Introduction
Oncogenic KRAS mutations can be detected in
approximately 40% of human colorectal cancers (CRCs) (1). Mutant KRAS remains activated because
of impaired GTPase activity (2).
Mutant KRAS has been demonstrated to contribute to the failure of
anti-epidermal growth factor receptor (EGFR) anti-bodies, and it is
associated with a poor prognosis in patients with CRC receiving
adjuvant chemotherapy (3);
however, the direct inhibition of mutant KRAS remains a
pharmacological challenge. Previous studies revealed that a KRAS
G12C-specific inhibitor (4-6) and
a Src homology phosphotyrosine phosphatase 2 inhibitor can
successfully inactivate mutant KRAS and inhibit tumorigenesis both
in vitro and in vivo (7-9).
However, to the best of our knowledge, clinically effective
targeted agents for KRAS-mutant CRC do not exist.
Structural and functional analyses indicated that
the MEK inhibitor trametinib can achieve superior efficacy in
KRAS-driven tumors by inhibiting ERK1 and phosphorylated
(p)-ERK1/2, as well as MEK1/2 phosphorylation and activation
(10,11). MEK is a serine/threonine kinase
that lies downstream of RAS in the RAS/MEK/ERK pathway, which
regulates key cellular activities, including differentiation,
proliferation and survival (12).
The downstream position of MEK in this cascade makes it an
attractive therapeutic target for patients whose tumors carry
upstream gain-of-function mutations. Studies of multiple allosteric
inhibitors of MEK in KRAS-mutant cancers have demonstrated targeted
inhibition (13); however, their
use has generally resulted in stable disease in early-stage
clinical trials (14-17). In contrast to BRAF-mutant
melanomas, which are highly sensitive to MEK inhibitors (18), this limited efficacy indicates that
different mechanisms of inhibition are required for optimal
antitumor activity against each phenotype.
MEK/ERK inhibitors have been demonstrated to induce
the pro-apoptotic BH3-only protein BIM by suppressing ERK-mediated
phosphorylation (19). BH3-only
proteins appear to be critical for the response to targeted
therapies, including EGFR and combined MEK/PI3K inhibitors
(20,21). BIM, the most potent BH3-only
protein, binds and neutralizes all anti-apoptotic B cell lymphoma 2
(Bcl2) proteins, such as Bcl-xL, whereas other BH3-only proteins
(BID, BIK and NOXA) have greater restrictions (22). Therefore, the upregulation of BIM
may offer the greatest potential to improve therapeutic efficacy.
However, the induction of BIM proteins by MEK/ERK inhibition
(23,24) is inhibited by the anti-apoptotic
proteins Bcl2 and Bcl-xL, which are frequently overexpressed in
solid tumors (25). In human CRC,
Bcl-xL is significantly upregulated compared with its expression in
normal mucosa and adenoma (26).
In a study using mutant KRAS cell lines, mutant KRAS induced the
upregulation of Bcl-xL protein expression (27).
We previously reported a proliferation screening
assay system using green-fluorescent protein (GFP)-labeled
transductants and fluorescence-activated cell sorting (FACS)
(28). The present study modified
this assay system to create the Mix Culture assay, which is an
experimental system that can be used to screen for effective
therapeutic targets in KRAS-mutant CRC cells. Through this
screening, it was identified that a MEK inhibitor (trametinib)
exerted a therapeutic effect on mutant KRAS (G12D, G12V,
G13D)-transduced CACO-2 cells.
Trametinib suppressed the proliferation of
KRAS-mutant cells by decreasing p-ERK expression, whereas Bcl-xL
expression was increased. To overcome this resistance to apoptosis
in KRAS mutants, the combination of trametinib and the Bcl-xL
antagonist ABT263 was assessed both in vitro and in
vivo. Trametinib was effective at low doses when used in
combination with ABT263. Therefore, this therapy may also reduce
the adverse effects of trametinib, such as liver injury. Trametinib
and ABT263 combination therapy is expected to become a specific
treatment for KRAS-mutant CRC.
Materials and methods
Cell culture
Human CACO-2 cells were purchased from RIKEN BRC
through the National Bio-Resource Project of the MEXT/AMED, Japan
and maintained in DMEM (Gibco; Thermo Fisher Scientific, Inc.).
Human SW48 cells were purchased from the American Type Culture
Collection and maintained in RPMI-1640 with high glucose (FUJIFILM
Wako Pure Chemicals Corporation). All cells were cultured in medium
supplemented with 10% FBS (Biowest) and 1% penicillin/streptomycin
at 37°C in 5% CO2.
Antibodies and reagents
The following antibodies were used: Monoclonal mouse
FLAG (cat. no. 014-22383; 1:1,000 for western blotting; FUJIFILM
Wako Pure Chemicals Corporation); mono-clonal rabbit ERK; cat. no.
4695; 1:1,000), monoclonal rabbit p-ERK (cat. no. 4376; 1:1,000),
monoclonal mouse MEK1/2 (cat. no. 4694; 1:1,000), monoclonal rabbit
p-MEK1/2 (cat. no. 9121; 1:1,000) and monoclonal rabbit Bcl-xL
(cat. no. 2764; 1:1,000 for western blotting and 1:50 for
immunostaining) all purchased from Cell Signaling Technology, Inc.;
monoclonal mouse ribosomal s6 kinase (RSK; cat. no. sc-3933417;
1:200), monoclonal mouse p-RSK (cat. no. sc-377526; 1:200),
mono-clonal mouse BIM (cat. no. sc-374358; 1:500), monoclonal mouse
caspase-3 (cat. no. sc-7272; 1:500) and monoclonal mouse β-actin
(cat. no. sc-47787; 1:2,000) purchased from Santa Cruz
Biotechnology, Inc.; monoclonal mouse Bcl2 (cat. no. M0887;
1:1,000) purchased from Dako; Agilent Technologies, Inc.; and
monoclonal rabbit Bcl2 (cat. no. ab32124; 1:100 for immunostaining)
purchased from Abcam. The secondary antibodies polyclonal goat
anti-mouse (cat. no. P0447; 1:5,000) IgG and poly-clonal goat
anti-rabbit (cat. no. P0448; 1:5,000) IgG conjugated with HRP were
obtained from Dako; Agilent Technologies, Inc. Annexin V conjugated
with APC and 7-aminoactinomycin D (7-AAD) was obtained from
BioLegend, Inc. Cetuximab, panitumumab, trametinib and palbociclib
were purchased from Merck KGaA, Takeda Pharmaceutical Company,
Ltd., Cayman Chemical and LC Laboratories, respectively.
Regorafenib, vemurafenib (cat. no. PLX4032), BEZ-235 and ABT263
were obtained from AdooQ Bioscience.
Construction and retroviral transduction
of the KRAS mutations
Total mRNA from CACO-2 cells was extracted using
NucleoSpin RNAplus (Takara Bio, Inc.), and cDNA was synthesized
using PrimeScript RT Master mix from the PrimeScript™ RT reagent
kit (Takara Bio, Inc.). KRAS-4B carrying a C-terminal FLAG was
amplified using PCR with PrimeSTAR® Max DNA Polymerase
(Takara Bio, Inc.) using CACO2 cDNA as a template. The amplified
KRAS-4B was inserted into the pMXs-IRES-GFP vector using the
In-Fusion® HD Cloning kit (Takara Bio, Inc.) by the
inverse PCR method. The thermocycling conditions were as follows:
Denaturation at 98°C for 1 min, followed by 35 cycles of 98°C for
10 sec and 68°C for 20 sec for insert fragment, denaturation at
98°C for 1 min, followed by 35 cycles of 98°C for 10 sec and 68°C
for 3 min for inverse vector. The following primers were used:
KRAS-FLAG fragment forward: 5′-AGA CTG CCG GAT CCA ATG ACT GAA TAT
AAA CTT GTG G-3′, and reverse, 5′-GCG CCG GCC CTC GAG CTC GAG TCA
CTT GTC GTC ATC GTC CTT GTA ATC GAT CAT AAT TAC ACA CTT TGT-3′; and
pMXs-IRES-GFP vector forward #1, 5′-CTC GAG GGC CGG CGC GCC GCG-3′,
and reverse, 5′-TGG ATC CGG CAG TCT AGA GG-3′. Next, pMXs-IRES-GFP
vector carrying KRAS wild-type gene was used as a template to
create vectors carrying the KRAS mutations (G12D, G12V, and G13D)
with C-termed FLAG using the In-Fusion® HD Cloning kit
by the inverse PCR method. The thermocycling conditions were the
same as aforementioned and the following primers were used: G12D
fragment forward, 5′-AAG TGT GTA ATT ATG GAT GGC GTA GGC AAG AGT
GCC-3′; G12V fragment forward, 5′-AAG TGT GTA ATT ATG GTT GGC GTA
GGC AAG AGT GCC-3′; G13D fragment forward, 5′-AAG TGT GTA ATT ATG
GGT GAC GTA GGC AAG AGT GCC-3′; mutants (G12D, G12V and G13D)
reverse, 5′-GTC CTT GTA ATC GAT CTC GAG TCA CTT GTC GTC ATC GTC-3′;
and pMXs-IRES-GFP vector forward #2, 5′-ATC GAT TAC AAG GAC GAT GAC
G-3′, and reverse, 5′-CAT AAT TAC ACA CTT TGT CTT TG-3′. Then,
using the pMXs-IRES-GFP (KRAS wild, G12D, G12V and G13D) vectors as
a template, pDON-5 Neo DNA vectors (Takara Bio, Inc.) carrying the
KRAS wild-type and its mutations were constructed using the
In-Fusion® HD Cloning kit by the inverse PCR method. The
thermocycling conditions were the same as aforementioned and the
following primers were used: pDON-5 Neo fragment forward, 5′-CTC
ACG TGG GCC CAA ATG ACT GAA TAT AAA CTT G-3′, and reverse, 5′-ACG
TCG ACG GAT CCT TCA CTT GTC GTC ATC GTC CTT G-3′; and pDON-5 Neo
vector forward, 5′-AGG ATC CGT CGA CGT TAA CGC-3′ and reverse,
5′-TTG GGC CCA CGT GAG ATC CGA GC-3′. The DNA sequences of all
constructs were confirmed by sequencing using a BigDye®
Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific,
Inc.) using sequence primers as follow: pMXs sequence, 5′-GAC GGC
ATC GCA GCT TGG ATA CAC-3′; and pDON-5 Neo sequence, 5′-ATC TTG GTT
CAT TCT CAA GCC TC-3′. For retroviral transduction, 5 µg
vectors were transfected into amphotropic packaging cells
Phoenix-AMPHO (American Type Culture Collection) at 80% confluence
on 60-mm cell dishes using 15 µl (1 µg/µl) PEI
MAX (Polysciences, Inc.). The virus-containing supernatants were
harvested after 24 and 48 h, and 50,000 CACO-2 and SW48 cells/well
were infected with the retroviral particles on plates coated with
RetroNectin (Takara Bio, Inc.). The transduction efficiency of
pMXs-IRES-GFP vectors was confirmed using the GFP-positive ratio as
measured using a flow cytometer and analyzed with Kaluza 2.1
software (Beckman Coulter, Inc.). And the cells from the 10th
passage were used for the Mix Culture assay. Following transduction
using pDON-5Neo DNA vectors, the transduced cells were selected via
culture with G418 for 10 days. The transduction efficiency of
pDON-5Neo DNA vectors was confirmed using western blotting.
Cell proliferation assay
CACO-2 cells were seeded at a density of
1.0×104/cm2 into 6-well plates and counted
using the TC20™ Automated Cell Counter (Bio-Rad Laboratories, Inc.)
after culture for 96 h. The Cell Counting Kit-8 (CCK8) assay was
performed using a Cell Counting Kit-8 (CCK8) assay system (Dojindo
Molecular Technologies, Inc.). Cells (5.0×103/well) were
seeded into a 96-well tissue culture plate and incubated at 37°C.
Following 24, 48, 72 and 96 h, CCK8 reagent (10 µl/well) was
added and incubated for 3 h. Absorbance was measured using a plate
reader at 450 nm. Cell growth was calculated as relative values
from day 0 absorbance.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was prepared from CACO-2 and SW48 cells,
and mice xenograft tumors, using NucleoSpin RNA Plus (Takara Bio,
Inc.) for RT. cDNA was synthesized using PrimeScript RT Master mix
(Takara Bio, Inc.) at 37°C for 15 min and 85°C for 5 sec. qPCR was
conducted using SYBER Premix Ex Taq™ II (Takara Bio, Inc.) and
performed using a Mastercycler realplex2 (Eppendorf).
The thermocycling conditions were as follows: Denaturation at 95°C
for 2 min, followed by 40 cycles of 95°C for 15 sec, 55°C for 15
sec, and 68°C for 20 sec. The relative expression level was
calculated using the 2−ΔΔCq method (29). All data were normalized to the mRNA
expression of TBP and presented as the fold increase relative to
that in control cells. The following primers were used: TBP
forward, 5′-GAT CAG AAC AAC AGC CTG CCA C-3′ and reverse, 5′-TGG
TGT TCT GAA TAG GCT GTG G-3′; Bcl-xL forward, 5′-GTG CGT GGA AAG
CGT AGA CAA G-3′ and reverse, 5′-GGC TGC TGC ATT GTT CCC ATA G-3′;
and Bcl2 forward, 5′-GTG GCC TTC TTTGAG TTC GGT G-3′ and reverse,
5′-GAG TCT TCA GAG ACA GCC AGG AG-3′.
Protein sample preparation and western
blotting
CACO-2 and SW48 cells were lysed in RIPA lysis
buffer (cat. no. sc-24948; Santa Cruz Biotechnology, Inc.)
containing 1 mM PMSF on ice for 30 min. The lysates were separated
by centrifugation at 10,000 × g for 10 min at 4°C and the resultant
supernatant was collected as the total cell lysate. Protein was
quantified using a Pierce BCA Protein assay kit (Thermo Fisher
Scientific, Inc.) and 10-12.5 µg protein was separated using
15% SDS-PAGE and then electroblotted onto a PVDF membrane. The
membrane was blocked with Tris-buffered saline containing 5%
non-fat dry milk and 1% Tween-20 for 1 h at room temperature and
then probed using the primary antibodies at 4°C overnight. The
membrane was then incubated with horseradish peroxidase-conjugated
secondary antibody for 1 h at 4°C, which was detected by enhanced
chemiluminescence using Immobilon Western HRP (Amersham ECL Prime
Western Blotting Detection Reagent; Cytiva). The density of the
target protein measured using Image Lab Software version 6.0.1
(Bio-Rad Laboratories, Inc.) was divided by the density of each
β-actin to obtain the actual density. Relative densities of various
treatments were calculated with the actual density of wild type as
1.
Annexin V/7-AAD assay
CACO-2 cells were incubated with 50 nM trametinib
and/or 1 µM ABT263 for 48 h at 37°C. Trypsin was added to
detach adherent cells, which were then combined with floating
cells. Cells were incubated with Annexin V conjugated with APC and
7-AAD for 15 min at room temperature (25°C). The labeled cell
populations were quantitated by flow cytometry using Kaluza
Analysis Software v2.1 (Beckman Coulter, Inc.).
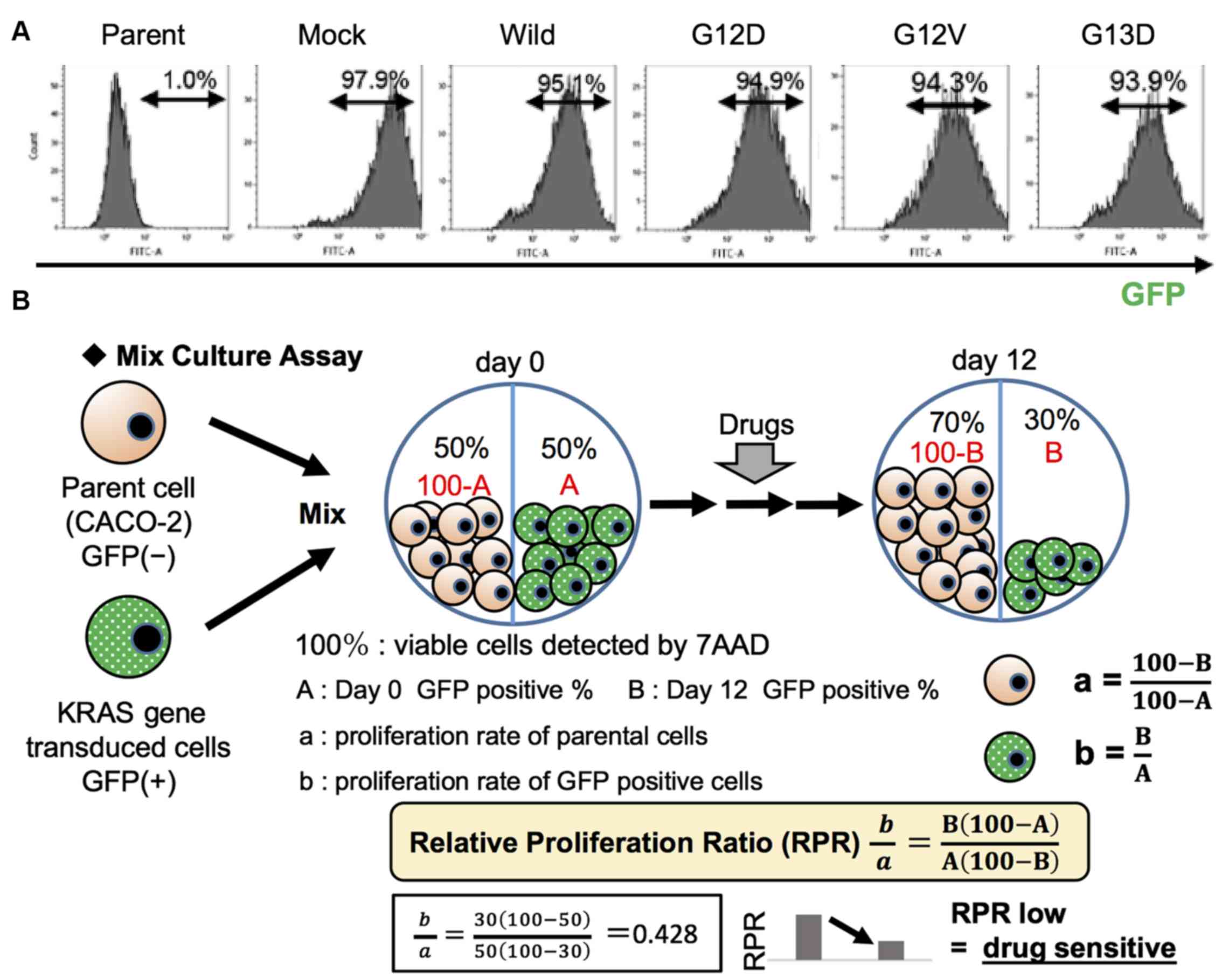
Mix Culture assay
In order to screen for effective therapeutic targets
stably and reliably, an in vitro Mix Culture assay
experimental system was developed using the pMX-IRES-GFP vector and
FACS. For this assay, wild-type and mutant KRAS genes (G12D, G12V
and G13D) inserted into the pMX-IRES-GFP vector were transduced
into CACO-2 cells retrovirally. A high gene transduction efficiency
of ≥90%, as determined by the GFP-positive rate (%) measured using
FACS, was obtained (Fig. 1A).
Parental cells (GFP-negative) and gene-transduced cells
(GFP-positive) were mixed at an ideal 1:1 ratio, as shown in
Fig. 1B. On the first day, the
mixed cells were seeded at 20% confluency into a 12-well plate and
cultured for 12 days with molecular targeted agents. They were then
passaged at a 5:1 ratio before reaching confluence. On day 12, the
cells were harvested and stained with 7-AAD. 7-AAD-negative
populations representing viable cells were gated, and the
GFP-positive ratio of these populations was determined using FACS.
The relative proliferation ratio (RPR) was calculated using the day
0 GFP-positive rate (%, A) and the day 12 GFP-positive rate (%, B).
RPR=B(100-A)/A(100-B). The outline of this experimental system, and
an experimental example are shown. A low RPR indicated that the
GFP-positive cell population was sensitive to the drug, whereas a
high RPR indicated drug resistance (Fig. 1B).
In vivo xenograft experiments
All animal experiments were carried out following
the national standard of the care and use of laboratory animals,
and the study was approved by the Animal Research Committee of
Shinshu University (Matsumoto, Japan; approval no. 019046). A total
of 30 mice were used in the present study. Mice were kept in a
pathogen-free room under standard conditions with a controlled
temperature (23±3°C), humidity and a 12-h light/dark cycle. Mice
had free access to water and food. The male 6-8-week-old BALB/c
nude mice (weight, 24-27 g) were purchased from CLEA Japan, Inc.
Mice were kept in a pathogen-free room with a 12-h light/dark cycle
and free access to water and food. For tumorigenesis assays,
xenograft tumors were generated via the subcutaneous injection of
CACO-2 cells (5×106) stably expressing wild-type or G12V
mutant KRAS in 200 µl solution [50% Hank's Balanced Salt
Solution (FUJIFILM Wako Pure Chemicals Corporation) + 50% Matrigel
(Corning, Inc.)] into the flanks of 6-8-week-old male BALB/c nude
mice. For the reagent experiment, vehicle (12.5% Cremophor, 12.5%
ethanol, 75%), trametinib (0.1 mg/kg) and/or ABT-263 (5 mg/kg) were
orally administered once daily for 10 consecutive days to mice (n=4
per group) that had been subcutaneously injected with CACO-2 cells
expressing the KRAS G12V gene. The tumor volume was measured twice
weekly according to the following formula: Volume
(mm3)=0.5× width2 (mm) × length (mm). All
mice were sacrificed by cervical dislocation under 3% sevoflurane
anesthesia on day 20 and were weighed.
Immunohistochemistry and TUNEL
staining
The resected xenograft tumors were fixed in 4%
paraformaldehyde over-night at RT and embedded in paraffin, and
then tissue sections (5-µm) were prepared and stained with a
primary antibodies against Bcl-xL or Bcl2. Antigen retrieval was
performed by boiling the sections at 98°C for 45 min in 0.05 M
citric acid buffer (pH 6.0) for Bcl-xL and 1 mM EDTA2Na solution
(pH 9.0) for Bcl2. The slides were then subjected to endogenous
peroxidase blocking with 3% H2O2. The primary
antibodies were added to the slides, which were incubated overnight
at 4°C Subsequently, the slides were visualized using Histofine
Simplestain Max PO kit (cat. no. 414142F; Nichirei Bioscience, Inc)
for 1 h at room temperature. HRP-conjugated streptavidin was used
to attach peroxidase to the antibodies, and DAB chromogen was used
for visualization. Then, hematoxylin was used for nuclear
counterstaining for 30-60 sec at room temperature.
The in situ detection of DNA fragmentation in
the tumor tissues was performed using TUNEL staining with an in
situ apoptosis detection kit (cat. no. MK500; Takara Bio,
Inc.). Deparaffinized sections (5-µm) were treated with 10
µg/ml proteinase K and left at room temperature for 15 min.
The endogenous peroxidase was inactivated by applying 3%
H2O2 for 5 min. The slides were treated with
50 µl labeling reaction mixture (consisting of 5 µl
TdT enzyme + 45 µl labeling safe buffer) and incubated at
37°C for 60-90 min. Then, the slides were incubated with 70
µl anti-FITC HRP conjugate at 37°C for 30 min. The slides
were stained with 3% methyl green for 1-2 min at room temperature
to visualize the nuclei. The numbers of TUNEL-positive cells were
counted in five random high-power fields using a light microscope
(magnifications, ×20 and ×400; Olympus Corporation). CellSense
Standard (v1.7.1; Olympus Corporation) was used to observe the
images. The obtained images were analyzed using ImageJ analysis
software v1.50i (National Institutes of Health) to calculate the
number of TUNEL-positive cells.
Statistical analysis
Data are expressed as the mean ± standard deviation
of 4-5 experiments for each assay. Statistical analysis was
performed using JMP® v14.2 (SAS Institute Inc.) for
analysis. Statistical significance was evaluated using an unpaired
Student's t-test or one-way ANOVA followed by Bonferroni's
correction. P<0.05 was considered to indicate a statistically
significant difference.
Results
Mutant KRAS promotes cell proliferation
via the upregulation of ERK phosphorylation
To investigate the effects of oncogenic mutations in
KRAS in CRC cells, wild-type and mutant KRAS genes (G12D, G12V and
G13D) carrying a C-terminal FLAG were inserted into pDON-5Neo DNA
vectors, which were retrovirally transduced into CACO-2 and SW48
human CRC cells expressing wild-type KRAS. Transduction was
confirmed using an anti-FLAG antibody (Fig. 2A). The effect of KRAS gene mutation
on cell proliferation were then examined. Mutant KRAS-transduced
CACO-2 cells exhibited a significantly higher rate of proliferation
compared with cells transduced with wild-type KRAS. This data was
supported by CCK8 assay (Fig. 2B).
The RAS/MEK/ERK signaling pathway is known to act downstream of the
KRAS gene and regulate key cellular activities including
differentiation, proliferation and survival. Therefore, p-MEK,
p-ERK and p-RSK expression was examined using western blotting, and
the ratios of p-protein/total protein were significantly higher in
all mutant KRAS cells compared with in wild-type cells (Figs. 2C and S1A). Subsequently, other pathways
associated with the RAS/MEK/ERK pathway were examined using the Mix
Culture assay system.
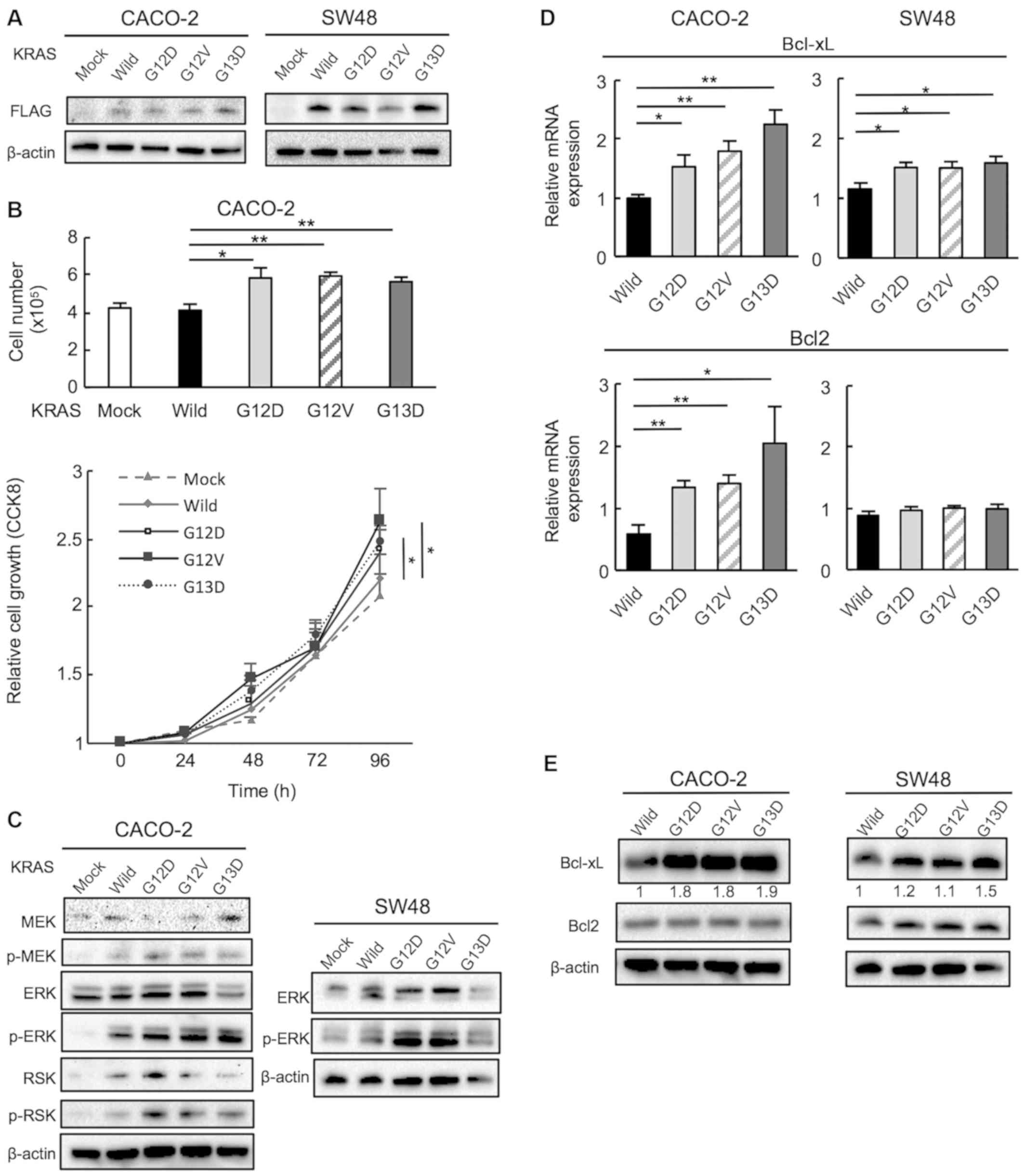 | Figure 2Mutant KRAS promotes CACO-2 and SW48
cell proliferation via the upregulation of ERK phosphorylation. (A)
The transduction of KRAS wild and mutant (G12D, G12V and G13D)
genes was confirmed using western blotting with a FLAG-specific
antibody. (B) Proliferation was evaluated by counting the cell
number at 96 h and use of CCK8 at 24, 48, 72 and 96 h after cell
seeding. n=5. *P<0.05, **P<0.01. (C)
MEK, ERK and RSK phosphorylation in CACO-2 cells and ERK
phosphorylation in SW48 cell was examined using western blotting.
Mutant KRAS upregulates anti-apoptotic Bcl-xL and Bcl2 expression.
The expression levels of Bcl-xL and Bcl2 in cells transduced with
wild and mutant KRAS (G12D, G12V and G13D) were examined by (D)
reverse transcription-quantitative PCR and (E) western blotting.
n=5. *P<0.05, **P<0.01. Wild,
wild-type; CCK8, Cell Counting Kit-8; p-, phosphorylated. |
Mutant KRAS upregulates the
anti-apoptotic markers Bcl-xL and Bcl2
MEK inhibitors are known to induce apoptosis via the
pro-apoptotic protein, Bim, and it has been reported that when
Bim-mediated apoptosis is induced by MEK inhibitors, Bcl-xL and
Bcl2 are also induced, which contributes to the resistance of MEK
inhibitors (25). Therefore, the
present study examined the expression of Bcl-xL and Bcl2 in
KRAS-transduced CACO-2 and SW48 cells to evaluate whether the
expression of these molecules differed in cells with mutant KRAS.
The mRNA levels of Bcl-xL were significantly higher in all mutant
KRAS-transduced CACO-2 and SW48 cells compared with the wild-type
cells. Bcl2 was significantly higher only in mutant
KRAS-transfected CACO-2 cells but not in SW48 cells (Fig. 2D). Previously it was reported that
KRAS mutations affect Bcl-xL expression more strongly than Bcl2
(27). Therefore, it can be
hypothesized that KRAS strongly affects Bcl-xL but not Bcl2 in SW48
cells. The protein expression of Bcl-xL was also significantly
upregulated in all KRAS-mutant cells, except for in G13D SW48
cells. The expression of Bcl2 protein was not higher in the KRAS
mutants compared with the wild-type (Figs. 2E and S1B). These results suggested that Bcl-xL
may be a therapeutic target in KRAS-mutant CRCs.
Mix Culture assay can detect MEK
inhibitor (trametinib) sensitivity in KRAS gene-mutant CACO-2 cells
with suppressed ERK phosphorylation
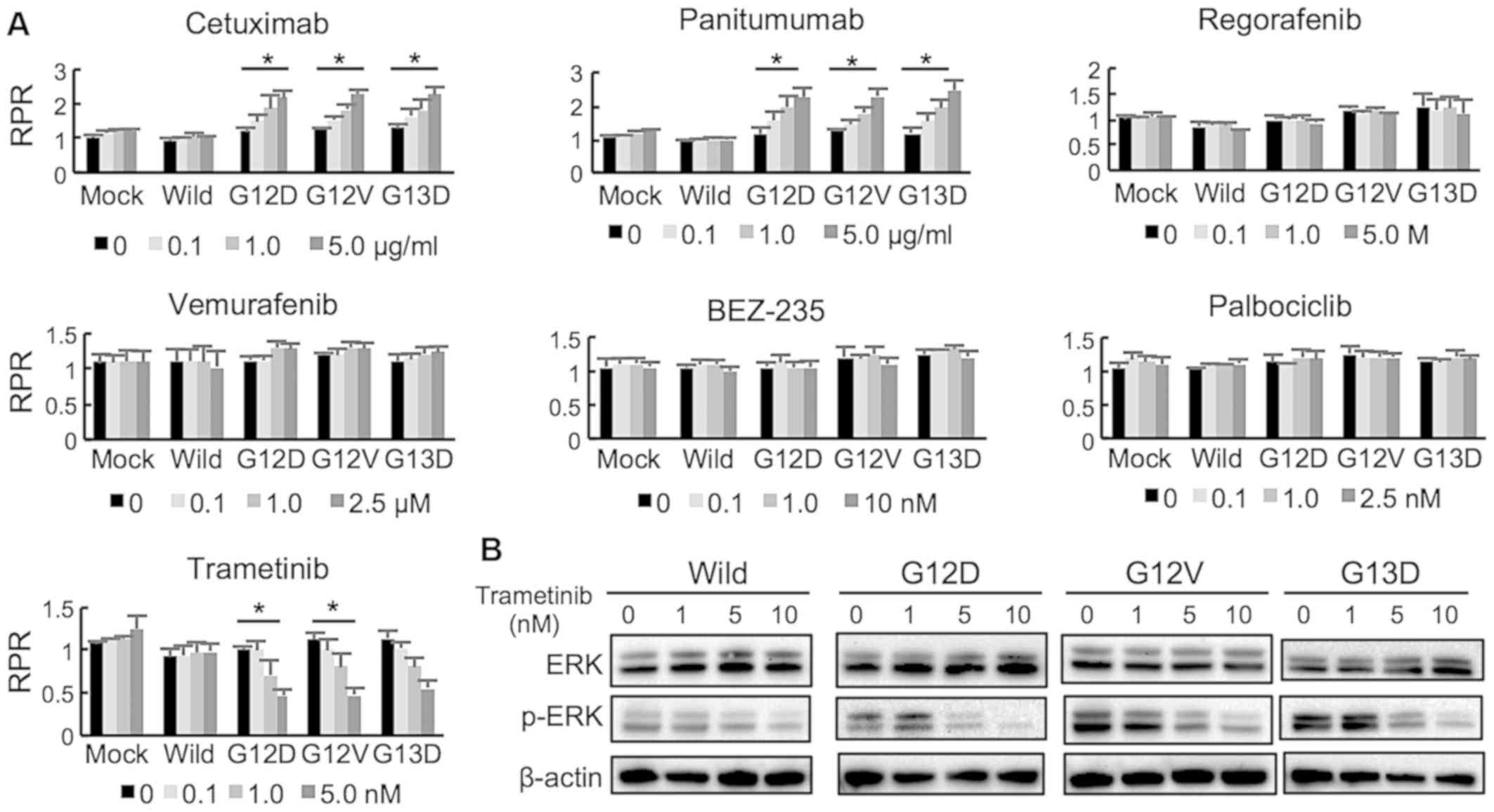
Using a Mix Culture assay system, the drug
sensitivities of KRAS-transduced cells to several drugs were
examined. EGFR inhibitors (cetuximab and panitumumab)
dose-dependently induced a significantly high RPR in KRAS-mutant
CACO-2 cells, indicating that KRAS mutations (G12D, G12V and G13D)
led to resistance to EGFR inhibitors (Fig. 3A). A multi-kinase inhibitor
(rego-rafenib), BRAF inhibitor (vemurafenib), PIK3CA inhibitor
(BEZ234) and CDK4/6 inhibitor (palbociclib) did not change the RPR.
By contrast, a MEK inhibitor (trametinib) reduced the RPR in
KRAS-mutant cells in a dose-dependent manner significantly in G12D
and G12V (Fig. 3A). Treatment with
trametinib (1-10 nM) suppressed ERK phosphorylation both in KRAS
wild type and mutant cells in a dose-dependent manner, however the
concentration of trametinib that suppressed p-ERK level was
different between the KRAS mutants and wild type (Figs. 3B and S2).
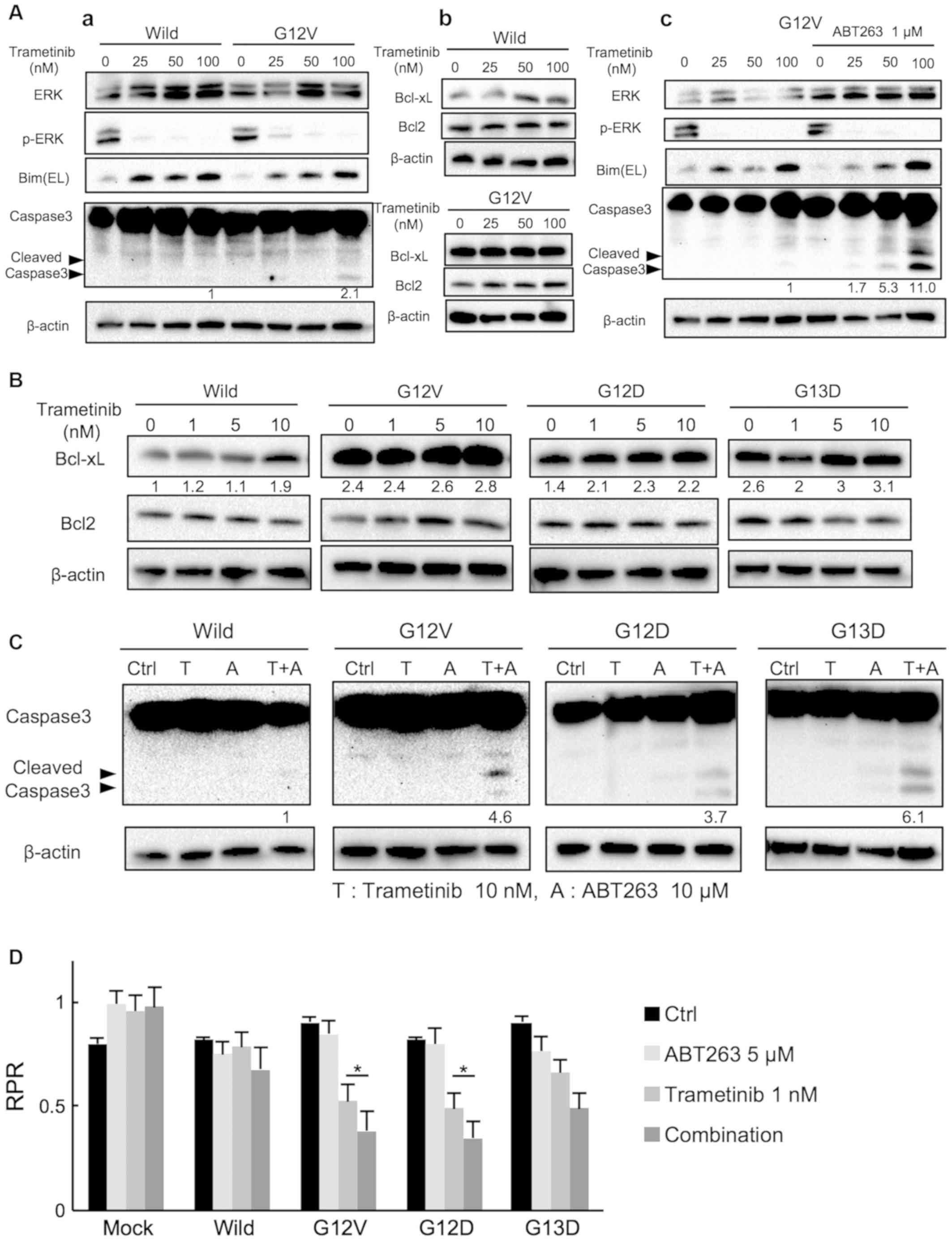
Combined treatment with ABT263 and
low-dose trametinib effectively induces apoptosis in mutant
KRAS-transduced CACO-2 cells
According to Zaanan et al (27), the treatment of HCT116 and SW620
cells with the MEK inhibitor GDC-0623 at clinically relevant doses
has no effect on Bcl-xL expression; however, inhibition of Bcl-xL
synergistically enhances GDC-0623-induced apoptosis. To investigate
the influence of trametinib on apoptosis in wild-type and mutant
KRAS (G12V)-transduced CACO-2 cells, trametinib (100 nM) was
administered according to the methodology described by Yamaguchi
et al (10). As a result,
100 nM trametinib increased cleaved caspase 3 expression, with a
significant dose- dependent increase in BIM, expression observed in
mutant cells The increase in BIM was also observed in wild-type
cells. (Figs. 4A-a and S3A). A similar increase in BIM
expression was observed; however, cleaved caspase 3 expression was
hardly detected in wild-type cells (Figs. 4A-a and S3A). Trametinib (100 nM) did not alter
Bcl-xL or Bcl2 protein levels in the mutant cells (Figs. 4A-b and S3B), similar to the results reported by
Zaanan et al (27). Next,
the effect of the combined treatment of ABT263 and trametinib was
examined in mutant KRAS transduced cells. Combined with ABT263 (1
µM), trametinib significantly increased cleaved caspase 3
expression in a dose-dependent manner at with a large increase in
BIM protein levels also observed indicating the dose-dependent
increase in apoptosis. Trametinib (100 nM) alone demonstrated a
sufficient effect of suppressing p-ERK expression, and the effect
was similar when ABT263 was used in combination. The effect of
combination treatment on cleaved caspase 3 was more significant at
trametinib doses of 50 or 100 nM compared with trametinib alone
(Figs. 4A-c and S3C).
Using our Mix Culture assay, the effects of low-dose
trametinib on anti-apoptotic protein expression in KRAS-mutant
CACO-2 cells were then investigated. Notably, Bcl-xL expression was
increased in a dose-dependent manner by low doses of trametinib
(0-10 nM) in G12D and G12V mutants (Figs. 4B and S4). Accordingly, it was hypothesized
that ABT263 can suppress Bcl-xL function and induce apoptosis when
combined with low-dose (10 nM) trametinib to a similar extent as
high dose (100 nM) trametinib. In fact, combined treatment with
ABT263 and low-dose trametinib significantly enhanced the cleavage
of caspase 3 in KRAS G12V cells compared with in wild-type cells,
and this combination therapy induced apoptosis in cells carrying
other KRAS variants (G12D and G13D) in the same manner (Figs. 4C and S5A).
These apoptosis data were supported by results of
the FACS assay. These results demonstrated that apoptosis was
enhanced by combination therapy in cells carrying KRAS variants,
whereas this effect was not observed in wild-type cells (Fig. S5B and C). Furthermore, the Mix
Culture assay data supported these results, indicating that
combined low-dose trametinib (1 nM) and ABT263 (5 µM)
therapy significantly suppressed the RPR in G12D and G12V mutant
KRAS-transduced cells compared with cells treated with trametinib,
a result that was not seen in wild-type cells (Fig. 4D). Thus, the effectiveness of
combination therapy consisting of low-dose trametinib and ABT263
was for facilitating apoptosis and suppressing the proliferation of
G12D and G12V KRAS-mutant CACO-2 cells was confirmed.
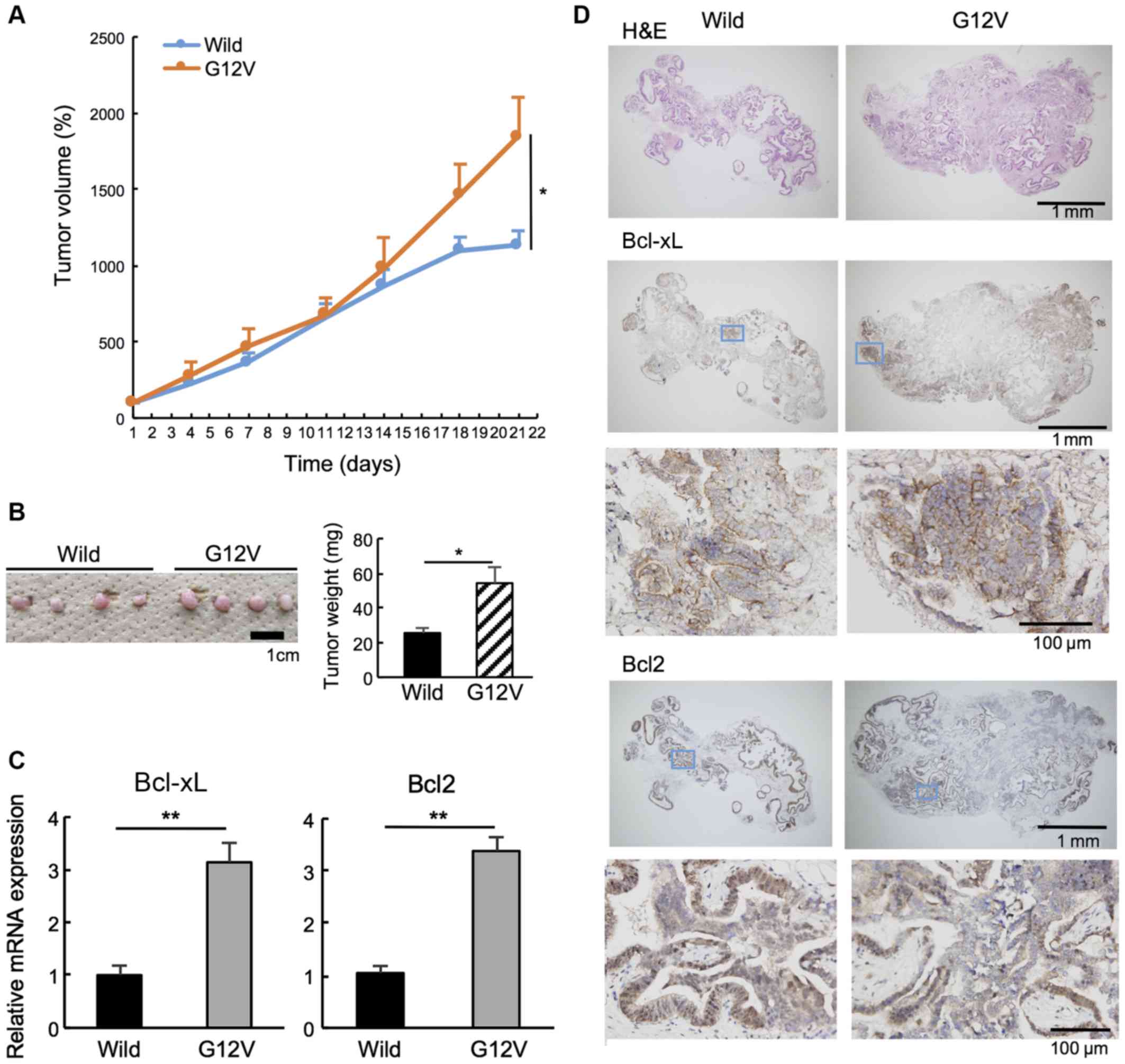
KRAS mutation (G12V) is associated with
tumorigenesis by upregulating Bcl-xL and Bcl2 expression in
vivo
The present study examined the effects of KRAS
mutation on tumorigenicity in vivo using CACO-2 cells
transduced with wild-type and mutant KRAS (G12V). As presented in
Fig. 5A, the tumor volume of
mutant KRAS-bearing murine subcutaneous xenografts was
significantly larger than that of mice bearing wild-type xenografts
on day 21. The tumor weight was also significantly larger for the
KRAS-mutant xenografts (Fig. 5B).
Bcl-xL and Bcl2 mRNA expression was significantly higher in
KRAS-mutant xenografts compared with in wild-type xenografts
(Fig. 5C). Furthermore,
immunohistochemistry revealed that Bcl-xL expression was higher in
KRAS-mutant xenografts, whereas Bcl2 expression was not different
between wild-type and mutant xenografts (Fig. 5D). These results suggest that the
KRAS G12V mutation promotes tumorigenesis by increasing Bcl-xL
expression in vivo.
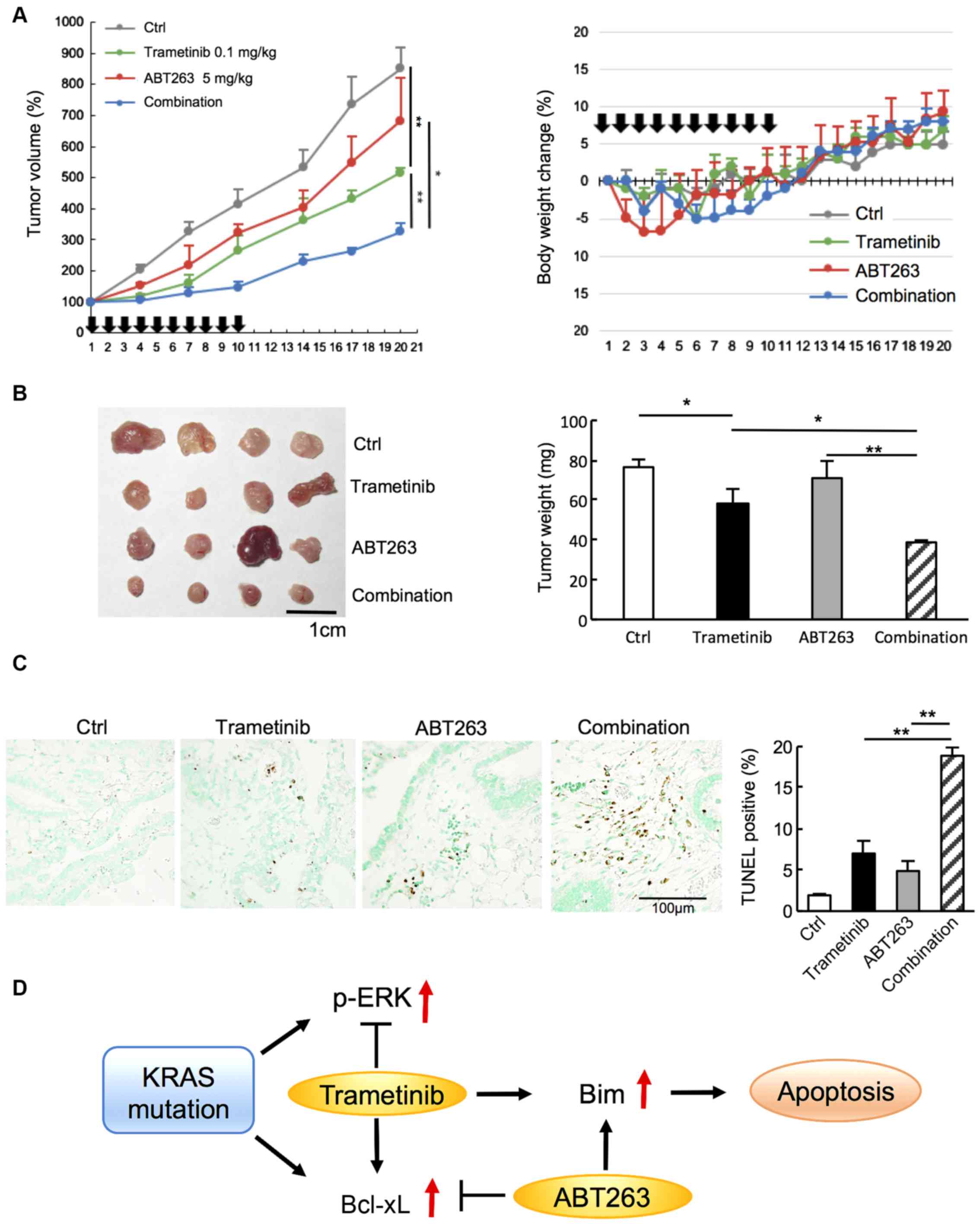
Combined treatment with low-dose
trametinib and ABT263 effectively inhibits tumor growth and induces
apoptosis in murine xenografts of mutant KRAS (G12V)-transduced
CACO-2 cells
Low-dose trametinib (0.1 mg/kg), rather than the
usual dose of 1-3 mg/kg (10,11),
was used in this experiment based on the results of the in
vitro experiments to efficiently and effectively suppress tumor
progression.
Trametinib and ABT263 monotherapy suppressed tumor
growth, but combined treatment with both agents significantly
reduced tumor growth on day 20 compared with the individual
treatments alone (Fig. 6A). The
fluctuation in body weight was within 10% in all mice throughout
the experiments, and the weights of the mice on day 20 ranged
between 26-28 g (Fig. 6A) The
combined treatment resulted in a significantly smaller tumor weight
(Fig. 6B) and an increase in cell
death, as visualized using TUNEL staining (Fig. 6C), compared with the individual
treatments on day 20. Accordingly, combination therapy with
low-dose trametinib and ABT263 was found to be more effective
against mutant KRAS (G12V)-transduced tumors than trametinib or
ABT263 alone in vivo. A schematic of the mechanism by which
trametinib and ABT263 combination therapy is considered to induce
apoptosis is shown in Fig. 6D.
KRAS mutation results in p-ERK and Bcl-xL overexpression.
Trametinib suppresses p-ERK expression, while increases Bcl-xL
expression and may induce resistance to apoptosis. Therefore,
ABT263 combined with trametinib effectively induces apoptosis
probably via increased BIM expression in KRAS-mutant CRC.
Discussion
In basic research, numerous reports have
demonstrated the effect of MEK inhibitors against mutant KRAS
cancers (10,11); however, to the best of our
knowledge, no clinical trials have demonstrated the efficacy of MEK
inhibitors. Our drug screening system, the Mix Culture assay,
suggested that the MEK inhibitor trametinib, among several tested
agents, had a therapeutic effect on KRAS-mutant CACO-2 cells. Thus,
experiments were performed to identify the therapeutic target(s) of
trametinib in KRAS-mutant CRC. It was revealed that the low-dose
trametinib inhibited tumor cell proliferation and suppressed p-ERK
expression, while simultaneously increasing Bcl-xL protein
expression in mutant KRAS transductants. The combination of
low-dose trametinib and the anti-Bcl-xL/Bcl2 agent ABT263 increased
the efficacy of tumor suppression. The efficacy of this combination
therapy against KRAS transductants was validated using both in
vitro and in vivo xenograft models.
Anti-EGFR agents are effective for the treatment of
certain patients with CRC (30-32);
however, anti-EGFR treatments are ineffective in patients with
KRAS-mutant CRC (33,34). As part of the search for new
therapeutic targets in KRAS-mutant CRC, numerous reports have used
KRAS-mutant cell lines, such as DLD-1, HCT116 and SW620 (25,27,35).
To examine the effects of KRAS gene mutation on several signaling
pathways, the present study introduced KRAS gene mutations (G12D,
G12V and G13D) into CACO-2 and SW48 cells expressing wild-type RAS
(KRAS, HRAS, NRAS) (36). Mutant
KRAS transductants exhibited increased proliferation and p-ERK
protein overexpression due to RAS/MEK/ERK activation.
We previously reported a FACS-based assay system
using GFP-labeled transduced cells and parental cells (28). We have since modified this system
and developed a new drug screening assay named the Mix Culture
Assay, in which sensitivity/resistance is evaluated using the RPR
as an index. Because parental cells were mixed as an internal
control and the ratio of transfectants to parental cells was
evaluated using FACS as the GFP-positive rate, this assay is
considered to be relatively stable and reproducible compared with
conventional set-ups. Our assay also avoids the risk of selecting
champion clones, which limits the reliability of stable clone
assays. The present study identified the sensitivity of KRAS-mutant
transductants to the MEK inhibitor trametinib using this
system.
Trametinib is reportedly more effective against
KRAS-mutant cell lines (HCT-116 and SW620) than against wild-type
cells (10,11). The present study identified that
commonly used doses of trametinib (25-100 nM) can potently
downregulate p-ERK and upregulate BIM expression in KRAS
transductants. BIM is the most potent pro-apoptotic BH3-only
protein given its ability to neutralize all anti-apoptotic Bcl2
family proteins (22). The
activity of upregulated BIM was detected by the presence of cleaved
caspase 3 in KRAS-mutant cells in the current study.
Despite the induction of BIM, mutant KRAS
transductants exhibited resistance to the MEK inhibitor trametinib.
Zaanan et al (27) reported
that resistance to MEK inhibitors in DLD-1 cells (KRAS mutant) is
partly attributable to the upregulation of Bcl-xL, indicating that
MEK/ERK inhibition and related BIM induction are insufficient to
overcome mutant KRAS-mediated apoptosis resistance. Trametinib
(25-100 nM) could not suppress the expression of Bcl-xL in the G12V
transductants. These data may underlie the limited efficacy of MEK
inhibitors against KRAS-mutant tumors even when complete
suppression of the RAS/MEK/ERK signaling pathway has been achieved.
Although the mechanisms by which KRAS activation can suppress
apoptosis are poorly understood, it was identified that Bcl-xL mRNA
and protein expression was upregulated in all KRAS-mutant cells.
The clinical relevance of this finding can be observed in human
CRC, in which the Bcl-xL protein expression as determined using
immunohistochemistry was higher in KRAS-mutant tumors than in
wild-type tumors (37). Notably,
in the current study, Bcl-xL expression was further enhanced using
a low dose (1-10 nM) of trametinib in all KRAS-mutant cells,
significantly in G12D and G12V, in a dose-dependent manner. A
similar trend was obtained from all KRAS mutants G12D, G12V and
G13D. Among these mutants, G12V was selected for xenograft
experiments because it produced the most stable and reproducible
effects of trametinib with/or ABT263 on the proliferation (Figs. 3A, 4C
and D).
The potential for Bcl-xL antagonism to increase
apoptotic susceptibility during treatment with a MEK inhibitor in
KRAS transductants was then investigated. ABT263 (1 µM), a
BH3 mimetic, synergistically enhanced trametinib (100 nM)-induced
apoptosis in KRAS G12V transductants. Furthermore, it was
identified that low-dose trametinib (10 nM) and ABT263 (10
µM) synergistically induced apoptosis more effectively in
all KRAS-mutant transductants than in wild-type transductants.
These results provide important insights regarding KRAS-mutant
transduced cells as follows: i) Bcl-xL upregulation is considered a
novel mechanism for KRAS-mediated resistance to apoptosis; ii) the
interaction between trametinib and ABT263 synergistically promotes
apoptosis; and iii) even at a low trametinib dose, a sufficient
tumor-suppressive effect can be observed when combined with ABT263.
Taken together, these data suggest that Bcl-xL upregulation is an
important contributor to clinical resistance to MEK inhibitors in
KRAS-mutant CRC.
The synergistic effect of low-dose trametinib and
ABT263 on tumor suppression was supported by our Mix Culture Assay.
This assay system is expected to be useful for screening the
synergistic effects of multiple drugs targeting oncogenemutant
CRC.
For the murine in vivo xenograft model, KRAS
G12V transductants were selected because of their stability.
Tumorigenesis of the mutant cells was facilitated compared with the
findings for wild-type cells, similar to the results of the in
vitro experiments. The Bcl-xL and Bcl2 mRNA levels of the
mutant xenografts were significantly higher than those of the
wild-type xenografts, and this result was more noticeable than
observed in vitro. This result is presumably because the
expression of Bcl-xL and Bcl2 was further increased by resistance
to cell death in mutant KRAS transductants when exposed to the
stress of the in vivo environment because of immunity,
cytokines, hypoxia, malnutrition and other variables. In the in
vivo experiment examining combination therapy with trametinib
and ABT263 in KRAS-mutant xenografts, xenograft growth was
synergistically suppressed compared with effects of either agent
alone. The doses used in this experiment were ~10-fold lower (0.1
mg/kg) for trametinib and ~20-fold lower (5 mg/kg) for ABT263 than
reported previously (10,11,25,35).
Trametinib has a number of side effects such as rash, diarrhea,
fatigue and liver injury (38).
ABT263 has been reported to act selectively on
cancer cells in a Bcl-xL-dependent manner, and combination
treatment with ABT263 reduces the injury and inflammation induced
in normal tissue by anticancer agents (39). Low-dose trametinib and ABT263 are
expected to exert a synergistic effect and result in fewer adverse
events in the treatment of KRAS-mutant CRC.
A limitation of the present study is that the
difference in Bcl2 induction by KRAS mutations between CACO-2 and
SW48 cells was not further investigated, and SW48 cells were not
used for subsequent assays. CACO-2 cells were used in the mixed
culture and apoptosis assays without SW48 cells for the following
reasons. In the preliminary experiments, CACO-2 cells produced
clearer results compared with SW48 cells. In addition, SW48 cells
were considered as unusual CRC cells with mismatch repair
deficiency. Because mismatch repair-deficient colorectal cancer is
a rare type and exhibits different drug sensitivities compared with
other CRC types, it was expected that using SW48 cells in these
assays would likely not yield results consistent with clinical
practice. On the other hand, CACO-2 cells are considered to be
typical CRC cells with mismatch repair proficiency, TP53 mutation
and APC mutation. Therefore, CACO-2 cells were used in subsequent
experiments.
In conclusion, low-dose trametinib and ABT263
combination therapy targeting p-ERK and Bcl-xL, which are elevated
by KRAS gene mutation, is expected to become a specific treatment
modality for KRAS-mutant CRC.
Supplementary Data
Funding
This study was supported by the Japan Society for
the Promotion of Science KAKENHI (grant no. JP18K15238).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MKi, MT, SMiyag YS and TM contributed to the design
of the study and editing of the manuscript. MKo, SN, SMiyaz, NH and
TE performed the experiments, contributed to data analysis and
wrote the manuscript. MT contributed to critical revision of the
article. YM, FM, STo, MO, STa and YY participated in the
experimental design, and interpreted the acquired data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were carried out following
the national standard of the care and use of laboratory animals,
and the study was approved by the Animal Research Committee of
Shinshu University (Matsumoto, Japan; approval no. 019046).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Stintzing S, Stremitzer S, Sebio A and
Lenz HJ: Predictive and prognostic markers in the treatment of
metastatic colorectal cancer (mCRC): Personalized medicine at work.
Hematol Oncol Clin North Am. 29:43–60. 2015. View Article : Google Scholar
|
|
2
|
Adjei AA: Blocking oncogenic Ras signaling
for cancer therapy. J Natl Cancer Inst. 93:1062–1074. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yoon HH, Tougeron D, Shi Q, Alberts SR,
Mahoney MR, Nelson GD, Nair SG, Thibodeau SN, Goldberg RM, Sargent
DJ, et al: KRAS codon 12 and 13 mutations in relation to
disease-free survival in BRAF wild-type stage III colon cancers
from an adjuvant chemo-therapy trial (N0147 alliance). Clin Cancer
Res. 20:3033–3043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patricelli MP, Janes MR, Li LS, Hansen R,
Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al:
Selective inhibition of oncogenic KRAS output with small molecules
targeting the inactive state. Cancer Discov. 6:316–329. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lito P, Solomon M, Li LS, Hansen R and
Rosen N: Allele-specific inhibitors inactivate mutant KRAS G12C by
a trapping mechanism. Science. 35:604–608. 2016. View Article : Google Scholar
|
|
6
|
Janes MR, Zhang J, Li LS, Hansen R, Peters
U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al:
Targeting KRAS mutant cancers with a covalent G12C-specific
inhibitor. Cell. 172:578–589.e17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garcia Fortanet J, Chen CH, et al:
Allosteric inhibition of SHP2: Identification of a potent,
selective, and orally efficacious phosphatase inhibitor. J Med
Chem. 59:7773–7782. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen YN, LaMarche MJ, Chan HM, Fekkes P,
Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke
VG, et al: Allosteric inhibition of SHP2 phosphatase inhibits
cancers driven by receptor tyrosine kinases. Nature. 535:148–52.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu H, Liu C, Velazquez R, Wang H, Dunkl
LM, Kazic-Legueux M, Haberkorn A, Billy E, Manchado E, Brachmann
SM, et al: SHP2 Inhibition overcomes RTK-mediated pathway
reactivation in KRAS-mutant tumors treated with MEK inhibitors. Mol
Cancer Ther. 18:1323–1334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y
and Sakai T: Antitumor activities of JTP-74035(GSK1120212), a novel
MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in
vivo. Int J Oncol. 39:23–21. 2011.PubMed/NCBI
|
|
11
|
Gilmartin AG, Bleam MR, Groy A, Moss KG,
Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang
J, et al: GSK1120212(JTP-74057) is an inhibitor of MEK activity and
activation with favorable pharmacologic properties for sustained in
vivo pathway inhibition. Clin Cancer Res. 17:989–1000. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kirkwood JM, Bastholt L, Robert C, Sosman
J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V,
Kemsley K and Dummer R: Phase II, open-label, randomized trial of
the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide
in patients with advanced melanoma. Clin Cancer Res. 18:555–567.
2012. View Article : Google Scholar
|
|
14
|
Adjei AA, Cohen RB, Franklin W, Morris C,
Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, et al:
Phase I pharmacokinetic and pharmacodynamics study of the oral,
small-molecule mitogen-activated protein kinase kinase 1/2
inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers.
J Clin Oncol. 26:2139–2146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Infante JR, Fecher LA, Nalllapareddy S,
Gordon MS, Flaherty KT, Cox DS, DeMarini DJ, Morris SR, Burris HA
and Messersmith WA: Safety and efficacy results from the
first-in-human study of the oral MEK 1/2 inhibitor GSK1120212. J
Clin Oncol. 28(15_Suppl): S25032010. View Article : Google Scholar
|
|
16
|
Bennouna J, Lang I, Valladares-Ayerbes M,
Boer K, Adenis A, Escudero P, Kim TY, Pover GM, Morris CD and
Douillard JY: A phase II, open-label, randomised study to assess
the efficacy and safety of the MEK1/2 inhibitor AZD6244
(ARRY-142886) versus capecitabine monotherapy in patients with
colorectal cancer who have failed one or two prior chemotherapeutic
regimens. Invest New Drugs. 29:1021–1028. 2011. View Article : Google Scholar
|
|
17
|
Zhao Y and Adjei AA: The clinical
development of MEK inhibitors. Nat Rev Clin Oncol. 11:385–400.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ascierto PA, Ferrucci PF, Fisher R, Del
Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV,
Di Giacomo AM, et al: Darafenib, trametinib and pembrolizumab or
placebo in BRAF-mutant melanoma. Nat Med. 25:941–946. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan N, Wong M, Nannini MA, Hong R, Lee LB,
Price S, Williams K, Savy PP, Yue P, Sampath D, et al: Bcl-2/Bcl-xl
inhibition increases the efficacy of MEK inhibition alone and in
combination with PI3 kinase inhibition in lung and pancreatic tumor
models. Mol Cancer Ther. 12:853–864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gong Y, Somwar R, Politi K, Balak M,
Chmielecki J, Jiang X and Pao W: Induction of BIM is essential for
apoptosis triggered by EGFR kinase inhibitors in mutant
EGFR-dependent lung adenocarcinomas. PLoS Med. 4:e2942007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hata AN, Yeo A, Faber AC, Lifshits E, Chen
Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, et al:
Failure to induce apoptosis via BCL-2 family proteins underlies
lack of efficacy of combined MEK and PI3K inhibitors for
KRAS-mutant lung cancers. Cancer Res. 74:3146–3156. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Willis SN, Wei A, Smith BJ,
Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC:
Differential targeting of prosurvival Bcl-2 proteins by their
BH3-only ligands allows complementary apoptotic function. Mol Cell.
17:393–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawabata T, Tanimura S, Asai K, Kawasaki
R, Matsumaru Y and Kohno M: Up-regulation of pro-apoptotic protein
Bim and downregulation of anti-apoptotic protein Mcl-1
cooperatively mediate enhanced tumor cell death induced by the
combination of ERK kinase (MEK) inhibitor and microtubule
inhibitor. J Biol Chem. 287:10289–10300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meng J, Fang B, Liao Y, Chresta CM, Smith
PD and Roth JA: Apoptosis induction by MEK inhibition in human lung
cancer cells is mediated by Bim. PLoS One. 5:e130262010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Corcoran RM, Cheng KA, Hata AN, Faber AC,
Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et
al: Synthetic lethal interaction combined BCL-XL and MEK inhibition
promotes tumor regressions in KRAS mutant cancer models. Cancer
Cell. 14:121–128. 2013. View Article : Google Scholar
|
|
26
|
Scherr AL, Gdynia G, Salou M,
Radhakrishnan P, Duglova K, Heller A, Keim S, Kautz N, Jassowicz A,
Elssner C, et al: Bcl-xl is an oncogenic driver in colorectal
cancer. Cell Death Dis. 7:e23422016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zaanan A, Okamoto K, Kawakami H, Khazaie
K, Huang S and Sinicrope FA: Mutant KRAS upregulates BCL-XL via
STAT3 to confer apoptosis resistance that is reversed by BIM
induction and BCL-XL antagonism. J Biol Chem. 290:23838–23849.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kitazawa M, Hida S, Fujii C, Taniguchi S,
Ito K, Matsumura T, Okada N, Sakaizawa T, Kobayashi A, Takeoka M
and Miyagawa SI: ASC induces apoptosis via activation of caspase-9
by enhancing gap junction-mediated intercellular communication.
PLoS One. 12:e01693402017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real time quantitative PCR and
the 2(Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Jonker DJ, O'Callaghan CJ, Karapetis CS,
Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, et
al: Cetuximab for the treatment of colorectal cancer. N Engl J Med.
357:2040–2048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Van Cutsem E, Köhne CH, Hitre E, Zaluski
J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G,
et al: Cetuximab and chemotherapy as initial treatment for
metastatic colorectal cancer. N Engl J Med. 360:1408–1417. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Soikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
et al: K-ras mutations and benefit from cetuximab in advanced
colorectal cancer. N Engl J Med. 359:1757–1765. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Douillard JY, Oliner KS, Siena S,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Panitumumab-FOLFOX4 treatment and RAS mutations
in colorectal cancer. N Engl J Med. 369:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cho SY, Han JY, Na D, Kang W, Lee A, Kim
J, Lee J, Min S, Kang J, Chae J, et al: A novel combination
treatment targeting BCL-XL and MCL1 for KRAS/BRAF-mutated and
BCL2L1-amplified colorectal cancers. Mol Cancer Ther. 16:2178–2190.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ahmed D, Eide PW, Eilertsen IA, Danielsen
SA, Eknæs M, Hektoen M, Lind GE and Lothe RA: Epigenetic and
genetic features of 24 colon cancer cell lines. Oncogenesis.
16:e712013. View Article : Google Scholar
|
|
37
|
Kasper S, Breitenbuecher F, Reis H,
Brandau S, Worm K, Köhler J, Paul A, Trarbach T, Schmid K and
Schuler M: Oncogenic RAS simultaneously protects against anti-EGFR
antibody-dependent cellular cytotoxicity and EGFR signaling
blockade. Oncogene. 32:2873–2881. 2013. View Article : Google Scholar
|
|
38
|
Welsh SJ and Corrie PG: Management of BRAF
and MEK inhibitor toxicities in patients with metastatic melanoma.
Ther Adv Med Oncol. 7:122–1236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tutusaus A, Stefanovic M, Boix L, et al:
Antiapoptotic BCL-2 proteins determine Sorafenib/regorafenib
resistance and BH3-mimetic efficacy in hepatocellular carcinoma.
Oncotarget. 30:16701–16717. 2018. View Article : Google Scholar
|