Introduction
Acute myeloid leukaemia (AML) has the highest
mortality rate compared to the other major leukaemia types and
accounts for 64% of deaths caused by leukaemia (1), due in part to its rapid progression.
Mixed lineage leukaemia (MLL), a genetically distinct form of AML,
is considered particularly aggressive with sufferers having poorer
prognoses and earlier relapse-onset (2,3).
Although not as common as other types of cancer (4) relapsed AML cells may acquire
additional mutations that render them refractory (5,6). In
the case of such aggressive relapsed leukaemia, it is desirable
that alternative treatment should be obtained which can kill these
cells. The current standard chemotherapy treatment regime for AML
includes 7 days of cytarabine infusion combined with anthracycline
drugs, such as doxorubicin or daunorubicin for the first 3
days.
Apoptosis (also known as programmed cell death I) is
one of the main targets for the induction of cancer cell death with
chemotherapeutic drugs. However, cancer cells have the potential to
develop a mechanism with which to counteract apoptotic cell death,
which contributes to their chemoresistance (7,8). It
has been suggested that some cancer cells develop chemoresistance
through the modulation of autophagy. Autophagy is a cellular
conservative process, using a catabolic 'self-cannibalising'
mechanism, to either enhance cell survival or promote cell death
(9,10). Initially, the activation of
autophagy was considered to be an adaptive response of malignant
cells in cancer therapy, to develop resistance and promote survival
(14). However, the overactivation
of autophagy may lead to non-apoptotic cell death, known as
programmed cell death II (10).
Thus, under different conditions, autophagy acts as a guardian or
executioner of tumour cells, via the promotion of chemoresistance
and chemosensitivity, respectively (9,10).
The link between autophagy and other cell death types has been
elucidated (11,12) and in certain settings, autophagy
inhibits apoptosis (13),
facilitates/cooperates with apoptosis (14), or acts as an alternative cell death
when apoptosis is blocked (9).
There is reportedly an interplay between Bcl-2 (an
anti-apoptotic protein) and Beclin 1 (an autophagy marker), which
can determine whether cells undergo apoptosis and/or autophagy
(15-17). Blocking the Bcl-2/Beclin 1 complex
at the endoplasmic reticulum level via the downregulation of Bcl-2
leads to the upregulation of Beclin 1 with the activation of
autophagy (17). Thus, Bcl-2 is
involved in the regulation of both apoptosis and autophagy. The
deregulation of the Bcl-2 protein in leukaemia may be one of the
contributing factors of drug resistance against apoptotic cell
death. Therefore, compounds targeting the Bcl-2 protein family may
increase patients' response to chemotherapy. In addition, bypassing
apoptotic death by inducing other alternative cell death, such as
autophagy, may prove to be a valuable mechanism in fighting
refractory cancer cells.
The present study aimed to determine aspects of
selectively induced cell death in AML by doxorubicin (Dox) through
the modulation of apoptosis and autophagy. To the best of our
knowledge, the present study reports for the first time that a
p15-20-Bcl-2 protein is expressed in the AML MOLM-13 cell line in
addition to the normal p26-Bcl-2-α protein, but is absent in the
leukaemic cell lines, OCI-AML2, U-937 and CML K562, which only
express p26-Bcl-2-α. The level of this novel p15-20-Bcl-2 isoform,
but not the p26-Bcl-2-α level was inversely proportional to the
concentration of Dox during the drug treatment. It was reported
that Dox was found to exhibit a level of selectivity in inducing
apoptotic cell death in the MOLM-13 and that this effect may be in
part through the selective inhibition of the novel
p15-20-Bcl-2.
Materials and methods
Cells and cell culture
SC (ATCC CRL-9855) cells were purchased from the
American Type Culture Collection and maintained in Iscove's
modified Dulbecco's medium (IMDM, Sigma-Aldrich; Merck KGaA). SC
cells were originally monocytes from human peripheral blood, but
have since been cross-contaminated by U-937 cells (ATCC
CRL-1593.2). U-937 cells are derived from histiocytic lymphoma;
leukaemia of the mononuclear phagocyte system consisting of
mono-cyte/macrophage cells, which are myeloid in nature (18). Therefore, this cell line is
referred to as the U-937 cell line in the present study.
Authenticated leukaemic MOLM-13, OCI-AML2 and K562 cell lines
(purchased from the European Collection of Cell Cultures, Public
Health England) were maintained in Roswell Park Memorial Institute
(RPMI)-1640 medium (Sigma-Aldrich; Merck KGaA). The MOLM-13 cell
line is an immortalised AML cell line derived from MLL-rearranged
cells from a relapsed patient with FLT3 mutation, which has evolved
from myelodysplastic syndrome (19). This cell line allowed for the study
of relapsed AML. OCI-AML2 is a cell line established from a patient
acute myelomonocytic leukaemia, which carries DNMT3A R635W
mutation. K562 was established from a patient in the blast crisis
stage of chronic myelogenous leukaemia and positive for
Philadelphia chromosome. Each type of complete culture medium was
supplemented with 1% L-glutamine, 1% penicillin-streptomycin
antibiotic and 10% fetal bovine serum (FBS, Sigma-Aldrich; Merck
KGaA). All cell lines were tested regularly for mycoplasma using
polymerase chain reaction. Cells were cultured and grown in a
humidified incubator at 5% CO2 environment and 37°C. The
leukaemic cell medium was changed every 48-72 h through cell
pelleting by centrifugation (at room temperature at 260 × g for 5
min) followed by removing the old medium and replenishing with a
fresh complete medium. Only cells in logarithmic growth were used
in experiments. Trypan blue HyClone® dye assay was used
to determine cell viability and cell density prior to proceeding
with other assays. The cells were stained at a 1:1 ratio with the
dye and manually counted using a haemocytometer under light
microscopy. Cell viability of at least 97% was accepted as
appropriate for experiments.
Cell death population assays using Alexa
Fluor® 488 Annexin V and PI: Flow cytometry
Cells (1.0×106 cells/ml) were treated
with Dox (final concentration, 0.5, 1 and 5 µM dissolved in
0.05% DMSO) for 48 h. Cells treated with 0.05% DMSO only were used
as the vehicle control. Following the incubation period, cells were
harvested, washed in cold PBS and then stained with Alexa
Fluor® 488 Annexin V (Thermo Fisher Scientific, Inc.)
for 10 min followed by PI for a further 10 min. They were analysed
using a FACSCalibur flow cytometer (BD CellQuest™ Pro P software,
BD Biosciences) at a fluorescent emission at 525 nm (FL1; Alexa
Fluor®) and 575 nm (FL3; PI) using 488 nm excitation
wavelength. Alexa Fluor® uses Annexin V and propidium
iodide (PI) to differentially stain and estimate cell population of
viable and dead (necrotic and apoptotic) cells.
Determination of cell proliferation using
5(6)-carboxyfluorescein diacetate
succinimidyl ester (CFSE)
Labelling cells with CFSE fluorescent
dye
MOLM-13 cells were labelled with a cell membrane
permeable fluorescent dye, CFSE prior to cell treatment. The cells
were harvested, washed and re-suspended in phosphate buffer saline
(PBS). A stock solution of CFSE (2 µM in DMSO) was freshly
diluted 100-fold in PBS for cell labelling. The cells were
incubated in the dark with CFSE at room temperature for 20 min with
shaking at 5 min intervals. The reaction was terminated by the
addition of complete RPMI medium followed by pelleting of the
cells. The cells were then re-suspended in fresh complete RPMI
medium and incubated in the dark at 37°C for 10 min before being
harvested and re-suspended at 1.0×106 cells/ml with
fresh complete RPMI medium. Cells not labelled with CFSE and not
stained with PI were used as a blank control.
Cell treatments and PI staining
CFSE-labelled MOLM-13 cells (at 1.0×106
cells/ml; 950 µl) were treated with Dox (final conc. 0.5 and
1 µM, 50 µl) in a 24-well plate. The treatments were
incubated for 24, 48 and 72 h in 5% CO2 at 37°C.
Immediately before harvesting, 50 µl fixed MOLM-13 cells
were added to the CFSE-labelled cells and data obtained from the
fixed cells were used for normalisation. For fixation, exponential
growing unlabelled cells were harvested and washed in PBS and fixed
in 0.4% paraformaldehyde (0.5 ml) for 10 min, washed and then
re-suspended in PBS. Fixed cells (50 µl; equivalent to
1.0×105 cells/ml) were added into each cell treatment.
The treated cells containing fixed cells were then harvested,
washed in flow buffer [PBS, 2% FBS, 2 mM ethylenediaminetetraacetic
acid (EDTA), 0.1% sodium azide], pelleted and stained with 0.5
µg/ml PI for 10 min in the dark at room temperature.
Finally, flow buffer was added and the samples were analysed using
FACSCalibur flow cytometry (BD CellQuest™ Pro software; BD
Biosciences). Fluorescent detection was made at an emission
wavelength of 480 nm (FL1 for CFSE) and 630 nm (FL3 for PI) using
488 nm excitation wavelength. Fixed cells stained only with PI were
used as control.
Investigation of proteins involved in
cell death: Western blot analysis SDS-PAGE electrophoresis and
membrane transfer
Protein concentrations in RIPA whole cell lysates
from 48 h treatments with Halt Protease Inhibitor Cocktail (100X)
(Thermo Fisher Scientific, Inc.) were quantified by Bradford assay.
The lysates containing sample buffer (2X Laemmli loading buffer and
a reducing agent, 5% β-mercaptoethanol) were heated at 95°C for 5
min. The reduced lysates (30 µg) were separated by sodium
dodecyl sulfate polyacrylamide electrophoresis (SDS-PAGE) using
Mini-PROTEAN® TGX™ (4-15%) Precast gels (Bio-Rad
Laboratories, Inc.) for 30-40 min at 100 volts. This was followed
by protein transfer onto a nitrocellulose membrane by
Trans-Blot® Turbo™ Transfer System (Bio-Rad
Laboratories, Inc.).
Immunoblotting and visualization
The transferred blots were blocked with 5% BSA in
0.01% Tween-20 in PBS (PBS-T) with rocking for 1 h at room
temperature and then washed 5 times in PBS-T. The blots were
incubated with rocking overnight at 4°C with the following primary
rabbit monoclonal antibodies in PBS-T with 5% BSA: Anti-Bcl-2
(1:1,000; cat. no. ab32124), anti-Bax (1:1,000; cat. no. ab32503),
or anti-Beclin 1 (1:2,000; cat. no. ab207612) (all from Abcam).
Following the incubation period, the blots were washed 5 times in
PBS-T and then probed with goat anti-rabbit IgG (H+L) horseradish
peroxidase secondary antibody in PBS-T with 5% BSA (1:3,000; cat.
no. 170-6515; Bio-Rad Laboratories, Inc.) for 1 h at room
temperature and then washed 5 times in PBS-T. β-actin was used as a
housekeeping protein control and detected using anti-β-actin mouse
monoclonal (1:5,000 in 5% BSA; cat. no. ab8226; Abcam) followed by
goat anti-mouse IgG (H+L) horseradish peroxidase-labelled secondary
antibody (1:3,000 in 5% BSA; cat. no. 170-6516; Bio-Rad
Laboratories, Inc.). The blots were then incubated with ECL
(Pierce™ ECL Western Blotting Substrate) and the proteins
visualised using ODYSSEY® FC imager (Li-Cor,
Bioscience). A semi-quantitative comparison of protein levels was
determined by densitometry using Image Studio™ Lite software.
Statistical analysis
Statistical analyses were performed using
Minitab18® statistical software. Assumptions of
homogeneity of variance and underlying normality of distributions
(all with 95% coefficient interval) were tested using standard
equal variance testing and the Anderson-Darling normality test as
appropriate. A two-sample t-test was used to compare the means of
two populations. One-way ANOVA was used to compare multiple means.
Tukey (honest significant difference) post-hoc tests were then
applied. Statistical significance was accepted with P-value ≤0.05.
The IC50 values of the drug were calculated by using
GraphPad Prism 6 software, based on the mean ± SE cell
viability.
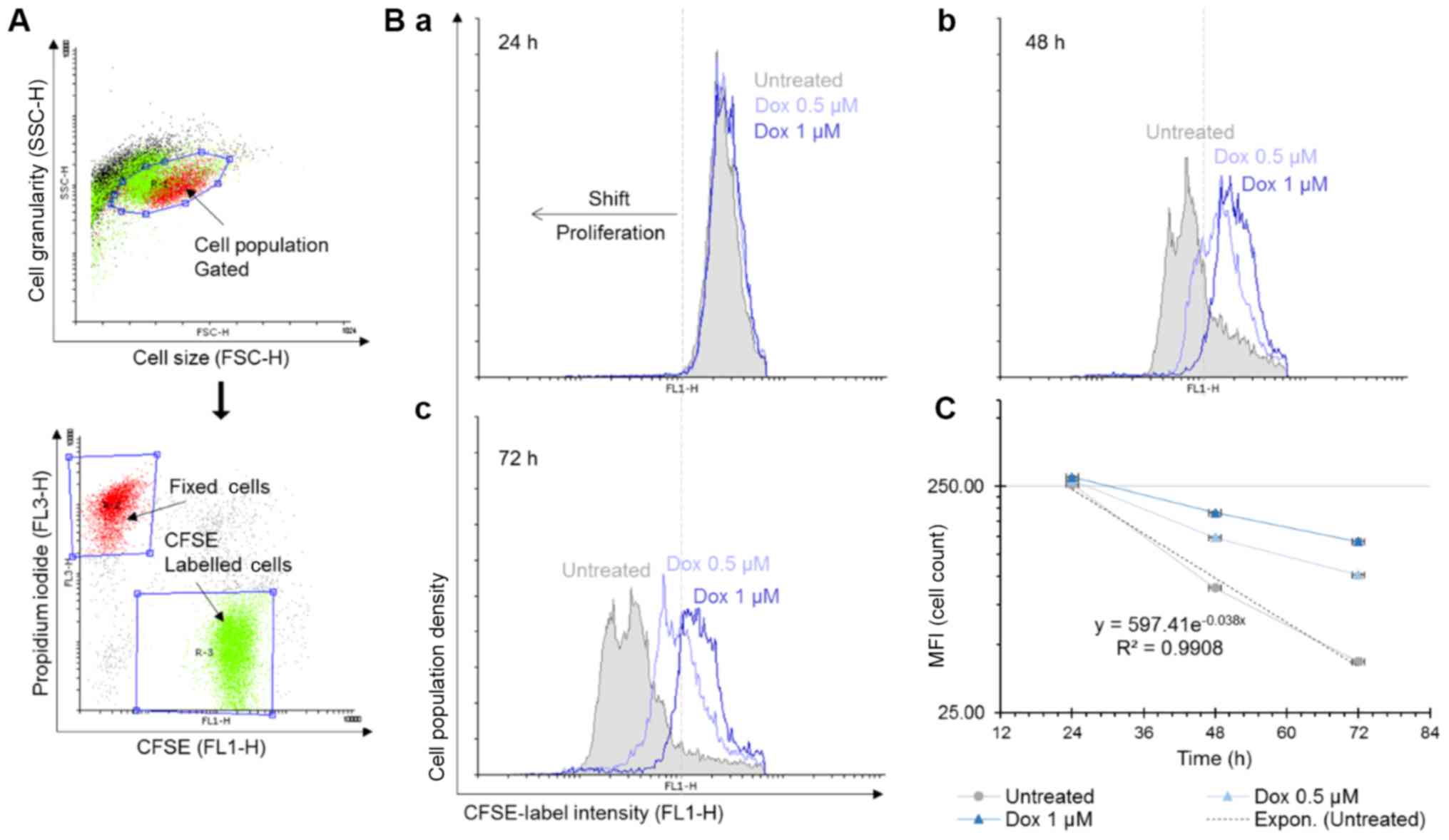
Data acquired from flow cytometric analyses of CFSE
cell proliferation assays were analysed using Flowing Software
(free flow cytometry data analysis software created by Perttu
Terho, version 2.5.1). Cell population events were gated
distinctively and back-gated to compensate for colour of Dox and
auto-fluorescence. Distribution (cell population, y-axis) of the
mean fluorescent intensity (MFI) shift within CFSE labelled cells
(FL1-H, x-axis) was visualised using histograms. Since CFSE binds
irreversibly to the mammalian cells it distributes 50% to each
daughter cell every time a cell divides. MFI is therefore directly
related to cell number. Thus, the cell population can be expressed
as (Eqn. 1):
MFI(t) = MFI(0) exp(αt)
where MFI(t) represents the measure of MFI at time t, MFI(0) is the
fluorescence at the time of labelling (t=0) and α is the growth
rate when the cell population grows exponentially.
Results
Doxorubicin exhibits a level of
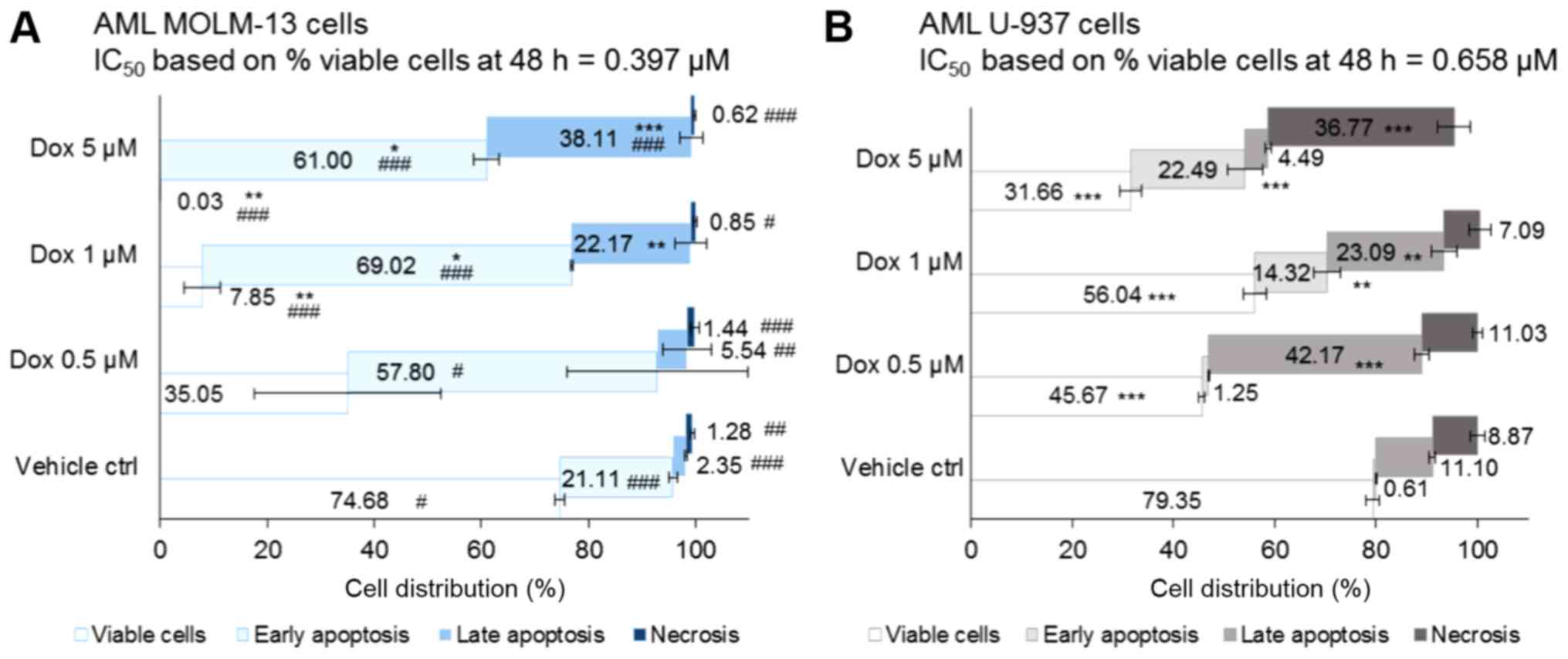
selectivity in the induction of leukaemic cell death
The Dox-induced death of leukaemic monocytic cells
was examined by flow cytometry. Apoptotic and necrotic cells were
differentiated by the double-staining of MOLM-13 and U-937 cells
with Annexin V and PI at following 48 h co-incubation of the cells
with or without Dox (Fig. 1). The
effect of therapeutically relevant concentrations of Dox (0.5 and 1
µM) was examined against the vehicle control (DMSO 0.05%). A
supraclinical dose of 5 µM of Dox was used as a positive
control.
Following 48 h co-incubation of cells with the
treatments, the vehicle control treated-cells exhibited comparable
cell death, with 25 and 21% dead MOLM-13 and U-937 cells,
respectively (with no statistically significant differences). In
the treated MOLM-13 cells, including the vehicle control, the cell
death populations resided more in early apoptosis (P=0.03 for Dox
0.5 µM and P<0.001 for all other treatments compared to
the U-937 cells) (Fig. 1A). By
contrast, compared to the MOLM-13 cells, dead cells were
predominantly found in late apoptosis in the U-937 cells
(P<0.01) apart from the cells treated with 1 µM Dox,
which exhibited a non-significant effect compared to the MOLM-13
cells, but was shown to be highly significant (P<0.01) compared
to the vehicle control (Fig.
1B).
Cell death by necrosis was <2% in all of the
treated MOLM-13 cells, whereas it ranged from 7-9% (in the 1
µM Dox- and vehicle control-treated cells) to 37% (in the 5
µM Dox-treated cells) in the U-937 cells (all P<0.05
compared to the MOLM-13 cells). Thus, Dox induced MOLM-13 cell
death in a dose-dependent manner through mainly early apoptotic
death with minimal necrosis. However, it is noteworthy that the
different concentrations of Dox induced the death of the U-937
cells by apparently different mechanisms. Dox 5 µM induced
death mainly by necrosis. A lower concentration of Dox, 0.5
µM, induced greater late apoptotic cell death and was more
potent than treatment with 1 µM Dox; the reason for this is
currently uncertain. At therapeutically relevant concentrations,
the MOLM-13 cells treated with Dox at 0.5 and 1 µM exhibited
53% (P<0.05) and 89% (P<0.001) dead cells, respectively,
compared to the vehicle control (Fig.
1). Comparatively, the U-937 monocytic cells exhibited fewer
dead cells with a 1.3-fold decrease in the 1 µM-treated
cells (P<0.001 compared to the MOLM-13 cells). The
IC50 value of Dox following 48 h of co-incubation with
MOLM-13 cells (based on the percentage of viable cell population)
was lower compared to that for the U-937 monocytic cells (Fig. 1).
Doxorubicin decreases the proliferative
rate of MOLM-13 cell lines
To further investigate the molecular mechanisms of
Dox-induced cell death, a single-cell assay based on CFSE
tracker-labelled MOLM-13 cells was performed. CSFE binds covalently
and irreversibly to free amino groups on proteins within cells and
the fluorescence intensity of the dye halves with each cell
division. Combined with live-dead staining, this allows not only to
determine whether the drug affects the rate of cell division but
also how the cells are dying. Fixed cells were positive for PI
staining and were used to normalise the data (Fig. 2A). The number of MOLM-13 cells in
the untreated cell samples doubled every 18 h (Fig. 2C). Following the 24-h incubation
period, the control (untreated cells) and the Dox-treated samples
all exhibited similar mean fluorescence intensity (MFI) values. The
changes in the MFI at 48 and 72 h demonstrated that the rate of
MOLM-13 cell proliferation was affected by Dox in a time- and
concentration-dependent manner (Fig.
2B-b and -c). In the untreated cells, there was a shift to
lower MFI values between the 24- and 72-h incubation time due to
halving of the amount of the CFSE stain during each cell division
cycle.
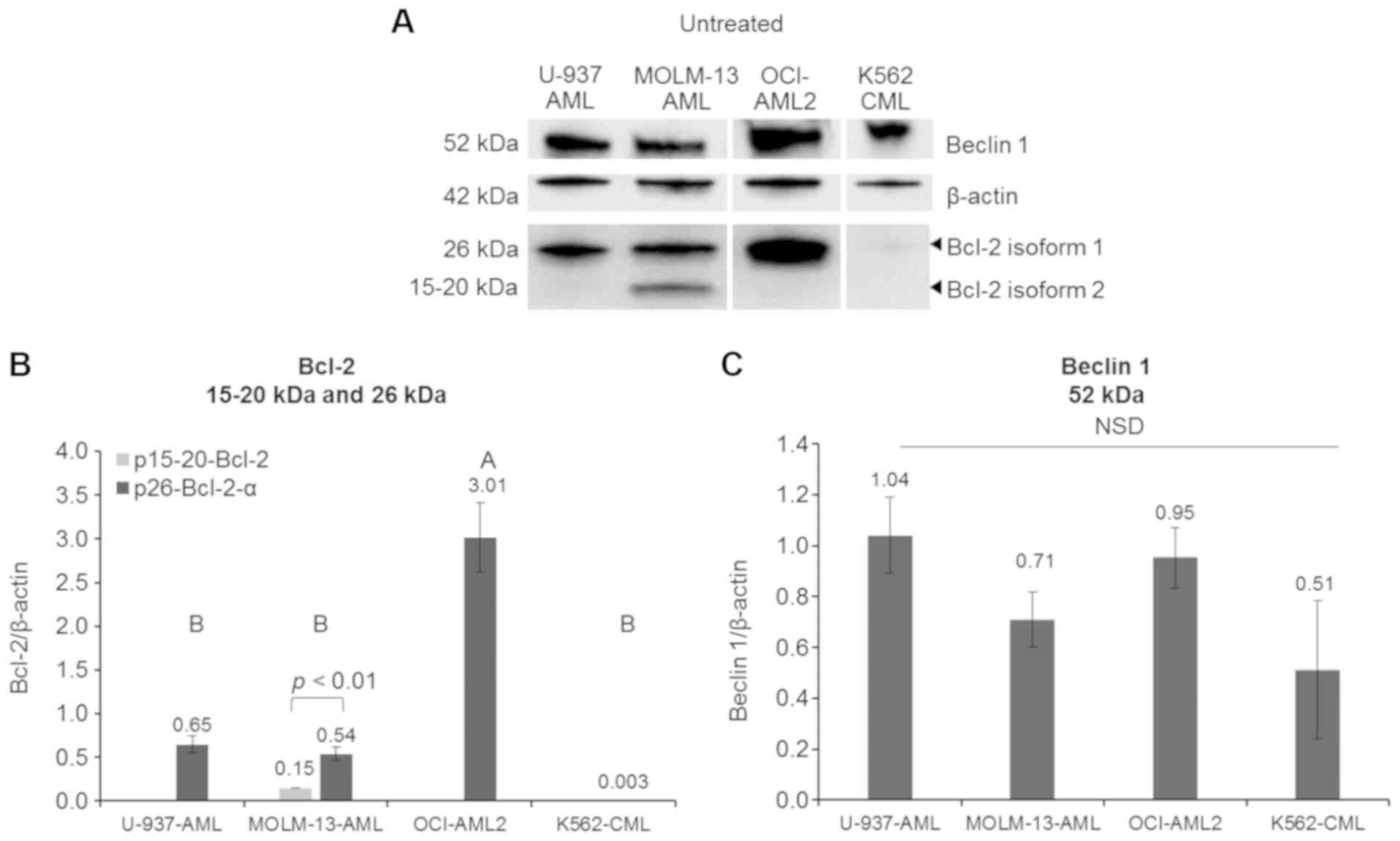
Doxorubicin selectively inhibits Beclin 1
and a novel Bcl-2 isoform exclusively expressed in the AML MOLM-13
cell line
To determine the molecular mechanisms of Dox-induced
cyto-toxicity in MOLM-13 cells, Bcl-2 and Beclin 1 protein levels
were quantified by western blot analysis. Initial experiments on
the expression of Bcl-2 on MOLM-13 revealed two isoforms of the
protein, estimated from the molecular weight of the proteins from
the ladder control, to be 26 kDa (p26-Bcl-2-α) and 15-20 kDa
(p15-20-Bcl-2). As p26-Bcl-2-α is the functional Bcl-2 protein
known to have anti-apoptotic effect and p15-20-Bcl-2 appear to be a
novel isoform in MOLM-13, further experiments were conducted to
determine if they were both present in other leukaemic cell lines
containing different mutations. Unstressed AML cell lines (MOLM-13,
OCI-AML2 and U-937 cells), as well as CML K562, all exhibited basal
protein expression levels of the usually reported isoform of Bcl-2,
p26-Bcl-2-α (Fig. 3). However, the
expression of the protein was minimal in CML K562 cells. The
OCI-AML2 cells exhibited the highest expression (P<0.001) of
p26-Bcl-2-α when compared to the other cells. Of the cells tested,
only the MOLM-13 cells contained the p15-20-Bcl-2 protein, but at
lower levels compared to the p26-Bcl-2-α protein (P<0.01). Bcl-2
is an anti-apoptotic protein involved in cell death and interacts
with an autophagy pathway through regulating Beclin 1 (16). The differences in the basal
expression levels of Beclin 1 protein were not statistically
significant (P>0.05) between the MOLM-13, OCI-AML2, CML K562 and
U-937 cells.
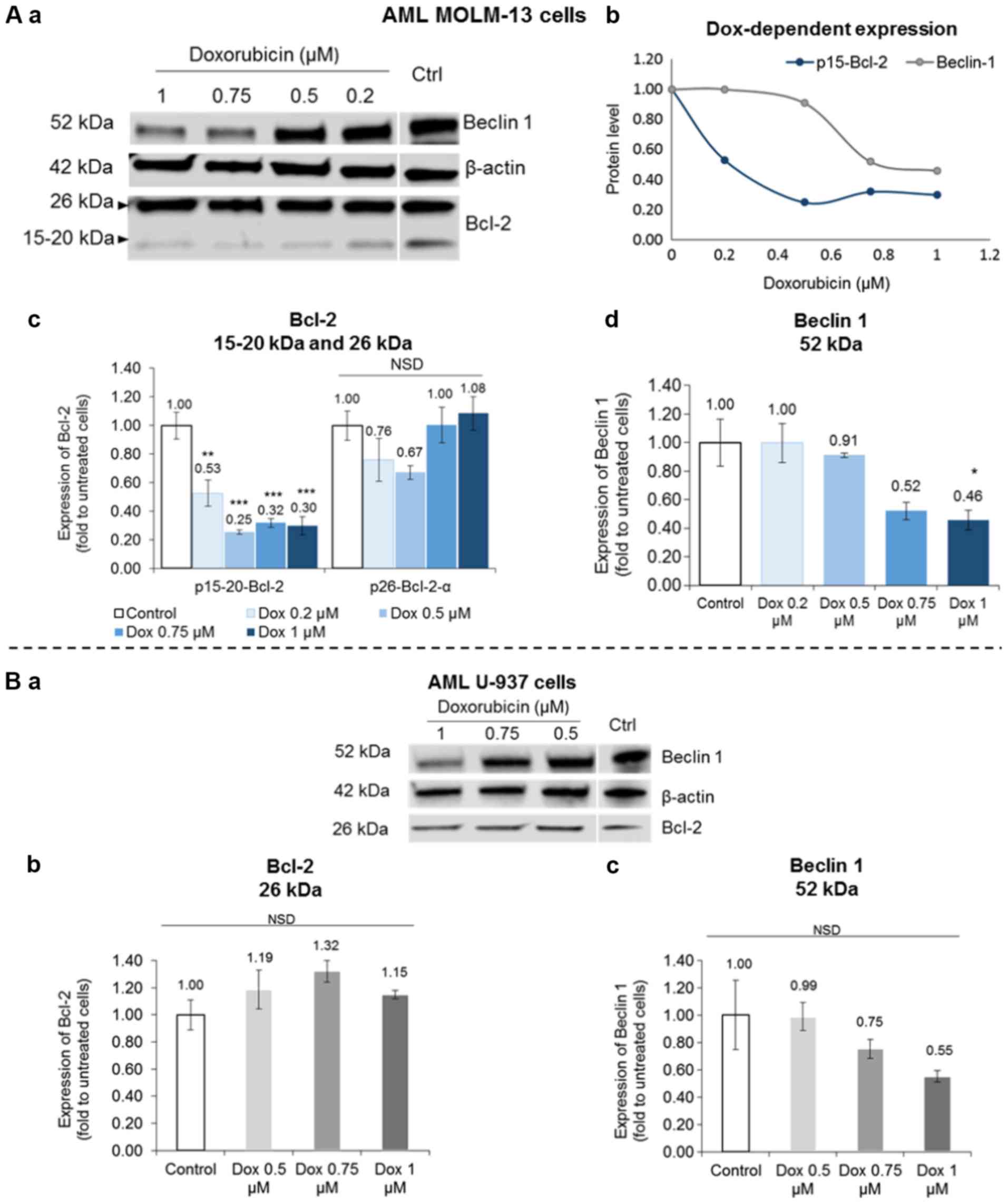
Further experiments were conducted to determine if
the p15-20-Bcl-2 isoform could be selectively targeted by drug
therapy to induce cell death by comparing the effects of Dox on
MOLM-13 and the U-937 cells. The expression of this novel
p15-20-Bcl-2 protein was selectively inhibited in the Dox-treated
MOLM-13 cells without any appreciable drug-specific effect on the
expression of the p26-Bcl-2-α isoform in either MOLM-13 or U-937
cells (Fig. 4A-c and B-b). The
p15-20-Bcl-2 protein levels in the MOLM-13 cells were significantly
reduced by Dox (P<0.05) compared to the untreated control cells.
Conversely, Dox inhibited the autophagic Beclin 1 protein levels by
almost half at the higher concentrations tested with statistical
significance shown for the effect of the 1 µM concentration
(P<0.05) (Fig. 4A-d). However,
a non-significant (P>0.05) dose-dependent reduction trend of
Beclin 1 was observed in the treated U-937 cells (Fig. 4B-c).
Discussion
The deregulation of the Bcl-2 proteins in leukaemia
may be one of the contributing factors of drug resistance against
apoptotic cell death. Treatment of haematological malignancies have
been shown to benefit from BH3-mimetics, which are compounds that
promote the release of pro-apoptotic proteins from anti-apoptotic
Bcl-2 effects (20). This leads to
the induction of cell apoptosis. For instance, BH3-mimetic drugs
such as venetoclax (ABT-199) enhance the effectiveness of available
therapies for AML by inhibiting the overexpression of Bcl-2 seen in
refractory leukaemia (21). Beclin
1 (an autophagy protein), is also a BH3-containing protein and,
similar to some pro-apoptotic Bcl-2 members, it binds to the
hydrophobic groove of Bcl-2, making this interaction one of the
regulatory mechanisms of autophagy (15). In the present study, Dox was
evaluated on its potential to induce selective apoptotic cell death
in AML MOLM-13 cells and to modulate autophagy through Bcl-2 and
Beclin 1 protein expression levels.
Doxorubicin inhibits cell proliferation
and exhibits a level of selective cell-death induction in MOLM-13
cells
The concentration of Dox in a patient's plasma
within approximately 2 h of intravenous administration of the drug
is 0.1-1 µM (22). The
documented effect of Dox as an anti-proliferative agent (23) was confirmed in the present study at
0.5 and 1 µM. However, Dox may not have a major effect on
MOLM-13 during the first 24 h. Dox-treated MOLM-13 population
decreased (relative to drug-free cell population) through a
drug-induced reduction in the rate of proliferation in a time- and
concentration-dependent manner (Fig.
2). It was also reported that Dox induced more cell death in
the MOLM-13 than in U-937 cells (P<0.05; Fig. 1) indicating a certain level of
selectivity of Dox toxicity on MOLM-13 cells, and this may be due
to cell-dependent action of Dox.
In the literature, Dox has been reported to
demonstrate a pleiotropic effect on cells ranging from affecting
cell proliferation by binding to topoisomerase II (24) to inducing apoptosis by activating
caspases (25-27) and disrupting mitochondrial membrane
potential (28). In the search for
the molecular mechanisms of Dox, further experiments were conducted
to investigate the effect of the drug on members of the Bcl-2
family, the anti-apoptotic Bcl-2 and Beclin 1, an initiator of
autophagy.
Doxorubicin inhibits the expression of a
novel Bcl-2 protein variant in MOLM-13 cells
The BCL-2 gene has been reported to be upregulated
in 84% of AML patients at diagnosis and the cases increase at
relapse to 95% (29). MOLM-13, the
cell line investigated in the present study, is derived from cells
of a patient with relapsed AML (19). It was found that a novel Bcl-2
isoform variant of 15-20 kDa (p15-20-Bcl-2) was expressed in
unstimulated leukaemic MOLM-13 cells (in addition to the usual 26
kDa Bcl-2 protein), but not in other AML and CML cells tested in
the present study (which only expressed p26-Bcl-2-α) (Fig. 3). Anti-apoptotic Bcl-2 protein
reduction by Dox has also been reported in HeLa cells (30) and breast cancer cells (31). In previous studies, the apoptosis
inhibitory properties of Bcl-2 have been credited to the main
p26-Bcl-2-α isoform, consisting of hydrophobic transmembrane region
that can regulate mitochondria permeability (32,33).
The phosphorylation status of Bcl-2 protein family
members defines the ability of the protein to regulate the
death/survival of cells. Depending on the site of phosphorylation,
the activation of the protein can be either enhanced or inhibited
and these post-translational modifications can be modified by
certain drugs. Alternative splicing variants of p22-Bcl-2 creates
distinct isoforms (32). Several
alternative protein variants or isoforms of the Bcl-2 family
members have been identified. However, the two main Bcl-2 proteins,
encoded by the BCL-2 gene, have been documented as being
p26-Bcl-2-α and p22-Bcl-2-β which differ mainly in their carboxyl
tails (33,34). Despite the gene expression of
Bcl-2-α remaining dominant, the Bcl-2-β isoform levels have been
found to be higher in chronic lymphocytic leukaemia patients
compared to healthy patients (35). Alternative spliced Bcl-2 proteins
have also been observed in other cancer cells. A study on thyroid
tumours identified a protein isoform of Bcl-2 at 21 kDa alongside
the standard Bcl-2 phenotype (36). Another truncated Bcl-2 isoform,
Bcl-2 Ψ (psi) was found in invasive prostate cancer. This Bcl-2 Ψ
lacked a BH-3 domain which prevents dimerization with pro-apoptotic
molecules (Bax and Bid) and thus protects cancer cells from cell
death (37). In the present study,
an isoform of Bcl-2 at 15-20 kDa was detected selectively in
MOLM-13 cells. Messingerova et al (2015) also reported a
Bcl-2 protein band at approximately 19 kDa in an AML
cross-resistance MOLM-13 cells (resistant to azacytidine), but the
26 kDa isoform was absent (38).
The authors of that study explained this finding as resulting from
a Bcl-2 protein shift. However, they did not report further on the
protein alteration linked to function. The present study reports a
Bcl-2 isoform similar in size as that reported by Messingerova
et al (2015) and demonstrates that the isoform is a
functional protein, which is selectively sensitive to Dox treatment
in MOLM-13 cells. It is our opinion that the proteomic diversity of
anti-apoptotic Bcl-2 in MOLM-13 cell lines may contribute to the
oncogenic behaviour of the cancer. Understanding the different
isoforms of Bcl-2, particularly those that are preferentially
expressed in cancer cells, may be useful for developing specific
drugs to target cells to induce cancer cell death.
Doxorubicin reduces Beclin 1, leading to
cell death
The present study reported that the protein
expression of Beclin 1 was reduced by Dox, but only at
concentrations >0.5 µM, with statistical significance for
1 µM (P<0.001) (Fig.
4A-d). It has been reported that depleting Beclin 1 by gene
silencing augments mitochondrial permeabilisation and enhances
Dox-induced apoptosis (39) and
treatment with pharmacological and genetic inhibitors of autophagy
augmented Dox-induced apoptotic cell death (40). In the present study, treatment with
Dox led to a greater number of apoptotic MOLM-13 compared to U-937
cells, with an inhibition of p15-20-Bcl-2 and a concurrent
inhibition of Bcl-2.
The interplay between Beclin 1 and Bcl-2 to regulate
apoptosis and autophagy has also been documented by other
researchers. The dissociation of this complex allows the activation
of Beclin 1 to bind and form a complex with various proteins in
initiating pre-autophaghosmal structure (17). Due to the ability of Bcl-2 protein
to form a complex with Beclin 1, it also performs a role in
regulating autophagy (16). Beclin
1 plays a crucial role in autophagy by forming vesicle nucleation
and promoting the isolation membrane development through a complex
of phosphatidyl inositol-3 kinase (PI3K) class III. Bcl-2 blocks
autophagy by preventing Beclin 1 from facilitating the formation of
the pro-autophagosome promoting complex. This leads to a decreased
PI3K activity of Beclin 1-associated binding partner (41). Beclin 1 is also regulated by other
autophagy proteins, such as Rubicon and Ambra 1 (9,16).
Similar to the findings in the present study, some researchers have
reported that the reduction in autophagic proteins causes an
increase in cell apoptosis (42).
By contrast, Smuder et al (2011) reported that Dox
treatments increased markers of autophagy, including Beclin 1 mRNA
and protein levels in muscle tissues, which may have contributed to
Dox-induced muscle toxicity (43).
In addition, Beclin 1 levels increased time-dependently in multiple
myeloma cell lines when treated by Dox (40). Therefore, increases in autophagy
proteins in some cells could be an adaptive response to
drug-induced stress for survival initiated by dying cells and
inhibition of these proteins result in death (40). Although the role of autophagy in
cancer is yet to be confirmed, there is a possibility of its
modulation and usefulness in cancer therapy.
The present study reports initial findings of a
larger project examining the interplay between autophagic and
apoptotic proteins and how they can be modulated by drug treatments
to induce selective cell death in cancer cells. In the present
study, the AML cell line, MOLM-13, expressed a Dox-regulated
p15-20-Bcl-2 isoform in addition to the usual p26-Bcl-2-α isoform
of which expression levels are unaffected. The induction of cell
death in MOLM-13 by Dox may also be due to its modulation of Beclin
1. Further studies are warranted to determine if p15-20-Bcl-2 can
be selectively targeted by drugs to induce cell death in MOLM-13
cells.
Studies are currently underway using apoptosis or
autophagy inhibitors for further verification of the association
between Dox-induced apoptosis and autophagy. Other studies include
the investigation of a wider panel of autophagic and apoptotic
proteins in different cell lines, as well as primary patient cells
and non-leukaemic cells to study the interplay between the two
pathways. The study of Bcl-2 in these cells is a matter of
priority. Recommended future work will also investigate Beclin
1/Bcl-2 complexes by immunoprecipitation with anti Beclin-1
followed by western blot analysis with anti-Bcl-2 to provide some
insight into the interactions of the two proteins. In addition,
other studies are warranted, including proteomic and genomic
studies to provide more accurate determination of the novel Bcl-2
variant in MOLM-13. Confirmation studies, such as sodium dodecyl
sulfate protein separation with Coomassie staining followed by
time-of-flight mass spectrometry could validate the distinct
isoform. Other studies may include immuno-precipitation followed by
proteo-lytic fragmentation and time-of-flight mass spectrometry to
identify deletion and altered splicing. Knockout experiments, as
well as, cloning the p15-20-Bcl-2 isoform, transfecting it into
suitable cell lines and purifying to analyse association with other
proteins using western blot analysis are also proposed to evaluate
the role of the protein. qPCR analysis with different primer sets
may be used to study the relative gene expression levels of BCL-2
in MOLM-13 and other cell lines.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MV and SA conceived, designed, performed the
experiments, analysed, interpreted and wrote the manuscript with
signifi-cant contributions from NK and CB. RO and TL were involved
in the design, acquisition, analysis, interpretation and revision
of the manuscript. All the authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Huw Jones and Dr
Steve Kett both of Middlesex University (UK), for proofreading the
manuscript.
References
|
1
|
Cancer Research UK: Leukaemia (all
subtypes combined) statistics. Cancer Research UK; Oxford: 2016,
https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/leukaemia.
|
|
2
|
Lun Y, Yang JJ and Wu Y: Complete
molecular remission in relapsed and refractory acute myeloid
leukaemia with MLL-AF9 treated with chidamide-based chemotherapy. J
Clin Pharm Ther. 42:786–789. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Daigle SR, Olhava EJ, Therkelsen CA, Majer
CR, Sneeringers CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao
Y, et al: Selective killing of mixed lineage leukemia cells by a
potent small-molecule DOT1L inhibitor. Cancer Cell. 20:53–65. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hassan C, Afshinnekoo E, Li S, Wu S and
Mason CE: Genetic and epigenetic heterogeneity and the impact on
cancer relapse. Exp Hematol. 54:26–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Madanat YF, Kalaycio ME and Nazha A:
Advances in acute myeloid leukemia genomics, where do we stand in
2018? Acta Med Acad. 48:35–44. 2019.PubMed/NCBI
|
|
6
|
Ding L, Ley TJ, Larson DE, Miller CA,
Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan
MD, et al: Clonal evolution in relapsed acute myeloid leukaemia
revealed by whole-genome sequencing. Nature. 481:506–510. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zahreddine H and Borden KLB: Mechanisms
and insights into drug resistance in cancer. Front Pharmacol.
4:282013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu X and Xuan Y: Bypassing cancer drug
resistance by activating multiple death pathways - a proposal from
the study of circumventing cancer drug resistance by induction of
necroptosis. Cancer Lett. 259:127–137. 2008. View Article : Google Scholar
|
|
9
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Radogna F, Dicato M and Diederich M:
Cancer-type-specific crosstalk between autophagy, necroptosis and
apoptosis as a pharmacological target. Biochem Pharmacol. 94:1–11.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goodall ML, Fitzwalter BE, Zahedi S, Wu M,
Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD and
Thorburn A: The autophagy machinery controls cell death switching
between apoptosis and necroptosis. Dev Cell. 37:337–349. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang J, Shao SH, Xu ZX, Hennessy B, Ding
Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al:
The energy sensing LKB1-AMPK pathway regulates p27(kip1)
phosphorylation mediating the decision to enter autophagy or
apoptosis. Nat Cell Biol. 9:218–224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar
|
|
15
|
Decuypere JP, Parys JB and Bultynck G:
Regulation of the autophagic bcl-2/beclin 1 interaction. Cells.
1:284–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marquez RT and Xu L: Bcl-2:Beclin 1
complex: multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
17
|
Lian J, Karnak D and Xu L: The
Bcl-2-Beclin 1 interaction in (-)-gossypol-induced autophagy versus
apoptosis in prostate cancer cells. Autophagy. 6:1201–1203. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Usmani S, Sivagnanalingam U, Tkachenko O,
Nunez L, Shand JC and Mullen CA: Support of acute lymphoblastic
leukemia cells by nonmalignant bone marrow stromal cells. Oncol
Lett. 17:5039–5049. 2019.PubMed/NCBI
|
|
19
|
Matsuo Y, MacLeod RA, Uphoff CC, Drexler
HG, Nishizaki C, Katayama Y, Kimura G, Fujii N, Omoto E, Harada M,
et al: Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13
and MOLM-14) with interclonal phenotypic heterogeneity showing
MLL-AF9 fusion resulting from an occult chromosome insertion,
ins(11;9)(q23;p22p23). Leukemia. 11:1469–1477. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gomez-Bougie P, Maïga S, Tessoulin B,
Bourcier J, Bonnet A, Rodriguez MS, Le Gouill S, Touzeau C, Moreau
P, Pellat-Deceunynck C, et al: BH3-mimetic toolkit guides the
respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment.
Blood. 132:2656–2669. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campos EdV: Pinto R: Targeted therapy with
a selective BCL-2 inhibitor in older patients with acute myeloid
leukemia. Hematology Transfus Cell Ther. 41:169–177. 2019.
View Article : Google Scholar
|
|
22
|
McHowat J, Swift LM, Arutunyan A and
Sarvazyan N: Clinical concentrations of doxorubicin inhibit
activity of myocardial membrane-associated, calcium-independent
phospholipase A(2). Cancer Res. 61:4024–4029. 2001.PubMed/NCBI
|
|
23
|
Rudolfová P, Hanušová V, Skálová L,
Bártíková H, Matoušková P and Boušová I: Effect of selected
catechins on doxorubicin anti-proliferative efficacy and
hepatotoxicity in vitro. Acta Pharm. 64:199–209. 2014. View Article : Google Scholar
|
|
24
|
Fornari FA, Randolph JK, Yalowich JC,
Ritke MK and Gewirtz DA: Interference by doxorubicin with DNA
unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 45:649–656.
1994.PubMed/NCBI
|
|
25
|
Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC,
Han S, Ganesan YT, Ishizawa K, Jebiwott S, Dong Y, Pietanza MC, et
al: Targeting the differential addiction to anti-apoptotic BCL-2
family for cancer therapy. Nat Commun. 8:160782017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Florou D, Patsis C, Ardavanis A and
Scorilas A: Effect of doxorubicin, oxaliplatin, and methotrexate
administration on the transcriptional activity of BCL-2 family gene
members in stomach cancer cells. Cancer Biol Ther. 14:587–596.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Panaretakis T, Pokrovskaja K, Shoshan MC
and Grandér D: Activation of Bak, Bax, and BH3-only proteins in the
apoptotic response to doxorubicin. J Biol Chem. 277:44317–44326.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gamen S, Anel A, Pérez-Galán P, Lasierra
P, Johnson D, Piñeiro A and Naval J: Doxorubicin treatment
activates a Z-VAD-sensitive caspase, which causes deltapsim loss,
caspase-9 activity, and apoptosis in Jurkat cells. Exp Cell Res.
258:223–235. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moon JH, Sohn SK, Lee MH, Jang JH, Kim K,
Jung CW and Kim DH: BCL2 gene polymorphism could predict the
treatment outcomes in acute myeloid leukemia patients. Leuk Res.
34:166–172. 2010. View Article : Google Scholar
|
|
30
|
Bien S, Rimmbach C, Neumann H, Niessen J,
Reimer E, Ritter CA, Rosskopf D, Cinatl J, Michaelis M, Schroeder
HW, et al: Doxorubicin-induced cell death requires cathepsin B in
HeLa cells. Biochem Pharmacol. 80:1466–1477. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pilco-Ferreto N and Calaf GM: Influence of
doxorubicin on apoptosis and oxidative stress in breast cancer cell
lines. Int J Oncol. 49:753–762. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akgul C, Moulding DA and Edwards SW:
Alternative splicing of Bcl-2-related genes: Functional
consequences and potential therapeutic applications. Cell Mol Life
Sci. 61:2189–2199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guillem V, Amat P, Collado M, Cervantes F,
Alvarez-Larrán A, Martínez J, Tormo E, Eroles P, Solano C and
Hernández-Boluda JC: BCL2 gene polymorphisms and splicing variants
in chronic myeloid leukemia. Leuk Res. 39:1278–1284. 2015.
View Article : Google Scholar
|
|
34
|
Tanaka S, Saito K and Reed JC:
Structure-function analysis of the Bcl-2 oncoprotein. Addition of a
heterologous transmembrane domain to portions of the Bcl-2 beta
protein restores function as a regulator of cell survival. J Biol
Chem. 268:10920–10926. 1993.PubMed/NCBI
|
|
35
|
Ghassemifar R, Forster L, Finlayson J,
Calogero T, Augustson B, Joske D and Cull G: Differential
expression of the Bcl-2 and Bax isoforms in CD19 positive
B-lymphocytes isolated from patients diagnosed with chronic
lymphocytic leukaemia. Pathology. 44:632–637. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Manetto V, Lorenzini R, Cordon-Cardo C,
Krajewski S, Rosai J, Reed JC and Eusebi V: Bcl-2 and Bax
expression in thyroid tumours. An immunohistochemical and western
blot analysis. Virchows Arch. 430:125–130. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang JM, Lin TY, Chang D, Lin SL and Ying
SY: Truncated Bcl-2, a potential pre-metastatic marker in prostate
cancer. Biochem Biophys Res Commun. 306:912–917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Messingerova L, Imrichova D, Kavcova H,
Turakova K, Breier A and Sulova Z: Acute myeloid leukemia cells
MOLM-13 and SKM-1 established for resistance by azacytidine are
crossresistant to P-glycoprotein substrates. Toxicol In Vitro.
29:1405–1415. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Daniel F, Legrand A, Pessayre D, Vadrot N,
Descatoire V and Bernuau D: Partial Beclin 1 silencing aggravates
doxorubicin- and Fas-induced apoptosis in HepG2 cells. World J
Gastroenterol. 12:2895–2900. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang
Y, Dong B and Chen X: Targeting autophagy augments in vitro and in
vivo anti-myeloma activity of DNA-damaging chemotherapy. Clin
Cancer Res. 17:3248–3258. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Boya P, González-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Smuder AJ, Kavazis AN, Min K and Powers
SK: Exercise protects against doxorubicin-induced markers of
autophagy signaling in skeletal muscle. J Appl Physiol (1985).
111:1190–1198. 2011. View Article : Google Scholar
|