Introduction
Hepatocellular carcinoma (HCC) was the sixth most
commonly diagnosed cancer and the third leading cause of
cancer-associated mortality worldwide in 2018, with a 5-year
relative survival rate of <20% (1). The incidence of HCC is increasing
faster than for any other cancer type in both men [5-year average
annual percent change (AAPC) = 2.8%] and women (5-year AAPC = 3.8%)
(2). Currently, surgical resection
or liver transplantation are the only curative treatments for
early-stage HCC; however, only a small portion of patients are
candidates for these curative treatments as most patients are
diagnosed at an advanced stage (3). Transcatheter arterial
chemoembolization or systemic chemotherapy have been reported to
improve the survival of patients with advanced HCC (4). However, the clinical benefits remain
unfavorable, most likely due to HCC malignant progression, such as
acquired drug resistance, metastasis and recurrence (5). Therefore, further understanding of
the underlying mechanisms is critical for the development of more
effective therapies.
Inhibitor of differentiation 1 (ID1), a
dominant-negative inhibitor of the basic helix-loop-helix
transcription factor, has been reported to mediate diverse cellular
functions, including inhibition of differentiation, as well as
promotion of cancer neo-angiogenesis and metastasis (6). Increased ID1 expression has been
demonstrated to be associated with both cancer progression and poor
prognosis in several types of solid human tumors, such as breast,
lung, brain and pancreatic cancer (7-11). A
previous study proposed that ID1 mediates androgen-stimulated HCC
cell migration and invasion (12).
In addition, it was previously identified that ID1 confers drug
resistance to oxaliplatin or sorafenib in HCC cells (13,14),
indicating an important role of ID1 in HCC malignant progression
and suggesting ID1 may be a potential target for the treatment of
advanced HCC. However, to the best of our knowledge, the precise
biological function of ID1 in HCC progression remains largely
unknown and there is no effective therapeutic strategy targeting
ID1 currently available.
Aurora kinase A (AURKA), a serine/threonine kinase,
is a crucial regulator of cell cycle progression and mitosis
(15). A number of studies have
reported that AURKA can function as an oncoprotein and therefore
may be a promising therapeutic target in various types of malignant
tumor, including HCC (15-17). An aberrant high expression of AURKA
has been observed in HCC tissues and is significantly correlated
with poor prognosis (17). It has
been demonstrated that AURKA is involved in uncontrolled
proliferation, anti-apoptotic properties and the
epithelial-mesenchymal transition in cancer cells, as well as the
self-renewal cancer stem cell phenotype (16). Therefore, a number of AURKA
inhibitors (AKI) have been produced in the past decades, some of
which are already being evaluated in clinical trials (18,19).
However, to the best of our knowledge, the mechanism that is
responsible for the dysregulation of AURKA expression remains
unknown, and no literature has reported the association between ID1
and AURKA in cancer, particularly HCC. Notably, previous studies
have reported that ID1 can be localized at centrosomes and is
implicated in mitotic regulation, while AURKA is a
centrosome-localized mitotic kinase (20-22).
Furthermore, overexpression of either ID1 or AURKA results in
similar centrosome amplification, suggesting a potential
association between ID1 and AURKA (20,23).
A previous study investigating nasopharyngeal epithelial cells
reported that knockdown of AURKA largely rescued the ID1-induced
abnormal mitotic phenotype (24),
further supporting the hypothesis that AURKA may participate in the
biological function of ID1 in HCC.
The present study identified that ID1 protein is
upregulated in HCC and closely associated with tumor recurrence and
poor survival in patients with HCC. In addition, the expression
levels of ID1 and AURKA were positively correlated. Patients with
high expression of both ID1 and AURKA exhibited poor
recurrence-free survival and overall survival rates. Further
mechanistic studies revealed that the novel downstream AURKA/Myc
signaling pathway mediated ID1-induced HCC cell growth, migration,
invasion, colony formation and drug resistance, which may promote
understanding of HCC progression and provide a novel target to
improve the therapeutic treatment strategies for patients with
advanced HCC.
Materials and methods
Patients
The present study included 81 patients (67 males and
14 females; mean age, 52±12.6 years) who underwent HCC resection
between April 2008 and January 2011 at Fudan University Affiliated
Zhongshan Hospital (Shanghai, China). Written informed consent was
obtained from each patient. No patient had received any
preoperative anticancer therapy. HCC was diagnosed based on the
diagnostic criteria issued by the American Association for the
Study of Liver Diseases (25), and
was staged according to the Barcelona Clinic Liver Cancer staging
system (26). The clinical data
were collected, including age, sex, Child-Pugh classification,
HBsAg, TNM grade, tumor number and tumor diameter (Table I). All patients were followed up
until May 2013, with a median follow-up time of 46 months. The
present study was approved by the Clinical Research Ethics
Committee of Fudan University Affiliated Zhongshan Hospital and
carried out in accordance with the Declaration of Helsinki.
 | Table IClinical and pathological
characteristics of recruited patients with hepatocellular
carcinoma. |
Table I
Clinical and pathological
characteristics of recruited patients with hepatocellular
carcinoma.
| Characteristic | No. of patients
(%) |
|---|
| Age, years | |
| <60 | 59 (72.8) |
| ≥60 | 22 (27.2) |
| Sex | |
| Male | 67 (82.7) |
| Female | 14 (17.3) |
| Child-Pugh
classification | |
| A | 81 (100) |
| HBsAg | |
| Positive | 68 (84.0) |
| Negative | 13 (16.0) |
| TNM grade | |
| I | 38 (46.9) |
| II | 21 (25.9) |
| III | 22 (27.2) |
| Tumor thrombus | |
| Yes | 32 (39.5) |
| No | 49 (60.5) |
| Tumor number | |
| Single | 67 (82.7) |
| Multiple | 14 (17.3) |
| Tumor diameter,
cm | |
| ≤5 | 35 (43.2) |
| >5 | 46 (56.8) |
Antibodies and reagents
The following antibodies were used: Anti-ID1 (cat.
no. ab134163; Abcam), anti-ID1 (cat. no. sc-734; Santa Cruz
Biotechnology, Inc.), anti-AURKA rabbit mAb (cat. no. 14475; Cell
Signaling Technology, Inc.), anti-AURKA mouse mAb (cat. no.
ab13824; Abcam), anti-Myc (cat. no. 9402; Cell Signaling
Technology. Inc.), anti-APC/CCdh1 (cat. no. ab3242;
Abcam), anti-ubiquitin (cat. no. ab7780; Abcam) and
anti-human/mouse GAPDH (cat. no. 60004-1-Ig; ProteinTech Group,
Inc.). VX689 (cat. no. S2770) was purchased from Selleck Chemicals.
Cycloheximide (CHX; cat. no. 2112) was obtained from Cell Signaling
Technology, Inc. BioCoat Matrigel invasion Chambers were obtained
from BD Biosciences. Primers for reverse transcription-quantitative
PCR (RT-qPCR) were generated by Sangon Biotech Co., Ltd.
PrimeScript RT reagent kit (cat. no. DRR037A) and SYBR Premix Ex
Taq (cat. no. DRR081) were purchased from Takara Bio, Inc. All
chemicals were of reagent grade or higher.
Cell culture, plasmids and
transfection
The human HCC cell line MHCC-97H was established at
our Liver Cancer Institute in Fudan University Affiliated Zhongshan
Hospital (27) and tested for
mycoplasma contamination before use. MHCC-97H cells in passages
12-15 were used in the experiments in the present study. Cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% antibiotic
mixture (penicillin, streptomycin and amphotericin B;
Sigma-Aldrich; Merck KGaA) at 37°C in a 5% CO2
incubator.
MHCC-97H cells with stable ID1 transfection
(97H-ID1) were generated using ID1-overexpressing lentiviruses
(Shanghai GeneChem Co., Ltd.). MHCC-97H cells were prepared and
infected at a multiplicity of infection (MOI) of 10 with empty
vector, ID1-overexpressing lentivirus or 3xFlag-tagged
AURKA-overexpressing lentivirus (Shanghai GeneChem Co., Ltd.) at
37°C for 24 h. For AURKA-knockdown, short hairpin RNA (shRNA)
targeting AURKA was delivered by lentiviral infection into MHCC-97H
cells with a MOI of 10 (Shanghai GeneChem Co., Ltd). After 3 days
of transfection, cells were used for subsequent experiments. The
sequence of AURKA shRNA was 5′-CATTCCTTTGCAAGCACAA-3′. A scrambled
shRNA (5′-TTCTCCGAACGTGTCACGT-3′) was used as the control (Shanghai
GeneChem Co., Ltd.).
Immunohistochemistry (IHC) and H-Scoring
of tissue microarray (TMA)
IHC staining was performed with a TMA consisting of
the 81 HCC cases. The TMA was prepared as described previously
(28). The TMA section (thickness,
4-µm) was deparaffinized and rehydrated, followed by
heat-induced antigen retrieval and blocking with 5% goat serum
(Thermo Fisher Scientific, Inc.) at room temperature for 2 h. The
section was then incubated with anti-ID1 rabbit antibodies (1:500)
or anti-AURKA mouse antibodies (1:200) overnight at 4°C. The
following day, the section was washed and incubated with
biotin-conjugated anti-mouse (cat. no. B0529; 1:300; Sigma-Aldrich;
Merck KGaA) or anti-rabbit secondary antibodies (cat. no. B8895;
1:300; Sigma-Aldrich; Merck KGaA) for 1 h at room temperature.
Subsequently, the ABC enhanced Vectastain kit system (Vector
Laboratories, Inc.; Maravai LifeSciences), a DAB peroxidase
substrate kit (Thermo Fisher Scientific, Inc.) and hematoxylin
solution (cat. no. ab220365; Abcam) were used for detection and
visualization. Sections were stained with hematoxylin for 2 min at
room temperature, Images were obtained using a light microscope
(magnification, ×200).
The IHC staining was quantified as the H-score,
which has been validated in breast cancer (29). Typical images (4 random images per
sample) were analyzed and divided into four intensity levels: 0,
negative; 1, weak; 2, intermediate; and 3, strong brown staining.
The H-score (between 0 and 300) for each sample was calculated as
follows: H-score = (% of cells stained at intensity 1 × 1) + (% of
cells stained at intensity 2 × 2) + (% of cells stained at
intensity 3 × 3). Using the median value of the H-score as a cutoff
value, patients with HCC were divided into low and high ID1 or
AURKA expression groups. For further analysis, patients were also
divided into three groups based on H-scores: i)
ID1highAURKAhigh group; ii)
ID1highAURKAlow/ID1lowAUKRAhigh
group; and iii) ID1lowAURKAlow group.
Cell proliferation assay
Cell proliferation was analyzed using a Cell
Counting Kit-8 (CCK-8; Abcam) assay. HCC cells were plated in
96-well plates at a density of 3×103 cells/well in DMEM
supplemented with 10% FBS and cultured for 1, 2, 3, 4, 5 and 6
days. CCK-8 reagent was then added to each well, according to the
manufacturer's instructions. The absorbance at 450 nm of each well
was measured using a microplate spectrophotometer.
Tumor cell viability assay
Currently, an oxalipl-atin-containing regimen
(FOLFOX4), sorafenib and lenvatinib are the first-line chemotherapy
drugs for advanced HCC (30).
Among them, due to its high efficiency and lower cost,
oxaliplatin-containing regimen is the most widely used chemotherapy
in the treatment of advanced HCC, particularly in China (31). Furthermore, our previous study
demonstrated that ID1 was highly expressed in oxaliplatin-treated
HCC tumors and was critical for the chemoresistance of HCC cells to
oxaliplatin (13) Therefore, to
investigate the mechanism underlying ID1-promoted chemoresistance,
the present study used oxaliplatin to analyze the drug sensitivity
of HCC cells. Cells were plated in 96-well plates at a density of
1×104 cells/well and exposed to oxaliplatin
(Sigma-Aldrich; Merck KGaA) at increasing concentrations (0, 5, 10,
50, 100 and 150 µmol/l) at 37°C for 48 h. The CCK-8 reagent
was then added to each well according to the manufacturer's
instructions. The absorbance of each well was measured at 450 nm
using a microplate. Based on results of three independent
experiments, the half-maximal inhibitory concentration
(IC50) was calculated for each group.
Tumor cell invasion assay
Invasion of tumor cells was examined by Transwell
assay with BioCoat Matrigel invasion Chambers (pore size,
8-µm). Cells (4×104 cells per chamber) were
seeded in serum-free medium in the upper chamber, and 10% FBS was
used as chemoattractant in the lower chamber. Cells were then
cultured for 24 h, and the invasive cells were stained with 0.05%
crystal violet at room temperature for 20 min and images were
obtained using a light microscope (magnification, ×200).
Experiments were repeated at least three times.
Tumor cell migration assay
The wound healing assays were performed according to
the methods described by Slevin et al (32). MHCC-97H and 97H-ID1 cells were
separately grown on cover slips in 6-well plates to 100%
confluence, and monolayers were then scratched to form a wound area
using 200-µl sterile pipette tips. MHCC-97H cells were then
cultured with DMEM containing 1% FBS, and 97H-ID1 cells were
cultured with solvent control or 250 nM VX689 for a further 24 h.
Cells were fixed with 4% formalin at room temperature for 1 h, and
images were captured by light microscope (magnification, ×100).
Experiments were repeated at least three times.
Cell colony formation assay
HCC cells were plated at a density of
1×103 cells/well in 6-well plates and cultured with DMEM
containing 1% FBS. Culture medium was changed every 3 days, and the
colonies were stained with 5% Giemsa stain at room temperature for
20 min and images were obtained using a camera. Quantification was
performed using Image-Pro plus v5.0 (Media Cybernetics, Inc.).
Experiments were repeated at least three times.
RT-qPCR
Total RNA was isolated from HCC cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse-transcribed to cDNA using PrimeScript RT reagent kit
according to the manufacturer's instructions. For qPCR, SYBR Premix
Ex Taq was used according to the manufacturer's instructions. The
thermocycling conditions were as follows: 95°C for 30 sec; followed
by 40 cycles of 95°C for 5 sec and 60°C for 20 sec; and 95°C for 15
sec. The primers used for qPCR are listed in Table II. The 2−ΔΔCq method
was used for quantification (33).
GAPDH was used as the reference gene.
| Table IIPrimers for reverse
transcription-quantitative PCR. |
Table II
Primers for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| Forward | Reverse |
|---|
| ID1 |
ACACAAGATGCGATCGTCC |
GGAATCCGAAGTTGGAACC |
| AURKA |
TTCAGGACCTGTTAAGGCTACA |
ATTTGAAGGACACAAGACCCG |
| MYC |
GGAGGCTATTCTGCCCATTTG |
CGAGGTCATAGTTCCTGTTGGTG |
| GAPDH |
GGTGGTCTCCTCTGACTTCAACA |
GTTGCTGTAGCCAAATTCGTTGT |
Western blotting, pulse-chase analysis,
ubiquitination analysis and co-immunoprecipitation assay
Western blot analysis was performed as previously
described (34). A BCA kit (Thermo
Fisher Scientific, Inc.) was used for protein determination, and 30
µg cell lysates were separated by 10% SDS-PAGE and
transferred to nitrocellulose membranes (Thermo Fisher Scientific,
Inc.) using the iBlot gel transfer device (Thermo Fisher
Scientific, Inc.). The membranes were separately blotted with
primary antibodies (1:1,000) against ID1, AURKA, Myc or
APC/CCdh1 at 4°C overnight, and were subsequently
incubated with HRP-conjugated anti-mouse or anti-rabbit secondary
antibodies (cat. nos. ab205719 and ab205718; Abcam; 1:5,000) at
room temperature for 1 h. For pulse-chase analysis, CHX was used to
inhibit protein production. Cells were treated with 25 mmol/l CHX
at 37°C for 0, 1, 2, 4, 6, and 8 h, then the cell lysates were
subjected to western blot analysis. Image J 1.43 software (National
Institutes of Health) was used for densitometric analysis. For
ubiquitination analysis, cells were pretreated with 10
µmol/l MG132 at 37°C for 6 h to inhibit proteasome-mediated
proteolysis, followed by immunoprecipitation and western blot
analysis. For the co-immunoprecipitation assay, cells were directly
lysed with lysis buffer (Tris-HCl, 50 mmol/l, pH 8.0; NaCl, 150
mmol/l; EDTA, 5 mmol/l; 0.5% NP-40). Lysates (1 mg) were then
incubated with 1 µg primary antibodies against ID1 or AURKA
on a rotating wheel at 4°C overnight and then pulled down using
protein A/G agarose beads at 4°C for 6 h. Beads were collected by
centrifugation at 200 × g for 2 min at 4°C and washed 5 times with
ice-cold PBS buffer, and then boiled in 2 × SDS-PAGE
electrophoresis sample buffers, following by western blot
analysis.
Oncomine database analysis
The Oncomine 4.5 database (www.oncomine.org), a publicly available database of
published cancer gene expression profiles, was queried for
alterations in ID1 and AURKA gene expression in HCC. The Cancer
Genome Atlas (TCGA; www.cancer.gov), Guichard_Liver
(35) and Lamb JR_Liver DNA
(36) datasets were retrieved for
further analysis, comparing the difference between HCC and normal
tissues, as well as the correlation between ID1 and AURKA. Data
were log-transformed and median-centered, and all statistical
analyses were performed using functions implemented in
Oncomine.
Statistical analysis
All quantified data are presented as mean ± standard
deviation. Statistical analysis was performed using SPSS v22.0 (IBM
Corp.). Differences between two groups were analyzed using an
unpaired Student's t-test. Differences among three or more groups
were analyzed by one-way analysis of variance followed by Tukey's
multiple comparison test. In addition, data in the 2×2 contingency
table were analyzed by χ2 test. Pearson's correlation
analysis was performed to determine the correlation between ID1 and
AURKA DNA levels. Differences in survival curves were analyzed by
Kaplan-Meier analysis and a log-rank test. All data presented are
representative of three independent experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
Patients with HCC exhibit an
overexpression of ID1, which is associated with poor prognosis
To reveal the ID1 expression pattern in HCC, ID1
expression was first analyzed in TCGA, Guichard_Liver and Lamb
JR_Liver DNA datasets. Oncomine analysis revealed a significantly
higher expression level of ID1 gene in HCC compared with normal
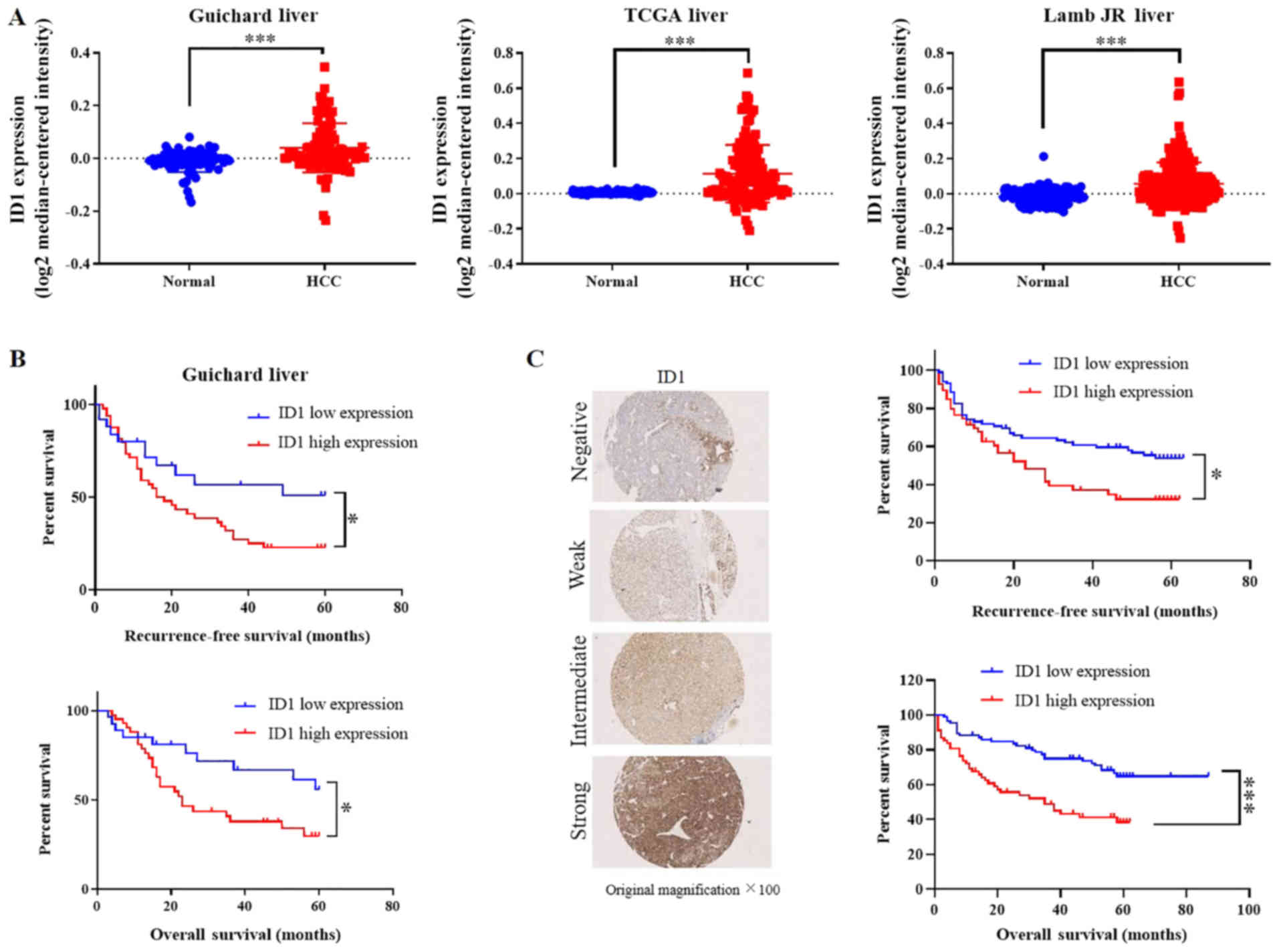
tissues (P<0.001; Fig. 1A).
Survival analysis using the Guichard_Liver dataset revealed that a
high expression of ID1 gene was significantly associated with a
poor recurrence-free survival rate [hazard ratio (HR), 1.865;
P<0.05] and low overall survival rate (HR, 2.035; P<0.05;
Fig. 1B). To determine whether
this association also existed at the protein level, IHC staining
was performed with a human HCC TMA, which included tissues obtained
from 81 patients with HCC undergoing surgical resection. The
clinical and pathological information of the patients is presented
in Table I. It was revealed that
ID1 was mostly located in the cytoplasm and sometimes located in
the nucleus and the area surrounding it (data not shown). According
to the expression intensity of ID1, IHC results were divided into
four intensity levels: Negative, weak, intermediate, and strong
(Fig. 1C). Survival analysis based
on the H-score was then performed. This demonstrated that the ID1
protein level was also significantly associated with a poor
recurrence-free survival rate (HR, 1.831; P<0.05) and low
overall survival rate (HR, 2.397; P<0.0001; Fig. 1C). These results implied that ID1
was a risk factor for poor prognosis and most likely facilitated
HCC progression.
Positive correlation of ID1 and AURKA in
patients with HCC and the clinical relevance
Given the close interaction between ID1 and AURKA in
nasopharyngeal epithelial cells (24), the present study investigated
whether this close association also exists in HCC by analyzing the
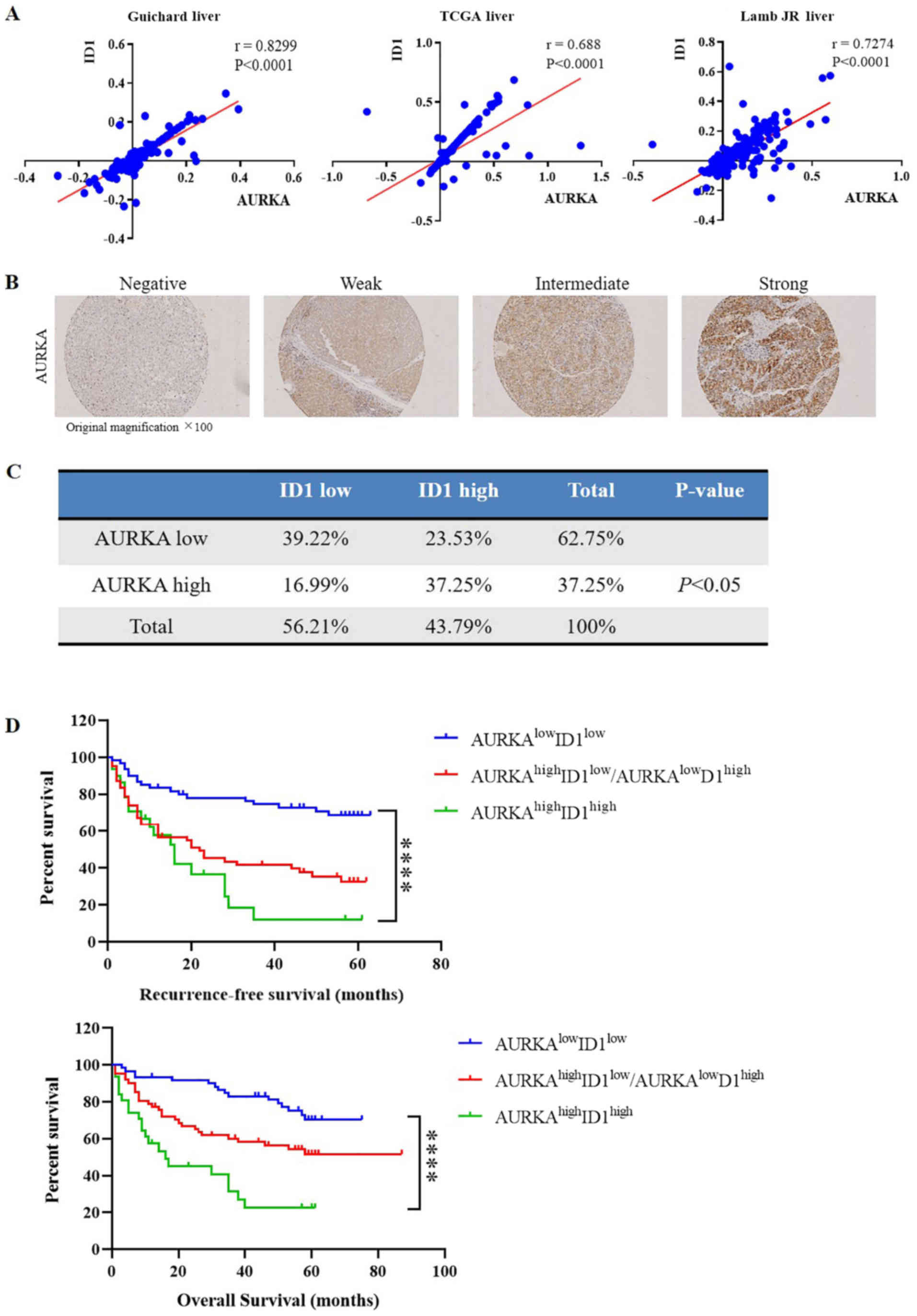
three aforementioned independent DNA datasets. Notably, a
significant positive correlation was identified between ID1 and
AURKA in all three datasets (P<0.0001; Fig. 2A). To confirm these results and
further elucidate the clinical relevance of ID1 and AURKA
expression in HCC, AURKA expression was measured in the 81 HCC
samples by IHC staining (Fig. 2B).
Similar to ID1, AURKA was mostly located in the cytoplasm (data not
shown). Consistently, survival analysis revealed that the protein
level of ID1 was significantly associated with AURKA expression
(P<0.05; Fig. 2C). Furthermore,
the patients with HCC were divided into three groups based on
H-scores: i) ID1highAURKAhigh group (n=29);
ii)
ID1highAURKAlow/ID1lowAUKRAhigh
group (n=30); and iii) ID1lowAURKAlow group
(n=22). Survival analysis demonstrated a significant difference
among all three groups (P<0.0001). Additionally, compared with
the ID1lowAURKAlow group, patients in
ID1highAURKAhigh group had a significantly
lower overall survival rate (HR, 4.801; P<0.0001) and
recurrence-free survival rate (HR, 4.084; P<0.0001), indicating
that high expression of both ID1 and AURKA was associated with an
unfavorable prognosis in HCC (Fig.
2D).
Overexpression of ID1 significantly
promotes HCC cell proliferation, migration, invasion, colony
formation and chemoresistance against oxaliplatin
To investigate the functional mechanism of ID1 in
HCC malignant progression, a stable ID1-overexpressing cell line
(97H-ID1) was established. The successful establishment of 97H-ID1
was confirmed by western blotting (Fig. 3A). Our previous study demonstrated
that silencing ID1 expression inhibits oxaliplatin-resistant HCC
cell proliferation, clone formation and chemoresistance (13). Consistently, the present study
demonstrated that, compared with the control cells, 97H-ID1 cells
were more resistant to oxaliplatin (Fig. 3B), with an IC50 value of
45.3±5.65 µmol/l in control cells compared with 165.4±14.12
µmol/l in 97H-ID1 cells (P<0.001; Fig. 3C). In addition, it was identified
that ID1 overexpression significantly promoted HCC cell
proliferation at days 4-6 (P<0.05; Fig. 3D). Furthermore, ID1 overexpression
markedly enhanced HCC cell migration (Fig. 3E), invasion (P<0.001; Fig. 3F) and colony formation (Fig. 3G). These results further indicated
that ID1 promotes HCC malignant progression, contributing to tumor
expansion, metastasis and recurrence, which partially explains the
close association of ID1 with adverse clinical prognosis.
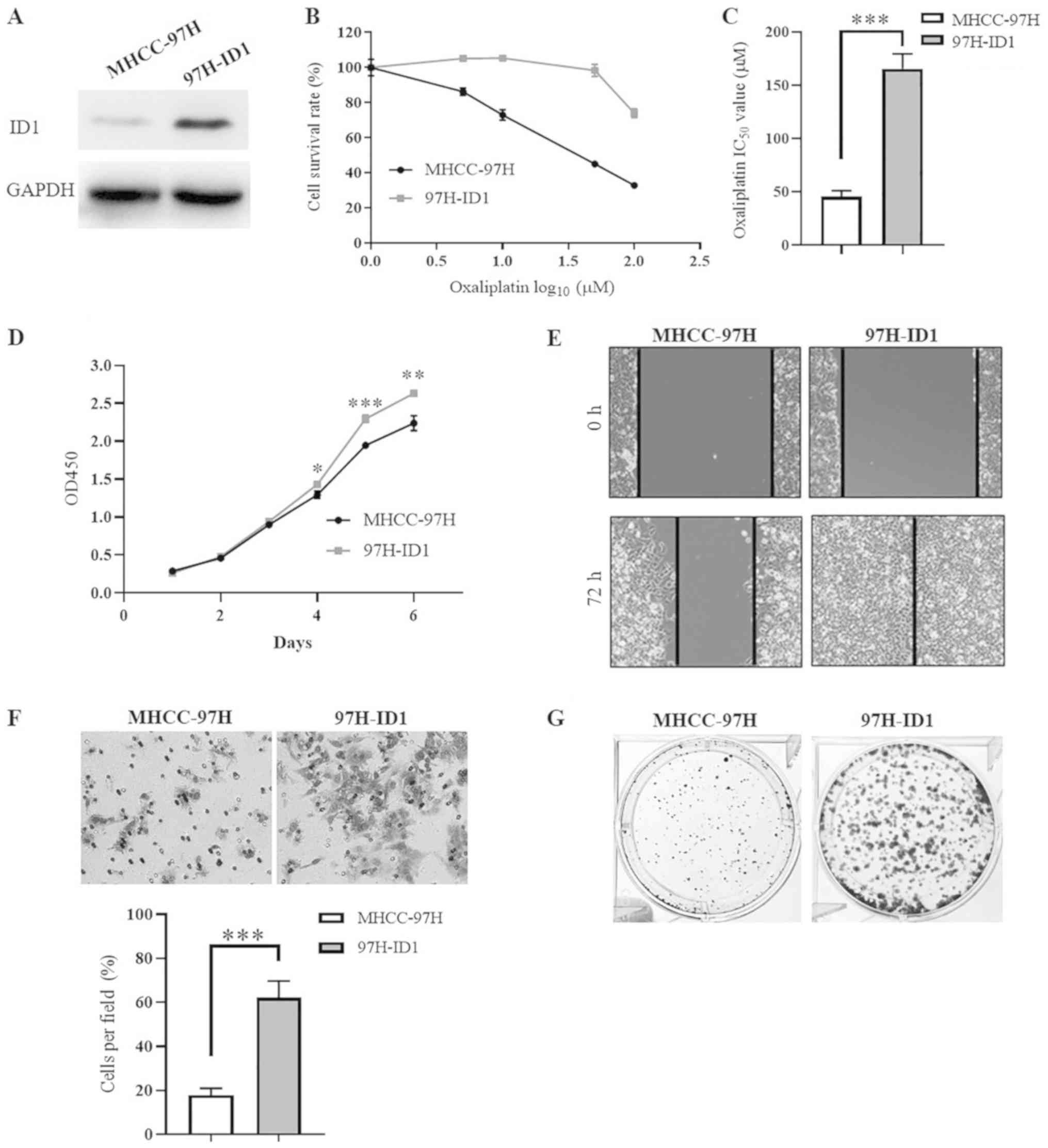 | Figure 3ID1 promotes HCC cell proliferation,
migration, invasion, colony formation and chemoresistance. (A)
Western blot analysis confirmed the successful establishment of
ID1-overexpressing HCC cells (97H-ID1). (B) Results of CCK-8 assay
demonstrated the different cytotoxic effects of oxaliplatin on
97H-ID1 cells and the parental cells MHCC-97H. (C) Statistical
analysis revealed a significant difference in the IC50
of oxaliplatin in 97H-ID1 and MHCC-97H cells.
***P<0.001. (D) CCK-8 assay revealed that ID1
overexpression significantly promoted HCC cell proliferation at
days 4-6. *P<0.05, **P<0.01,
***P<0.001 vs. MHCC-97H cells. (E) Wound healing
assay demonstrated an increase of cell migration induced by ID1
overexpression. Magnification, ×100. (F) Transwell invasion assay
showed that ID1 overexpression significantly enhanced HCC cell
invasion through Matrigel. ***P<0.001. Magnification,
×200. (G) ID1 over-expression facilitated the ability of HCC cells
to form colonies. Each experiment was repeated independently at
least three times. HCC, hepatocellular carcinoma; ID1, inhibitor of
differentiation 1; CCK-8, Cell Counting Kit-8; IC50,
half-maximal inhibitory concentration; OD, optical density. |
AURKA mediates the ID1-induced HCC cell
malignant progression
Based on the positive correlation identified between
ID1 and AURKA in HCC, it was speculated that AURKA may be involved
in ID1-induced HCC cell progression. Notably, it was identified
that, along with an increased ID1 expression, AURKA expression was
also increased in 97H-ID1 cells compared with MHCC-97H cells
(Fig. 4A). To verify if AURKA
mediates ID1-induced HCC malignant progression, a highly selective
AKI VX689 was used (Fig. 4B).
Compared with the control, 250 nM VX689 significantly reversed the
ID1-induced resistance to oxaliplatin in 97H-ID1 cells (Fig. 4C), with the IC50 of
oxaliplatin in 97H-ID1 cells decreasing from 162.1±10.63 to
54.73±8.45 µmol/l following treatment with VX689
(P<0.001; Fig. 4C). To address
the chemosensitizer role of AKI, different concentrations were
evaluated (data not shown) and a low dose (250 nM) of VX689 was
selected. This dose alone had no cytotoxic effect on HCC cell
viability (P>0.05; Fig. 4D),
which excludes the possibility that the increased sensitivity to
oxaliplatin was due to a direct cytotoxic effect of VX689 itself.
In addition, it was identified that VX689 significantly inhibited
97H-ID1 cell proliferation at concentrations ≥1,500 nM
(P<0.0001; Fig. 4E), while
concentrations ≤500 nM had no significant effect at day 5. However,
treatment with 250 nM VX689 impaired the ID1-promoted HCC cell
migration (Fig. 4F), invasion
(P<0.001; Fig. 4G) and colony
formation (Fig. 4H), suggesting
that the malignant biological phenotypes induced by ID1 are largely
dependent on the activation of AURKA.
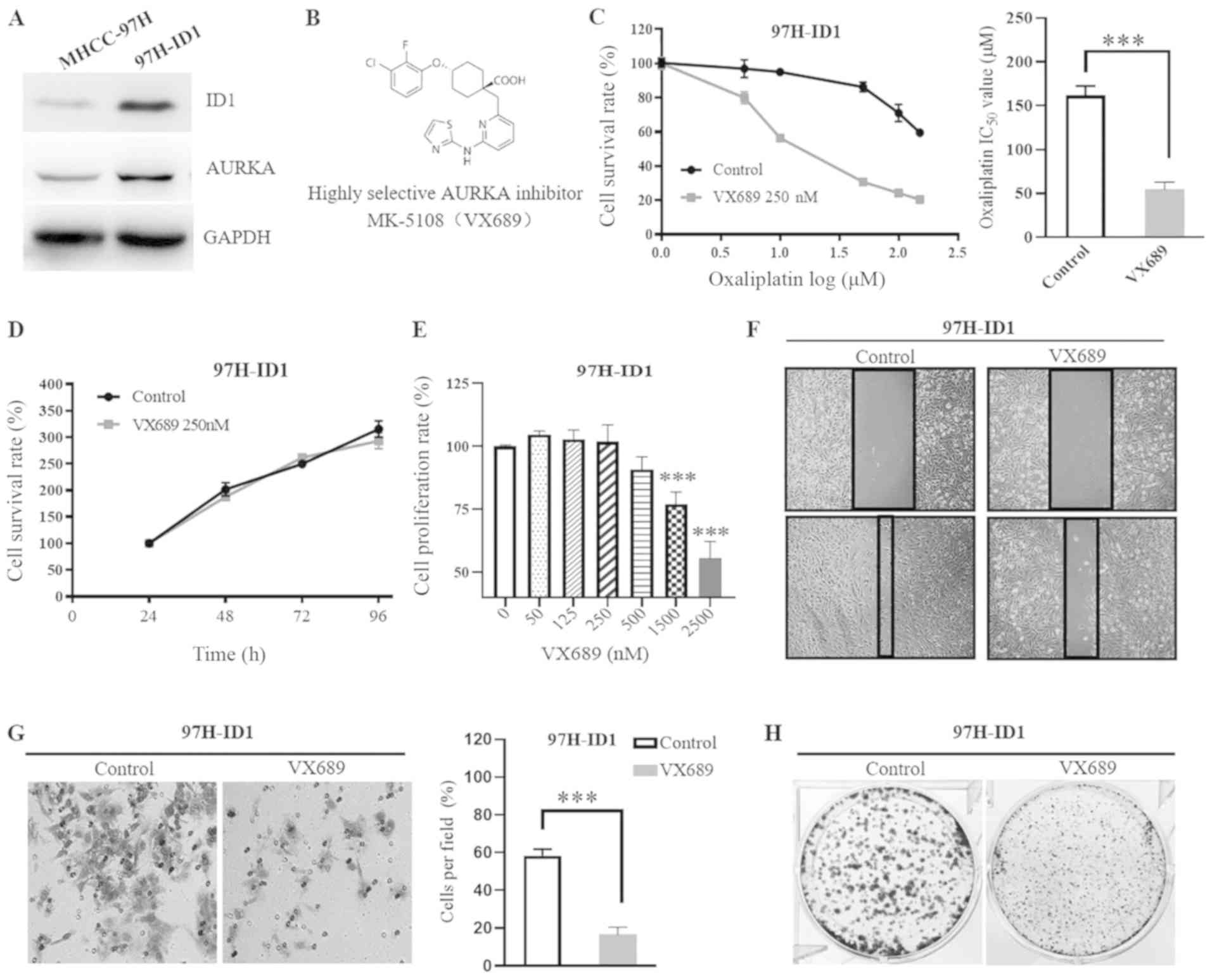 | Figure 4AURKA mediates ID1-induced HCC cell
malignant progression. (A) Western blotting revealed an increased
expression of AURKA protein in 97H-ID1 cells. (B) Structural
formula of the highly selective AURKA inhibitor MK-5108, also known
as VX689. (C) Co-treatment with 250 nM AURKA inhibitor VX689 for 48
h reversed the ID1-induced chemoresistance to oxaliplatin.
Statistical analysis revealed that addition of 250 nM VX689
significantly decreased the IC50 value of oxaliplatin in
97H-ID1 cells. ***P<0.001. (D) Treatment with 250 nM
VX689 alone had no cytotoxic effect on 97H-ID1 cells. (E) Cell
Counting Kit-8 assay revealed the effects of different
concentrations of VX689 on HCC cell proliferation. A low dose of
VX689 (≤500 nM) had no significant effect on HCC cell
proliferation, while a high dose (≥1,500 nM) significantly
inhibited cell growth. ***P<0.001 vs. 0 nM VX689.
VX689 at a low concentration (250 nM) could inhibit ID1-enhanced
HCC (F) migration (magnification, ×100), (G) colony formation and
(H) invasion (magnification, ×200). ***P<0.001.
AURKA, aurora kinase A; ID1, inhibitor of differentiation 1; HCC,
hepatocellular carcinoma; 97H-ID1 cells, ID1-overexpressing
MHCC-97H cells; IC50, half-maximal inhibitory
concentration. |
ID1 upregulates AURKA via inhibition of
AURKA proteolysis degradation
Since ID1 expression was correlated with AURKA
protein expression in HCC, the present study further analyzed
whether ID1 could transcriptionally activate AURKA. Notably, no
significant change in AURKA mRNA level was observed in 97H-ID1
cells compared with control cells (Fig. 5A), indicating that the regulation
of ID1 on AURKA may be at the protein level. Pulse-chase analysis
results demonstrated that ID1 overexpression significantly impeded
AURKA protein degradation (P<0.01; Fig. 5B). As E3 ubiquitin ligase-mediated
lysine 48 (K48) polyubiquitination and subsequent proteasomal
degradation predominantly controls AURKA protein turnover (37,38),
the present study analyzed the K48 ubiquitination level of AURKA
protein. Consistently, ID1 markedly decreased the ubiquitination of
AURKA protein (Fig. 5C). A
previous study reported that anaphase-promoting complex/cyclosome
Cdh1 (APC/CCdh1) directly binds with AURKA, initiating
the process of ubiquitination-mediated AURKA degradation; and
depletion of APC/CCdh1 expression results in
stabilization of AURKA protein, indicating a crucial role of
APC/CCdh1 in AURKA protein degradation (37). Since ID1 has been reported to bind
with APC/C-associated protein Cdc20 (39), it was speculated that ID1 may also
interact with APC/CCdh1. A co-immunoprecipitation assay
revealed that, similar to AURKA, ID1 was a direct binding partner
of APC/CCdh1 in HCC cells (Fig. 5D), while ID1 and AURKA did not bind
with each other, suggesting that ID1 may stabilize AURKA indirectly
by competitive binding with APC/CCdh1. Taken together,
these results revealed a novel mechanism in which excessive ID1
competitively inhibits APC/CCdh1-mediated AURKA protein
degradation, leading to upregulation of AURKA.
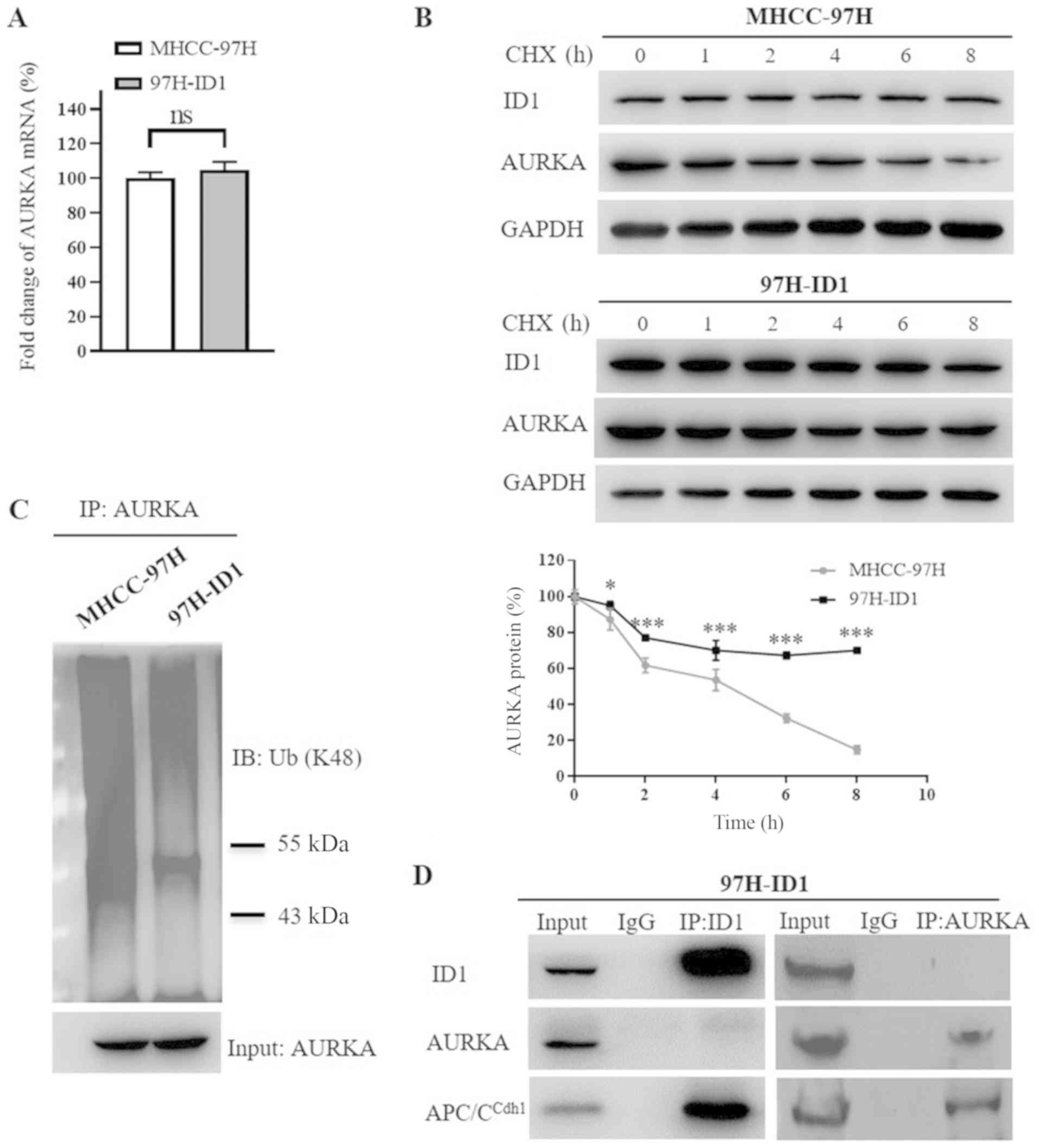 | Figure 5ID1 upregulates AURKA expression by
competitively binding with APC/CCdh1, and thus inhibiting AURKA
protein degradation. (A) Reverse transcription-quantitative PCR
demonstrated that ID1 overexpression had no significant effect on
AURKA mRNA expression level. (B) Pulse-chase analysis demonstrated
that ID1 overexpression inhibited the proteolysis degradation of
AURKA protein. The degradation profile of AURKA protein was
examined by blocking protein synthesis with CHX for the indicated
times. The AURKA protein levels were examined by western blotting,
and the plot represents the AURKA protein degradation rates.
*P<0.05, ***P<0.001 vs. MHCC-97H cells.
(C) Decreased K48 ubiquitination of AURKA protein induced by ID1
overexpression. To detect the levels of ubiquitinated AURKA,
proteasome inhibitor MG132 was used to inhibit AURKA proteolysis,
then AURKA protein was pulled down by specific a AURKA antibody.
The cell pellets were analyzed using antibodies against K48
ubiquitin. The input level of AURKA protein was shown as reference.
(D) Co-immunoprecipitation assay revealed that both ID1 and AURKA
directly interact with APC/CCdh1, which is responsible for the
ubiquitin-mediated proteasomal-dependent degradation of AURKA. No
direct binding between ID1 and AURKA was found. ns, no
significance; AURKA, aurora kinase A; ID1, inhibitor of
differentiation 1; HCC, hepatocellular carcinoma; 97H-ID1 cells,
ID1-overexpressing MHCC-97H cells; APC/CCdh1, anaphase-promoting
complex/cyclosome Cdh1; ns, no significance; CHX, cycloheximide;
IB, immunoblotting. |
AURKA promotes Myc expression by
facilitating its mRNA expression and simultaneously inhibiting its
protein degradation
The oncoprotein Myc, which represents a central hub
of pro-tumorigenic signaling networks, is frequently hyperactivated
and implicated in the pathogenesis of several types of human
cancer, including HCC (40). In
light of the close association of AURKA with Myc family members
(41,42), and our previous study that
indicated that knockdown of ID1 downregulates Myc expression
(13), it was speculated that
AURKA may activate the Myc pathway to induce the biological effects
of ID1 in HCC. The present results demonstrated that, accompanied
with increased AURKA levels, ID1 overexpression also led to a
marked upregulation of Myc expression in HCC cells (Fig. 6A). To reveal whether Myc is
involved in the biological function of ID1, the Myc inhibitor
(MYCI) 10058-F4 was used. Notably, addition of 100 µM MYCI
impaired ID1-mediated HCC cell chemoresistance (Fig. 6B). The IC50 value of
oxaliplatin in 97H-ID1 cells decreased from 160.8±17.19 to
107.6±5.97 µmol/l following the addition of MYCI (P<0.01;
Fig. 6C), verifying that Myc
activation is a critical downstream signaling pathway of ID1.
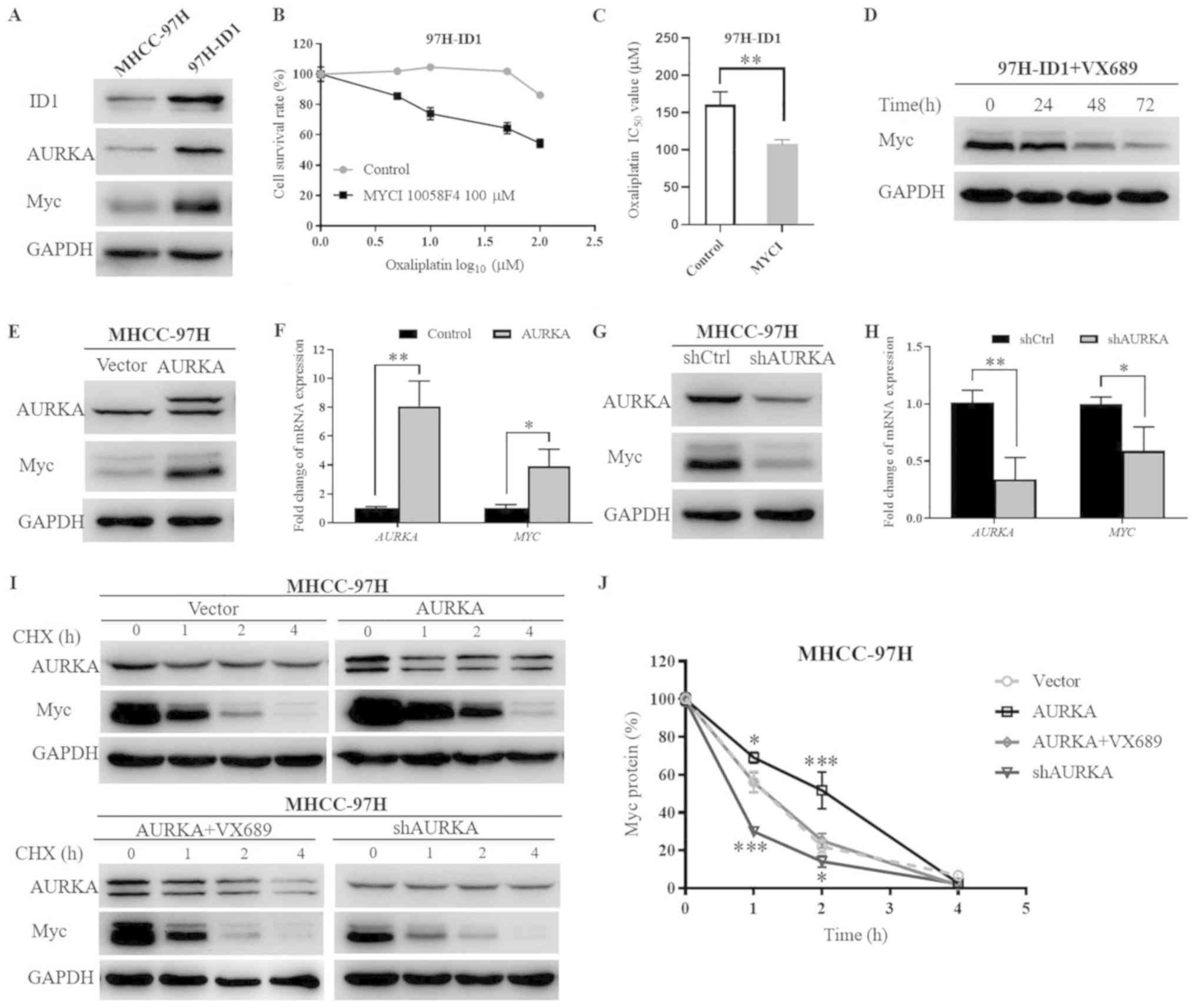 | Figure 6ID1-upregulates the AURKA-induced Myc
signaling pathway at both the transcriptional and
post-transcriptional levels. (A) Western blotting demonstrated that
ID1 overexpression increased AURKA and Myc levels. (B) Addition of
100 µM MYCI (10058-F4) for 48 h impeded ID1-induced HCC
chemoresistance to oxaliplatin. (C) Statistical analysis of the
IC50 values of oxaliplatin in 97H-ID1 cells with or
without MYCI treatment. **P<0.01. (D) AURKA inhibitor
VX689 reserved ID1-upregulated Myc expression. (E) Western blotting
demonstrated the effect of AURKA overexpression on Myc protein
levels. The extra larger-size band of AURKA in AURKA-overexpressing
cells was due to the transfection of a 3xFlag-tagged
AURKA-overexpression lentivirus. (F) Reverse
transcription-quantitative PCR revealed the effect of AURKA
overexpression on Myc mRNA expression. (G) Effect of
AURKA-knockdown on Myc protein levels. (H) Effect of AURKA
knockdown on Myc mRNA expression. *P<0.05,
**P<0.01. (I) AURKA expression effected the protein
stability of Myc. Pulse-chase analysis of Myc protein stability was
performed under conditions of AURKA-overexpression alone,
AURKA-overexpression with AURKA inhibitor VX689, or
AURKA-knockdown. Parental cells MHCC-97H were used as a control.
(J) Statistical analysis of Myc protein degradation rates.
*P<0.05, ***P<0.001 vs. vector. AURKA,
aurora kinase A; ID1, inhibitor of differentiation 1; HCC,
hepatocellular carcinoma; 97H-ID1 cells, ID1-overexpressing
MHCC-97H cells; MYCI, Myc inhibitor; IC50, half-maximal
inhibitory concentration; shRNA, short hairpin RNA. |
Notably, it was identified that treatment with AKI
VX689 markedly decreased Myc expression over time in 97H-ID1 cells,
implying that ID1 regulates Myc expression via modulating AURKA
(Fig. 6D). To further reveal how
AURKA regulates Myc expression, MHCC-97H cells with AURKA
overexpression or knockdown were established. The extra larger-size
band of AURKA protein in AURKA-overexpressing cells was due to the
transfection of a 3xFlag-tagged AURKA vector. The molecular size of
the 3xFlag-tag was ~3 kD. Consistent with the previous hypothesis,
it was identified that AURKA overexpression upregulated Myc, while
knockdown of AURKA decreased its protein levels (Fig. 6E and G). RT-qPCR demonstrated
similar trends that AURKA overexpression resulted in a significant
increase of Myc mRNA level, whereas knockdown of AURKA
significantly reduced the mRNA level of Myc (Fig. 6F and H). Notably, pulse-chase
analysis revealed that AURKA also affected Myc protein stability.
Specifically, overexpression of AURKA retarded Myc protein
degradation, which could be reversed by addition of 250 nM VX689.
In addition, knockdown of AURKA significantly facilitated the
degradation of Myc protein (Fig. 6I
and J). Collectively, these results demonstrated a novel
downstream AURKA/Myc signaling pathway that mediates ID1-promoted
HCC cell malignant progression.
Discussion
Acquired chemoresistance and metastasis account for
the majority of the treatment failure in HCC (3,43).
However, the precise molecular mechanism underlying HCC malignant
progression remains largely unknown, which severely hinders the
improvement of therapeutic efficacy. The present study demonstrated
that ID1 protein was upregulated in HCC tumor tissues and
positively associated with early relapse and poor prognosis of
patients with HCC. Furthermore, it was identified that the novel
downstream AURKA/Myc signaling played a role in ID1-promoted HCC
cell growth, migration, invasion and chemoresistance. Blocking
either AURKA or Myc could largely reverse ID1-induced HCC malignant
progression. Further mechanistic studies revealed that ID1
competitively bound with APC/CCdh1 and inhibited
APC/CCdh1-mediated AURKA protein degradation, thus
resulting in increased AURKA expression, which subsequently
enhanced the Myc-activated protumorigenic signaling networks.
Aberrant overexpression of ID1 has previously been
reported in multiple human cancers and is closely associated with
poor survival (11,39). However, little is known regarding
the expression pattern of ID1 and its biological function in HCC.
Our previous study based on TCGA database revealed that a higher
gene expression of ID1 predicted a poorer prognosis in patients
with HCC (13). In the present
study, by using a human HCC TMA, it was demonstrated that ID1
protein levels were upregulated in HCC and positively associated
with tumor recurrence and unfavorable survival. TMA, which was
first reported by Kononen et al (44), has been long demonstrated to be an
effective and efficient tool for immunohistochemical and molecular
studies. Although few researchers claim that TMA may not be
indicative of the entire sample due to the small size of tissue
sections, numerous studies have reported good concordance between
tissue microarray spots and whole sections in IHC studies of
multiple tumor types (45-47). Generally, the present results
largely support that ID1 is an important prognostic factor and is
closely involved in HCC pathology.
Abnormal proliferation, migration and invasion are
important hallmarks of cancer (48). Our previous study clarified that
silencing ID1 inhibits oxaliplatin-resistant HCC cell
proliferation, clone formation and chemoresistance (13). In addition, Cho et al
(49) reported that downregulation
of ID1 by small interfering RNA significantly decreases HCC cell
invasion. The present study identified that ID1 overexpression
significantly promoted aggressive behaviors of MHCC-97H cells,
including increased proliferation, migration, invasion, and colony
formation. In addition, it was demonstrated that ID1 reduced the
sensitivity of HCC cells to oxaliplatin, which partially explained
the close association of ID1 with adverse clinical outcomes in
patients with HCC. These results further supported the hypothesis
that ID1 plays a pivotal role in priming the aggressive behaviors
of HCC cells. However, to the best of our knowledge, currently no
effective inhibitor targeting ID1 is available. Therefore, further
understanding of the functional mechanisms and the downstream
signaling of ID1 is urgently required
A number of studies have reported that AURKA serves
an important role in regulating the phenotypes of numerous
different cancer cells (16,50-53).
However, the regulatory mechanism of AURKA is largely unknown.
Notably, the current study identified that ID1 overexpression
significantly increased AURKA expression in HCC cells, and the AKI
VX689 reversed ID1-induced HCC cell growth, migration, invasion,
colony formation and chemoresistance, indicating that AURKA
mediates the ID1-induced HCC cell malignant progression. A previous
study reported that ID1 can weekly activate the AURKA promoter in
nasopharyngeal epithelial cells (24), whereas, in the present study, no
significant change in AURKA mRNA levels was observed in
ID1-overexpressing HCC cells, which may be due to different cell
types. In addition to the transcriptional activation, AURKA level
is also reported to be controlled by ubiquitination-mediated
proteasomal degradation. Previous studies have shown that in
somatic cells, AURKA is degraded during mitotic exit by the
anaphase-promoting complex, which requires APC/Ccdh1as
the coactivator (37,38). APC/Ccdh1 recognizes and
binds with its substrates, such as AURKA, and then initiates
ubiquitination, leading to ubiquitin-mediated proteasomal
degradation (37). Notably, ID1
has been reported to bind with APC/C-associated protein Cdc20
(39), and a previous report
claimed that ID1 and APC/CCdh1 may be binding partners
in nasopharyngeal epithelial cells (24). Notably, the present study revealed
that ID1 competitively binds with APC/CCdh1, leading to
reduced interaction of APC/CCdh1 with AURKA, which then
inhibited APC/Ccdh1-mediated AURKA protein degradation
and upregulated AURKA expression. Of note, certain studies have
claimed that ID1 can also bind to various components of the 26S
proteasome, such as the S5A subunit, which is important in the
recognition and shuttling of the polyubiquitinated substrates to
the proteasome (54-56). These effects may also block the
AURKA proteasomal degradation, which needs further
confirmation.
Myc is a crucial proto-oncogene that has been
implicated in both tumor initiation and maintenance in most types
of human cancer, and a number of studies have shown that HCC is
dependent on Myc (40-42). Previous findings reported that
AURKA regulates proteolytic turnover of MYCN in neuroblastoma
(42), and a more recent study
suggested that AURKA and Myc interact physically in TP53-altered
cells (41). These previous
studies encouraged us to clarify if Myc is the critical downstream
molecule of the ID1/AURKA signaling pathway. It was identified that
ID1 upregulated Myc activation in an AURKA-dependent manner, which
was largely abolished by AKI. Further analysis demonstrated that
AURKA transcriptionally activated Myc, which was consistent with
previous findings that AURKA activates the Myc promoter (53). Furthermore, the present study
demonstrated that AURKA also affected Myc protein turnover,
implying that AURKA can regulate Myc expression at both the
transcriptional and post-transcriptional levels, subsequently
amplifying the Myc signaling pathway. Notably, unlike the AKI, MYCI
did not completely reverse ID1-induced chemoresistance, suggesting
other mechanisms downstream of AURKA may be involved and need to be
determined in the future.
However, there were several limitations of the
current study. First, only oxaliplatin was used to investigate the
role of ID1/AURKA/Myc signaling in HCC chemoresistance. Since
sorafenib and lenvatinib are also first-line chemotherapy drugs for
advanced HCC (30,57,58),
future studies are required to explore the mechanisms underlying
chemoresistance against sorafenib and lenvatinib, and whether
ID1/AURKA/Myc signaling is involved. Secondly, a larger sample size
would be helpful to further confirm the association of ID1/AURKA
with HCC prognosis. Thirdly, cells should be serum-starved for
wound healing assays; however, 1% FBS was used in the present
study. Furthermore, further studies are required to clarify other
unknown downstream signaling underlying ID1/AURKA, which is ongoing
in our laboratory.
In conclusion, the present study revealed the
critical role of a novel ID1/AURKA/Myc signaling pathway in HCC
malignant progression. Increase expression of ID1 induced HCC cells
to exhibit highly malignant phenotypes, with enhanced metastatic
ability and drug resistance, leading to HCC progression and poor
prognosis. Specifically, ID1 upregulated AURKA expression by
competitively binding with APC/CCdh1, thus inhibiting
APC/CCdh1-mediated AURKA protein degradation. Increased
AURKA subsequently amplified the Myc oncogenic signaling pathway at
both the transcriptional and post-transcriptional levels. Based on
the present results, targeting the ID1/AURKA/Myc regulatory axis
could provide a novel promising therapeutic target that could
improve the clinical outcome of advanced HCC.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
ID1
|
inhibitor of differentiation 1
|
|
AURKA
|
aurora kinase A
|
|
CHX
|
cycloheximide
|
|
HR
|
hazard ratio
|
|
IC50
|
half-maximal inhibitory
concentration
|
|
AKI
|
AURKA inhibitor
|
|
MYCI
|
Myc inhibitor
|
|
APC/CCdh1
|
anaphase-promoting complex/cyclosome
Cdh1
|
|
TMA
|
tissue microarray
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81272565, 81502491
and 81600095) and the State Scholarship Fund of China (grant no.
201706100120).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZR and MW conceived and designed this study. MW, YZ
and CF performed all the experiments. XY, LZ, and TC collected the
data and performed statistical analyses. XY and LZ provided optimal
experimental protocols and materials. ZR and MW prepared the
manuscript, with input from all authors. All authors provided
critical feedback and helped shape the research, analysis and
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Zhongshan Hospital Affiliated with Fudan
University, and all research strictly abided to the Declaration of
Helsinki. All individuals involved in the study provided written
informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cronin KA, Lake AJ, Scott S, Sherman RL,
Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, et
al: Annual Report to the Nation on the Status of Cancer, part I:
National cancer statistics. Cancer. 124:2785–2800. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang ZY: Hepatocellular carcinoma--cause,
treatment and metastasis. World J Gastroenterol. 7:445–454. 2001.
View Article : Google Scholar
|
|
4
|
Llovet JM and Bruix J: Systematic review
of randomized trials for unresectable hepatocellular carcinoma:
Chemoembolization improves survival. Hepatology. 37:429–442. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Craig AJ, von Felden J, Garcia-Lezana T,
Sarcognato S and Villanueva A: Tumour evolution in hepatocellular
carcinoma. Nat Rev Gastroenterol Hepatol. 17:139–152. 2020.
View Article : Google Scholar
|
|
6
|
Perk J, Iavarone A and Benezra R: Id
family of helix-loop-helix proteins in cancer. Nat Rev Cancer.
5:603–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sachdeva R, Wu M, Smiljanic S, Kaskun O,
Ghannad-Zadeh K, Celebre A, Isaev K, Morrissy AS, Guan J, Tong J,
et al: ID1 is critical for tumorigenesis and regulates
chemoresistance in glioblastoma. Cancer Res. 79:4057–4071. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang YH, Hu J, Chen F, Lecomte N, Basnet
H, David CJ, Witkin MD, Allen PJ, Leach SD, Hollmann TJ, et al: ID1
mediates escape from TGFβ tumor suppression in pancreatic cancer.
Cancer Discov. 10:142–157. 2020. View Article : Google Scholar
|
|
9
|
Gumireddy K, Li A, Kossenkov AV, Cai KQ,
Liu Q, Yan J, Xu H, Showe L, Zhang L and Huang Q: ID1 promotes
breast cancer metastasis by S100A9 regulation. Mol Cancer Res.
12:1334–1343. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Román M, López I, Guruceaga E, Baraibar I,
Ecay M, Collantes M, Nadal E, Vallejo A, Cadenas S, Miguel ME, et
al: Inhibitor of differentiation-1 sustains mutant KRAS-driven
progression, maintenance, and metastasis of lung adenocarcinoma via
regulation of a FOSL1 network. Cancer Res. 79:625–638. 2019.
View Article : Google Scholar
|
|
11
|
Fong S, Debs RJ and Desprez PY: Id genes
and proteins as promising targets in cancer therapy. Trends Mol
Med. 10:387–392. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ao J, Meng J, Zhu L, Nie H, Yang C, Li J,
Gu J, Lin Q, Long W, Dong X, et al: Activation of androgen receptor
induces ID1 and promotes hepatocellular carcinoma cell migration
and invasion. Mol Oncol. 6:507–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yin X, Tang B, Li JH, Wang Y, Zhang L, Xie
XY, Zhang BH, Qiu SJ, Wu WZ and Ren ZG: ID1 promotes hepatocellular
carcinoma proliferation and confers chemoresistance to oxaliplatin
by activating pentose phosphate pathway. J Exp Clin Cancer Res.
36:1662017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niu LL, Cheng CL, Li MY, Yang SL, Hu BG,
Chong CCN, Chan SL, Ren J, Chen GG and Lai PBS: ID1-induced p16/IL6
axis activation contributes to the resistant of hepatocellular
carcinoma cells to sorafenib. Cell Death Dis. 9:8522018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marumoto T, Zhang D and Saya H: Aurora-A -
a guardian of poles. Nat Rev Cancer. 5:42–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang A, Gao K, Chu L, Zhang R, Yang J and
Zheng J: Aurora kinases: Novel therapy targets in cancers.
Oncotarget. 8:23937–23954. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jeng YM, Peng SY, Lin CY and Hsu HC:
Overexpression and amplification of Aurora-A in hepatocellular
carcinoma. Clin Cancer Res. 10:2065–2071. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tayyar Y, Jubair L, Fallaha S and McMillan
NAJ: Critical risk-benefit assessment of the novel anti-cancer
aurora a kinase inhibitor alisertib (MLN8237): A comprehensive
review of the clinical data. Crit Rev Oncol Hematol. 119:59–65.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beltran H, Oromendia C, Danila DC,
Montgomery B, Hoimes C, Szmulewitz RZ, Vaishampayan U, Armstrong
AJ, Stein M, Pinski J, et al: A phase II trial of the Aurora Kinase
A inhibitor Alisertib for patients with castration-resistant and
neuroendocrine prostate cancer: Efficacy and biomarkers. Clin
Cancer Res. 25:43–51. 2019. View Article : Google Scholar
|
|
20
|
Hasskarl J, Duensing S, Manuel E and
Münger K: The helix-loop-helix protein ID1 localizes to centrosomes
and rapidly induces abnormal centrosome numbers. Oncogene.
23:1930–1938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cowley DO, Rivera-Pérez JA, Schliekelman
M, He YJ, Oliver TG, Lu L, O'Quinn R, Salmon ED, Magnuson T and Van
Dyke T: Aurora-A kinase is essential for bipolar spindle formation
and early development. Mol Cell Biol. 29:1059–1071. 2009.
View Article : Google Scholar :
|
|
22
|
Nikonova AS, Astsaturov I, Serebriiskii
IG, Dunbrack RL Jr and Golemis EA: Aurora A kinase (AURKA) in
normal and pathological cell division. Cell Mol Life Sci.
70:661–687. 2013. View Article : Google Scholar
|
|
23
|
Meraldi P, Honda R and Nigg EA: Aurora-A
overexpression reveals tetraploidization as a major route to
centrosome amplifi-cation in p53-/- cells. EMBO J. 21:483–492.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Man C, Rosa J, Yip YL, Cheung AL, Kwong
YL, Doxsey SJ and Tsao SW: Id1 overexpression induces
tetraploidization and multiple abnormal mitotic phenotypes by
modulating aurora A. Mol Biol Cell. 19:2389–2401. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marrero JA, Kulik LM, Sirlin CB, Zhu AX,
Finn RS, Abecassis MM, Roberts LR and Heimbach JK: Diagnosis,
staging, and management of hepatocellular carcinoma: 2018 practice
guidance by the american association for the study of liver
diseases. Hepatology. 68:723–750. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Llovet JM, Fuster J and Bruix J;
Barcelona-Clínic Liver Cancer Group: The Barcelona approach:
Diagnosis, staging, and treatment of hepatocellular carcinoma.
Liver Transpl. 10(Suppl 1): S115–S120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, Tian B, Yang J, Zhao L, Wu X, Ye SL,
Liu YK and Tang ZY: Stepwise metastatic human hepatocellular
carcinoma cell model system with multiple metastatic potentials
established through consecutive in vivo selection and studies on
metastatic characteristics. J Cancer Res Clin Oncol. 130:460–468.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jawhar NM: Tissue Microarray: A rapidly
evolving diagnostic and research tool. Ann Saudi Med. 29:123–127.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Detre S, Saclani Jotti G and Dowsett M: A
'quickscore' method for immunohistochemical semiquantitation:
Validation for oestrogen receptor in breast carcinomas. J Clin
Pathol. 48:876–878. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang P, Wen F and Li Q: FOLFOX4 or
sorafenib as the first-line treatments for advanced hepatocellular
carcinoma: A cost-effectiveness analysis. Dig Liver Dis.
48:1492–1497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin S, Bai Y, Lim HY, Thongprasert S, Chao
Y, Fan J, Yang TS, Bhudhisawasdi V, Kang WK, Zhou Y, et al:
Randomized, multicenter, open-label study of oxaliplatin plus
fluoro-uracil/leucovorin versus doxorubicin as palliative
chemotherapy in patients with advanced hepatocellular carcinoma
from Asia. J Clin Oncol. 31:3501–3508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Slevin M, Kumar S and Gaffney J:
Angiogenic oligosaccharides of hyaluronan induce multiple signaling
pathways affecting vascular endothelial cell mitogenic and wound
healing responses. J Biol Chem. 277:41046–41059. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Li H, Li CW, Li X, Ding Q, Guo L, Liu S,
Liu C, Lai CC, Hsu JM, Dong Q, et al: MET inhibitors promote liver
tumor evasion of the immune response by stabilizing PDL1.
Gastroenterology. 156(2019): 1849–1861e13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lamb JR, Zhang C, Xie T, Wang K, Zhang B,
Hao K, Chudin E, Fraser HB, Millstein J, Ferguson M, et al:
Predictive genes in adjacent normal tissue are preferentially
altered by sCNV during tumorigenesis in liver cancer and may rate
limiting. PLoS One. 6:e200902011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Taguchi S, Honda K, Sugiura K, Yamaguchi
A, Furukawa K and Urano T: Degradation of human Aurora-A protein
kinase is mediated by hCdh1. FEBS Lett. 519:59–65. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Walter AO, Seghezzi W, Korver W, Sheung J
and Lees E: The mitotic serine/threonine kinase Aurora2/AIK is
regulated by phosphorylation and degradation. Oncogene.
19:4906–4916. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li B, Xu WW, Guan XY, Qin YR, Law S, Lee
NPY, Chan KT, Tam PY, Li YY, Chan KW, et al: Competitive binding
between Id1 and E2F1 to Cdc20 regulates E2F1 degradation and
thymidylate synthase expression to promote esophageal cancer
chemoresistance. Clin Cancer Res. 22:1243–1255. 2016. View Article : Google Scholar
|
|
40
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dauch D, Rudalska R, Cossa G, Nault JC,
Kang TW, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L,
et al: A MYC-aurora kinase A protein complex represents an
actionable drug target in p53-altered liver cancer. Nat Med.
22:744–753. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Otto T, Horn S, Brockmann M, Eilers U,
Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R,
Christiansen H, et al: Stabilization of N-Myc is a critical
function of Aurora A in human neuroblastoma. Cancer Cell. 15:67–78.
2009. View Article : Google Scholar
|
|
43
|
Chuma M, Terashita K and Sakamoto N: New
molecularly targeted therapies against advanced hepatocellular
carcinoma: From molecular pathogenesis to clinical trials and
future directions. Hepatol Res. 45:E1–E11. 2015. View Article : Google Scholar
|
|
44
|
Kononen J, Bubendorf L, Kallioniemi A,
Bärlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G
and Kallioniemi OP: Tissue microarrays for high-throughput
molecular profiling of tumor specimens. Nat Med. 4:844–847. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Alkushi A: Validation of tissue microarray
biomarker expression of breast carcinomas in Saudi women. Hematol
Oncol Stem Cell Ther. 2:394–398. 2009. View Article : Google Scholar
|
|
46
|
Graham AD, Faratian D, Rae F and Thomas
JS: Tissue microarray technology in the routine assessment of HER-2
status in invasive breast cancer: A prospective study of the use of
immunohistochemistry and fluorescence in situ hybridization.
Histopathology. 52:847–855. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Drev P, Grazio SF and Bracko M: Tissue
microarrays for routine diagnostic assessment of HER2 status in
breast carcinoma. Appl Immunohistochem Mol Morphol. 16:179–184.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cho Y, Cho EJ, Lee J-H, Yu SJ, Kim YJ, Kim
CY and Yoon JH: Fucoidan-induced ID-1 suppression inhibits the in
vitro and in vivo invasion of hepatocellular carcinoma cells.
Biomed Pharmacother. 83:607–616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yan M, Wang C, He B, Yang M, Tong M, Long
Z, Liu B, Peng F, Xu L, Zhang Y, et al: Aurora-A Kinase: A potent
oncogene and target for cancer therapy. Med Res Rev. 36:1036–1079.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cammareri P, Scopelliti A, Todaro M,
Eterno V, Francescangeli F, Moyer MP, Agrusa A, Dieli F, Zeuner A,
Stassi G, et al: Aurora-A is essential for the tumorigenic capacity
and chemoresistance of colorectal cancer stem cells. Cancer
Research. 70:4655–4665. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu
L, Zhao Y, Huang XF, Wang XR, Zhu XF, et al: Inhibition of Aurora-A
suppresses epithelial-mesenchymal transition and invasion by
downregulating MAPK in nasopharyngeal carcinoma cells.
Carcinogenesis. 29:1930–1937. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zheng F, Yue C, Li G, He B, Cheng W, Wang
X, Yan M, Long Z, Qiu W, Yuan Z, et al: Nuclear AURKA acquires
kinase-independent transactivating function to enhance breast
cancer stem cell phenotype. Nat Commun. 7:101802016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hasskarl J, Mern DS and Münger K:
Interference of the dominant negative helix-loop-helix protein ID1
with the proteasomal subunit S5A causes centrosomal abnormalities.
Oncogene. 27:1657–1664. 2008. View Article : Google Scholar
|
|
55
|
Geng F, Wenzel S and Tansey WP: Ubiquitin
and proteasomes in transcription. Annu Rev Biochem. 81:177–201.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bruix J and Sherman M; American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Peck-Radosavljevic M: Drug therapy for
advanced-stage liver cancer. Liver Cancer. 3:125–131. 2014.
View Article : Google Scholar : PubMed/NCBI
|