Introduction
Hepatitis B virus (HBV) infection is a major health
problem, which causes acute and chronic hepatitis, and progresses
to cirrhosis, and hepatocellular carcinoma (HCC) (1-4).
More than 800,000 people succumb to HBV infection or related
complications each year (5).
Approximately 25-40% of cases of liver fibrosis result in cirrhosis
or HCC (6,7). Moreover 70-90% of clinical HCC cases
are related to advanced liver fibrosis or cirrhosis (8).
Liver fibrosis is a common wound healing process
response to chronic liver injury via excessive production and
deposition of extracellular matrix (ECM) (9). Hepatic stellate cells (HSCs) are
major producers of matrix components and play pivotal roles in
regulating the production and secretion of the ECM (10). Typically, HSCs remain in a
quiescent state and function in the storage of vitamin A. Upon
liver injury, HSCs may undergo transdifferentiation, transform into
highly proliferative myofibroblast-like cells, and acquire
fibrogenic properties, including expression of α-smooth muscle
actin (α-SMA), type I collagen, and type III collagen, which are
vital components of the ECM (11).
Although inhibition of HSC activation has been proposed as a
therapeutic strategy in anti-fibrosis treatment of fibrosis
(12), novel approaches to reveal
the mechanisms of liver fibrosis and for the development of
antifibrotic treatments remain challenging.
Transforming growth factor (TGF)-β is a critical
mediator that plays important roles in human fibrogenesis (13). Numerous studies have revealed that
TGF-β signaling through the Smad pathway and reactive oxygen (ROS)
imbalance are responsible for liver fibrosis (14-17).
TGF-β has also been revealed to inhibit the antioxidant system and
hence induce oxidative stress or redox imbalance (18-20).
Redox imbalance has been revealed to significantly contribute to
TGF-β-related fibrosis (16).
Therapeutics targeting TGF-β-induced ROS-dependent cellular
signaling may be a new therapeutic method in the treatment of
fibrotic disorders. However, the mechanisms underlying liver
fibrosis associated with redox-sensitive targets remain
unclear.
Peroxisome proliferator activated receptors (PPARs)
including PPAR-α, PPAR-β/δ, and PPAR-γ, are ligand-activated
transcription factors belonging to the nuclear hormone receptor
family (21-25). Previous studies have revealed that
PPAR-γ is predominantly present in adipose tissue, and plays an
important role in numerous biological processes, such as
adipogenesis, cell differentiation, cell growth regulation and
inflammatory reactions (26,27).
Activation of PPAR-γ has been revealed to retard the progression of
liver fibrosis, and its activation promotes insulin sensitivity and
inhibits the trans-formation of HSCs from a quiescent to activated
state (28-31). Previous studies have indicated that
the activation of PPAR-γ can reduce connective tissue growth factor
expression induced by TGF-β1 in HSCs (32) and that the PPAR-γ agonist
rosiglitazone can enhance PPAR-γ expression in activated HSCs,
leading to reduced oxidative stress and decreased expression of
α-SMA and collagen I (33).
The transcriptional regulator nuclear factor (NF)-κB
is an important mediator of inflammatory signals in response to
stimulation (34-36). Numerous studies have revealed that
upregulation of NF-κB stimulates HSC proliferation and inhibits HSC
apoptosis, playing a key role in fibrogenesis (36-38).
Moreover, NF-κB can induce the expression of inflammatory factors
[TGF-β, interleukin (IL)-6, and tumor necrosis factor-α], which
play pivotal roles in the development of liver fibrosis (36,37,39,40).
Excessive production of ROS also can induce phosphorylation of
NF-κB, which then migrates to the nucleus to increase the
transcription of pro-inflammatory cytokines and results in HSC
activation (41). Accordingly,
reducing the activation of NF-κB can lead to inhibition of HSC
activation and ECM production (42,43).
Recent studies have revealed numerous mechanisms
that mediate liver fibrosis. However, no highly effective
antifibrotic therapies are currently available. In our previous
study, we found that adeno-associated virus (AAV) short hairpin
RNAs (shRNAs) targeting HBV and TGF-β inhibited HBV replication and
liver fibrosis in an HBV-induced liver fibrosis mouse model
(44). Removal of the causative
agent (HBV) by RNA interference (RNAi) was an effective strategy
for treating HBV-induced liver fibrosis, whereas inhibition of the
TGF-β pathway alone was not effective. Our previous studies
revealed the advantages of the combinatorial use of shRNAs against
both HBV and TGF-β in alleviating liver fibrosis (44,45).
The mechanisms through which RNAi protects against HBV are unclear,
and the inhibiting or activating responses of host factors remain
elusive. Isobaric tags for relative and absolute quantification
(iTRAQ) has been widely applied to identify differentially
expressed proteins in numerous diseases (46-48)
including liver fibrosis (49,50).
As a potent new technique in comparative proteomics analysis, iTRAQ
has relatively high sensitivity and allows the determination of
diverse proteins compared with traditional proteome approaches
(51).
In the present study, liver proteins were analyzed
in AAV-shRNA-treated mice using iTRAQ-based quantitative proteomics
in order to identify differentially expressed proteins and to
elucidate the therapeutic mechanisms of liver fibrosis.
Materials and methods
Animal study
Nine normal C57BL/6 male mice (aged 6-8 weeks;
weighing 16-18 g; Beijing Vital River Laboratory Animal Technologly
Co., Ltd.) were housed and maintained at the Laboratory Animal
Facility of the Institute of Laboratory Animal Sciences, Chinese
Academy of Medical Sciences. The protocols for the care and use of
laboratory animals were approved by the Institutional Animal Care
and Use Committee of the Chinese Academy of Medical Sciences, and
all animal care procedures and experiments were performed in
accordance with these protocols. Briefly, mice were housed at room
temperature (20-25°C) and relative humidity (45-60%) with a 12-h
light/dark cycle under individually ventilated cage (IVC) systems.
AAV8-HBV1.2 vector [2×1011 vector genome (vg)
equivalents] was injected into mice via the tail vein to construct
an HBV persistent replication model as previously described
(44,52). Serum samples were obtained by
collection of blood from the tail vein into heparinized capillary
tubes using standard methods 1 month after injection and then
subjected to enzyme-linked immunosorbent assays (ELISAs) and
quantitative polymerase chain reaction (qPCR). After dilution with
PBS, the serum HBV surface antigen (HBsAg) and HBV e antigen
(HBeAg) concentrations were measured using an Auszyme Monoclonal
Diagnostic ELISA kit according to the manufacturer's instructions
(Abbott Laboratories). Serum or cellular DNA was extracted using
DNeasy Blood and Tissue Kits (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions and stored at
−80°C prior to PCR analyses.
Total RNA was isolated using a NucleoSpinRNA II kit
(Macherey Nagal, GmbH & Co., KG) and reverse transcribed using
a First Strand cDNA Synthesis Kit (Toyobo Life Science). A qPCR
standard curve was generated using 10-fold dilutions of the
SSV9-1.2HBV plasmid (1.0×103-1.0×109
copies/ml). All of the qPCR reactions were performed in triplicate
in 96-well optical reaction plates using an ABI 7900 Sequence
Detection System (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and SYBR Green I PCR mix (Roche Diagnostics) as previously
described (52). PCR was performed
under the following conditions: One cycle at 95°C for 10 min; 40
cycles at 95°C for 15 sec, 55°C for 30 sec, and 72°C for 30 sec and
then a dissolution curve was produced.
HBsAg-, HBeAg- and HBV DNA-positive mice were
injected with AAV-shRNA1+3 and AAV-shRNA-TGF-β, or AAV-scrambler
(2×1011 vg), respectively. Normal C57BL/6 mice were
injected with phosphate-buffered saline (PBS) as a negative control
and designated as HBV(-) mice. HBV-positive mice were injected with
AAV-scrambler and designated as HBV(+) mice. AAV-shRNA1+3 targeted
S and X coding regions of HBV. AAV-shRNA-TGF-β vectors targeted the
coding region of TGF-β. The AAV vector containing the shRNA
sequence not targeted to the HBV genome was designated as
AAV-scrambler as previously described (44). All mice were sacrificed at 6 months
after injection. Serum and liver samples were collected and frozen
at -80°C in a freezer or liquid nitrogen. All shRNA sequences
targeting HBV and TGF-β are listed Table SI.
At the end of the experiment, mice were anesthetized
with 2.5% avertin and perfused with cold PBS (pH 7.4)
transcardially, followed by 4% paraformaldehyde (over 2 h at 25°C)
in PBS (0.1 M, pH 7.4) (Boster Biological Technology Co., Ltd.) to
fix tissues for immunohistochemistry (IHC). Livers were then
collected for IHC analysis. Intrahepatic HBV core antigen (HBcAg)
and HBsAg were evaluated by IHC staining of paraffin-embedded
tissues (6-µm thickness of sections) incubated with rabbit
anti-HBc (1:100 dilution; product code ab115992; Abcam) and goat
anti-HBs antibodies (1:200 dilution; cat. no. PA1-73084, Thermo
Fisher Scientific, Inc.), respectively, at 37°C and 45 min and
developed with the Envision HRP (diaminobenzidine) system (Dako;
Agilent Technologies, Inc.). Liver sections were examined with
light microscopy after Masson's trichrome staining and Sirius Red
staining (scale bar, 250 µm). Sirius red staining of liver
sections was also observed by polarizing microscope (scale bar, 250
µm). Total collagen in the liver was determined using a
Hydroxyproline Colorimetric Assay Kit according to the
manufacturer's instructions (BioVison, Inc.). Image-Pro Plus
software 6.0 supplied by Media Cybernetics, Inc. was used for
analysis.
Treatment groups
The various treatment groups were as follows:
Treated mice, HBV-positive mice were infected with AAV-shRNA1+3 and
AAV-shRNA-TGF-β; AAV-shRNA1+3, the combination of two shRNAs
against S and X coding regions of HBV by a self-complementary AAV
vector; AAV-shRNA-TGF-β, vector carrying shRNA against TGF-β;
HBV(+) mice, HBV-positive mice were infected with AAV-scrambler;
AAV-scrambler, AAV vector containing shRNA sequence not targeted to
the HBV genome; HBV(−) mice, normal mice were injected with PBS;
AAV-shRNA-treated mice, treated mice.
Protein preparation and iTRAQ
labeling
Total protein extraction was performed using a kit
(FOCUS Mammalian Proteome; G-Biosciences) in accordance with the
manufacturer's instructions. Protein samples were stored at −80°C
for proteomic analysis and western blotting. The iTRAQ method used
was previously described (53).
Briefly, total protein concentrations were determined using an EZQ
Protein Quantitation Kit (Invitrogen; Thermo Fisher Scientific,
Inc.), and protein samples from treated mice, HBV(+) mice and
HBV(−) mice were reconstituted in dissolution buffer, denatured,
reduced, and trypsinized. Next, tryptic digests of the samples were
labeled with iTRAQ reagents (Table
SII). All samples were balanced, mixed, and pre-separated for
liquid chromatography (LC)-mass spectrometry (MS)/MS analysis.
Nano-LC-MS/MS analysis
LC-MS/MS analysis was performed with an Easy-nLC1000
(Thermo Fisher Scientific, Inc.) and Q Exactive MS (Thermo Fisher
Scientific, Inc.). A reversed-phase ReproSil-PurC18-AQ column
(column, 3 µm; 120 Å, 100 µm ×10 cm) was used to
separate the peptides at a flow rate of 600 nl/min. The LC linear
gradient elution was performed from 6 to 9% B (0.1% formic acid in
acetonitrile) for 15 min, 9 to 14% B for 20 min, 14 to 30% B for 60
min, 30 to 40% B for 15 min, and 40 to 95% B for 3 min, followed by
elution with 95% B for 7 min. A precursor scan was performed using
an Orbitrap instrument by scanning from m/z 300-1800 for detection
with Q Exactive MS. The MS resolution was 60,000 at 400 m/z. The
parameters of MS/MS settings were as follows: The product ion scan
range started at 100 m/z; the activation type was collision-induced
dissociation (CID); the minimum signal required was 1500; the
isolation width was 3; the normalized collision energy was 40; the
default charge state was 6; the activation Q was 0.25; the
activation time was 30 sec. Data were acquired using a
data-dependent acquisition mode in which, for each cycle, the most
abundant multiply-charged peptides with an m/z between 300 and 1800
were selected for MS/MS with the 15 sec dynamic exclusion
setting.
Functional analysis of differentially
expressed proteins
In order to reduce false positives of differentially
expressed proteins, an additional cut off of fold change greater
than 1.30 or less than 0.77 (1/1.3) was exploited for all iTRAQ
ratios (54). Proteins with iTRAQ
ratios >1.30 or <0.77 were considered upregulated or
downregulated, respectively. Gene Ontology (GO) annotations
(46,55), pathway enrichment, and
protein-protein interaction (PPI) networks for all the identified
proteins and differentially expressed proteins were evaluated with
OmicsBean (http://www.omicsbean.cn). GO
annotations were classified into three major categories, including
biological processes (BPs), cell components (CCs), and molecular
functions (MFs). Pathway enrichment analysis was performed with
Kyoto Encyclopedia of Genes and Genomes (KEGG) mapping (47,49).
PPI networks were applied to obtain key nodes, such as degree
centrality, betweenness, closeness, and cluster coefficient and
Venn diagrams were used to reveal mathematical or logical
associations between the HBV(+) vs. HBV(−) groups and the treated
vs. HBV(+) groups.
Immunoblotting
For immunoblotting, 10 µg protein from liver
tissue was separated by sodium dodecyl sulfate poly-acrylamide gel
electrophoresis (SDS-PAGE) on 4-12% gels. The separated proteins
were blotted onto polyvinylidene difluoride membranes, and the
membranes were then washed with TBST and then incubated with
blocking buffer containing 5% skimmed milk in TBST for 2 h at 25°C.
The membranes were washed again with TBST and incubated overnight
at 4°C with primary antibodies diluted in TBST. The primary
antibodies were rabbit anti-mouse antibodies targeting glutathione
S-transferase Pi 1 (GSTP1; 1:2,000 dilution; cat. no. 15902-1-AP),
peroxiredoxin-1 (PRDX1; 1:10,000 dilution; cat. no. 15816-1-AP),
acetyl-CoA acyltransferase 1 (ACAA1; 1:1,000 dilution; cat. no.
12319-2-AP), malic enzyme 1 (ME1; 1:2,000 dilution; cat. no.
16619-1-AP), fatty acid binding protein 1 (FABP1; 1:1,000 dilution;
cat. no. 13626-1-AP), PPAR-α (1:500 dilution; cat. no. 15540-1-AP)
and PPAR-γ (1:1,000 dilution; cat. no. 16643-1-AP) (all from
ProteinTech Group), α-SMA (1:4,000 dilution; product code ab124964;
Abcam), TGF-β (1:1,000 dilution; cat. no. 21898-1-AP; ProteinTech
Group), NF-κB p65 (1:1,000 dilution; product no. 8242), and
phospho-NF-κB p65 Ser468; (1:1,000 dilution; product no. 3039; both
from Cell Signaling Technology, Inc.), and glyceraldehyde
3-phosphate dehydrogenase (GAPDH; 1:10,000 dilution; product code
ab181602; Abcam Inc.). The membranes were then incubated with goat
anti-rabbit secondary antibodies, HRP (1:10,000 dilution; cat. no.
31460; Thermo Fisher Scientific, Inc.) for 1 h at room temperature.
Finally, the signal was visualized using an electrochemiluminescent
reagent kit (EMD Millipore), and blots were imaged using X-ray
film. ImageJ software v.1.31 was used for densitometric
analysis.
Cell line
LX-2 cells (obtained from the Chinese Academy of
Medical Sciences and Peking Union Medical College) were maintained
in Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum and 1% penicillin-streptomycin solution (all from
Thermo Fisher Scientific, Inc.) at 37°C in a humidified incubator
with 5% CO2. LX-2 cells were seeded into 6-well plates
at 4×105 cells/well and cultured. pSSV9-HBV1.2 was
transfected into LX-2 cells using Lipofectamine 2,000 (Thermo
Fisher Scientific, Inc.) with or without pAAV-shRNAs (pAAV-shRNA1+3
and pAAV-shRNA-TGF-β or pAAV-scrambler) (3 µg) according to
the manufacturer's instructions. Lipofectamine was used as a
negative control. The supernatants and transfected cells were
collected 72 h after transfection and subjected to protein
extraction with RIPA Lysis and Extraction Buffer (Thermo Fisher
Scientific, Inc.). The concentration of protein was determined with
a Thermo Scientific Pierce BCA Protein Assay Kit. Transfection
efficiency was evaluated using HBsAg and HBeAg ELISA kits according
to the manufacturer's instructions (Shanghai Kehua Bio-Engineering,
Co., Ltd.).
Statistical analysis
The data are reported as the means ± standard
deviations. One-way analysis of variance (ANOVA) with GraphPad
Prism 5.0 (GraphPad Software, Inc.) was used to determine
statistically significant differences between groups. P-values
<0.05 were considered to indicate statistically significant
differences.
Results
Baseline characteristics of the study
mice
Our previous study demonstrated that
co-administration of shRNAs targeting HBV and TGF-β decreased HBV
antigens, HBV DNA, and liver fibrosis markers in the serum and
livers of HBV-replicated mice (44). In order to explore the mechanisms
underlying the antiviral and antifibrotic effects, AAV-shRNA1+3 and
AAV-shRNA-TGF-β co-injection was evaluated. HBV(+) and HBV(−) mice
were used as positive and negative controls, respectively. All
three treated mice exhibited lower HBsAg and HBV DNA levels in the
serum compared with that in untreated mice (Table I). HBsAg and HBcAg levels were
significantly decreased in the livers of treated mice, as
demonstrated by IHC staining (Fig.
1). The sequence of shRNAs for HBV and TGF-β were available in
supplementary Table SI.
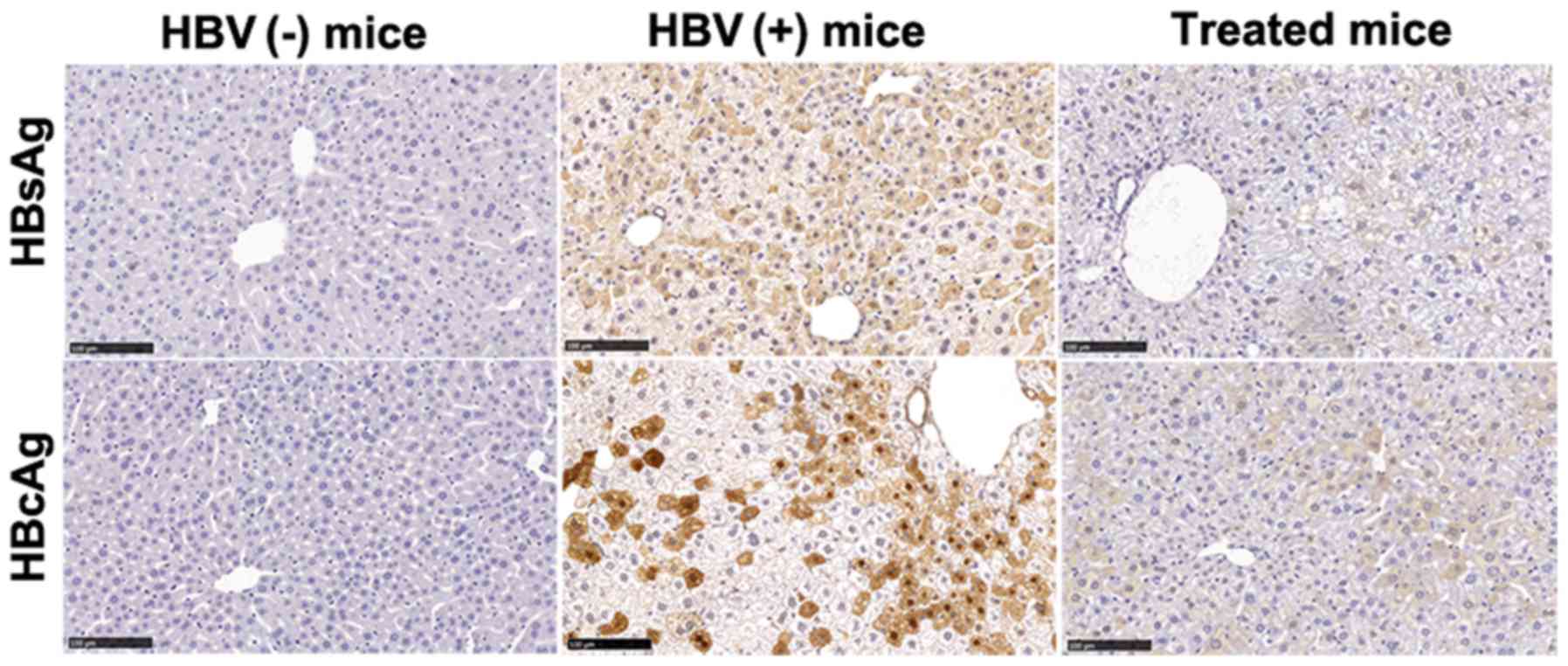 | Figure 1AAV-shRNA treatment decreases HBsAg
and HBcAg in hepatocytes from HBV-replicated mice. Liver samples
from mice treated with AAV-shRNAs were collected at 6 months after
injection. Liver sections were fixed and stained for HBsAg and
HBcAg using IHC staining. HBsAg- and HbcAg-positive hepatocytes
were stained brown. Treated mice, HBV-positive mice were infected
with AAV-shRNA1+3 and AAV-shRNA-TGF-β; HBV(+) mice, HBV-positive
mice were infected with AAV-scrambler; HBV(−) mice, normal mice
were injected with PBS. Scale bar, 100 µm. Three mice were
detected in each group. The representative sections are presented.
AAV, adeno-associated virus; shRNA, short hairpin RNA; HBsAg, HBV
surface antigen; HBcAg, HBV core antigen; HBV, hepatitis B virus;
IHC, immunohistochemical. |
 | Table IBaseline characteristics of mice used
in this study. |
Table I
Baseline characteristics of mice used
in this study.
| Mice | Sex
(Male/Female) | HBsAg serum
(IU/ml) | HBV-DNA serum
(copies/ml) | HBV-DNA liver
(copies/g) | Collagen liver
(µg/mg) | Collagen I serum
(pg/ml) | Collagen III serum
(pg/ml) |
|---|
| Treated N=3 | M | 125.80 |
1.80×104 |
2.01×108 | 107.48 | 141.34 | 0.51 |
| M | 383.70 |
2.51×104 |
2.00×108 | 112.24 | 159.45 | 0.75 |
| M | 0.40 |
1.04×104 |
1.60×108 | 104.55 | 143.51 | 0.62 |
| HBV(+) N=3 | M | >2,500.00 |
1.70×105 |
2.15×108 | 241.29 | 332.12 | 1.31 |
| M | >2,500.00 |
4.24×104 |
4.81×108 | 236.29 | 349.49 | 1.45 |
| M | >2,500.00 |
5.02×104 |
5.01×108 | 375.75 | 365.75 | 1.75 |
| HBV(−) N=3 | M | - | - | - | 95.50 | 131.14 | 0.30 |
| M | - | - | - | 100.21 | 145.49 | 0.52 |
| M | - | - | - | 94.01 | 132.42 | 0.27 |
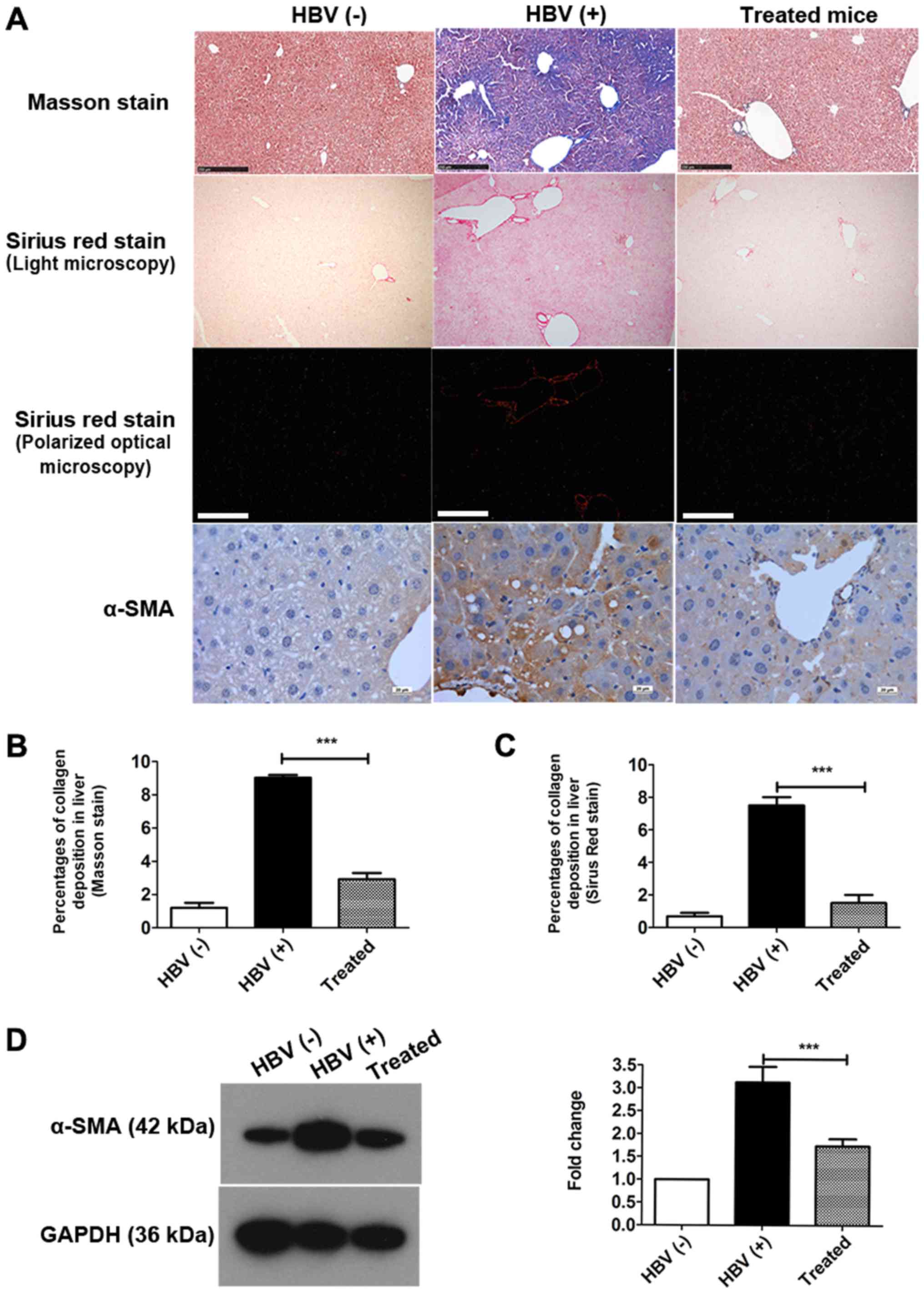
Collagen levels were significantly decreased in the
livers of treated mice compared with those in HBV(+) mice (Fig. 2A, Table I). Total collagen was also
quantitatively assessed using hydroxyproline assays; lower collagen
levels were observed in the livers of treated mice and HBV(−) mice
than in those of HBV(+) mice (Table
I). Masson staining and Sirius Red staining revealed that the
percentages of collagen deposition in hepatocytes were decreased by
approximately 67.71 and 80.01%, respectively, after treatment
(Fig. 2B and C). Collagen I and
III levels in serum were also significantly reduced in the treated
group compared with that in HBV(+) mice (Table I).
Next, the expression of α-SMA, a marker of fibrosis,
was detected in the liver by IHC staining (Fig. 2A) and western blotting (Fig. 2D). As indicated by IHC staining,
α-SMA expression was markedly reduced in the treated group compared
with that in the HBV(+) group (Fig.
2A). The percentage of α-SMA expression was decreased by over
45% in the livers of treated mice compared with that in HBV(+)
mice, as demonstrated by western blotting (Fig. 2D). Collectively, these data
indicated that the mouse model in this study was appropriate.
Proteomic analysis of AAV-shRNA-treated
HBV-replicated mice by iTRAQ-based quantitative proteomics
Next, differentially expressed proteins and
potential pathways were investigated for attenuating liver fibrosis
using iTRAQ-based quantitative proteomics by comparing these three
groups of mice in order to elucidate the potential antifibrotic
mechanisms. AAV-shRNA1+3- and AAV-shRNA-TGF-β-treated groups were
analyzed by iTRAQ-based quantitative proteomics, as revealed in the
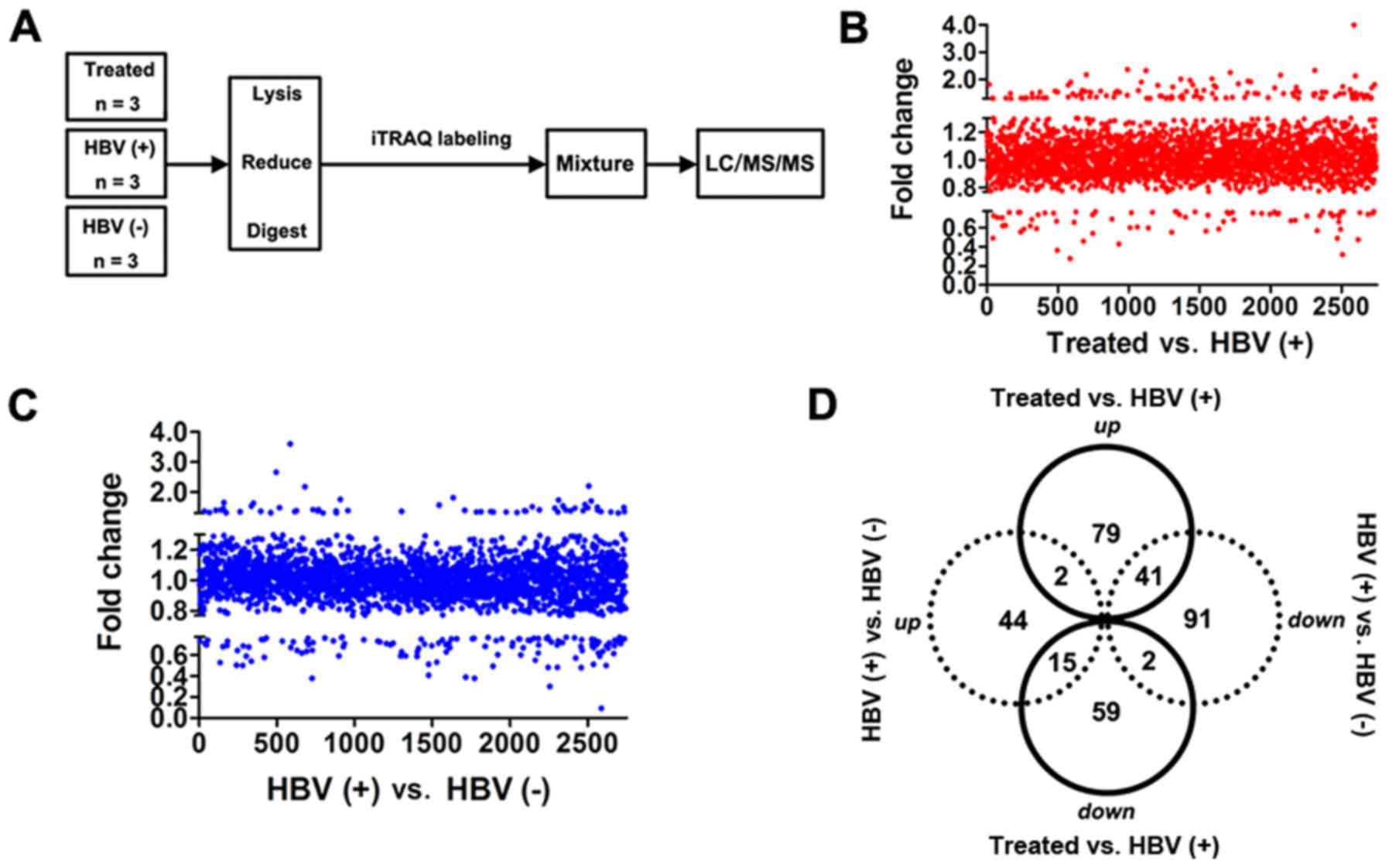
flowchart in Fig. 3A. HBV(+) mice
were used as a positive control, and HBV(-) mice were used as a
negative control. In total, 2,743 proteins were identified in all
groups (Fig. 3B and C). Notably,
76 downregulated and 122 upregulated proteins were revealed in the
treated group compared with that in the HBV(+) group (Table SIII). Sixty-one proteins were
upregulated, and 134 proteins were downregulated in HBV(+) mice
compared with that in HBV(−) mice (Table SIV). We also evaluated the
differentially expressed proteins in all three groups using
Venn-Euler diagrams (Fig. 3D) and
found 41 upregulated and 15 downregulated proteins in the treated
group vs. the HBV(+) group compared with the HBV(+) group vs. the
HBV(−) group (Table SV). Two
proteins (Abcb7 and Chil3) were upregulated in both comparisons,
and two proteins (Opa1 and Eml2) were downregulated in both
comparisons (Fig. 3D; Tables SV, SVI and SVII).
In order to obtain an overall functional view of the
differentially expressed proteins, GO functional annotations and
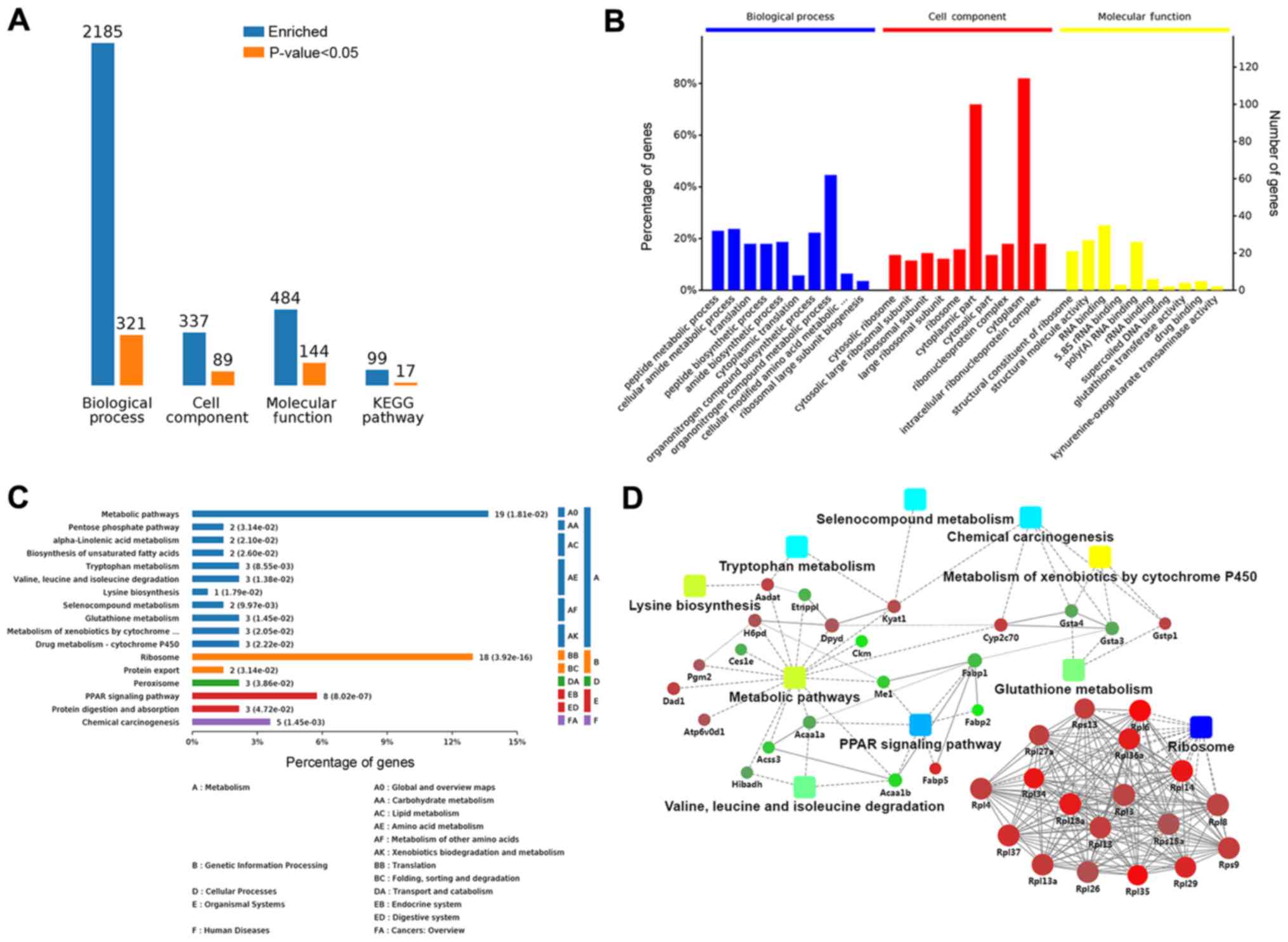
KEGG metabolic pathway analyses were used. Comparison of the
treated group and HBV(+) group revealed enrichment of 2,185 BPs;
321 of these BPs were significant according to analysis of
P-values. Additionally, 89 CCs were significantly altered among 337
enriched CCs, and 484 MFs were enriched, among which 144 MFs were
significant. Seventeen KEGG terms among 99 enriched KEGG terms were
significant (Fig. 4A). In order to
clarify the functions and features of the identified proteins, we
annotated protein functions and features based on GO and KEGG
analyses. An overview of the GO analysis is presented in Fig. 4B. There were 10 distinctly enriched
categories of BPs, CCs, and MFs. The top proteins enriched in BPs
were involved in organ-nitrogen compound metabolic process (45%),
and some proteins enriched in BPs were related to liver fibrosis,
e.g., lipid metabolic process (11%), oxidation-reduction (11%),
response to oxidative stress (5%), negative regulation of cell
adhesion (4%), and cellular oxidant detoxification (2%; Fig. S1A). The main MF category of
enriched proteins was cytoplasm (85%). Proteins involved in hepatic
fibrosis and oxidative stress were also observed in MFs, including
adherens junction (7%), endoplasmic reticulum membrane (7%),
complex of collagen trimers (2%), and the TRAF2-GSTP1 complex (1%;
Fig. S1B). Proteins enriched in
CCs were involved in nucleic acid binding (40%), hydro-lase
activity (8%), oxidoreductase activity (4%), organic acid binding
(3%), transferase activity (3%), vitamin binding (2%), and vitamin
B6 binding (2%; Fig. S1C).
These proteins were also mapped to KEGG pathways
based on their KEGG gene IDs. There were seventeen significant KEGG
pathways presented, including metabolic pathways (14%), ribosome
(13%), PPAR signaling pathway (6%), chemical carcinogenesis (3%),
protein digestion and absorption (2%), protein export (1%),
tryptophan metabolism (2%), and valine, leucine, and isoleucine
degradation (2%; Fig. 4C). The
significant (P<0.05) pathways were ribosome, PPAR signaling
pathway, and chemical carcinogenesis (Fig. S2A).
To clarify the functional relationships of the
identified proteins, a PPI network was created using OmicsBean. In
the PPI network, GSTP1, which participated in glutathione
metabolism, chemical carcinogenesis, and metabolism of xenobiotics
by cytochrome P450, and ribosomal proteins, including Rpl13, Rpl37,
and Rpl27a, were upregulated. Additionally, FABP1, ME1, and ACAA1,
which were relevant to the PPAR signaling pathway and metabolic
pathways, were downregulated in the treatment group compared with
that in HBV(+) mice (Figs. 4D and
S2B).
Verification of proteins associated with
oxidative stress and the PPAR signaling pathway by western
blotting
In order to identify the therapeutic mechanisms of
liver fibrosis by AAV-shRNA treatment, we next focused on
differentially expressed proteins related to oxidative stress, the
PPAR signaling pathway, lipid metabolism, and inflammation, which
are involved in hepatic fibrosis. In fact, in our previous study,
oxidative stress was revealed to play an important role in liver
fibrosis (45). Additionally,
differentially expressed proteins related to oxidative stress,
including GSTP1 and PRDX1, were identified by iTRAQ-based
quantitative proteomics. Thus, in order to verify changes in
oxidative stress after treatment, the expression of GSTP1, PRDX1,
and TGF-β, which are involved in oxidative stress and the redox
imbalance, were evaluated by western blotting (Fig. 5). GSTP1 (Fig. 5A) was significantly upregulated in
the treated group compared with that in the HBV(+) group (increased
1.57-fold) and was significantly downregulated in HBV(+) mice
compared with that in HBV(−) mice (decreased 0.54-fold). PRDX1
(Fig. 5B) and TGF-β (Fig. 5C) were significantly downregulated
in the treated group compared with that in the HBV(+) group
(decreased 0.35-fold and 0.74-fold, respectively) and were
upregulated in the HBV(+) group compared with that in the HBV(-)
group (increased 3.31-fold and 4.20-fold, respectively). Changes in
the expression levels of GSTP1 and PRDX1 verified by western
blotting were consistent with the alterations determined by
iTRAQ-based quantitative proteomics analysis.
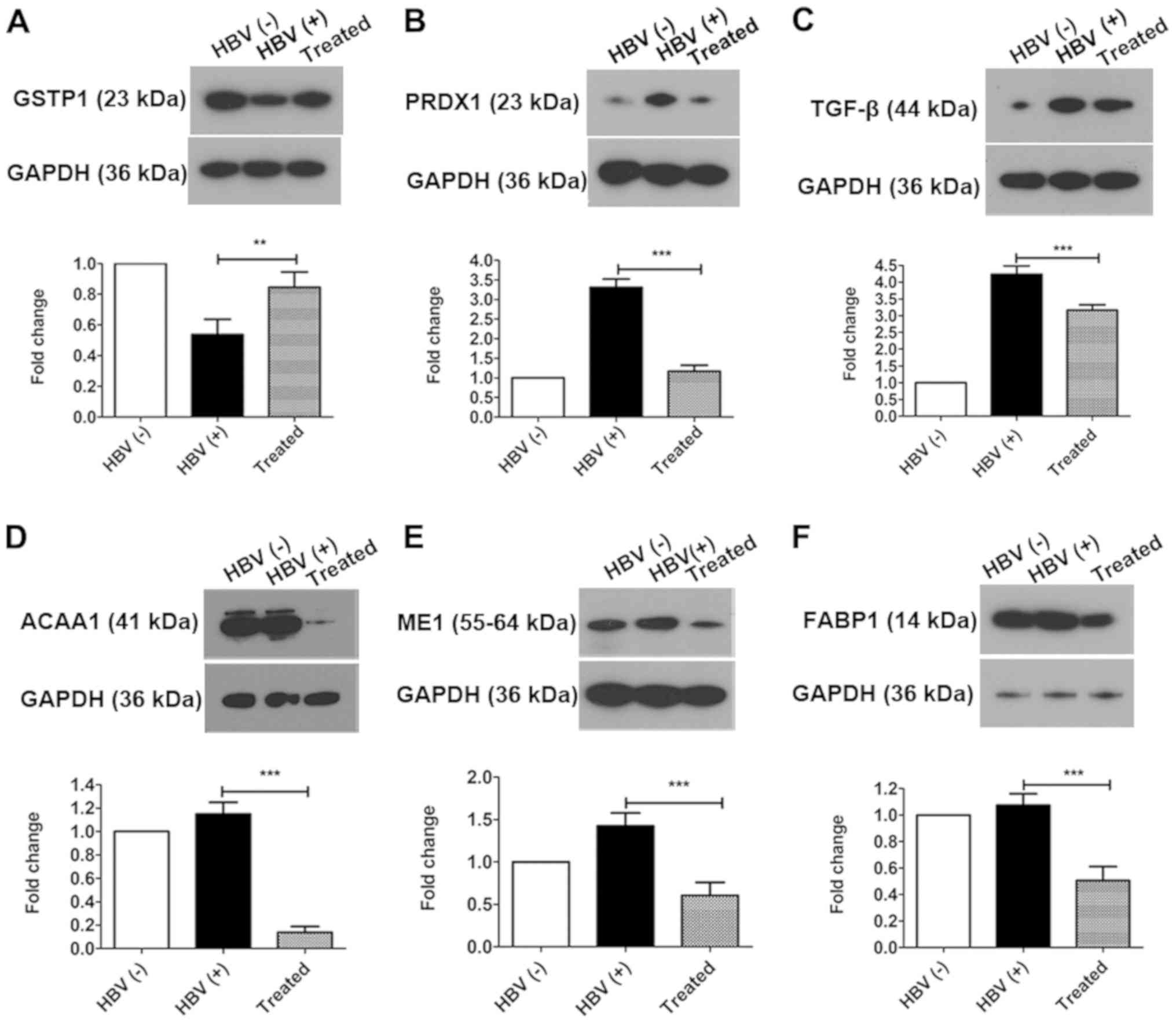 | Figure 5Oxidative stress is alleviated and
downstream proteins in the PPAR signaling pathway are altered by
AAV-shRNA treatment. The differentially expressed proteins (A)
GSTP1, (B) PRDX1 and (C) TGF-β were identified and confirmed by
western blotting. Downstream proteins in the PPAR signaling
pathway, including (D) ACAA1, (E) ME1 and (F) FABP1, were
downregulated in the treated group compared with that in the HBV(+)
group. **P<0.01 and ***P<0.001. Protein
data are expressed as the means ± SD (n=3). PPAR, peroxisome
proliferator-activated receptor; AAV, adeno-associated virus;
shRNA, short hairpin RNA; GSTP1, glutathione S-transferase Pi 1;
PRDX1, peroxiredoxin-1; TGF, transforming growth factor; ACAA1,
acetyl-CoA acyltransferase 1; ME1, malic enzyme 1; FABP1, fatty
acid binding protein 1; HBV, hepatitis B virus. |
Bioinformatics analysis revealed that the PPAR
signaling pathway was activated in the treated group, as
demonstrated by downregulation of ACAA1, ME1, and FABP1. Therefore,
the differential expression of these proteins regulated by the PPAR
signaling pathway in the liver was next investigated by western
blotting. The three proteins were significantly downregulated to
11.90% (ACAA1; Fig. 5D), 42.50%
(ME1; Fig. 5E), and 47.10% (FABP1;
Fig. 5F) in the treated group
compared with that in the HBV(+) group. In a comparison of the
HBV(+) with HBV(−) groups, it was determined that the expression
levels of ACAA1 and FABP1 were not significantly altered, whereas
ME1 was significantly upregulated (increased 1.43-fold; Fig. 5E).
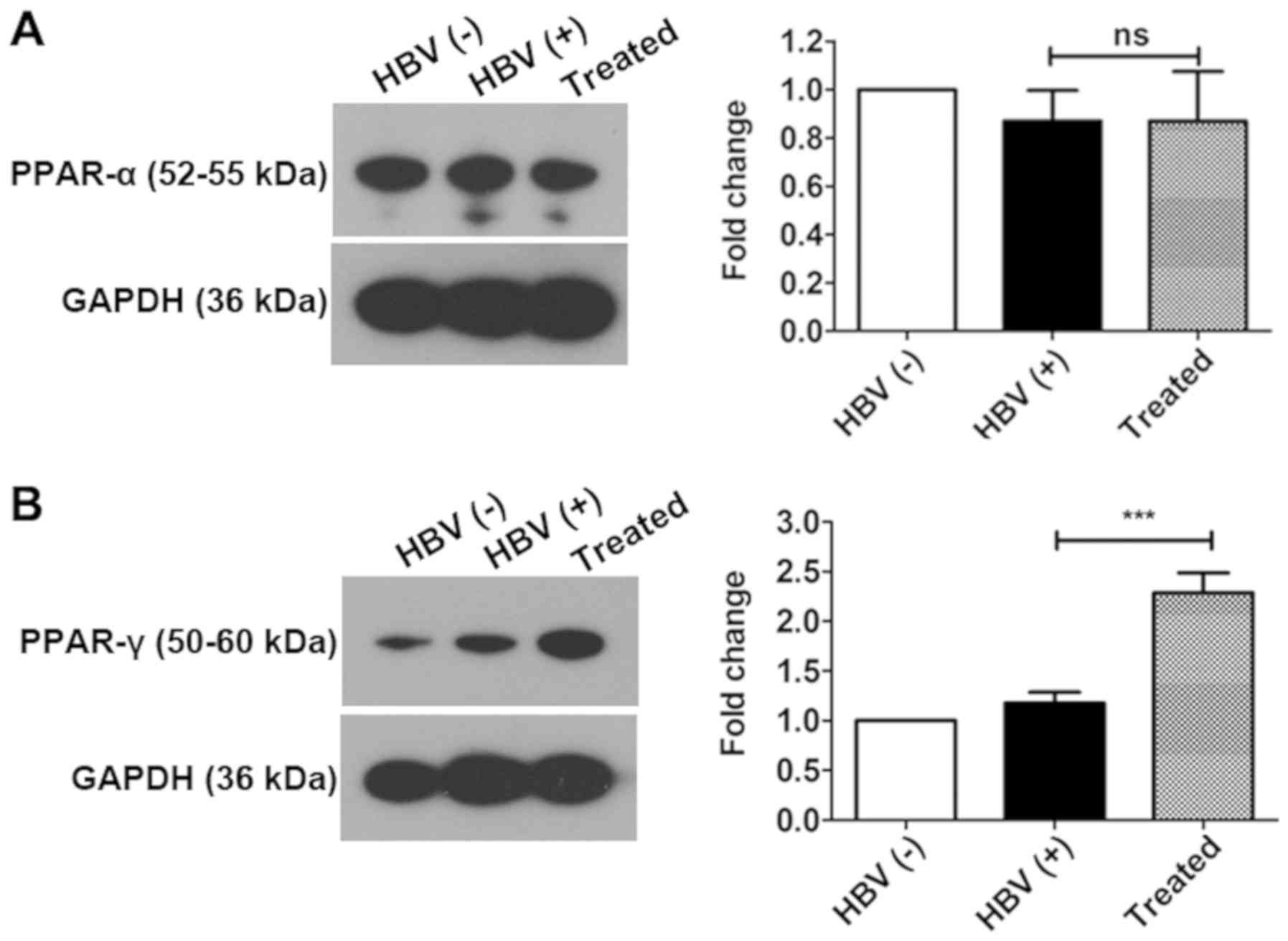
PPAR-γ plays key roles in activating the
PPAR signaling pathway
There are three different isoforms of PPARs, i.e.,
PPAR-α, PPAR-β/δ, and PPAR-γ (56). PPAR-α is mainly expressed in the
liver, and PPAR-γ is expressed in adipose and liver tissues.
Therefore, PPAR-α and PPAR-γ expression was evaluated by western
blotting. The results revealed that PPAR-α expression was not
altered in all three experimental groups. PPAR-γ was significantly
upregulated by 3.20-fold in the livers of treated mice compared
with those of HBV(+) mice; however, no significant changes were
observed in the livers of HBV(+) and HBV(−) groups (Fig. 6). These findings suggest that
PPAR-γ may play an important role inactivating the PPAR signaling
pathway following AAV-shRNA treatment and that PPAR-α may not have
an important a role as PPAR-γ in activating the PPAR signaling
pathway.
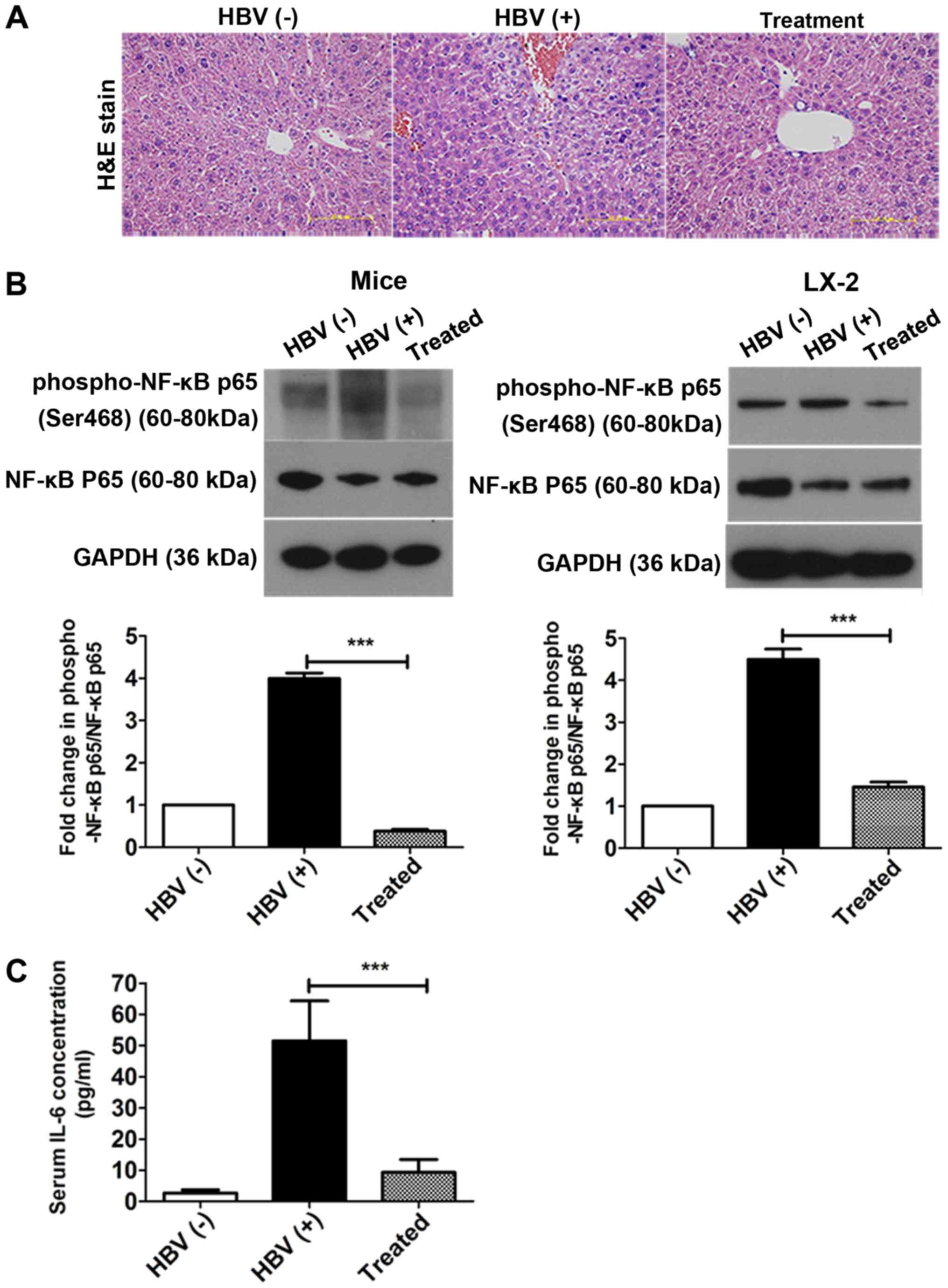
AAV-shRNA treatment attenuates NF- κB p65
phosphorylation in the liver and decreases IL-6 secretion into the
serum
H&E staining was used to investigate the
pathological process of liver fibrosis. Although most hepatocytes
appeared histologically normal in all three groups, some hepatic
necrosis was observed at 6 months in the HBV(+) mice (Fig. 7A). NF-κB is a key mediator of
inflammatory signaling and plays important roles in liver
fibrogenesis (38). Thus, the
levels of NF-κB p65 and phosphorylated NF-κB p65 were then examined
in vivo and in vitro using western blotting. The
levels of phosphorylated NF-κB p65/NF-κB p65 were significantly
reduced in the treatment group compared with that in the HBV(+)
group in livers and in LX-2 cells after transfection with
pAAV-shRNA; 90.5 and 69% decreases were observed in vivo and
in vitro after treatment, respectively (Fig. 7B, Fig. S3). The expression of the
inflammatory factor IL-6 was also measured by ELISA in serum. IL-6
levels in the serum were decreased by over 92% after treatment
compared to the HBV (+) group (Fig.
7C).
Discussion
Chronic HBV infection is a major health problem in
developing countries, including China, and up to one-third of
chronically HBV-infected individuals will progress to fibrosis,
cirrhosis, and even HCC (57-59).
Liver fibrosis involves inflammation induced by a vicious circle of
hepatic damage, driving HSC activation and worsening liver damage
(9,10). Liver fibrosis is a reversible
process that represents the pivotal early stage of hepatic
cirrhosis (60), and few therapies
for liver fibrosis have been developed. Thus, it is necessary to
elucidate the mechanisms of hepatic fibrosis and develop new
medicines for blocking and reversing hepatic fibrosis. Our previous
study revealed that AAV-shRNAs had anti-hepatic fibrosis effects in
HBV-replicated mice with liver fibrosis (44). Moreover, fibrotic markers,
including α-SMA, collagen I, and III, were significantly reduced.
However, the mechanisms mediating the antifibrotic effects of
AAV-shRNAs remain unclear. In the present study, ITRAQ-based
quantitative proteomics was used to elucidate the antifibrotic
mechanism of AAV-shRNAs. Through a comprehensive analysis comparing
the treatment group and HBV(+) mice, it was determined that
ribosomal proteins, downstream proteins of the PPAR signaling
pathway, and inflammation- and oxidative stress-related proteins
were significantly enriched in the AAV-shRNA-treated group. In
order to elucidate the mechanisms of liver fibrosis, the
involvement of oxidative stress, the PPAR signaling pathway, and
inflammation, which are closely associated with liver fibrosis,
were investigated.
Previously studies have suggested that TGF-β, GSTP1,
and PRDX1 are correlated with oxidative stress or ROS imbalance
(18,61-63).
In the present study, it was also determined that these proteins
were altered in treated mice compared with that in HBV(+) mice.
Numerous studies have demonstrated that TGF-β can inhibit the
antioxidant system and cause oxidative stress or redox imbalance
(5,18,44,64).
Additionally, PPAR-γ activation can block the TGF-β signaling
pathway (65). Hence, disruption
of TGF-β expression can relieve oxidative stress. In the present
study, reduction of TGF-β expression was observed following
treatment with AAV-shRNA-TGF-β by direct inhibition of TGF-β mRNA
at the transcript level, resulting in upregulation of PPAR-γ. The
findings indicated that AAV-shRNA treatment alleviated oxidative
stress by reducing TGF-β expression. PRDXs, as redox-regulating
proteins, function to eliminate various ROS and maintain cellular
redox homeostasis (66). PRDX1 can
be easily overoxidized on its catalytically active cysteine upon
stimulation with various stimuli (62). PRDX1 was significantly upregulated
in HBV(+) mice compared with that in HBV(-) mice and was
downregulated after treatment, indicating that oxidative stress was
reduced. As an important phase II enzyme, GSTP1 can protect cells
from oxidative stress in human cancers (61,67).
In accordance with a previous study (61,67),
it was deter-mined that GSTP1 was increased to alleviate oxidative
stress and played a critical role in antioxidant defense after
AAV-shRNA treatment. Collectively, these findings revealed that
AAV-shRNA treatment could prevent oxidative stress by suppressing
the oxidative stress inducers TGF-β and PRDX1 and enhancing GSTP1
expression.
In the PPI network, proteins up- or downstream of
the PPAR signaling pathway (including ACAA1, ME1, and FABP1) were
found to be regulated, suggesting activation of the PPAR signaling
pathway. Notably, FABP1 and ME1 were downregulated in the PPAR
signaling pathway, as demonstrated by KEGG analysis. These proteins
also played pivotal roles in fatty acid synthesis and transport.
ACAA1 is broadly expressed in humans and animals and can catalyze
free cholesterol and long-chain fatty acids to synthesize
esterified cholesterol (68).
ACAA1 is also a marker of β-oxidation (69,70).
ME1 is the cytoplasmic component of the NADPH pool and is used by
fatty acid synthase as a primary lipogenic enzyme. ME1 is also
dysregulated in numerous types of cancers and is involved in
tumorigenesis and metastasis (71,72).
FABP1 is a liver-specific fatty acid-binding protein with key roles
in intracellular metabolism (73).
Overexpression of FABP1 significantly promotes hepatocyte fatty
acid uptake (74), de novo
lipogenesis (75), and VLDL
secretion (73,76). In addition, knockdown of FABP1
significantly suppressed lipid accumulation in hepatocytes
(76) and markedly reduced liver
weight and hepatic triacylglycerol accumulation (75). Consistent with previous research
(69-72,75,76),
it was found that ACAA1, ME1, and FABP1 were downregulated in
treated mice compared with that in HBV(+) mice. These results
indicated that AAV-shRNA treatment inhibited lipogenesis and
improved lipid metabolism. Hepatocyte steatosis was observed in
HBV(+) mice, consistent with our previous study (52), and was alleviated after AAV-shRNA
treatment (44). These findings
suggested that AAV-shRNA alleviated hepatocyte steatosis and liver
fibrosis by decreasing hepatocyte fatty acid uptake and de
novo lipogenesis via attenuation of FABP1 and ME1
expression.
PPAR-γ has broad anti-inflammatory effects and plays
important roles in controlling fibrogenesis and reducing oxidative
stress (31,33,56).
The present data indicated that AAV-shRNA activated the PPAR
signaling pathway by upregulating PPAR-γ expression directly,
resulting in decreased expression of liver fibrosis markers (α-SMA
and ECM) and inflammatory factors (TGF-β and IL-6), consistent with
previous studies (31,33). Recent investigations have revealed
that NF-κB is a crucial mediator of inflammatory signals and that
activation of NF-κB promotes liver fibrogenesis (37). Therefore, inhibition of the NF-κB
pathway may have therapeutic effects on liver fibrosis. In the
present study, it was also revealed that NF-κB p65 phosphorylation
was inhibited in treated mice compared with that in HBV(+) mice and
in cells transfected with pAAV-shRNA. Overall, these data suggested
that AAV-shRNA inhibited liver fibrosis by blocking the NF-κB
pathway.
There are some limitations to the present study. On
one hand, although proteomics is a powerful technology to identify
proteins, combining this method with other analyses such as
transcriptomics and metabolomics may increase the significance of
proteomics data and eventually aid in elucidating the HBV-induced
liver disease in a more systematic manner. On the other hand,
although we found that removal of causative factors and direct
knockdown of TGF-β using short hairpin RNAs are realistic
therapeutic strategies, which enhanced the reversibility of liver
by regulating the PPAR-γ and NF-κB pathways in HBV-induced liver
fibrosis in mice, the detailed signaling factors in these pathways
and the mechanism of action of these molecules remains unclear.
Future and ongoing study will explore the mechanism of
anti-fibrosis in liver by using an integrative approach.
Based on these findings, we proposed an antifibrotic
model for AAV-shRNA (Fig. 8).
AAV-shRNA induced PPAR-γ expression and inhibited TGF-β expression
and NF-κB phosphorylation. TGF-β was downregulated after AAV-shRNA
treatment in HBV-replicated mice, leading to relief of oxidative
stress by upregulation of GSTP1 and down-regulation of PRDX1. TGF-β
was also downregulated due to upregulation of PPAR-γ. Upregulation
of PPAR-γ resulted in activation of the PPAR-γ signaling pathway.
The PPAR signaling pathway influenced lipid metabolism by
decreasing the expression of FABP1 and ME1 and reducing hepatocyte
steatosis. Concurrently, upregulation of PPAR-γ inhibited
inflammation by blocking the NF-κB signaling pathway by decreasing
NF-κB p65 phosphorylation.
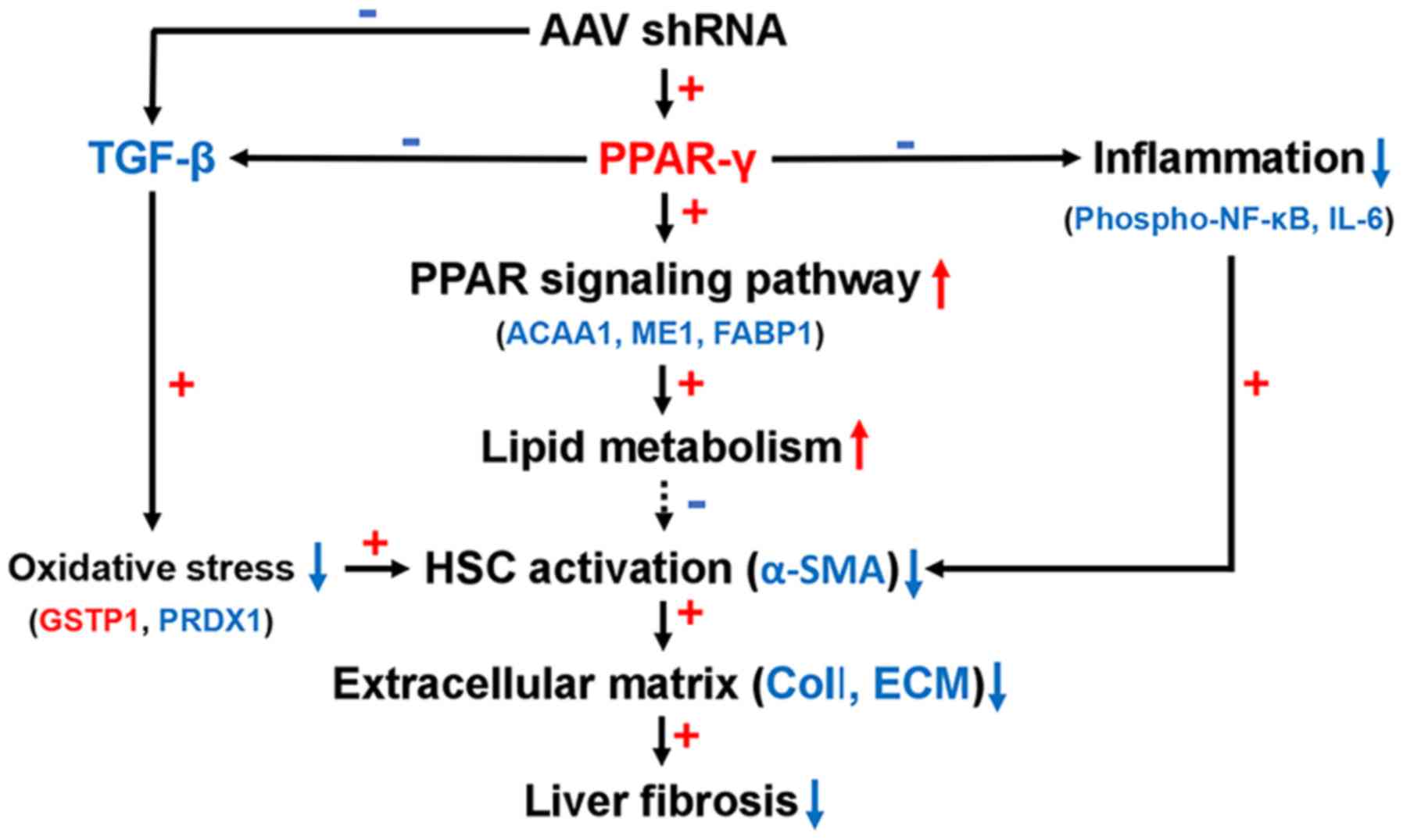 | Figure 8A proposed model showing the
mechanism of reduced oxidative stress, inflammation, and PPAR-γ
signaling activation, resulting in antifibrotic effects of
AAV-shRNA treatment. Red color up arrow and plus sign indicate
upregulated proteins and positive correlations. Blue color down
arrow and minus sign indicate down regulated proteins and negative
correlations. AAV, adeno-associated virus; shRNA, short hairpin
RNA; PPAR, peroxisome proliferator-activated receptor; TGF,
transforming growth factor; NF-κB, nuclear factor-κB; IL-6,
interleukin-6; ACAA1, acetyl-CoA acyltransferase 1; ME1, malic
enzyme 1; FABP1, fatty acid binding protein 1; GSTP1, glutathione
S-transferase Pi 1; PRDX1, peroxiredoxin-1; HPC, hepatic stellate
cell; α-SMA, α-smooth muscle actin; ECM, extracellular matrix. |
Supplementary Data
Funding
The present study was supported by a grant to WL and
LY from the CAMS Innovation Fund for Medical Sciences (CIFMS; grant
no. 2016-I2M-3-020).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LY and TC contributed equally to this work. LY, WL
and CZ conceived and designed the experiments. LY, JC, LS, and TC
performed the experiments. LY analyzed the data. LY and WL wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study of HBV-related liver fibrosis in mice was
performed in accordance with the Guide for the Care and Use of
Laboratory Animals, which was approved by the Institutional Animal
Care and Use Committee of the Chinese Academy of Medical
Sciences.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
AAV
|
adeno-associated virus
|
|
shRNA
|
short hairpin RNA
|
|
HBV
|
hepatitis B virus
|
|
TGF
|
transforming growth factor
|
|
iTRAQ
|
isobaric tags for relative and
absolute quantitation
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
ROS
|
reactive oxygen species
|
|
NF-κB
|
nuclear factor-κB
|
|
GO
|
Gene ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
CC
|
cell component
|
|
MF
|
molecular function
|
|
BP
|
biological process
|
|
TRAF2-GSTP1
|
tumor necrosis factor
receptor-associated factor 2-glutathione S-transferase Pi
|
|
PPI
|
protein-protein interaction
|
|
GSTP1
|
glutathione S-transferase Pi 1
|
|
Rpl
|
ribosome large subunit protein
|
|
FABP1
|
fatty acid binding protein 1
|
|
ME1
|
malic enzyme 1
|
|
ACAA1
|
acetyl-CoA acyltransferase 1
|
|
PRDX1
|
peroxiredoxin-1
|
|
LC-MS/MS
|
liquid chromatography-mass
spectrometry/mass spectrometry
|
Acknowledgments
We would like to thank Professor Jianhua Zheng at
the Institute of Pathogen Biology, Chinese Academy of Medical
Sciences and Peking Union Medical College for expert advice on
proteomics.
References
|
1
|
Ringelhan M, Heikenwalder M and Protzer U:
Direct effects of hepatitis B virus-encoded proteins and chronic
infection in liver cancer development. Dig Dis. 31:138–151. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McMahon BJ: The natural history of chronic
hepatitis B virus infection. Hepatology. 49(5 Suppl): S45–S55.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin CL and Kao JH: Risk stratification for
hepatitis B virus related hepatocellular carcinoma. J Gastroenterol
Hepatol. 28:10–17. 2013. View Article : Google Scholar
|
|
4
|
Bonilla Guerrero R and Roberts LR: The
role of hepatitis B virus integrations in the pathogenesis of human
hepatocellular carcinoma. J Hepatol. 42:760–777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karayiannis P: Hepatitis B virus:
Virology, molecular biology, life cycle and intrahepatic spread.
Hepatol Int. 11:500–508. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Poynard T, Mathurin P, Lai CL, Guyader D,
Poupon R, Tainturier MH, Myers RP, Muntenau M, Ratziu V, Manns M,
et al: A comparison of fibrosis progression in chronic liver
diseases. J Hepatol. 38:257–265. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar
|
|
8
|
Alkofer B, Lepennec V and Chiche L:
Hepatocellular cancer in the non-cirrhotic liver. J Visc Surg.
148:3–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Friedman SL, Roll FJ, Boyles J and Bissell
DM: Hepatic lipocytes: The principal collagen-producing cells of
normal rat liver. Proc Natl Acad Sci USA. 82:8681–8685. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Senoo H, Mezaki Y and Fujiwara M: The
stellate cell system (vitamin A-storing cell system). Anat Sci Int.
92:387–455. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Popov Y and Schuppan D: Targeting liver
fibrosis: Strategies for development and validation of antifibrotic
therapies. Hepatology. 50:1294–1306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gressner AM, Weiskirchen R, Breitkopf K
and Dooley S: Roles of TGF-beta in hepatic fibrosis. Front Biosci.
7:d793–d807. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rahimi RA and Leof EB: TGF-beta signaling:
A tale of two responses. J Cell Biochem. 102:593–608. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shek FW and Benyon RC: How can
transforming growth factor beta be targeted usefully to combat
liver fibrosis? Eur J Gastroenterol Hepatol. 16:123–126. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weidinger A and Kozlov AV: Biological
activities of reactive oxygen and nitrogen species: Oxidative
stress versus signal transduction. Biomolecules. 5:472–484. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim YM and Cho M: Activation of NADPH
oxidase subunit NCF4 induces ROS-mediated EMT signaling in HeLa
cells. Cell Signal. 26:784–796. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schafer FQ and Buettner GR: Redox
environment of the cell as viewed through the redox state of the
glutathione disulfide/glutathione couple. Free Radic Biol Med.
30:1191–1212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dayer R, Fischer BB, Eggen RI and Lemaire
SD: The peroxiredoxin and glutathione peroxidase families in
Chlamydomonas reinhardtii. Genetics. 179:41–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wheeler MD, Kono H, Yin M, Nakagami M,
Uesugi T, Arteel GE, Gäbele E, Rusyn I, Yamashina S, Froh M, et al:
The role of kupffer cell oxidant production in early
ethanol-induced liver disease. Free Radic Biol Med. 31:1544–1549.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Issemann I and Green S: Activation of a
member of the steroid hormone receptor superfamily by peroxisome
proliferators. Nature. 347:645–650. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Greene ME, Blumberg B, McBride OW, Yi HF,
Kronquist K, Kwan K, Hsieh L, Greene G and Nimer SD: Isolation of
the human peroxisome proliferator activated receptor gamma cDNA:
Expression in hematopoietic cells and chromosomal mapping. Gene
Expr. 4:281–299. 1995.PubMed/NCBI
|
|
23
|
Dreyer C, Krey G, Keller H, Givel F,
Helftenbein G and Wahli W: Control of the peroxisomal
beta-oxidation pathway by a novel family of nuclear hormone
receptors. Cell. 68:879–887. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xing G, Zhang L, Zhang L, Heynen T,
Yoshikawa T, Smith M, Weiss S and Detera-Wadleigh S: Rat PPAR delta
contains a CGG triplet repeat and is prominently expressed in the
thalamic nuclei. Biochem Biophys Res Commun. 217:1015–1025. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen F, Law SW and O'Malley BW:
Identification of two mPPAR related receptors and evidence for the
existence of five subfamily members. Biochem Biophys Res Commun.
196:671–677. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu J, Fu Y and Chen A: Activation of
peroxisome proliferator-activated receptor-gamma contributes to the
inhibitory effects of curcumin on rat hepatic stellate cell growth.
Am J Physiol Gastrointest Liver Physiol. 285:G20–G30. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ahmadian M, Suh JM, Hah N, Liddle C,
Atkins AR, Downes M and Evans RM: PPARγ signaling and metabolism:
The good, the bad and the future. Nat Med. 19:557–566. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leclercq IA, Da Silva Morais A, Schroyen
B, Van Hul N and Geerts A: Insulin resistance in hepatocytes and
sinusoidal liver cells: Mechanisms and consequences. J Hepatol.
47:142–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao X, Xue J, Wang XL, Zhang Y, Deng M
and Xie ML: Involvement of hepatic peroxisome
proliferator-activated receptor α/γ in the therapeutic effect of
osthole on high-fat and high-sucrose-induced steatohepatitis in
rats. Int Immunopharmacol. 22:176–181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo YT, Leng XS, Li T, Peng JR, Song SH,
Xiong LF and Qin ZZ: Effect of ligand of peroxisome
proliferator-activated receptor gamma on the biological characters
of hepatic stellate cells. World J Gastroenterol. 11:4735–4739.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Anty R and Lemoine M: Liver fibrogenesis
and metabolic factors. Clin Res Hepatol Gastroenterol. 35(Suppl 1):
S10–S20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun K, Wang Q and Huang XH: PPAR gamma
inhibits growth of rat hepatic stellate cells and TGF beta-induced
connective tissue growth factor expression. Acta Pharmacol Sin.
27:715–723. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang L, Chan CC, Kwon OS, Liu S, McGhee J,
Stimpson SA, Chen LZ, Harrington WW, Symonds WT and Rockey DC:
Regulation of peroxisome proliferator-activated receptor-gamma in
liver fibrosis. Am J Physiol Gastrointest Liver Physiol.
291:G902–G911. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brasier AR: The NF-kappaB regulatory
network. Cardiovasc Toxicol. 6:111–130. 2006. View Article : Google Scholar
|
|
35
|
Calzado MA, Bacher S and Schmitz ML:
NF-kappaB inhibitors for the treatment of inflammatory diseases and
cancer. Curr Med Chem. 14:367–376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luedde T and Schwabe RF: NF-κB in the
liver-linking injury, fibrosis and hepatocellular carcinoma. Nat
Rev Gastroenterol Hepatol. 8:108–118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kong D, Zhang F, Wei D, Zhu X, Zhang X,
Chen L, Lu Y and Zheng S: Paeonol inhibits hepatic fibrogenesis via
disrupting nuclear factor-κB pathway in activated stellate cells:
In vivo and in vitro studies. J Gastroenterol Hepatol.
28:1223–1233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu M, Wu Q, Chen P, Büchele B, Bian M,
Dong S, Huang D, Ren C, Zhang Y, Hou X, et al: A boswellic
acid-containing extract ameliorates schistosomiasis liver granuloma
and fibrosis through regulating NF-κB signaling in mice. PLoS One.
9:e1001292014. View Article : Google Scholar
|
|
39
|
Bromberg J and Wang TC: Inflammation and
cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Naugler WE and Karin M: The wolf in
sheep's clothing: The role of interleukin-6 in immunity,
inflammation and cancer. Trends Mol Med. 14:109–119. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wobser H, Dorn C, Weiss TS, Amann T,
Bollheimer C, Büttner R, Schölmerich J and Hellerbrand C: Lipid
accumulation in hepato-cytes induces fibrogenic activation of
hepatic stellate cells. Cell Res. 19:996–1005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Montiel-Duarte C, Ansorena E,
López-Zabalza MJ, Cenarruzabeitia E and Iraburu MJ: Role of
reactive oxygen species, glutathione and NF-kappaB in apoptosis
induced by 3,4-methylenedioxymethamphetamine ('Ecstasy') on hepatic
stellate cells. Biochem Pharmacol. 67:1025–1033. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hernández E, Bucio L, Souza V, Escobar MC,
Gómez-Quiroz LE, Farfán B, Kershenobich D and Gutiérrez-Ruiz MC:
Pentoxifylline downregulates alpha (I) collagen expression by the
inhibition of Ikappabalpha degradation in liver stellate cells.
Cell Biol Toxicol. 24:303–314. 2008. View Article : Google Scholar
|
|
44
|
Ye L, Kan F, Yan T, Cao J, Zhang L, Wu Z
and Li W: Enhanced antiviral and antifibrotic effects of short
hairpin RNAs targeting HBV and TGF-β in HBV-persistent mice. Sci
Rep. 7:38602017. View Article : Google Scholar
|
|
45
|
Kan F, Ye L, Yan T, Cao J, Zheng J and Li
W: Proteomic and transcriptomic studies of HBV-associated liver
fibrosis of an AAV-HBV-infected mouse model. BMC Genomics.
18:6412017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
The Gene Ontology Consortium: The gene
ontology resource: 20 Years and still GOing strong. Nucleic Acids
Res. 47(D1): D330–D338. 2019. View Article : Google Scholar
|
|
47
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47(D1): D590–D595. 2019. View Article : Google Scholar :
|
|
48
|
Dillon ST, Bhasin MK, Feng X, Koh DW and
Daoud SS: Quantitative proteomic analysis in HCV-induced HCC
reveals sets of proteins with potential significance for racial
disparity. J Transl Med. 11:2392013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
50
|
Yin L, Qi Y, Xu Y, Xu L, Han X, Tao X,
Song S and Peng J: Dioscin inhibits HSC-T6 cell migration via
adjusting SDC-4 expression: Insights from iTRAQ-based quantitative
proteomics. Front Pharmacol. 8:6652017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wiese S, Reidegeld KA, Meyer HE and
Warscheid B: Protein labeling by iTRAQ: A new tool for quantitative
mass spectrometry in proteome research. Proteomics. 7:340–350.
2007. View Article : Google Scholar
|
|
52
|
Ye L, Yu H, Li C, Hirsch ML, Zhang L,
Samulski RJ, Li W and Liu Z: Adeno-associated virus vector mediated
delivery of the HBV genome induces chronic hepatitis B virus
infection and liver fibrosis in mice. PLoS One. 10:e01300522015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang X, Li Y, Xu G, Liu M, Xue L, Liu L,
Hu S, Zhang Y, Nie Y, Liang S, et al: Mechanism study of peptide
GMBP1 and its receptor GRP78 in modulating gastric cancer MDR by
iTRAQ-based proteomic analysis. BMC Cancer. 15:3582015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gan CS, Chong PK, Pham TK and Wright PC:
Technical, experimental, and biological variations in isobaric tags
for relative and absolute quantitation (iTRAQ). J Proteome Res.
6:821–827. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium Nat Genet. 25:25–29. 2000.
|
|
56
|
Desvergne B and Wahli W: Peroxisome
proliferator-activated receptors: Nuclear control of metabolism.
Endocr Rev. 20:649–688. 1999.PubMed/NCBI
|
|
57
|
Venook AP, Papandreou C, Furuse J and de
Guevara LL: The incidence and epidemiology of hepatocellular
carcinoma: A global and regional perspective. Oncologist. 15(Suppl
4): S5–S13. 2010. View Article : Google Scholar
|
|
58
|
Wang D, Cai H, Yu WB and Yu L:
Identification of hepatitis B virus X gene variants between
hepatocellular carcinoma tissues and pericarcinoma liver tissues in
Eastern China. Int J Clin Exp Pathol. 7:5988–5996. 2014.PubMed/NCBI
|
|
59
|
Ringelhan M, O'Connor T, Protzer U and
Heikenwalder M: The direct and indirect roles of HBV in liver
cancer: Prospective markers for HCC screening and potential
therapeutic targets. J Pathol. 235:355–367. 2015. View Article : Google Scholar
|
|
60
|
Atta HM: Reversibility and heritability of
liver fibrosis: Implications for research and therapy. World J
Gastroenterol. 21:5138–5148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li T, Zhao XP, Wang LY, Gao S, Zhao J, Fan
YC and Wang K: Glutathione S-transferase P1 correlated with
oxidative stress in hepatocellular carcinoma. Int J Med Sci.
10:683–690. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ding C, Fan X and Wu G: Peroxiredoxin 1-an
antioxidant enzyme in cancer. J Cell Mol Med. 21:193–202. 2017.
View Article : Google Scholar
|
|
63
|
Ginguay A, Cynober L, Curis E and Nicolis
I: Ornithine amino-transferase, an important glutamate-metabolizing
enzyme at the crossroads of multiple metabolic pathways. Biology
(Basel). 6:182017.
|
|
64
|
Chávez E, Castro-Sánchez L, Shibayama M,
Tsutsumi V, Moreno MG and Muriel P: Sulfasalazine prevents the
increase in TGF-β, COX-2, nuclear NFκB translocation and fibrosis
in CCl4-induced liver cirrhosis in the rat. Hum Exp Toxicol.
31:913–920. 2012. View Article : Google Scholar
|
|
65
|
Bitencourt S, de Mesquita FC, Caberlon E,
da Silva GV, Basso BS, Ferreira GA and de Oliveira JR: Capsaicin
induces de-differentiation of activated hepatic stellate cell.
Biochem Cell Biol. 90:683–690. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Rhee SG, Chae HZ and Kim K:
Peroxiredoxins: A historical over-view and speculative preview of
novel mechanisms and emerging concepts in cell signaling. Free
Radic Biol Med. 38:1543–1552. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Halliwell B: Oxidative stress and cancer:
Have we moved forward? Biochem J. 401:1–11. 2007. View Article : Google Scholar
|
|
68
|
Reza JZ, Doosti M, Salehipour M, Packnejad
M, Mojarrad M and Heidari M: Modulation peroxisome proliferators
activated receptor alpha (PPAR alpha) and acyl coenzyme A:
Cholesterol acyltransferase1 (ACAT1) gene expression by fatty acids
in foam cell. Lipids Health Dis. 8:382009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Guo Y, Jolly RA, Halstead BW, Baker TK,
Stutz JP, Huffman M, Calley JN, West A, Gao H, Searfoss GH, et al:
Underlying mechanisms of pharmacology and toxicity of a novel PPAR
agonist revealed using rodent and canine hepatocytes. Toxicol Sci.
96:294–309. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
van der Leij FR, Bloks VW, Grefhorst A,
Hoekstra J, Gerding A, Kooi K, Gerbens F, te Meerman G and Kuipers
F: Gene expression profiling in livers of mice after acute
inhibition of beta-oxidation. Genomics. 90:680–689. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Murai S, Ando A, Ebara S, Hirayama M,
Satomi Y and Hara T: Inhibition of malic enzyme 1 disrupts cellular
metabolism and leads to vulnerability in cancer cells in
glucose-restricted conditions. Oncogenesis. 6:e3292017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang G, Bonkovsky HL, de Lemos A and
Burczynski FJ: Recent insights into the biological functions of
liver fatty acid binding protein 1. J Lipid Res. 56:2238–2247.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wu YL, Peng XE, Zhu YB, Yan XL, Chen WN
and Lin X: Hepatitis B virus X protein induces hepatic steatosis by
enhancing the expression of liver fatty acid binding protein. J
Virol. 90:1729–1740. 2016. View Article : Google Scholar :
|
|
75
|
Mukai T, Egawa M, Takeuchi T, Yamashita H
and Kusudo T: Silencing of FABP1 ameliorates hepatic steatosis,
inflammation, and oxidative stress in mice with nonalcoholic fatty
liver disease. FEBS Open Bio. 7:1009–1016. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wolfrum C, Buhlmann C, Rolf B, Börchers T
and Spener F: Variation of liver-type fatty acid binding protein
content in the human hepatoma cell line HepG2 by peroxisome
proliferators and antisense RNA affects the rate of fatty acid
uptake. Biochim Biophys Acta. 1437:194–201. 1999. View Article : Google Scholar : PubMed/NCBI
|