Introduction
Activin A is a member of the transforming growth
factor-β superfamily, which elicits pleiotropic effects in various
biological systems, such as morphogenesis, organogenesis, growth,
differentiation and regulation of reproduction (1,2).
Activin A signaling involves binding to type II transmembrane
serine-threonine kinase receptors (ActRIIA or ActRIIB), which leads
to the recruitment, phosphorylation and activation of type I
activin receptors (ActRIA or ActRIB) (3,4). The
downstream signal transduction of activin A occurs via SMAD2/3,
which form a complex with SMAD4 and translocate to the nucleus
leading to transcription of target genes (4). The effects of activin A have also
been associated with non-canonical pathways, including AKT/PI3K,
MAPK/ERK and WNT/β-catenin pathways (4,5).
The effects of activin A are context-dependent and
its dysregulation is associated with the development and
progression of certain cancer types, including colorectal,
prostate, lung and breast cancer (6-9).
More than a basic marker of cancer progression, activin A also
appears to infuence survival by contributing to the development of
cachexia and loss of skeletal muscle mass (8,9). In
previous studies, it was demonstrated that both oral squamous cell
carcinoma (OSCC) cells and OSCC-associated fbroblasts express high
levels of activin A, which in an autocrine and paracrine manner,
respectively, regulate apoptosis, proliferation and invasiveness of
the tumor cells (10,11). Furthermore, overexpression of
activin A is clinically associated with lymph node metastasis,
tumor differentiation and poor survival of patients with OSCC
(11), and immunoexpression of
activin A is a useful predictor of occult lymph node metastasis in
patients with OSCC of the tongue (12). Tumor growth and metastasis depend
on angiogenesis triggered by secreted factors from tumor and cells
of the tumor microenvironment. The effects of activin A on
angiogenesis remain controversial, with studies describing
pro-angiogenic actions and associating its expression with poor
prognosis of several cancer types, such as liver and breast cancer
(13-15); however, other reports are
contradictory (16-18). Increased production of activin A by
hepatocellular and breast cancer cells that are no longer growth
inhibited by activin A leads to increased angiogenesis by inducing
vascular endothelial growth factor (VEGF) expression (14,15).
Nonetheless, to the best of our knowledge, whether activin A
regulates angiogenesis in oral cancer remains unknown.
The present study investigated the prognostic
significance of activin A immunoexpression in both blood vessels
and tumor cells in a number of OSCC cases, and evaluated the impact
of activin A on angiogenesis in vitro by assessing
endothelial cell proliferation, migration and tube formation.
Furthermore, to decipher its effects on angiogenesis, cDNA
generated from activin A-treated human umbilical vein endothelial
cells (HUVECs) and HUVECs stably expressing short hairpin RNA
(shRNA) targeting activin A were subjected to a human angiogenesis
quantitative PCR (qPCR) array, revealing enhanced amounts of total
VEGFA and of its pro-angiogenic isoform 121. Collectively, the
present study demonstrated that the assessment of activin A has a
reliable prognostic value in OSCC, which may be, in part, due to
the pro-angiogenic effects of activin A via stimulation of VEGFA
production.
Materials and methods
Immunohistochemistry
To investigate the expression of activin A in blood
vessels and in tumor cells, immunohistochemical analyses with
anti-activin A and anti-CD34 antibodies were performed with 95
cases of primary OSCC from The Oncology Center of Cascavel (Paraná,
Brazil) and The UOPECCAN Cancer Hospital (Paraná, Brazil), which
were previously described (19).
The samples were obtained between December 2012 and October 2013.
The age range of the patients was 31-84 years, with a median of 56
years. OSCC consecutive slices from paraffin-embedded samples were
subjected to immunostaining for activin A and CD34. Briefly, after
dewaxing in xylene for 30 min and hydration in graded alcohol
solutions (100, 90, 80, 70, 60 and 50%; 2 min/wash), the
3-µm sections were treated with 3% hydrogen peroxide
followed by antigen retrieval with 10 mM citrate buffer pH 6.0 in a
pressure cooker for 15 min. Following washing with PBS, the
sections were treated with 1% bovine serum albumin in PBS at room
temperature for 1 h and then incubated overnight at 4°C with
polyclonal rabbit activin A antibody (cat. no. HPA020031; 1:100;
Sigma Aldrich-Aldrich) or monoclonal mouse CD34 antibody (cat. no.
M7165; 1:400, clone QBEnd-10, Dako; Agilent Technologies, Inc.) at
room temperature for 1 h followed by LSAB kit (LSAB+ system-HRP
kit; Dako; Agilent Technologies, Inc.) or Advance HRP kit (Dako;
Agilent Technologies, Inc.), respectively. Reactions were developed
by incubating the sections with 0.6 mg/ml 3,3'-diami-nobenzidine
tetrahydrochloride (Dako; Agilent Technologies, Inc.) and
counterstained with hematoxylin at room temperature for 2 min.
Control reactions were performed by omission of the primary
antibodies.
Activin A immunoexpression was assessed in blood
vessels and tumor cells by two independent pathologists, who were
not aware of any clinical data. Activin A expression in blood
vessels was determined by counting of activin A-positive and
activin A-negative blood vessels in three magnifcation, x200 fields
per sample with a light microscope, and samples were categorized
into two groups based on median expression: Low (≤53% of activin
A-positive blood vessels) and high (>53% of activin A-positive
blood vessels). For tumor cells, the number of positive cells was
graded according to the following: 1, 1-25% staining; 2, 26-50%
staining; 3, 51-75% staining; and 4, 76-100% staining); and the
intensity of staining was scored as follows: 0, negative; 1, weak
staining; 2, moderate staining; and 3, strong staining. The two
grades were added together, producing scores from 0 to 7 that were
classified as low (scores 0-4) and high (scores 5-7) expression for
comparative analysis. Written informed consent was obtained from
each patient according to the declaration of Helsinki, and the
study was approved by the Human Research Ethics Committee of the
School of Dentistry, University of Campinas (approval no. 100/2012,
October 10, 2012).
Cell culture
HUVECs were obtained from American Type Culture
Collection and cultured as recommended in a 1:1 mixture of
Dulbecco's modified Eagle's medium and Ham's F12 medium (DMEM/F12;
Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; Cultilab), 400 ng/ml hydrocortisone
(Sigma-Aldrich; Merck KGaA) and antibiotic-antimycotic (penicillin,
streptomycin and amphotericin B solution; Thermo Fisher Scientific,
Inc.). The SCC-9 ZsGreen LN-1 cells, isolated from a meta-static
cervical lymph node (20), were
cultured in DMEM/F-12 (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS, 400 ng/ml hydrocortisone (Sigma-Aldrich;
Merck KGaA) and antibiotic-antimycotic. shINHBA OSCC cells (SCC-9
ZsGreen LN-1 cells constitutively expressing a shRNA sequence
against activin A) and the respective control (shControl OSCC
cells; cells stably transfected with the same vector encoding a
non-targeting scrambled shRNA sequence) were generated as
previously described (11). All
cells were cultured at 37°C in a humidifed atmosphere of 5%
CO2.
Treatments
Lyophilized recombinant activin A (Sigma-Aldrich;
Merck KGaA) and follistatin (R&D Systems, Inc.) were dissolved
in culture medium, aliquoted and stored at ‑80°C. To assess the
effect of activin A, cells were cultured in DMEM/F-12 containing 0
or 1 ng/ml of recombinant activin A for 24 h at 37°C. For
follistatin treatment, cells were cultured at 100 ng/ml for 24 h at
37°C. Concentrations of activin A and follistatin were selected
based on our previous study (11).
Activin A stable knockdown in HUVECs
HUVECs grown in 12-well plates at a confiuence of
50% were incubated with control shRNA lentiviral particles
(MISSION® pLKO.1-puro Non-Mammalian shRNA Control;
Sigma-Aldrich; Merck KGaA) or INHBA shRNA lentiviral particles
(INHBA MISSION® shRNA Lentiviral Transduction Particles,
NM_002192; 5'A-GACCCATGTCCATGTTGT-3'; Sigma-Aldrich; Merck KGaA) at
a multiplicity of infection of 1.5 in culture medium containing 8
µg/ml polybrene (Sigma-Aldrich; Merck KGaA) for 8 h. After washing
with PBS, cells were cultured in fresh medium for an additional
period of 48 h. Cells were then split in a 1:5 dilution, and
cultured for 10 days in the presence of 1 µg/ml puromycin
dihy-drochloride (Sigma-Aldrich; Merck KGaA) to select resistant
cells. The effcacy of activin A‑knockdown was determined by reverse
transcription-qPCR (RT-qPCR) and enzyme-linked immunosorbent assay
(ELISA).
RT- qPCR
Total RNA was extracted using the RNeasy mini kit
(Qiagen, Inc.), according to the manufacturer's protocol. Following
DNase I treatment in order to eliminate genomic DNA contamination,
1 µg total RNA per sample was used to generate cDNA using Oligo-dT
(Invitrogen; Thermo Fisher Scientifc, Inc.) and reverse
transcriptase (Superscript II RT enzyme; Invitrogen; Thermo Fisher
Scientifc, Inc.), according to the manufacturer's protocol. The
resulting cDNAs were subjected to qPCR using specifc primers and
SYBR Green PCR master mix (Applied Biosystems; Thermo Fisher
Scientifc, Inc.) in a Real Time PCR thermal cycler (Applied
Biosystems; Thermo Fisher Scientifc, Inc.). The thermocycling
conditions were: 95°C for 1 min followed by 40 cycles 95°C for 15
sec and 60°C for 30 sec. Gene expression was determined using the
2-ΔΔCq method (21) and
PPIA was used as reference gene for data normalization. All
reactions were performed in triplicate. Primer sequences are
provided in Table SI.
ELISA
HUVECs were plated at concentration of 25,000 cells
per well in a 24-well culture plate in triplicate and cultured for
24 h. After washing, the cells were cultured in serum-free medium
for an additional 24 h. The medium was then collected, centrifuged
to remove foating cells, and used for analysis. The concentration
of activin A was determined using a human activin A ELISA kit (cat.
no. RAB0324; Sigma-Aldrich; Merck KGaA), according to the
manufacturer's instructions.
Endothelial cell tube formation
assay
HUVECs (40,000 cells per well) were seeded into
wells of a 96-well plate coated with 50 µl Matrigel (BD
Biosciences). After 12 h of incubation, tube formation was observed
under an inverted microscope (magnifcation, x40; Nikon
Corporation), photographed and analyzed using Motic Images Plus 2.0
(Nikon Corporation), as previously described (22).
Proliferation analysis
HUVECs were plated in 96-well plates at a density of
10,000 cells per well in 100 µl medium containing 10% FBS. After 16
h, the cells were washed with PBS and cultured in serum-free medium
for an additional 24 h to reach cell cycle synchronism. Following
serum starvation, cells were treated for 24 h and proliferation
rates were determined by measuring bromodeoxyuridine (BrdU)
incorporation into DNA using a cell proliferation ELISA, BrdU
(colorimetric) kit (cat. no. 11647229001; Roche Applied Science;
Merck KGaA).
Migration analysis
Transwell migration assays were performed in 6.5-mm
inserts with 8-µm pore size (Corning, Inc.). Serum-starved cells
(80,000 cells/well) were plated into the upper chamber in 200 µl
serum-free DMEM/F12. As a chemoattractant in the lower chamber,
depending on the assay, serum-free medium containing activin A or
follistatin, and shINHBA OSCC- and shControl OSCC-conditioned
medium were tested. Activin A or follistatin were also added in the
upper chamber, in direct contact with the HUVEC cells, and in those
situations 10% FBS was used in the lower chamber as
chemoattractant. After 24 h, assessment of migration was performed
by gently removing cells in the interior part of the insert with a
cotton swab. Cells on the underside of the membrane were fxed in
10% formalin for 15 min and stained with 1% toluidine blue in 1%
borax solution, with both procedures at room temperature. The
excess dye was washed out and cells were then eluted in 1% SDS
solution for 5 min. Absorbance was measured at 650 nm.
qPCR array
To analyze the expression of pro- and
anti-angio-genic factors, RNA from HUVEC cells treated with 1 ng/ml
of activin A for 24 h, alongside untreated controls, and from
shControl-HUVECs and shINHBA-HUVECs were subjected to a human
angiogenesis cDNA-based qPCR array (TaqMan® Array Human
Angiogenesis; Applied Biosystems; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions.
Quantification of total VEGFA and
isoforms
HUVECs were cultured in the presence of 1 ng/ml
activin A for 24 h at 37°C and the expression of total VEGFA and of
isoforms 121, 165 and 189 were evaluated by RT-qPCR. To assess the
effects of activin A on the production and secretion of total VEGFA
and VEGFA isoform 121, ELISA was used. The concentration of VEGFA
was determined using the human VEGFA ELISA kit (cat. no. RAB0507;
Sigma-Aldrich; Merck KGaA) and the isoform 121 was evaluated with
the human VEGFA isoform 121 ELISA kit (cat. no. SEB851Hu;
Cloud-Clone Corp.), according to the manufacturers'
instructions.
Western blotting
Western blot analysis was used to determine the
abundance of phosphorylated SMAD2/3. Cells were washed with cold
PBS and lysed in cell lysis buffer (Cell Signaling Technology,
Inc.) containing protease and phosphatase inhibitors. Following
centrifugation, protein concentration was measured using a Bio-Rad
Protein assay (Bio-Rad Laboratories, Inc.) according to the
manufacturer's instructions. Total protein (30 µg per
sample) was resolved by 10% SDS-PAGE under reducing conditions, and
transferred to nitrocellulose membranes. The membranes were blocked
with 10% non-fat dry milk in PBS containing 0.1% Tween-20 at room
temperature for 2 h, rinsed in the same buffer, and incubated at
room temperature for 2 h with the following primary antibodies:
Polyclonal rabbit anti-phosphorylated SMAD2/3 (1:1,000; cat. no.
8828; Cell Signaling Technology, Inc.), polyclonal rabbit
anti-SMAD2/3 (1:1,000; cat. no. 3102; Cell Signaling Technology,
Inc.) and monoclonal rabbit anti-GAPDH (1:1,000; cat. no. 2118;
Cell Signaling Technology, Inc.). Following washing, the membranes
were incubated at room temperature for 1 h with polyclonal goat
anti-rabbit IgG HRP-linked antibody (1:2,000; cat. no. 7074; Cell
Signaling Technology, Inc.). Bands were detected using enhanced
chemiluminescence western blotting system (GE Healthcare) and
signals captured with an Alliance 9.7 instrument (UVITEC,
Ltd.).
Statistical analysis
Associations between immunohistochemical expression
of activin A in blood vessels and in tumor cells and
clinicopathological parameters of the tumors were analyzed using a
χ2 test. Survival curves were constructed based on the
Kaplan-Meier method and compared with the log-rank test. For
multivariate survival analysis, the Cox proportional hazard model
with a stepwise method including all parameters was employed.
Disease-specific survival (DSS) time was the time from treatment
initiation until death due to cancer or the last known date alive,
and disease-free survival (DFS) was the time from treatment
initiation until diagnosis of the first recurrence (local, regional
or distant) or the date of last follow up information for those
without recurrence. Spearman's correlation test was applied for
determine the correlation between activin A expression in blood
vessels and in tumor cells.
All in vitro assays were performed at least
three times in triplicate, and the results are presented as the
mean ± standard deviation. Differences were compared using
Mann-Whitney's U test or one-way analysis of variance with post-hoc
comparisons based on Tukey's multiple comparison test. All
statistical analyses were performed using GraphPad Prism v6.01
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of activin A in blood vessels
and tumor cells is associated with shortened survival time
To investigate whether activin A expression is
associated with clinicopathological features of patients with OSCC,
immunohistochemistry was performed in 95 cases of human OSCC. To
quantify activin A abundance in blood vessels, immunohistochemistry
for anti-CD34, a classical blood vessel marker, was performed
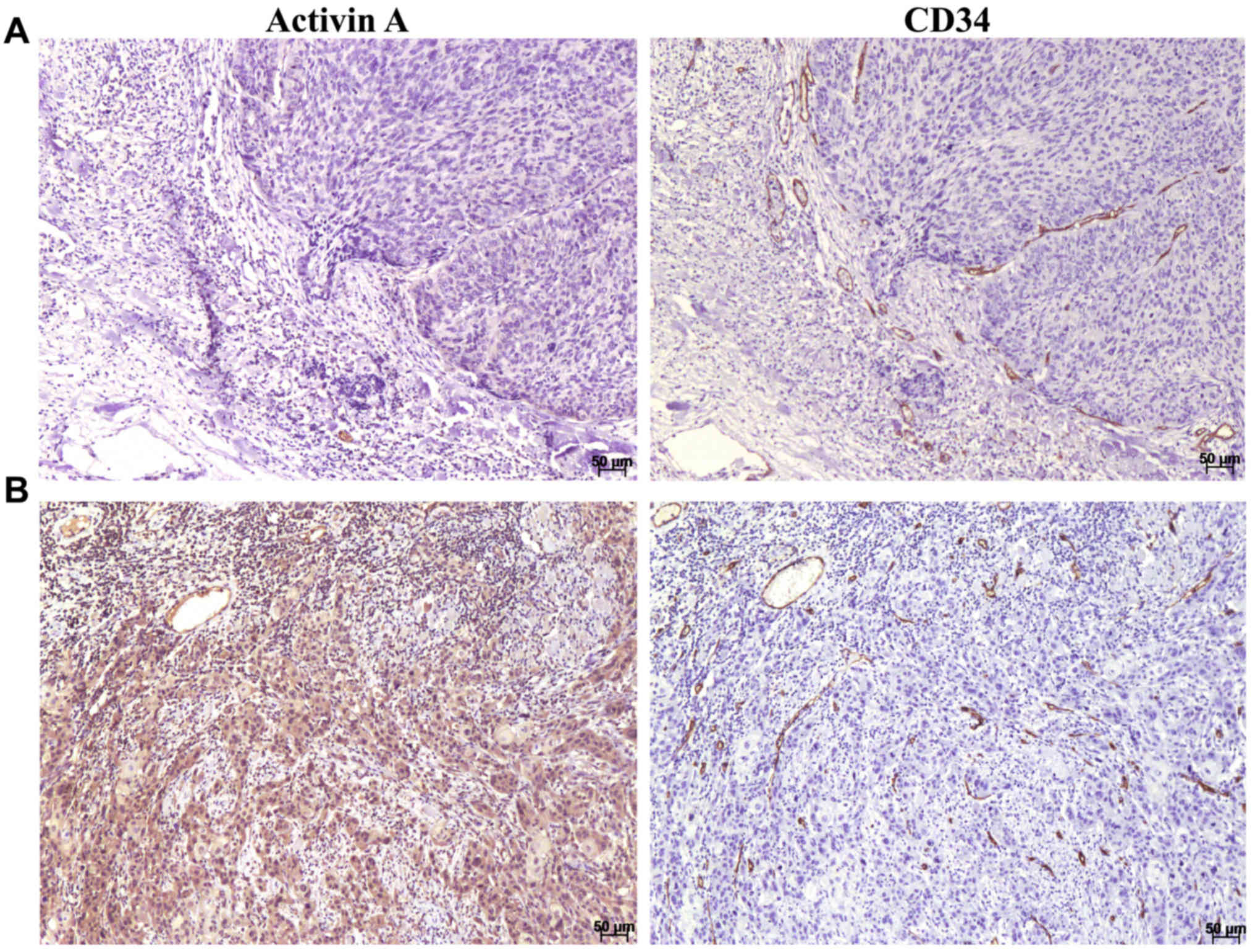
concurrent with anti-activin A in serial sections (Fig. 1). Activin A was observed as a
cytoplasmic stain with variable distribution and intensity in the
endothelial cells of the blood vessels and in the tumor cells
(Fig. 1). Expression was also
observed in some inflammatory cells and cancer-associated
fibroblasts. A significant correlation between activin A expression
in endothelial cells and in tumor cells was detected (r=0.6;
P<0.0001; data not shown). In order to categorize samples
regarding age and expression level, the median values were applied
to separate samples into two groups. As presented in Table I, no significant associations were
observed between activin A expression in blood vessels and the
clinicopathological features of patients with OSCC. However, a
significant association was identified between activin A expression
in tumor cells and margin status (P=0.02; Table I).
 | Table IAssociation of the
clinicopathological parameters of the oral squamous cell carcinoma
cases with the immunohisto-chemical expression of activin A in
blood vessels and tumor cells. |
Table I
Association of the
clinicopathological parameters of the oral squamous cell carcinoma
cases with the immunohisto-chemical expression of activin A in
blood vessels and tumor cells.
| Parameter | Activin A in blood
vessels
| Activin A in tumor
cells
|
|---|
| Low expression, n
(%) | High expression, n
(%) | P-value | Low expression, n
(%) | High expression, n
(%) | P-value |
|---|
| Age, years | | | | | | |
| <56 | 25 (55.6) | 23 (46.0) | | 27 (58.7) | 21 (42.9) | |
| ≥56 | 20 (44.4) | 27 (54.0) | 0.35 | 19 (41.3) | 28 (57.1) | 0.12 |
| Sex | | | | | | |
| Male | 39 (86.7) | 42 (84.0) | | 40 (87) | 41 (83.7) | |
| Female | 6 (13.3) | 8 (16.0) | 0.71 | 6 (13) | 8 (16.8) | 0.65 |
| Clinical stage | | | | | | |
| Early (I +
II) | 22 (48.9) | 22 (44.0) | | 21 (45.7) | 23 (46.9) | |
| Advanced (III +
IV) | 23 (51.1) | 28 (56.0) | 0.63 | 25 (54.3) | 26 (53.1) | 0.90 |
| Tumor site | | | | | | |
| Tongue | 26 (57.8) | 21 (42.0) | | 25 (54.3) | 22 (44.9) | |
| Others | 19 (42.2) | 29 (58.0) | 0.12 | 21 (45.7) | 27 (55.1) | 0.36 |
| Treatment | | | | | | |
| Surgery | 18 (40.0) | 18 (36.0) | | 20 (43.5) | 16 (32.7) | |
| Surgery + RTX | 25 (55.6) | 24 (48.0) | | 22 (47.8) | 27 (55.1) | |
| Surgery + RTX +
CTX | 2 (4.4) | 8 (16.0) | 0.18 | 4 (8.7) | 6 (12.2) | 0.53 |
| Histological
grade | | | | | | |
| WD/MD | 42 (93.3) | 43 (86.0) | | 42 (91.3) | 43 (87.8) | |
| PD | 3 (6.7) | 7 (14.0) | 0.25 | 4 (8.7) | 6 (12.2) | 0.57 |
| Margin status,
mma | | | | | | |
| ≥5 | 40 (90.9) | 49 (98.0) | | 40 (88.9) | 49 (100.0) | |
| <5 | 4 (9.1) | 1 (2.0) | 0.13 | 5 (11.1) | 0 (0.0) | 0.02 |
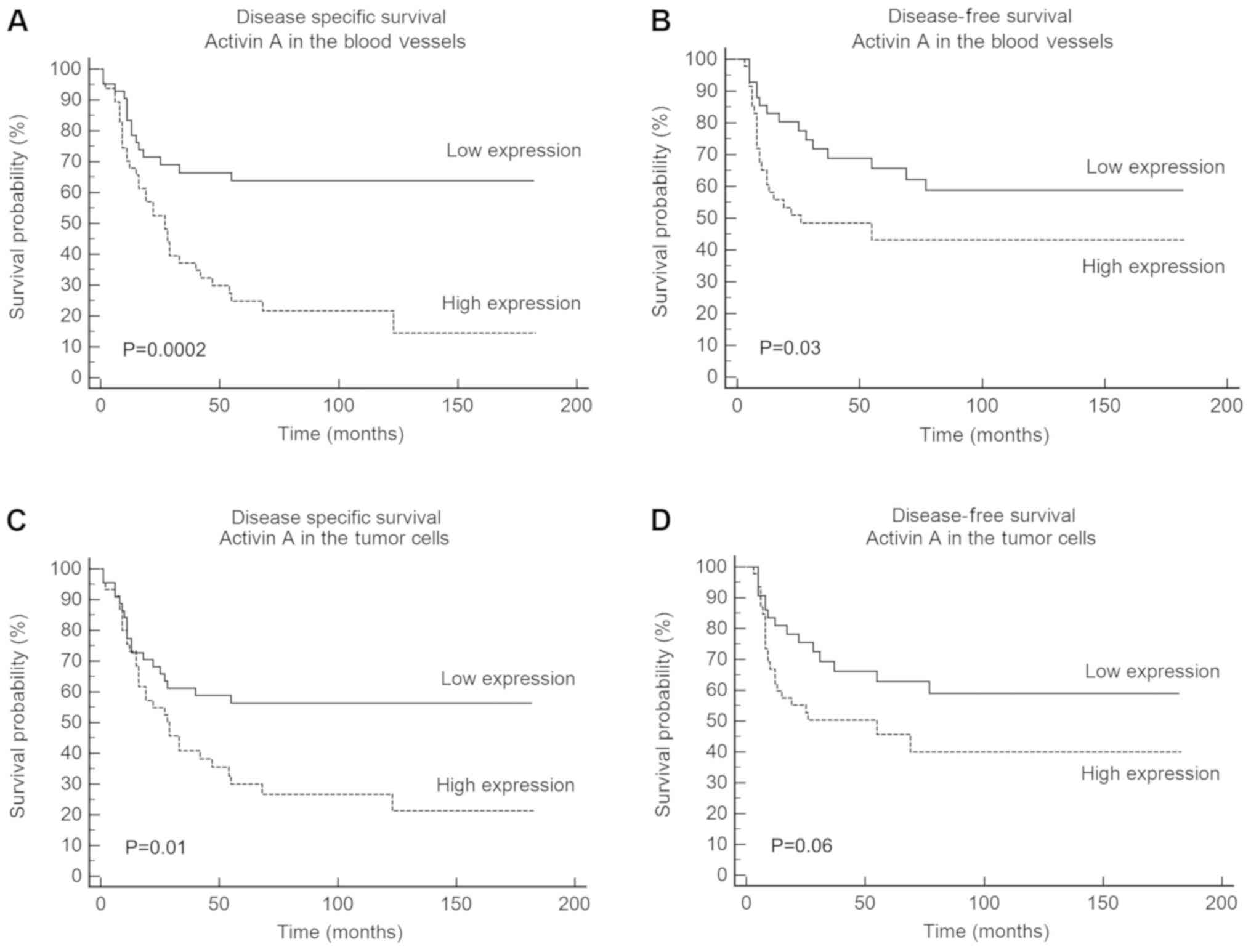
Univariate survival analysis based on log-rank test
revealed a significant association of DSS with clinical stage of
the tumor (P=0.001), histological grade (P=0.003), activin A
expression in blood vessels (P=0.0002) and activin A expression in
tumor cells (P=0.01; Table II).
High expression of activin A in tumor cells was a significant
marker of reduced DSS, with a 5-year survival of 30% for the
patients with high activin A expression compared with 56.3% for
those with low activin A expression (P=0.01; Fig. 2C). For activin A expression in the
blood vessels, the reduction in DSS was even more significant, with
a 5-year survival of 24.8% for patients with high activin A
expression compared with 63.8% for those with low activin A
expression (Fig. 2A). DFS revealed
a significant association with activin A expression in blood
vessels (P=0.03; Table II), but
not in tumor cells (Fig. 2D;
Table II). The expression of
activin A in the blood vessels was associated with a 5-year DFS of
43.1% for patients with high expression compared with 65.7% for
patients with low expression (Fig.
2B). Multivariate Cox regression analysis confirmed that
clinical stage [hazard ratio (HR), 2.02; 95% confidence interval
(CI), 1.09-3.73; P=0.03), activin A expression in blood vessels
(HR, 2.47; 95% CI, 1.30-4.71; P=0.006) and activin A expression in
tumor cells (HR, 1.63; 95% CI, 1.08-2.45; P=0.02) are independent
risk factors for DSS of patients with OSCC (Table III). Furthermore, high expression
of activin A in the blood vessels was determined to be an
independent indicator of DFS (HR, 2.09; 95% CI, 1.07-4.08; P=0.03;
Table III).
| Table IIUnivariate analysis for
disease‑specific survival and disease‑free survival of the oral
squamous cell carcinoma patients. |
Table II
Univariate analysis for
disease‑specific survival and disease‑free survival of the oral
squamous cell carcinoma patients.
| Parameter | Disease‑specific
survival rate
| Disease‑free
survival rate
|
|---|
| 5 years, % | HR (95% CI) | P-value | 5 years, % | HR (95% CI) | P-value |
|---|
| Age, years | | | | | | |
| <56 | 53.2 | 1 | | 55.8 | 1 | |
| ≥56 | 31.7 | 1.41
(0.81‑2.46) | 0.20 | 51.5 | 1.16
(0.62‑2.17) | 0.63 |
| Sex | | | | | | |
| Male | 45.4 | 1 | | 55.9 | 1 | |
| Female | 33.3 | 1.21
(0.54-2.71) | 0.61 | 46.2 | 1.25
(0.52-3.04) | 0.57 |
| Clinical stage | | | | | | |
| Early (I +
II) | 60.4 | 1 | | 62.1 | 1 | |
| Advanced (III +
IV) | 29.7 | 2.52
(1.45-4.36) | 0.001 | 46.7 | 1.67
(0.89-3.13) | 0.10 |
| Tumor site | | | | | | |
| Tongue | 38.3 | 1 | | 52.9 | 1 | |
| Others | 45.5 | 0.87
(0.50-1.50) | 0.61 | 55.4 | 1.08
(0.57-2.03) | 0.80 |
| Treatment | | | | | | |
| Surgery | 55.1 | 1 | | 67.5 | 1 | |
| Surgery + RTX | 33.5 | 1.47
(0.82-2.66) | | 47.3 | 1.64
(0.83-3.24) | |
| Surgery + RTX +
CTX | 33.3 | 1.22 (0.50
2.94) | 0.43 | 50.0 | 1.59
(0.51-4.65) | 0.38 |
| Histological
grade | | | | | | |
| WD/MD | 45.8 | 1 | | 55.5 | 1 | |
| PD | 22.2 | 3.09
(0.86-11.1) | 0.003 | 35.7 | 1.96
(0.49-7.79) | 0.18 |
| Margin status,
mm | | | | | | |
| ≥5 | 75.0 | 1 | | 75.0 | 1 | |
| <5 | 42.4 | 2.64
(0.75-9.30) | 0.31 | 53.6 | 2.16
(0.53-8.69) | 0.43 |
| Activin A in blood
vessels | | | | | | |
| Low
expression | 63.8 | 1 | | 65.7 | 1 | |
| High
expression | 24.8 | 2.86
(1.65-4.97) | 0.0002 | 43.1 | 1.93
(1.03-3.64) | 0.03 |
| Activin A in tumor
cells | | | | | | |
| Low
expression | 56.3 | 1 | | 62.9 | 1 | |
| High
expression | 30.0 | 1.95
(1.12-3.38) | 0.01 | 45.7 | 1.81
(0.96-3.39) | 0.06 |
| Table IIICox multivariate analysis for the
risk of mortality. |
Table III
Cox multivariate analysis for the
risk of mortality.
| Parameter | Disease‑specific
survival
| Disease‑free
survival
|
|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Clinical stage | | | | |
| Early (I + II) | 1 | | | |
| Advanced (III +
IV) | 2.02
(1.09-3.73) | 0.03 | | |
| Activin A in blood
vessels | | | | |
| Low expression | 1 | | 1 | |
| High
expression | 2.47
(1.30-4.71) | 0.006 | 2.09
(1.07-4.08) | 0.03 |
| Activin A in tumor
cells | | | | |
| Low expression | 1 | | | |
| High
expression | 1.63
(1.08-2.45) | 0.02 | | |
In order to strengthen the prognostic information
provided by these independent factors, activin A expression levels
in blood vessels and in tumor cells were combined and analyzed for
both univariate and multivariate survival. Combination of clinical
stage with activin A expression in blood vessels and with activin A
expression in the tumor cells were also evaluated. In all
combinations, the discriminatory ability to predict survival of
patients with OSCC was largely improved (Table IV). Activin A expression in tumor
cells was not individually associated with DFS in either univariate
or multi-variate analysis, but when combined with activin A
expression in the blood vessels and with clinical stage, a
significant association was observed, revealing a prognostic
discrimination. Collectively, these findings suggest that the
expression levels of activin A in both blood vessels and tumor
cells could be used as risk factor to predict poor prognosis of
OSCC.
| Table IVCox regression analysis for
disease-specific and disease-free survival for combination of
activin A expression and clinical stage. |
Table IV
Cox regression analysis for
disease-specific and disease-free survival for combination of
activin A expression and clinical stage.
| Parameters | Disease‑specific
survival
| Disease‑free
survival
|
|---|
Univariate
| Multivariate
| Univariate
| Multivariate
|
|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Activin A in blood
vessels and activin A in tumor cells | | | | | | | | |
| | | | | | | | |
| Low expression/low
expression | 1 | | 1 | | 1 | | 1 | |
| High
expression/high expression | 3.15
(1.67-5.92) | 0.0005 | 3.52
(1.69-7.32) | 0.0007 | 2.09
(1.05-4.15) | 0.03 | 2.29
(1.09-4.82) | 0.03 |
| Activin A in blood
vessels and clinical stage | | | | | | | | |
| Low
expression/early stage (I + II) | 1 | | 1 | | 1 | | 1 | |
| High
expression/advanced stage (III + IV) | 13.6
(6.09-30.5) | <0.0001 | 21.4
(3.97-115.3) | 0.0004 | 3.19
(1.36-7.48) | 0.006 | 3.40
(1.35-8.56) | 0.009 |
| Activin A in tumor
cells and clinical stage | | | | | | | | |
| Low
expression/early stage (I + II) | 1 | | 1 | | 1 | | 1 | |
| High
expression/advanced stage (III + IV) | 6.69
(2.98-15.1) | <0.0001 | 7.05
(2.38-20.8) | 0.0004 | 2.72
(1.21-6.13) | 0.01 | 2.81
(1.19-6.67) | 0.02 |
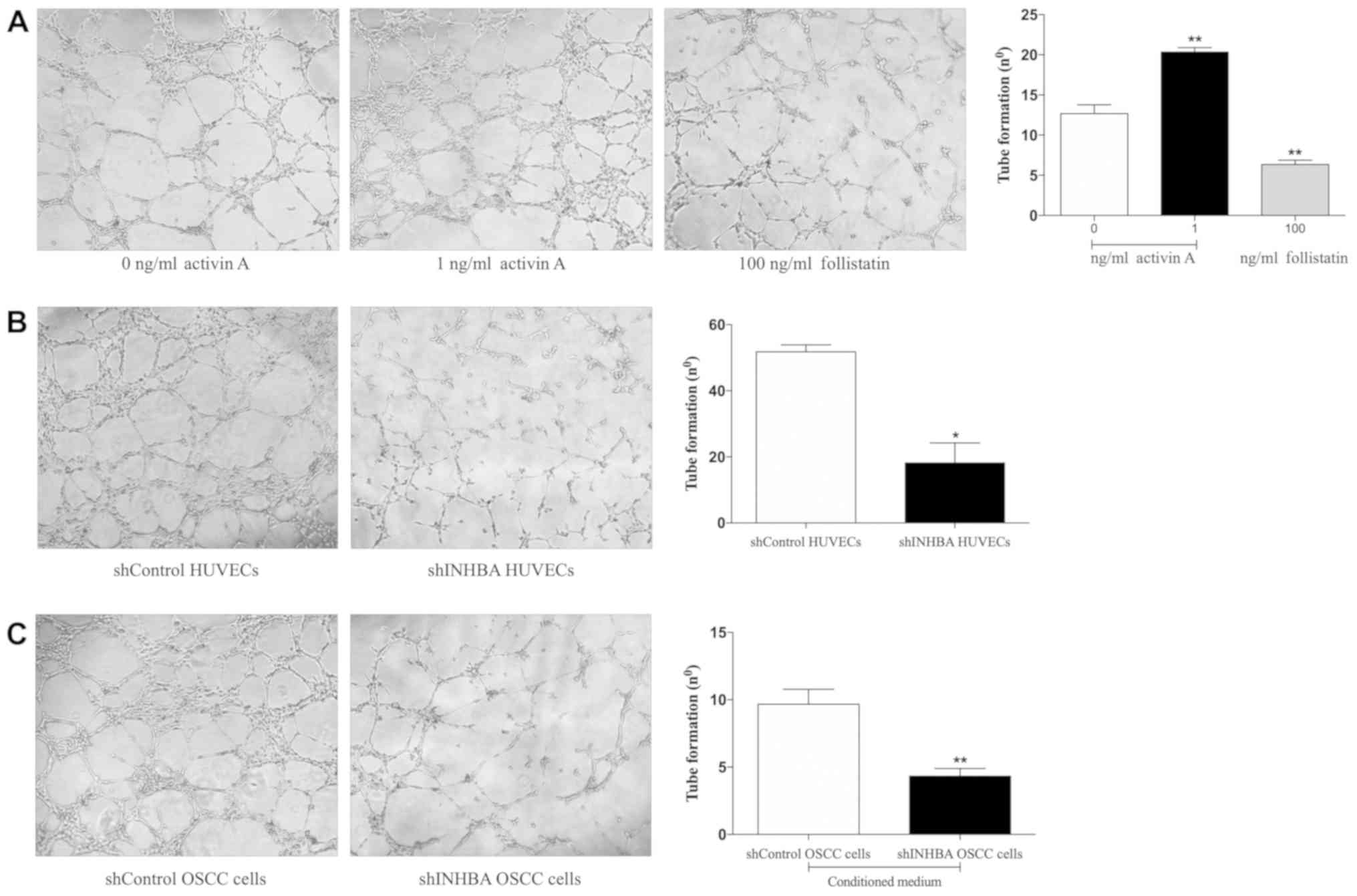
Activin A promotes tubulogenesis in
HUVECs
Since tube morphogenesis is a critical step in the
formation of blood vessels, the present study first assessed the
importance of activin A for the tubulogenesis of HUVECs. Cells were
treated with recombinant activin A, the activin A-antagonist
follistatin or were transduced with lentivirus carrying shINHBA.
shINHBA-transfected HUVECs demonstrated a significant reduction in
both activin A mRNA and protein levels in comparison with cells
transfected with shControl (Fig.
S1). A signifcant increase in the HUVEC tubulogenesis was
observed in cells treated with 1 ng/ml activin A (P<0.01;
Fig. 3A). Conversely, follistatin
significantly reduced the tubulogenic activity of HUVECs
(P<0.01; Fig. 3A). In addition,
shINHBA-transfected HUVECs generated a significantly lower number
of tubes compared with shControl-transfected HUVECs (P<0.05;
Fig. 3B). Similar inhibition was
observed when HUVECs were cultured in the presence of conditioned
medium harvested from shINHBA OSCC cells compared with shControl
OSCC cells (P<0.01: Fig.
3C).
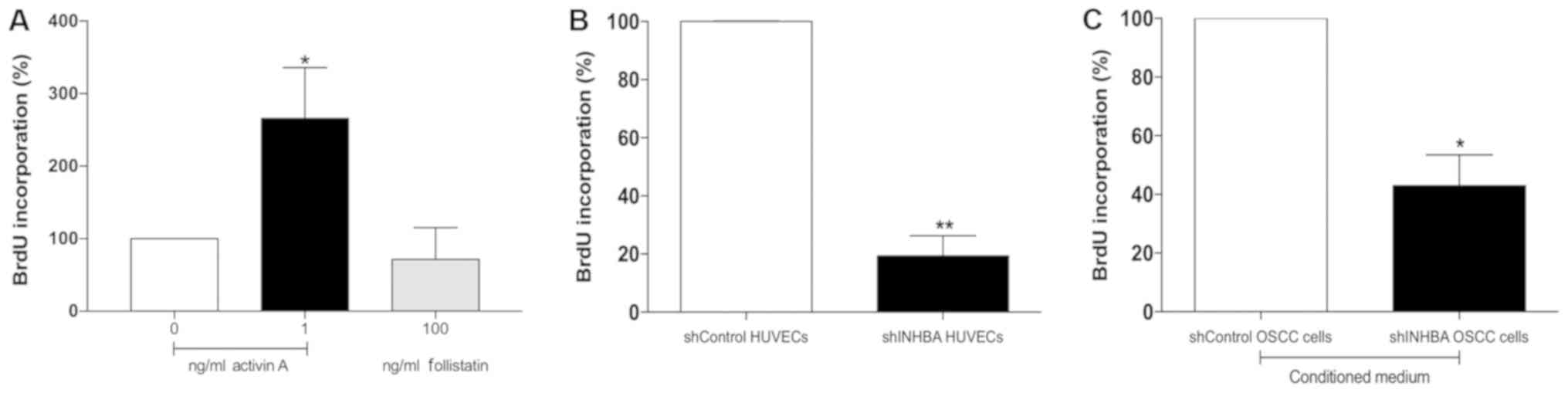
Activin A effects the proliferation and
migration of HUVECs
To improve understanding of the role of activin A in
the events that control tubulogenesis, the effects of activin A on
the proliferation and migration of HUVECs were evaluated. Compared
with untreated cells, activin A significantly increased the
proliferation of HUVECs (P<0.05; Fig. 4A). Conversely, the proliferation of
HUVECs was decreased, but not at significant level, after 24 h
treatment with 100 ng/ml follistatin (Fig. 4A), and by gene silencing of activin
A (P<0.01; Fig. 4B). Treatment
of HUVECs with conditioned medium collected from shINHBA OSCC cells
also significantly decreased HUVEC proliferation in comparison with
conditioned medium from shControl OSCC cells (P<0.05; Fig. 4C).
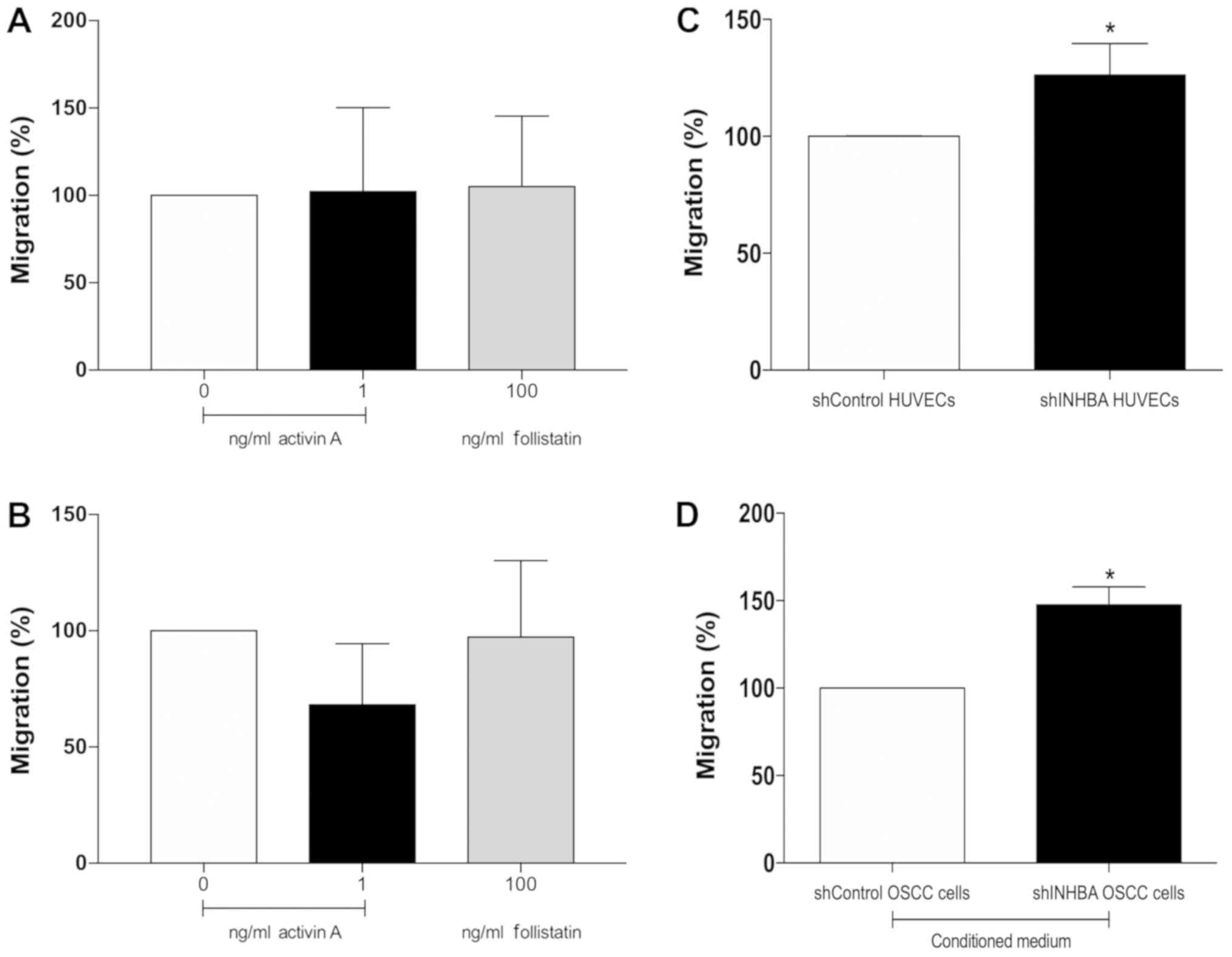
The migration of HUVECs was not affected by activin
A or follistatin directly in the upper chamber or when used as
chemoattractant in the lower chamber of the Transwell system
(Fig. 5A and B). However, activin
A-knockdown significantly increased the migration of HUVEC cells
(P<0.01; Fig. 5C). When
conditioned medium collected from shINHBA OSCC cells was used as a
chemoattractant, a significant increase in migration of HUVECs was
observed (P<0.01; Fig. 5D).
Activin A promotes differential
expression of genes related to angiogenesis
To identify putative activin A-target genes, the
expression of angiogenesis-related genes in HUVEC cells treated
with activin A and in shINHBA-transfected HUVECs was profiled. Up-
and downregulated genes were defined as those with an expression
level >2.0- or <2.0-fold different in an average of 3
independent experiments. Table
SII presents the up- and downregulated genes in HUVECs treated
with activin A compared with untreated HUVEC cells and in
shIN-HBA-transfected cells compared with shControl-transfected
cells. VEGFA was markedly upregulated by activin A treatment
(29.6-fold), whereas it was downregulated in HUVEC
shINHBA-transfected cells compared with shControl-transfected
HUVECs (-3.0-fold).
Effects of activin A on the expression of
VEGFA and VEGFA isoforms
Considering that VEGFA was markedly regulated by
activin A and given that VEGFA is the main regulator of
angiogenesis (13,14), RT-qPCR was performed to determine
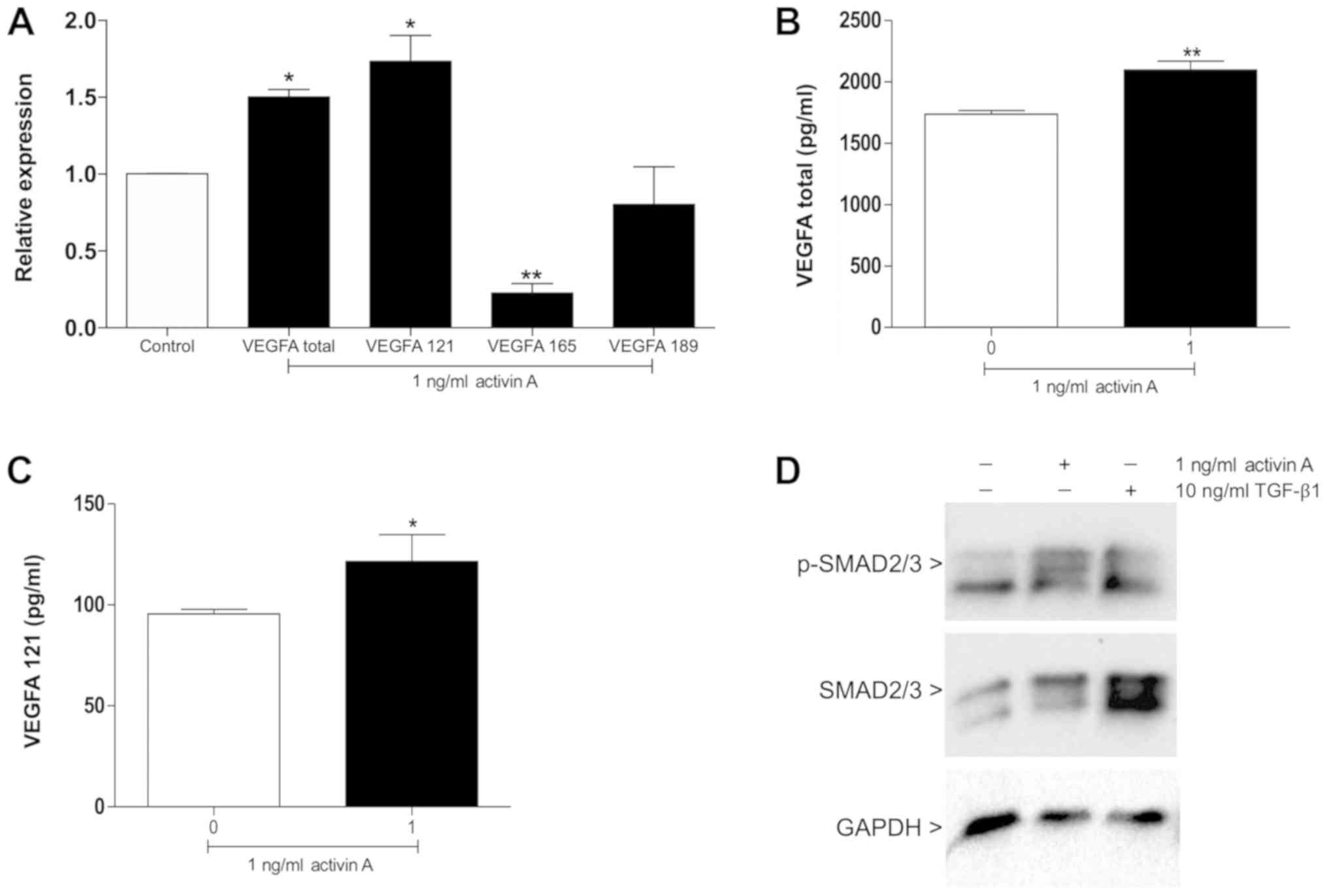
the effects of activin A on the expression of total VEGFA and its
major isoforms. The results demonstrated that compared with the
untreated control, activin A significantly increased the expression
of total VEGFA (P<0.01) and the pro-angiogenic isoform VEGFA121
(P<0.01; Fig. 6A). A
significant decrease of VEGFA165 was detected in cells treated with
activin A (P<0.001; Fig. 6A).
Accordingly, significantly higher levels of total VEGFA (P<0.01)
and VEGFA121 (P<0.05) were secreted by HUVECs following
treatment with activin A in comparison with the control (Fig. 6B and C).
SMAD2/3 signaling is activated by activin
A
Activin A signaling is mediated by the SMAD pathway
and VEGFA is positively regulated by SMAD2/3 (4,13,14).
The present study therefore examined whether activin A stimulates
SMAD2/3 phosphorylation in HUVECs. After 30 min of stimulation,
increased phosphorylated SMAD2/3 levels were observed in HUVECs
treated with 1 ng/ml activin A (Fig.
6D).
Discussion
Angiogenesis is an essential requirement for growth
and progression of solid tumors; however, the underlying molecular
mechanisms remain unclear (23).
The present study confirmed that activin A has a relevant
prognostic role for aggressive OSCC and this finding was supported
by in vitro evidence of a pro-angiogenic role for activin A
with both autocrine and paracrine (tumor-secreted) effects.
Initially it was observed that the expression of activin A in blood
vessels is a significant and independent risk factor to predict
relapse and shortened survival of patients with OSCC. The present
results also demonstrated that increased expression of activin A by
tumor cells is strongly associated with poor prognosis of patients
with OSCC, and the combination of activin A overexpression in both
tumor cells and blood vessels improved the discrimination of
patients at low-and high-risk of poor prognosis. Expression of
activin A has previously been described in tumors cells and in
components of the tumor microenvironment, including
cancer-associated fibroblasts, blood vessels and some
tumor-infiltrating inflammatory cells in OSCC (10-12,24,25).
The present study extends the reports of increased expression of
activin A in neoplastic and endothelial cells in OSCC and
demonstrates a significant association with shortened survival.
Besides the important role in the development of various types of
cancer (6-9), previous studies have indicated that
high levels of circulating activin A is an independent prognostic
factor of survival in pancreatic, lung, breast and colorectal
cancer (26-29), which may be associated with its
contribution to the development of cachexia and loss of skeletal
muscle mass (8). Altogether,
activin A has an important role in the development and progression
of numerous cancers, including OSCC.
The present study also revealed that activin A
regulates essential cell biology aspects associated with
angiogenesis, such as the proliferation, migration and tubulogenic
activity of the endothelial cells. Pro-angiogenic effects were
associated with stimulation of endothelial cells directly with
activin A or with conditioned medium from OSCC cells expressing
activin A, and these effects were reversed when endogenous
expression of activin A was inhibited in endothelial cells and when
these cells were treated with an activin A inhibitor (follistatin)
or with conditioned medium from tumor cells with shRNA-mediated
knockdown of activin A. These findings agree with other reports
reporting tubulogenic stimulation of activin A in vascular
endothelial cells (13,30) and with its association with breast
and skin cancer development and progression in vivo
(3,15,31).
In melanomas, paracrine activin A signaling stimulates tumor
vascularization and promotes tumor growth and metastasis through
mechanisms of immune evasion (32). However, some studies have
demonstrated that activin A inhibits proliferation and suppresses
tubulogenic activity of endothelial cells isolated from human
umbilical veins of newborns, bovine adrenal or brain capillary and
calf pulmonary artery, or in mice experimental neuroblastomas
(16-18,33-35).
The present results demonstrated that inhibition of activin A
promotes migration of HUVEC cells. In a recent study, activin A
neutralization promoted primary microvascular endothelial cell
migration as well as essential steps in the expansion and formation
of the vasculature (36). In
prostate cancer, activin A enhances cell migration through
increasing androgen receptor gene transcription, nuclear
translocation and interaction with SMADs (37). The formation of capillary-like
tubes in vitro on basement membrane matrix, as applied in
the present study, represents the later stages of the angiogenic
process in which the endothelial cells differentiate into tubes
simulating the in vivo situation. This assay is dependent on
a series of events, including the proliferation, migration and
differentiation of the cells (38). Thus, expansion and migration of
precursor cells prior to differentiation are essential for vessel
formation.
It was speculated that activin A influences a
network of other angiogenesis-related genes to promote angiogenesis
and tumor growth. Supporting this hypothesis, the present study
observed that activin A treatment induces the expression of several
genes in the angiogenesis pathway, most of them favoring
angiogenesis. Some these genes included the following: AMOT, which
has been shown to promote vascular development in vivo by
regulating motility of the endothelial cells (39); HSPG2, which promotes vascular
formation and angiogenesis by modulating FGF2 activity (40); and FGF2, a critical mediator of
capillary formation (30).
Notably, activin A was revealed to modulate the expression of
VEGFA. Expression of total VEFGA was significantly increased by
treatment of HUVEC cells with recombinant activin A, whereas
silencing activin A gene expression significantly reduced
expression of VEGFA. Treatment of endothelial cells with activin A
activated SMAD2/3, suggesting that this signaling may be involved
in the regulation of VEGFA by activin A in these cells. This is
consistent with previous evidence that activin A predominantly
mediates intracellular signaling via phosphorylation of SMADs, and
the oligomer of SMAD2/3 and SMAD4 acts as a transcription regulator
of various target genes (4,41).
In support of the present findings, activin A has been demonstrated
to be critically relevant for VEGF-induced tubulogenesis, and the
VEGF-induced pathway is almost completely inhibited when the
secretion of activin A is blocked (13). The functional relevance of SMAD2
for activin A-induced VEGF expression is also supported by
observations that SMA D2 overexpression significantly enhances V
EGF levels in human hepatocellular carcinoma cells, and SMAD2
dominant negative mutant inhibits activin A responsiveness,
abrogating VEGF stimulation (14).
Notably, activin A expression is a predictive marker of response to
treatment with the anti-VEGF antibody bevacizumab in metastatic
melanoma (42). However, a lack of
a specific assay connecting activin A, phosphorylation of SMADs and
regulation of VEGFA in the context of angiogenesis is a limitation
of the study that merits additional investigation.
VEGFA is considered a key mediator of angiogenesis
and its expression has been implicated in tumor development and
metastasis (23,43). Due to alternative splicing, VEGFA
gene is translated into a number of isoforms, with VEGFA121,
VEGFA165 and VEGFA189 being the most prevalent in humans (44). In OSCC, increased expression of
total VEGFA has been described as an independent prognostic
indicator of reduced survival and increased recurrence (45). VEGFA isoforms 121, 165 and 189 are
expressed at high levels in OSCC cells and increased expression of
isoform 165 is associated with poorer prognosis (46). Assessing the main pro-angiogenic
isoforms of VEGFA, the present study observed an enhanced
expression and secretion of isoform 121 and a downregulation of
isoform 165 in HUVECs stimulated with activin A. The balance in the
expression of VEGFA isoforms produced by alternative splicing seems
to be important for angiogenesis in physiological and pathological
conditions (44). VEGFA121 lacks
the heparin-binding motif and thus can diffuse relatively freely
through the extracellular matrix, inducing malformed and leaky
vessels (47). High concentrations
of VEGFA121 also promote the development of vessels with enhanced
diameter, whereas its lower concentration produces the growth of
long and thin vessels (48). The
abnormality of the tumor vasculature is already well documented in
the literature, constituting not only an obstacle to the efficacy
of the available chemotherapeutic agents but also providing a route
for metastatic dissemination (49,50).
The high intratumoral vascular permeability associated with the
absence of functional lymphatic vessels results in the elevation of
interstitial pressure, which constitutes a physiological barrier to
the delivery of chemotherapeutic agents inside the tumor. On the
other hand, the blockage of blood and oxygen supplies creates a
hypoxic and acidic microenvironment in the tumor, which fosters
tumor cells to become more aggressive and metastatic (50,51).
However, current tumor antiangiogenic and vascular normalizing
drugs display only moderate anticancer efficacy (50). Therefore, activin A blockade may be
a promising therapeutic approach as it would inhibit the formation
of new blood vessels, as well the development of a disorganized
tumor vasculature by isoform 121 of VEGFA.
Activin A serves an important role in tumorigenesis,
mostly acting by regulation of tumors cells but also by the
regulating tumor microenvironment, such as cancer-associated
fibroblast accumulation and angiogenesis. Collectively, the present
findings expand the knowledge regarding the prognostic value of
activin A expression in OSCC, and further suggest that activin A
signaling promotes angiogenesis, at least in part, by inducing
SMAD2/3 activation and the expression of VEGFA isoform 121.
Although other studies are required to clarify the precise
mechanisms by which activin A/SMAD/VEGFA121 signaling is implicated
in angiogenesis, the clinical potential of activin A as a
prognostic marker, a therapeutic target or a marker for
post-therapeutic monitoring of locoregional recurrence should be
considered in OSCC.
Supplementary Data
Funding
This study was supported by grants from the São
Paulo Research Foundation-FAPESP (grant nos. 2013/19856-2 and
2013/01607-6), and from the National Council for Scientific and
Technological Development-CNPq, Brasília, Brazil (grant no.
302964/2015-0).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CEO, CRJ, DL, TS, EG and RC conceived and designed
the experiments. CEO, MRD, MCM, NKC and JEL performed the
experiments. IS-C contributed with clinical samples, including the
generation and analysis of clinical, pathological and survival
data. CEO, MRD, CRJ, NKC and RC analyzed the data. CEO and RC wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from each
patient according to the declaration of Helsinki, and the study was
approved by the Human Research Ethics Committee of the School of
Dentistry, University of Campinas (approval no. 100/2012, Oct 10th,
2012).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Fabio Haach Teo
(University of Campinas, São Paulo, Brazil) for his excellent
technical assistance.
References
|
1
|
Matzuk MM, Kumar TR, Vassalli A,
Bickenbach JR, Roop DR, Jaenisch R and Bradley A: Functional
analysis of activins during mammalian development. Nature.
374:354–356. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ying SY, Zhang Z, Furst B, Batres Y, Huang
G and Li G: Activins and activin receptors in cell growth. Proc Soc
Exp Biol Med. 214:114–122. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Antsiferova M and Werner S: The bright and
the dark sides of activin in wound healing and cancer. J Cell Sci.
125:3929–3937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loomans HA and Andl CD: Intertwining of
Activin A and TGFβ Signaling: Dual roles in cancer progression and
cancer cell invasion. Cancers (Basel). 7:70–91. 2014. View Article : Google Scholar
|
|
5
|
McDowell N, Zorn AM, Crease DJ and Gurdon
JB: Activin has direct long-range signalling activity and can form
a concentration gradient by diffusion. Curr Biol. 7:671–681. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wildi S, Kleeff J, Maruyama H, Maurer CA,
Büchler MW and Korc M: Overexpression of activin A in stage IV
colorectal cancer. Gut. 49:409–417. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gold E and Risbridger G: Activins and
activin antagonists in the prostate and prostate cancer. Mol Cell
Endocrinol. 359:107–112. 2012. View Article : Google Scholar
|
|
8
|
Loumaye A, de Barsy M, Nachit M, Lause P,
van Maanen A, Trefois P, Gruson D and Thissen JP: Circulating
Activin A predicts survival in cancer patients. J Cachexia
Sarcopenia Muscle. 8:768–777. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seachrist DD and Keri RA: The Activin
social network: Activin, inhibin and follistatin in breast
development and cancer. Endocrinology. 160:1097–1110. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sobral LM, Bufalino A, Lopes MA, Graner E,
Salo T and Coletta RD: Myofbroblasts in the stroma of oral cancer
promote tumorigenesis via secretion of activin A. Oral Oncol.
47:840–846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bufalino A, Cervigne NK, de Oliveira CE,
Fonseca FP, Rodrigues PC, Macedo CC, Sobral LM, Miguel MC, Lopes
MA, Paes Leme AF, et al: Low miR-143/miR-145 cluster levels induce
activin A overexpression in oral squamous cell carcinomas, which
contributes to poor prognosis. PLoS One. 10:e01365992015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kelner N, Rodrigues PC, Bufalino A,
Fonseca FP, Santos-Silva AR, Miguel MC, Pinto CA, Leme AF, Graner
E, Salo T, et al: Activin A immunoexpression as predictor of occult
lymph node metastasis and overall survival in oral tongue squamous
cell carcinoma. Head Neck. 37:479–486. 2015. View Article : Google Scholar
|
|
13
|
Maeshima K, Maeshima A, Hayashi Y, Kishi S
and Kojima I: Crucial role of activin a in tubulogenesis of
endo-thelial cells induced by vascular endothelial growth factor.
Endocrinology. 145:3739–3745. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wagner K, Peters M, Scholz A, Benckert C,
Ruderisch HS, Wiedenmann B and Rosewicz S: Activin A stimulates
vascular endothelial growth factor gene transcription in human
hepato-cellular carcinoma cells. Gastroenterology. 126:1828–1843.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bashir M, Damineni S, Mukherjee G and
Kondaiah P: Activin-A signaling promotes epithelial-mesenchymal
transition, invasion, and metastatic growth of breast cancer. NPJ
Breast Cancer. 1:150072015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Panopoulou E, Murphy C, Rasmussen H, Bagli
E, Rofstad EK and Fotsis T: Activin A suppresses neuroblastoma
xenograft tumor growth via antimitotic and antiangiogenic
mechanisms. Cancer Res. 65:1877–1886. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krneta J, Kroll J, Alves F, Prahst C,
Sananbenesi F, Dullin C, Kimmina S, Phillips DJ and Augustin HG:
Dissociation of angio-genesis and tumorigenesis in follistatin- and
activin-expressing tumors. Cancer Res. 66:5686–5695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaneda H, Arao T, Matsumoto K, De Velasco
MA, Tamura D, Aomatsu K, Kudo K, Sakai K, Nagai T, Fujita Y, et al:
Activin A inhibits vascular endothelial cell growth and suppresses
tumour angiogenesis in gastric cancer. Br J Cancer. 105:1210–1217.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sawazaki-Calone I, Rangel A, Bueno AG,
Morais CF, Nagai HM, Kunz RP, Souza RL, Rutkauskis L, Salo T,
Almangush A, et al: The prognostic value of histopathological
grading systems in oral squamous cell carcinomas. Oral Dis.
21:755–761. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Agostini M, Almeida LY, Bastos DC, Ortega
RM, Moreira FS, Seguin F, Zecchin KG, Raposo HF, Oliveira HC,
Amoêdo ND, et al: The fatty acid synthase inhibitor orlistat
reduces the growth and metastasis of orthotopic tongue oral
squamous cell carcinomas. Mol Cancer Ther. 13:585–595. 2014.
View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Salo T, Sutinen M, Hoque Apu E, Sundquist
E, Cervigne NK, de Oliveira CE, Akram SU, Ohlmeier S, Suomi F,
Eklund L, et al: A novel human leiomyoma tissue derived matrix for
cell culture studies. BMC Cancer. 15:9812015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Palma M, Biziato D and Petrova TV:
Microenvironmental regulation of tumour angiogenesis. Nat Rev
Cancer. 17:457–474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chang KP, Kao HK, Liang Y, Cheng MH, Chang
YL, Liu SC, Lin YC, Ko TY, Lee YS, Tsai CL, et al: Overexpression
of activin A in oral squamous cell carcinoma: Association with poor
prognosis and tumor progression. Ann Surg Oncol. 17:1945–1956.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tsai CN, Tsai CL, Yi JS, Kao HK, Huang Y,
Wang CI, Lee YS and Chang KP: Activin A regulates the epidermal
growth factor receptor promoter by activating the PI3K/SP1 pathway
in oral squamous cell carcinoma cells. Sci Rep. 9:51972019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Togashi Y, Kogita A, Sakamoto H, Hayashi
H, Terashima M, de Velasco MA, Sakai K, Fujita Y, Tomida S, Kitano
M, et al: Activin signal promotes cancer progression and is
involved in cachexia in a subset of pancreatic cancer. Cancer Lett.
356(2 Pt B): 819–827. 2015. View Article : Google Scholar
|
|
27
|
Hoda MA, Rozsas A, Lang E, Klikovits T,
Lohinai Z, Torok S, Berta J, Bendek M, Berger W, Hegedus B, et al:
High circulating activin A level is associated with tumor
progression and predicts poor prognosis in lung adenocarcinoma.
Oncotarget. 7:13388–13399. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jueckstock J, Burkhardt N, Kuhn C,
Blankenstein T, Mahner S, Schindlbeck C, Janni W, Rack B and
Mylonas I: Expression of activin during and after chemotherapy in
peripheral blood of patients with primary breast cancer. Anticancer
Res. 36:2153–2159. 2016.PubMed/NCBI
|
|
29
|
Staudacher JJ, Bauer J, Jana A, Tian J,
Carroll T, Mancinelli G, Özden Ö, Krett N, Guzman G, Kerr D, et al:
Activin signaling is an essential component of the TGF-β induced
pro-metastatic phenotype in colorectal cancer. Sci Rep. 7:55692017.
View Article : Google Scholar
|
|
30
|
Hayashi Y, Maeshima K, Goto F and Kojima
I: Activin A as a critical mediator of capillary formation:
Interaction with the fbroblast growth factor action. Endocr J.
54:311–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kalli M, Mpekris F, Wong CK, Panagi M,
Ozturk S, Thiagalingam S, Stylianopoulos T and Papageorgis P:
Activin A signaling regulates IL13Rα2 expression to promote breast
cancer metastasis. Front Oncol. 9:322019. View Article : Google Scholar
|
|
32
|
Donovan P, Dubey OA, Kallioinen S, Rogers
KW, Muehlethaler K, Müller P, Rimoldi D and Constam DB: Paracrine
activin-A signaling promotes melanoma growth and metastasis through
immune evasion. J Invest Dermatol. 137:2578–2587. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McCarthy SA and Bicknell R: Inhibition of
vascular endothelial cell growth by activin-A. J Biol Chem.
268:23066–23071. 1993.PubMed/NCBI
|
|
34
|
Schramm A, von Schuetz V, Christiansen H,
Havers W, Papoutsi M, Wilting J and Schweigerer L: High activin
A-expression in human neuroblastoma: Suppression of malignant
potential and correlation with favourable clinical outcome.
Oncogene. 24:680–687. 2005. View Article : Google Scholar
|
|
35
|
Merfeld-Clauss S, Lu H, Wu X, March KL and
Traktuev DO: Hypoxia-induced activin A diminishes endothelial cell
vascu-logenic activity. J Cell Mol Med. 22:173–184. 2018.
View Article : Google Scholar
|
|
36
|
Fahmy-Garcia S, Farrell E, Witte-Bouma J,
Robbesom-van den Berge I, Suarez M, Mumcuoglu D, Walles H,
Kluijtmans SGJM, van der Eerden BCJ, van Osch GJVM, et al:
Follistatin effects in migration, vascularization, and osteogenesis
in vitro and bone repair in vivo. Front Bioeng Biotechnol.
7:382019. View Article : Google Scholar :
|
|
37
|
Kang HY, Huang HY, Hsieh CY, Li CF, Shyr
CR, Tsai MY, Chang C, Chuang YC and Huang KE: Activin A enhances
prostate cancer cell migration through activation of androgen
receptor and is overexpressed in metastatic prostate cancer. J Bone
Miner Res. 24:1180–1193. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tahergorabi Z and Khazaei M: A review on
angiogenesis and its assays. Iran J Basic Med Sci. 15:1110–1126.
2012.
|
|
39
|
Levchenko T, Veitonmäki N, Lundkvist A,
Gerhardt H, Ming Y, Berggren K, Kvanta A, Carlsson R and Holmgren
L: Therapeutic antibodies targeting angiomotin inhibit angiogenesis
in vivo. FASEB J. 22:880–889. 2008. View Article : Google Scholar
|
|
40
|
Douglass S, Goyal A and Iozzo RV: The role
of perlecan and endorepellin in the control of tumor angiogenesis
and endothelial cell autophagy. Connect Tissue Res. 56:381–391.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Zhu H, Klausen C, Peng B and Leung
PC: Vascular endothelial growth factor-A (VEGF-A) mediates activin
A-induced human trophoblast endothelial-like tube formation.
Endocrinology. 156:4257–4268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schuster C, Akslen LA, Stokowy T and
Straume O: Predictive value of angiogenic proteins in patients with
metastatic melanoma treated with bevacizumab monotherapy. J Pathol
Clin Res. 5:53–62. 2019. View Article : Google Scholar :
|
|
43
|
Caporarello N, Lupo G, Olivieri M,
Cristaldi M, Cambria MT, Salmeri M and Anfuso CD: Classical VEGF,
Notch and Ang signalling in cancer angiogenesis, alternative
approaches and future directions (Review). Mol Med Rep.
16:4393–4402. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vempati P, Popel AS and Mac Gabhann F:
Extracellular regulation of VEGF: Isoforms, proteolysis, and
vascular patterning. Cytokine Growth Factor Rev. 25:1–19. 2014.
View Article : Google Scholar :
|
|
45
|
Zhao SF, Yang XD, Lu MX, Sun GW, Wang YX,
Zhang YK, Pu YM and Tang EY: Prognostic significance of VEGF
immuno-histochemical expression in oral cancer: A meta-analysis of
the literature. Tumour Biol. 34:3165–3171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cai C, Böttcher MC, Werner JA and Mandic
R: Differential expression of VEGF121, VEGF165 and VEGF189 in
angiomas and squamous cell carcinoma cell lines of the head and
neck. Anticancer Res. 30:805–810. 2010.PubMed/NCBI
|
|
47
|
Ehrbar M, Djonov VG, Schnell C, Tschanz
SA, Martiny-Baron G, Schenk U, Wood J, Burri PH, Hubbell JA and
Zisch AH: Cell-demanded liberation of VEGF121 from fibrin implants
induces local and controlled blood vessel growth. Circ Res.
94:1124–1132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nakatsu MN, Sainson RC, Pérez-del-Pulgar
S, Aoto JN, Aitkenhead M, Taylor KL, Carpenter PM and Hughes CC:
VEGF(121) and VEGF(165) regulate blood vessel diameter through
vascular endothelial growth factor receptor 2 in an in vitro
angiogenesis model. Lab Invest. 83:1873–1885. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tozer GM, Kanthou C and Baguley BC:
Disrupting tumour blood vessels. Nat Rev Cancer. 5:423–435. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shang B, Cao Z and Zhou Q: Progress in
tumor vascular normalization for anticancer therapy: Challenges and
perspectives. Front Med. 6:67–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Goel S, Duda DG, Xu L, Munn LL, Boucher Y,
Fukumura D and Jain RK: Normalization of the vasculature for
treatment of cancer and other diseases. Physiol Rev. 91:1071–1121.
2011. View Article : Google Scholar : PubMed/NCBI
|