage by ADAM10. Mol Biol Cell. 25:3884–3899. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang J, Xiao L, Cui R, Li D, Zheng X, Zhu
L, Sun H, Pan Y, Du Y and Yu X: CX3CL1 increases invasiveness and
metastasis by promoting epithelial-to-mesenchymal transition
through the TACE/TGF-α/EGFR pathway in hypoxic androgen-independent
prostate cancer cells. Oncol Rep. 35:1153–1162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei LM, Cao S, Yu WD, Liu YL and Wang JT:
Overexpression of CX3CR1 is associated with cellular metastasis,
proliferation and survival in gastric cancer. Oncol Rep.
33:615–624. 2015. View Article : Google Scholar
|
|
23
|
Tardáguila M, Mira E, García-Cabezas MA,
Feijoo AM, Quintela-Fandino M, Azcoitia I, Lira SA and Mañes S:
CX3CL1 promotes breast cancer via transactivation of the EGF
pathway. Cancer Res. 73:4461–4473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yao X, Qi L, Chen X, Du J, Zhang Z and Liu
S: Expression of CX3CR1 associates with cellular migration,
metastasis, and prognosis in human clear cell renal cell carcinoma.
Urol Oncol. 32:162–170. 2014. View Article : Google Scholar
|
|
25
|
Zheng J, Yang M, Shao J, Miao Y, Han J and
Du J: Chemokine receptor CX3CR1 contributes to macrophage survival
in tumor metastasis. Mol Cancer. 12:1412013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang Y, Yi L, Liu P, Jiang L, Wang H, Hu
A, Sun C and Dong J: CX3CL1 involves in breast cancer metastasizing
to the spine via the Src/FAK signaling pathway. J Cancer.
9:3603–3612. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu P, Liang Y, Jiang L, Wang H, Wang S
and Dong J: CX3CL1/fractalkine enhances prostate cancer spinal
metastasis by activating the Src/FAK pathway. Int J Oncol.
53:1544–1556. 2018.PubMed/NCBI
|
|
28
|
Wang R, Yu Z, Chen F, Xu H, Shen S, Chen
W, Chen L, Su Q, Zhang L, Bi J, et al: miR-300 regulates the
epithelial-mesenchymal transition and invasion of hepatocellular
carcinoma by targeting the FAK/PI3K/AKT signaling pathway. Biomed
Pharmacother. 103:1632–1642. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen JS, Huang XH, Wang Q, Chen XL, Fu XH,
Tan HX, Zhang LJ, Li W and Bi J: FAK is involved in invasion and
metastasis of hepatocellular carcinoma. Clin Exp Metastasis.
27:71–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li D, Zhang Y, Zhang H, Zhan C, Li X, Ba
T, Qiu Z, E F, Lv G, Zou C, et al: CADM2, as a new target of
miR-10b, promotes tumor metastasis through FAK/AKT pathway in
hepatocellular carcinoma. J Exp Clin Cancer Res. 37:462018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang YL, Xing X, Cai LB, Zhu L, Yang XM,
Wang YH, Yang Q, Nie HZ, Zhang ZG, Li J, et al: Integrin α9
suppresses hepatocellular carcinoma metastasis by Rho GTPase
signaling. J Immunol Res. 2018:46025702018. View Article : Google Scholar
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Naito S, von Eschenbach AC, Giavazzi R and
Fidler IJ: Growth and metastasis of tumor cells isolated from a
human renal cell carcinoma implanted into different organs of nude
mice. Cancer Res. 46:4109–4115. 1986.PubMed/NCBI
|
|
34
|
Beyer H: The Wilcoxon, Mann and Whitney
U-test - a distribution-independent statistical procedure for the
comparison of 2 independent random samples. Z Arztl Fortbild
(Jena). 82:871–873. 1988.In German.
|
|
35
|
Stout MC, Narayan S, Pillet ES, Salvino JM
and Campbell PM: Inhibition of CX3CR1 reduces cell motility and
viability in pancreatic adenocarcinoma epithelial cells. Biochem
Biophys Res Commun. 495:2264–2269. 2018. View Article : Google Scholar
|
|
36
|
Ishida Y, Kimura A, Nosaka M, Kuninaka Y,
Hemmi H, Sasaki I, Kaisho T, Mukaida N and Kondo T: Essential
involvement of the CX3CL1-CX3CR1 axis in bleomycin-induced
pulmonary fibrosis via regulation of fibrocyte and M2 macrophage
migration. Sci Rep. 7:168332017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jamieson-Gladney WL, Zhang Y, Fong AM,
Meucci O and Fatatis A: The chemokine receptor CX(3)CR1 is directly
involved in the arrest of breast cancer cells to the skeleton.
Breast Cancer Res. 13:R912011. View Article : Google Scholar
|
|
38
|
Longo V, Brunetti O, D'Oronzo S, Ostuni C,
Gatti P and Silvestris F: Bone metastases in hepatocellular
carcinoma: An emerging issue. Cancer Metastasis Rev. 33:333–342.
2014. View Article : Google Scholar
|
|
39
|
Pascual J and Turner NC: Targeting the
PI3-kinase pathway in triple-negative breast cancer. Ann Oncol.
30:1051–1060. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang GX, Wang Y, Su J, Zhou P, Li B, Yin
LJ and Lu J: Up-regulation of Rho-associated kinase 1/2 by
glucocorticoids promotes migration, invasion and metastasis of
melanoma. Cancer Lett. 410:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song LJ, Liu Q, Meng XR, Li ShL, Wang LX,
Fan QX and Xuan XY: DLC-1 is an independent prognostic marker and
potential therapeutic target in hepatocellular cancer. Diagn
Pathol. 11:192016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Samarin J, Laketa V, Malz M, Roessler S,
Stein I, Horwitz E, Singer S, Dimou E, Cigliano A, Bissinger M, et
al: PI3K/AKT/mTOR-dependent stabilization of oncogenic far-upstream
element binding proteins in hepatocellular carcinoma cells.
Hepatology. 63:813–826. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cheng Y, Chen X, Ye L, Zhang Y, Liang J,
Liu W, Zhou B, Zheng S, Huang Y, Chen G, et al: The prognostic
significance of NEK2 in hepatocellular carcinoma: Evidence from a
meta-analysis and retrospective cohort study. Cell Physiol Biochem.
51:2746–2759. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Wang W, Wang Y, Huang X, Zhang Z,
Chen B, Xie W, Li S, Shen S and Peng B: NEK2 promotes
hepatocellular carcinoma migration and invasion through modulation
of the epithelial-mesenchymal transition. Oncol Rep. 39:1023–1033.
2018.PubMed/NCBI
|
|
45
|
De Sanctis F, Ugel S, Facciponte J and
Facciabene A: The dark side of tumor-associated endothelial cells.
Semin Immunol. 35:35–47. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bergmeier W, Piffath CL, Cheng G, Dole VS,
Zhang Y, von Andrian UH and Wagner DD: Tumor necrosis
factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding
from platelets in vitro and in vivo. Circ Res. 95:677–683. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Saad MI, Alhayyani S, McLeod L, Yu L,
Alanazi M, Deswaerte V, Tang K, Jarde T, Smith JA, Prodanovic Z, et
al: ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK
axis in KRAS-addicted lung cancer. EMBO Mol Med. 11:112019.
View Article : Google Scholar
|
|
48
|
Zhu H, Shao Q, Sun X, Deng Z, Yuan X, Zhou
X and Ding Y: Bone marrow cells: Important role on
neovascularization of hepatocellular carcinoma. J Gastroenterol
Hepatol. 27:1241–1251. 2012. View Article : Google Scholar
|
|
49
|
Pan Q, Wang L, Sun HC, Liu YK, Ye SL and
Tang ZY: Transcription factor activity profile of human
hepatocellular carcinoma cell lines with different metastatic
potentials. Zhonghua Gan Zang Bing Za Zhi. 14:37–40. 2006.In
Chinese. PubMed/NCBI
|
|
50
|
Kusumbe AP, Ramasamy SK and Adams RH:
Coupling of angiogenesis and osteogenesis by a specific vessel
subtype in bone. Nature. 507:323–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu W, Jiang L, Bian C, Liang Y, Xing R,
Yishakea M and Dong J: Role of CX3CL1 in diseases. Arch Immunol
Ther Exp (Warsz). 64:371–383. 2016. View Article : Google Scholar
|
|
52
|
Guarino M: Src signaling in cancer
invasion. J Cell Physiol. 223:14–26. 2010.PubMed/NCBI
|
|
53
|
Aligayer H, Boyd DD, Heiss MM, Abdalla EK,
Curley SA and Gallick GE: Activation of Src kinase in primary
colorectal carcinoma: An indicator of poor clinical prognosis.
Cancer. 94:344–351. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Van Slambrouck S, Grijelmo C, De Wever O,
Bruyneel E, Emami S, Gespach C and Steelant WF: Activation of the
FAK-src molecular scaffolds and p130Cas-JNK signaling cascades by
alpha1-integrins during colon cancer cell invasion. Int J Oncol.
31:1501–1508. 2007.PubMed/NCBI
|
|
55
|
Yoon H, Dehart JP, Murphy JM and Lim ST:
Understanding the roles of FAK in cancer: Inhibitors, genetic
models, and new insights. J Histochem Cytochem. 63:114–128. 2015.
View Article : Google Scholar :
|
|
56
|
Hu YL, Lu S, Szeto KW, Sun J, Wang Y,
Lasheras JC and Chien S: FAK and paxillin dynamics at focal
adhesions in the protrusions of migrating cells. Sci Rep.
4:60242014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sulzmaier FJ, Jean C and Schlaepfer DD:
FAK in cancer: Mechanistic findings and clinical applications. Nat
Rev Cancer. 14:598–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jeong KY: Inhibiting focal adhesion
kinase: A potential target for enhancing therapeutic efficacy in
colorectal cancer therapy. World J Gastrointest Oncol. 10:290–292.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Golubovskaya VM: Focal adhesion kinase as
a cancer therapy target. Anticancer Agents Med Chem. 10:735–741.
2010. View Article : Google Scholar
|
|
60
|
Schmidt TT, Tauseef M, Yue L, Bonini MG,
Gothert J, Shen TL, Guan JL, Predescu S, Sadikot R and Mehta D:
Conditional deletion of FAK in mice endothelium disrupts lung
vascular barrier function due to destabilization of RhoA and Rac1
activities. Am J Physiol Lung Cell Mol Physiol. 305:L291–L300.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Luo J, Yao JF, Deng XF, Zheng XD, Jia M,
Wang YQ, Huang Y and Zhu JH: 14, 15-EET induces breast cancer cell
EMT and cisplatin resistance by up-regulating integrin αvβ3 and
activating FAK/PI3K/AKT signaling. J Exp Clin Cancer Res.
37:232018. View Article : Google Scholar
|
|
63
|
Zheng H, Yang Y, Hong YG, Wang MC, Yuan
SX, Wang ZG, Bi FR, Hao LQ, Yan HL and Zhou WP: Tropomodulin 3
modulates EGFR-PI3K-AKT signaling to drive hepatocellular carcinoma
metastasis. Mol Carcinog. 58:1897–1907. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Neal CP, Fry AM, Moreman C, McGregor A,
Garcea G, Berry DP and Manson MM: Overexpression of the Nek2 kinase
in colorectal cancer correlates with beta-catenin relocalization
and shortened cancer-specific survival. J Surg Oncol. 110:828–838.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Akino T, Hida K, Hida Y, Tsuchiya K,
Freedman D, Muraki C, Ohga N, Matsuda K, Akiyama K, Harabayashi T,
et al: Cytogenetic abnormalities of tumor-associated endothelial
cells in human malignant tumors. Am J Pathol. 175:2657–2667. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Montes de Oca-B P: Ectdomain shedding and
regulated intracellular proteolysis in the central nervous system.
Cent Nerv Syst Agents Med Chem. 10:337–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dreymueller D, Martin C, Kogel T,
Pruessmeyer J, Hess FM, Horiuchi K, Uhlig S and Ludwig A: Lung
endothelial ADAM17 regulates the acute inflammatory response to
lipopolysaccharide. EMBO Mol Med. 4:412–423. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tsou CL, Haskell CA and Charo IF: Tumor
necrosis factor-alpha-converting enzyme mediates the inducible
cleavage of fractalkine. J Biol Chem. 276:44622–44626. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tang L, Hu HD, Hu P, Lan YH, Peng ML, Chen
M and Ren H: Gene therapy with CX3CL1/Fractalkine induces antitumor
immunity to regress effectively mouse hepatocellular carcinoma.
Gene Ther. 14:1226–1234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rades D, Stalpers LJ, Hulshof MC, Borgmann
K, Karstens JH, Koning CC and Alberti W: Comparison of 1×8 Gy and
10×3 Gy for functional outcome in patients with metastatic spinal
cord compression. Int J Radiat Oncol Biol Phys. 62:514–518. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li Z, Wu T, Zheng B and Chen L:
Individualized precision treatment: Targeting TAM in HCC. Cancer
Lett. 458:86–91. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Atanasov G, Dino K, Schierle K, Dietel C,
Aust G, Pratschke J, Seehofer D, Schmelzle M and Hau HM:
Immunologic cellular characteristics of the tumour microenvironment
of hepatocellular carcinoma drive patient outcomes. World J Surg
Oncol. 17:972019. View Article : Google Scholar : PubMed/NCBI
|