Introduction
Colorectal cancer is the third most commonly
diagnosed cancer in men and the second in women worldwide, with
~1.4 million cases and 693,000 mortalities in 2012. The incidence
rates are higher in Europe, Australia and Northern American
(1). The incidence of CRC is
likely to increase as the world becomes richer, as more individuals
shift to unhealthy diets and life styles (2). Currently, chemotherapy and surgery
are the main treatment strategies for malignant tumors that form in
the colon tissues (3).
Chemotherapy can be used either before surgery to shrink the tumor
before removal, or after surgery for patients with advanced CRC
(4). While the overall survival
duration of patients with advanced CRC has improved in the last
decade due to the various chemotherapy regimens, drug resistance
still occurs in nearly all patients with CRC and ultimately leads
to chemotherapy failure (5). Thus,
investigation of the molecular mechanisms of CRC progression and
the development of novel targeted drugs are essential.
Widespread-usage of high-throughput sequencing
allows for the identification of mutations present in CRC; however,
progress in targeted therapies for CRC has been slow (6). There has been significant progress in
the understanding of the molecular pathways regarding
tumorigenesis. For instance, several pathways are dysregulated by
common mutations that have been identified via CRC sequencing,
including the WNT, MAPK, PI3K and p53 pathways (7). In total, ~60% of sequenced CRC
tissues have potentially targetable genetic aberrations in the
PIK3CA, BRAF and PTEN signaling pathway, and early studies have
revealed promising effects for CRC by focusing on these markers
(8,9).
PI3K/AKT/mTOR (PAM) signaling acts as a pivotal
transduction pathway in tumor progression, which is involved in
controlling cancer cell proliferation, differentiation, motility
and survival (10). A mutation of
PI3Kα is present in >18% of metastatic CRC cases, and thus,
small-molecular compounds that can inhibit the PAM pathway may have
great potential in the treatment of CRC (11). VS-5584, a highly selective
PI3K/mTOR kinase inhibitor, demonstrates robust anti-tumor effects
both in vitro and in vivo (12). Moreover, VS-5584 has 10-fold
selectivity for cancer stem cells (13). These findings suggest that
medicinal chemists should identify and develop novel and potent
PI3K inhibitors for cancer treatment.
It has been reported that there is a close
connection between mTOR and autophagy (14). It was first discovered in yeast
that genetic or pharmacological inhibition of mTOR complex 1 could
induce autophagy (15). Autophagy
is the major cellular digestion process that is essential for
cellular development and homeostasis. In addition, it is critical
to provide energy in response to nutrient and environmental stress
by recycling macromolecules (16).
Autophagy induction could have pro-survival or pro-death properties
depending on tumor types or treatment strategies (17). For instance, autophagy may
facilitate tumor development when nutrients are limited (18). Thus, inhibition of autophagy may
sensitize cancer cells to stress, leading to cell death (19). On the other hand, autophagy may
induce autophagy-mediated cell death, which is also known as type
II programmed cell death (20).
Given the potential dual functions of autophagy in tumor
progression, elucidating the precise function of autophagy in
individual cancer types induced by PAM pathway inhibitors is
essential for cancer therapy.
Our previous study synthesized a compound W922
(refers to compound A7) based on the structure of PI3K/mTOR dual
inhibitor VS-5584 by replacing the imidazole ring with a triazine
skeleton (21). It has been
reported that triazine skeleton has potential anti-tumor
bioantivity (22). In the present
study aimed to evaluate the anti-proliferative effects of W922 both
in vitro and in vivo, and identify the underlying
mechanism of W922's anti-tumor effect. Data in this study will
provide a pharmacological basis for W922's potential application,
and will also provide information for further development of
PI3K/AKT pathway inhibitors and triazine skeletal derivatives.
Materials and methods
Chemicals
W922 was synthesized in Department of Pharmaceutical
Biochemistry, Xi'an Jiaotong University, and characterized using
1H NMR (nuclear magnetic resonance hydrogen spectrum),
13C NMR (nuclear magnetic resonance carbon spectrum) as
previously described (21).
VS-5584 (cat. no. A3927) and Choloroquine (CQ) diphosphate (cat.
no. A8628) were purchased from APExBIO Technology LLC.
For in vitro assays, chemicals were prepared
by dissolving into sterilized DMSO to a final concentration of 0.01
M and stored at −20°C before use. For the xenograft experiment,
W922 and VS-5584 were dissolved in a mixture containing DMSO,
PEG400 and ddH2O (ratio as 1:7:2).
Cell lines and cell culture
conditions
CRC cell lines HCT116, HT29, RKO, Colo205, SW620,
DLD1 and LOVO were purchased from American Type Culture Collection.
All cell lines were maintained in DMEM (HyClone; Cytiva)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin antibiotics (Beijing Solarbio
Science & Technology Co., Ltd.), and cultured at 37°C in a
humidified incubator with 5% CO2.
Reagents and antibodies
MTT, crystal violet solution, PI, RNAse A and DAPI
were purchased from Beijing Solarbio Science & Technology Co.,
Ltd. Western blotting reagents were obtained from Beyotime
Institute of Biotechnology. An Annexin V/PI apoptosis detection kit
was purchased from 7 Sea Biotech (http://www.7seapharmtech.com/).
Primary antibodies against the following proteins
were purchased from Cell Signaling Technology, Inc. (CST): AKT
(cat. no. 9272), phosphorylated (p)-AKT (Ser473; cat. no. 4060),
mTOR (cat. no. 2983), p-mTOR (Ser2448; cat. no. 5536), p70S6K (cat.
no. 9202), p-p70S6 kinase (p70S6K; Ser371; cat. no. 9208),
eukaryotic translation initiation factor 4E-binding protein 1
(4E-BP1; cat. no. 9644), p-4E-BP1 (Thr37/46; cat. no. 2855),
Beclin-1 (cat. no. 3495), p-Beclin-1 (Ser93; cat. no. 14717),
Cyclin D1 (cat. no. 2978), Cyclin E1 (cat. no. 4129), cleaved
caspase-3 (cat. no. 9664), autophagy related 5 (ATG5; cat. no.
9980) and GAPDH (cat. no. 5174). LC3-I/II (cat. no. ABC929) was
purchased from Sigma-Aldrich (Merck KGaA) and ki67 (cat. no.
ab15580) was obtained from Abcam. Secondary horseradish
peroxidase-linked antibodies against rabbit (cat. no. 7074) and
mouse (cat. no. 7076) were purchased from CST. Small interfering
RNA (si)ATG5 was purchased from Shanghai GenePharma Co., Ltd., and
Lipofectamine® 2000 was obtained from Thermo Fisher
Scientific, Inc.
Cell mutation search
The mutation data were obtained from the Catalogue
Of Somatic Mutations In Cancer website (https://cancer.sanger.ac.uk/cosmic).
Cell viability assay
The MTT assay was used to measure cell viability.
The CRC cells (HCT116, RKO, COLO205, SW620, DLD1, HT29 and LOVO)
were seeded in a 96-well plate (5,000 cells/well) and incubated at
37°C overnight. The following day, the cells were treated with W922
(10 µM) and cultured for 24 h at 37°C. Then, MTT solution (5
mg/ml; 20 µl) was added into each well, and the plate was
incubated for another 4 h at 37°C. The supernatant was removed and
the purple formazan crystals were dissolved in 150 µl DMSO.
The cell viability was calculated according to the absorbance
values at 570 nm, measured via a microplate reader (Thermo Fisher
Scientific, Inc.).
Colony formation assay
The long-term effect of W922 on cell proliferation
was evaluated using the colony formation assay in HCT116 cells.
Cells were plated in a 6-well plate (1,000 cells/well). After
overnight incubation, the cells were treated with W922 (0.1, 0.3, 1
and 3 µM) or DMSO and cultured at 37°C for ~3 weeks until
colonies were visible. The colonies in each well of the 6-well
plate were fixed with pure methanol (2 ml) for 20 min and stained
with crystal violet solution (2 ml) for 20 min at room temperature.
Then, the plates were washed with PBS three times, and the colonies
were counted.
Xenograft antitumor study
A total of 30 BALB/c nude mice (age, 4 weeks;
weight, 18 g; female) were purchased from Beijing Vital River
Laboratory Animal Technology Co, Ltd., and were fed in the
enviroument of 25°C, 60% humidity, 12 h light/dark cycle, and free
access to food and water in Xi'an Jiatong University Health Science
Center. HCT116 cells (1×106) resuspended in 100
µl PBS were subcutaneously injected into the right flanks of
4-week-old BALB/c nude mice. Once a palpable tumor was presented,
the mice were randomly grouped and treated daily intraperitoneally
with either vehicle solvent, W922 (10, 30 and 100 mg/kg) or VS-5584
(10 mg/kg) for 18 days. Tumor volumes were measured every 3 days
using a caliper and calculated as 0.5 × (length ×
width2). The mice were sacrificed 18 days after
transplanting, and the tumors were isolated and weighed.
Flow cytometric analysis
Cell cycle analysis was performed via PI staining.
The cells were seeded in 12-well plates (5×104/well) and
treated with W922 (0.1, 1 and 10 µM) for 24 h at 37°C. After
harvesting, the cells were fixed in 70% ethanol at 4°C overnight
and stained with PI solution (50 µl PI and 50 µl
RNAse A in 10 ml PBS) for 30 min at room temperature before
measurement. The data were obtained using a flow cytometer (Beckman
Coulter, Inc.) and analyzed using the ModFitLT software (Version
5.0; www.mybiosoftware.com).
Cell apoptosis was measured using a Annexin V/PI
double staining assay. The cells were seeded in 12-well plates
(5×104/well) overnight and treated with W922 (0.1, 1 and
10 µM) for 24 and 48 h at 37°C. Then, the cells were
harvested and subjected to the assay according to the
manufacturer's instruction. The data were collected from a flow
cytometer (Beckman Coulter, Inc.) and analyzed using FlowJo
software (Version 10; FlowJo LLC).
Western blotting
HCT116 cells were seeded in 6-well plates
(105 cells/well) and allowed to attach. The following
day, cells were treated with W922 (0.1, 0.3, 1, 3 and 10 µM)
at 37°C for 24 h. After treatment, the cells were harvested, and
the proteins were collected with RIPA buffer (Beyotime Institute of
Biotechnology) supplemented with protease inhibitor cocktail for 20
min on ice. The protein concentrations were determined using the
BCA method. The same amount of proteins (20 µg) were
separated via a 12% SDS-PAGE gel and transferred to a PVDF
membrane. Subsequently, the membrane was blocked in 5% skim milk in
room temperature for 2 h, and then incubated with primary
antibodies (antibody: 5% skim milk =1:1,000) at 4°C overnight. The
membranes were incubated with the secondary antibodies for 1 h. The
protein expression levels were visualized using an enhanced
chemiluminescence system (EMD Millipore), and the densitometry was
detected using Image J software (Version 1.48; imagej.net).
Immunofluorescence microscopy
The cells were grown on 13-mm coverslips
(10,000/well), placed in 24-well culture plate and treated with
W922 (0.1, 1 and 10 µM) at 37°C for 24 h. for desired time
periods. After treatment, the cells were rinsed in PBS, fixed with
pure methanol for 20 min on ice and washed twice with PBS.
Subsequently, the cells were blocked in PBS with 5% FBS at room
temperature for 1 h, and then incubated with anti-LC3-I/II
antibody, anti-Ki67 antibody and anti-Cleaved caspase-3 antibody
overnight at 4°C. Cells were washed with PBS and incubated with
Alexa Fluor 488-labeled and Alexa Fluor 555-labeled secondary
antibodies in room temperature for 2 h (Invitrogen; Thermo Fisher
Scientific, Inc.; cat. nos. A32790, A21208 and A32794; 1:1,000).
The nuclei were stained with Hoechst 33342 (1 µg/ml) at room
temperature for 30 min. After washing with PBS, the coverslips with
cells were mounted onto slides with aqueous mounting medium. The
cells were imaged using ZEISS LSM700 (Zeiss GmbH) confocal
microscope system (magnification, ×20).
CQ pretreatment
CQ was used to inhibit autophagy. CQ (10 and 20
µM) was added to cells at 37°C for 2 h, and then W922 (1
µM) was used to treat HCT116 cells at 37°C for 24 h to
distinguish whether W922 was an autophagy inhibitor or inducer. For
proteomics assay, 50 µM choloroquine was used to
pre-stimulate HCT116 cells for 2 h at 37°C before W922 (10
µM) treatment (24) to
examine the protective or suppressive role served by autophgy.
Transfection experiment
siRNA targeting the ATG5 cDNA sequencing
(5′-GACGUUGGUAACUGACAAATT-3′) was purchased from Shanghai
GenePharma Co., Ltd. And siRNA NC was also supplied by GenePharma
company and was taken as the negative control. HCT116 cells were
plated in 6-well plates at a density of 2×105/well and
cultured overnight. ATG5 siRNA at a concentration of 80 nM
(dissolved in FBS-free medium) was transfected into the cells using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. A total
of 6 h after transfection, the FBS-free medium was removed and
replaced with complete medium, and cells were cultured for another
24 h. Successfully transfected HCT116 cells were used in the
corresponding experiments.
Proteomics sample preparation
HCT116 cells were plated in 6-well plates
(3×105) and treated by W922 or treated with the
combination of choloroquine and W922 for 24 h at 37°C. After
treatment, the cells were collected and lysed in RIPA buffer
supplemented with PMSF and protease inhibitor on ice. The cell
lysate was then centrifugated at 15,500 × g for 15 min at 4°C and
the supernatant was collected into a new Eppendorf tube. After
protein reduction with 8 mM 1,4-dithiothreitol (Sigma-Aldrich;
Merck KGaA) at 55°C for 45 min and alkylation using 25 mM
iodoacetamide (Sigma-Aldrich; Merck KGaA) at room temperature for
30 min in the dark, the proteins were precipitated using pure
acetone at -20°C overnight. Subsequently, the proteins were
digested with Lys C (FUJIFILM Wako Pure Chemical Corporation; Lys
C:protein amount=1:75) at 30°C for 8 h and trypsin (Promega
Corporation) at 37°C overnight. Then, desalting of the peptides was
conducted using C18 Sep-pak (WAT054960; Waters Corportation) at
room temperature, and the peptides in each sample were analyzed via
nano liquid chromatography (LC)-mass spectrometry (MS)/MS, as
customary in shotgun proteomics (23).
LC-MS/MS analysis
LC-MS/MS analysis was performed on an Orbitrap Elite
mass spectrometer (Thermo Fisher Scientific, Inc.). The instrument
was equipped with an EASY ElectroSpray Source and connected to an
UltiMate 3000 RSLCnano UPLC system (Thermo Fisher Scientific,
Inc.). The injected sample fractions were preconcentrated and
desalted online using a PepMap C18 nanotrap column (Thermo Fisher
Scientific, Inc.) with a flow rate of 3 µl/min for 5 min.
Peptide separation was performed on an EASY-Spray C18
reversed-phase nano-LC column (Thermo Fisher Scientific, Inc.) at
55°C and a flow rate of 300 nl/min. Peptides were separated by a
binary solvent system, and were eluted with a gradient of 4-26% B
in 120 min and 26-95% B in 10 min. The instrument was operated in
the positive ion mode for data-dependent acquisition of MS/MS
spectra with a dynamic exclusion time of previously selected
precursor ions of 30 sec. Mass spectra were acquired in a
mass-to-charge (m/z) range of 375-1,500 with a resolution of
120,000 at m/z 200, and at a resolution of 60,000 with a target
value of 2×105 ions and a maximum injection time of 120
msec. The fixed first m/z was 100, and the isolation window was 1.2
m/z units.
The extracted data were searched via the Andromeda
search engine in MaxQuant 1.5.6.5 software (www.maxquant.org). Gene names corresponding to
upregulated and down-regulated proteins were submitted into STRING
website (http://string-db.org) to analyze the
potential network among proteins. The data of Gene Ontology (GO)
analysis was also collected from STRING website.
Statistical analysis
Each sample has three biological replicates. Data
are presented as the mean ± SEM. Statistical analyses were
performed using GraphPad Prism 8.0 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference. Data were analyzed using an unpaired Student's t-test,
and one-way or two-way ANOVA followed by Dunnett's test.
Results
W922 inhibits tumor growth in vivo via
the regulation of viability
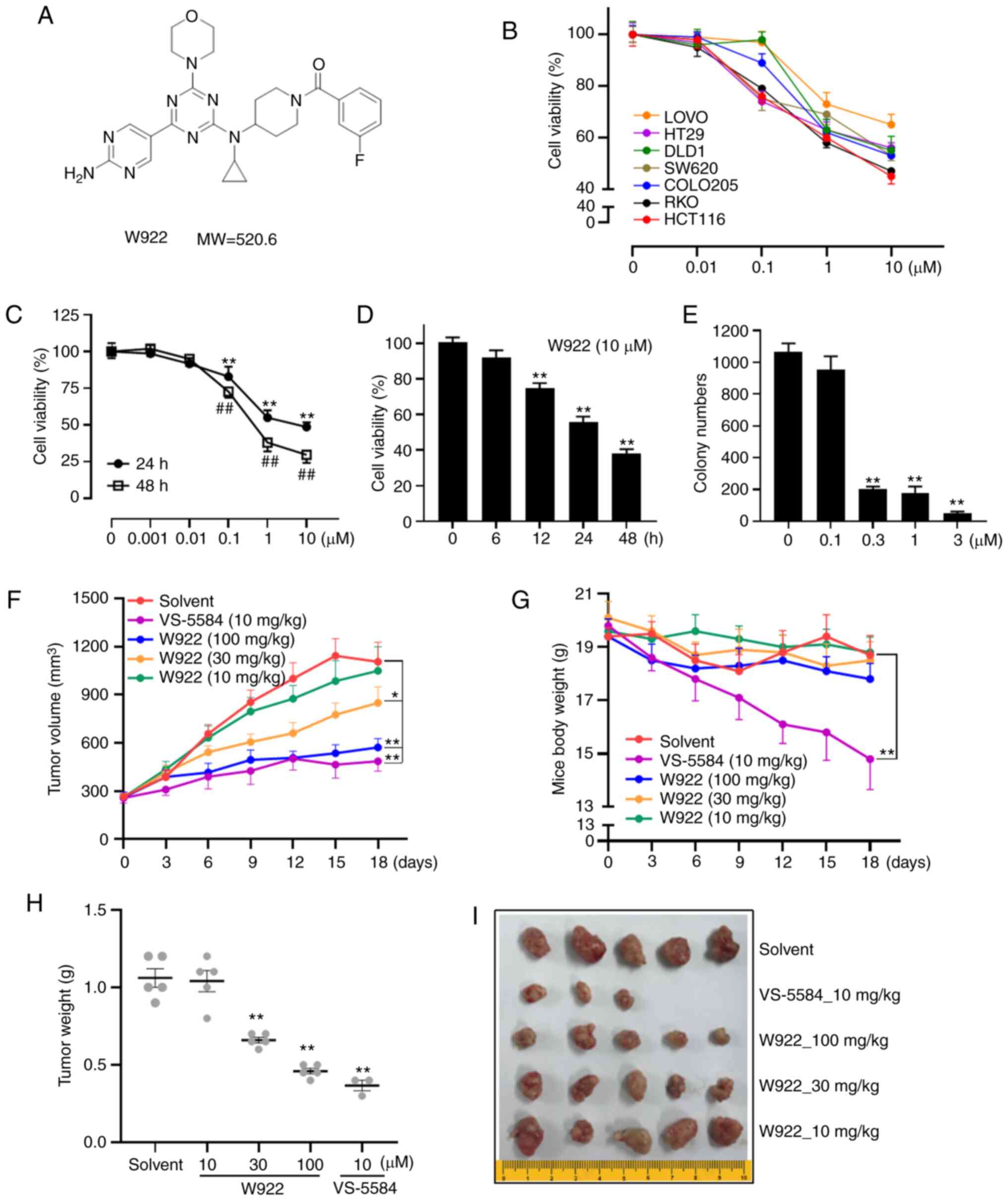
The chemical structure of W922 is presented in
Fig. 1A. The effects of W922 on
cell viability were first examined using seven different CRC cell
lines. The mutant sites in the tested seven cell lines are
summarized in Table I. HCT116 had
mutation sites on PIK3CA and KRAS. Among these CRC cell lines,
HCT116 was the most sensitive cell line to the drug treatment
(Fig. 1B). Thus, all subsequent
experiments were performed with this cell line.
 | Table IMutant sites in tested colorectal
cancer cell lines. |
Table I
Mutant sites in tested colorectal
cancer cell lines.
| Cell line | ATCC no. | Mutant gene |
|---|
| LOVO | CCL-229 | PIK3CB; RAF1;
KRAS |
| HT29 | HTB-38 | PIK3CA; PIK3R1;
BRAF; TP53 |
| DLD1 | CCL-221 | KRAS |
| SW620 | CCL-227 | KRAS |
| COLO205 | CCL-222 | BRAF; TP53 |
| RKO | CRL-2577 | PIK3CA; BRAF |
| HCT116 | CCL-247 | PIK3CA; KRAS |
W922 treatment markedly decreased the viability of
HCT116 cells in concentration- and time-dependent manners (Fig. 1C and D). Consistently, colony
formation assay results indicated that the cells treated with W922
formed significantly smaller and fewer colonies compared with the
untreated cells (Fig. 1E),
suggesting the long-term anti-tumor effect of W922.
Next, a HCT116 cell xenograft nude mouse model was
used to investigate the effect of W922 on CRC in vivo model.
Significant suppression of tumor growth was observed when the dose
of W922 was ≥30 mg/kg (Fig. 1F, H and
I). While VS-5584 exhibited robust anti-tumor potency, it had
significant toxicity, as the weight of the mice decreased
significantly and two mice died in the VS-5584 group (Fig. 1G and I).
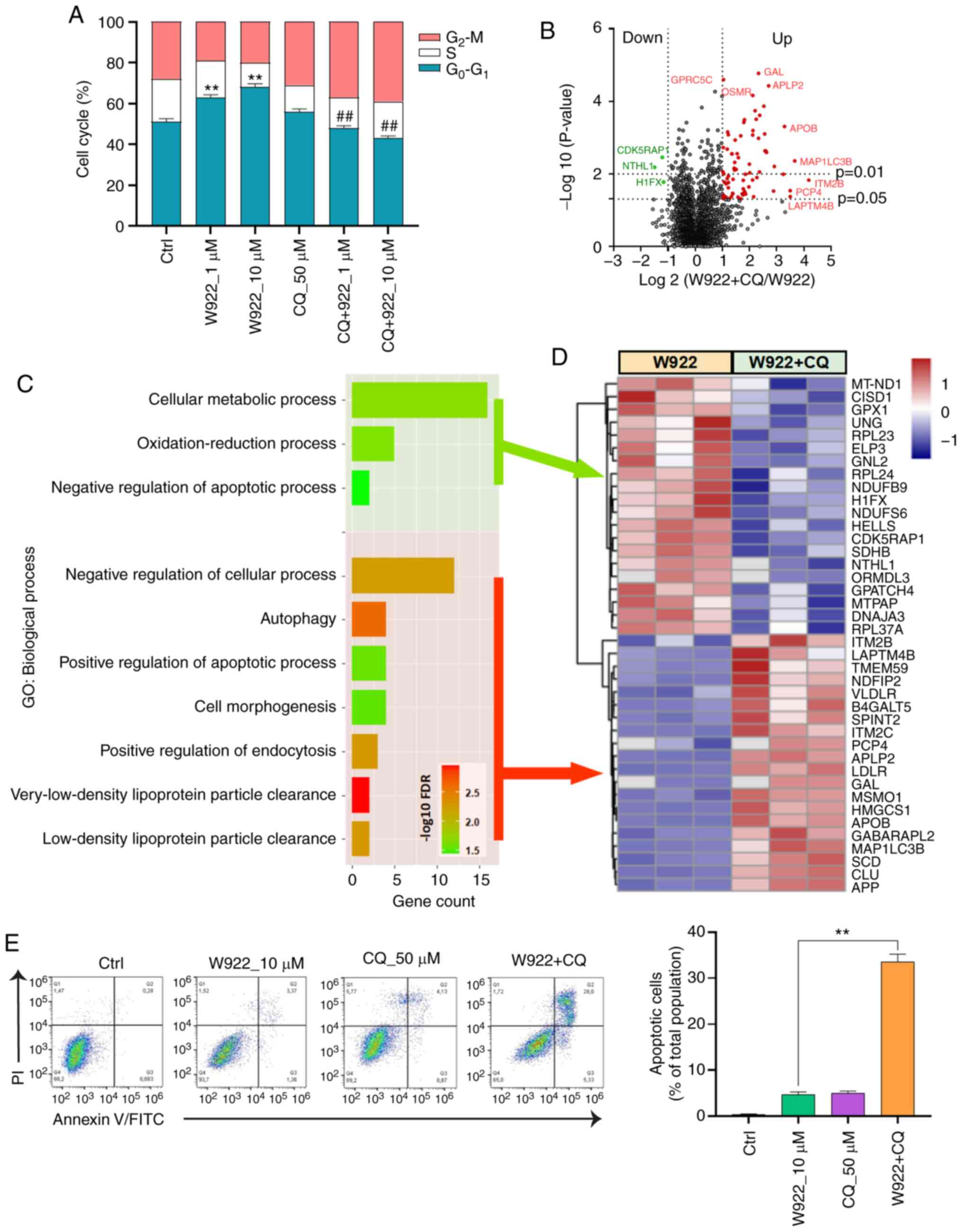
W922 negatively regulates the cell cycle
processes
In order to investigate the mechanism via which W922
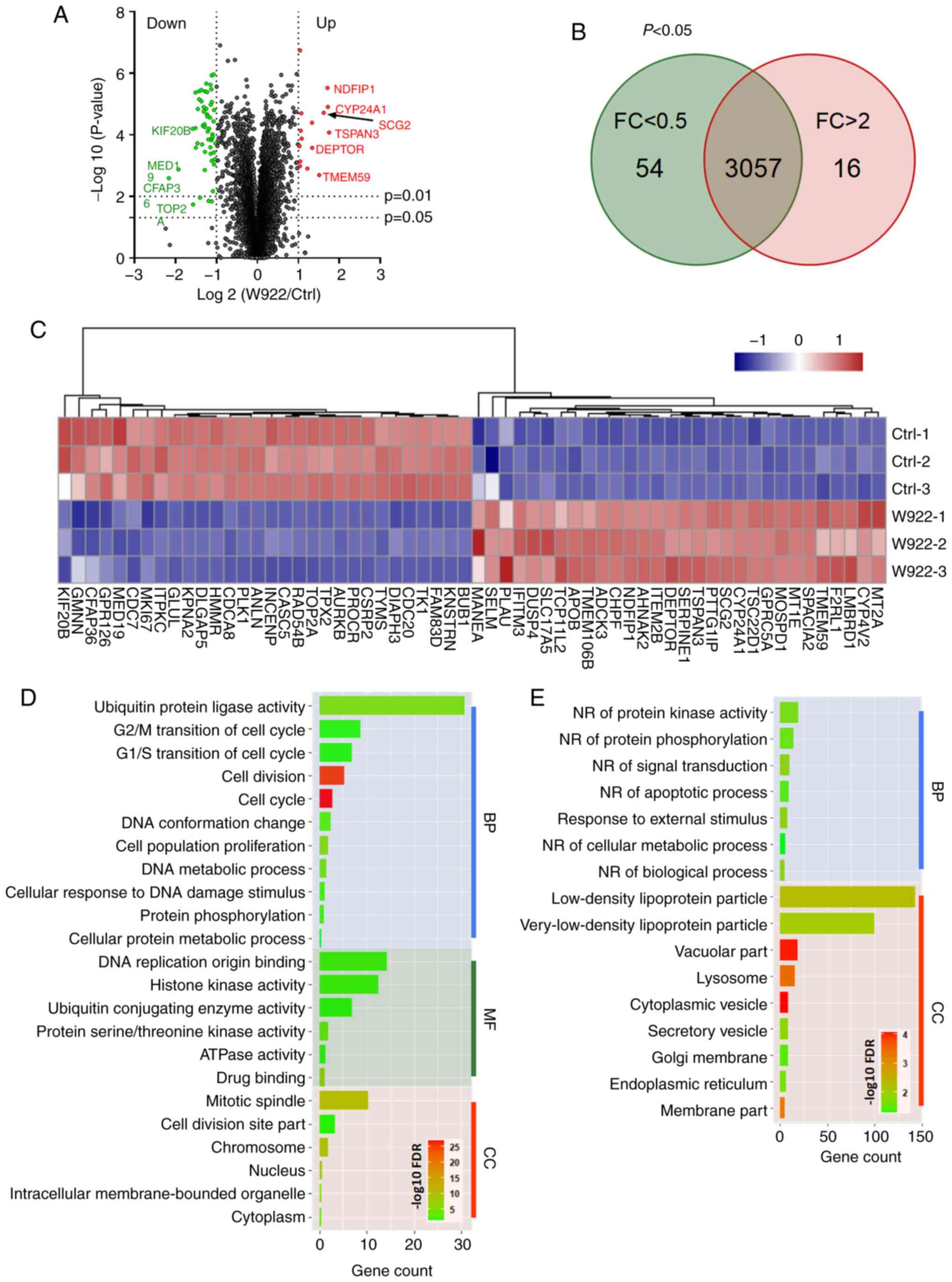
regulates tumor formation, MS analysis was performed. HCT116 cells
were treated with W922 at 10 µM for 24 h, and >7,000
proteins were identified and quantified in all the replicates of
treated and untreated cells (Fig.
2A). Among the common proteins in W922-treated and the control
cells, 54 proteins were significantly downregulated (fold change
<0.5) and 16 proteins were significantly upregulated (fold
change >2) after W922 treatment (Fig. 2B). The top 30 up- and downregulated
proteins are exhibited in Fig. 2C.
Subsequently, all the significantly down- or upregulated proteins
were classified using STRING online tool. W922 exhibited evident
inhibitory effect on cell cycle regulation. As presented in
Fig. 2C, >20 downregulated
proteins, such as CDC20, CDC7, CDCA8, polo like kinase 1 and TPX2,
caused by W922 treatment were associated with cell cycle
regulation. Besides, the cell cycle markers in G1/S and
G2/M transitions were found to be downregulated
(Fig. 2D). W922 negatively
regulated the signal transduction and cell biological process.
Moreover, upregulation of lysosome-related proteins were identified
after W922 treatment (Fig. 2E).
These results verified the function of W922 as a signal
transduction inhibitor, and it was suggested that the inhibitory
effects on tumor cells may be connected with cell cycle arrest.
W922 inhibits HCT116 cell proliferation
mainly via cell cycle arrest
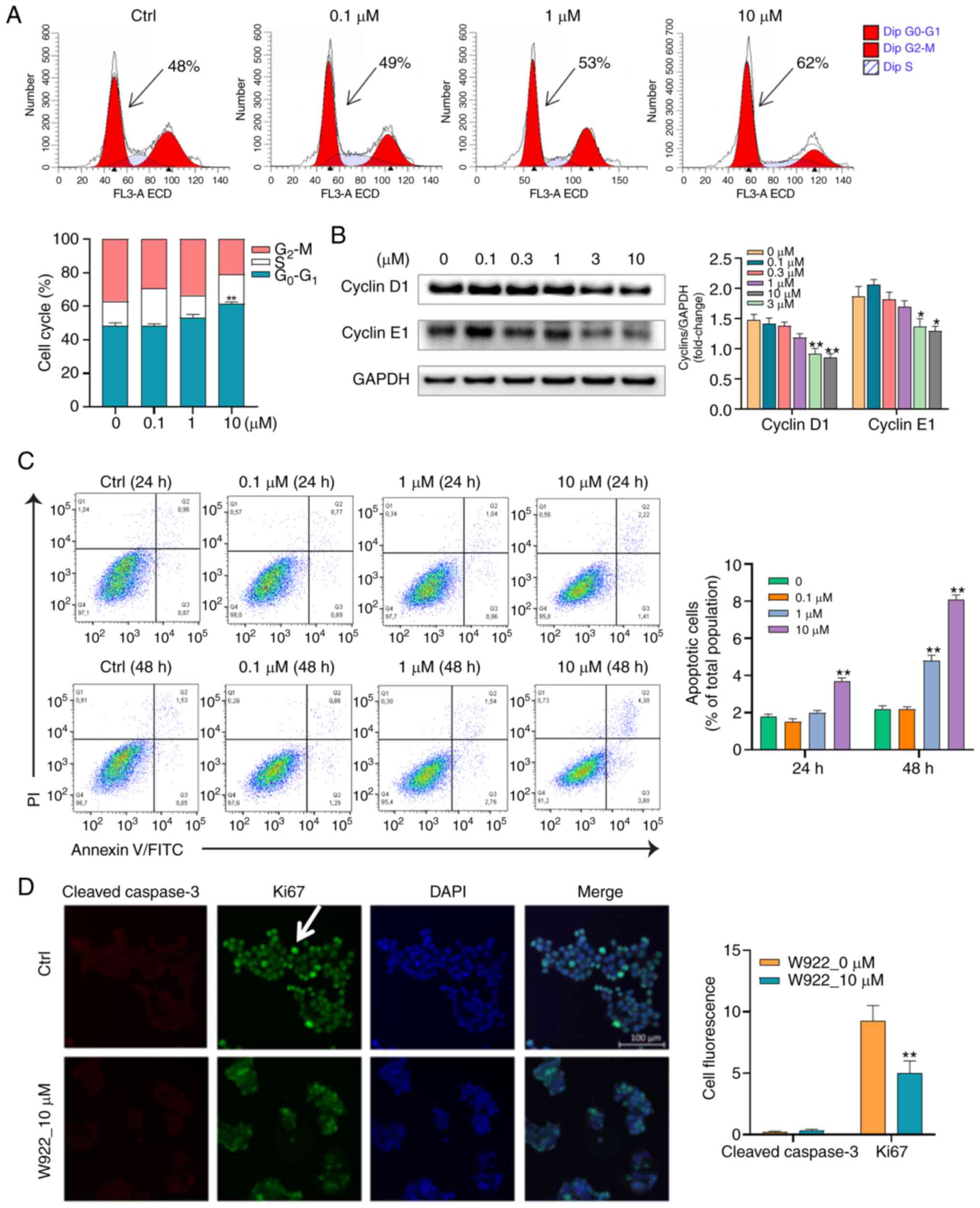
To further validate the proteomic results showing
that W922 regulated proteins are cell cycle-related, flow cytometry
was conducted to analyze the cell cycle distribution in
W922-treated HCT116 cells. W922 led to G0-G1
phase accumulation in a concentration-dependent manner (Fig. 3A). To evaluate the molecular
mechanism of the cell cycle arrest, immunoblot analysis was used to
investigate the expression of cell cycle regulatory proteins.
Cyclins are among the most important core cell cycle regulators.
For instance, downregulation of cyclin D1 and E1 expression levels
could block the cell cycle at G0-G1 stage and
stop cell progression turning into S phase (31). It was identified that the protein
expression levels of cyclin D1 and E1 were significantly
concentration- dependently decreased in W922-treated cells compared
with those in untreated cells (Fig.
3B).
Considering that apoptosis is the major form of cell
death induced by chemotherapeutic agents, it was examined whether
the anti-CRC effect of W922 was apoptosis dependent. Therefore,
apoptosis of W922-induced HCT116 cells was measured using Annexin
V/PI double staining method. Following the treatment of W922 (10
µM), the proportions of apoptotic HCT116 cells significantly
increased by up to 2 and 6% in 24 and 48 h-treatment, respectively
(Fig. 3C). Subsequently,
immunofluorescence was performed in order to detect the different
expression levels of ki67 and cleaved caspase-3 between
W922-treated and untreated cells. The ki67 protein is a marker of
proliferation and the expression of cleaved caspase-3 reflects the
apoptotic level (24). It was
found that the fluorescence intensity of ki67 in treated cells was
significantly lower compared with that in untreated cells; however,
no cleaved caspase-3 was identified in treated HCT116 cells
(Fig. 3D). These results indicated
that W922 inhibited HCT116 cell proliferation mainly by inducing
cell cycle arrest rather than apoptosis.
W922 suppresses the PAM signaling
pathway
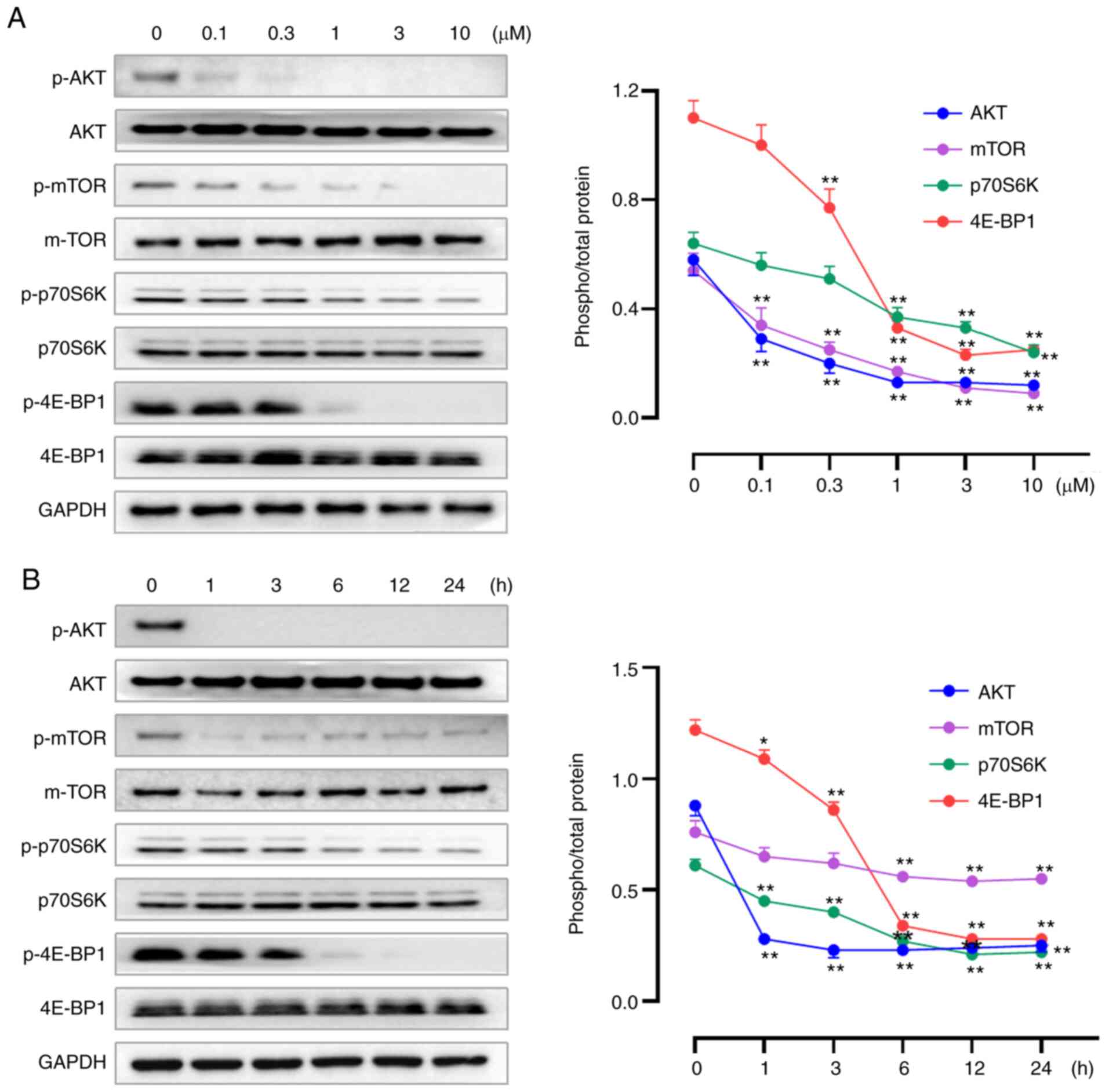
Our previous study reported W922 as an inhibitor
against PI3K and mTOR enzymes, demonstrating the selective
inhibition of W922 against PI3Kα extracellularly, with an
IC50 of 15.1 nM. Moreover, potent activities of W922
against PI3Kβ, PI3Kγ, PI3Kδ and mTOR were revealed (21). To verify the inhibition of W922 on
PAM pathway, western blotting assays were performed to determine
the expression levels of PAM-related proteins. W922 suppressed the
expression levels of p-AKT, p-mTOR, p-p70S6K and p-4E-BP1 in time-
and concentration-dependent manners, while no alteration was found
in the corresponding total proteins (Fig. 4). The proteomics results also
indicated that W922 has a negative effect on the regulation of
protein phosphorylation (Fig.
2E).
W922 induces autophagy in HCT116
cells
The PAM pathway is a well-established pathway that
acts as a key negative regulator of autophagy (14). Since W922 downregulates the
phosphorylation of mTOR, it was investigated whether W922 could
induce autophagy in HCT116 cells. The results demonstrated that
W922 treatment caused the induction of autophagy, as evidenced by
concentration-dependent increase of LC3-I to LC3-II conversion
(Fig. 5A). The phosphorylation of
Beclin, an autophagy marker, was examined after W922 treatment, and
it was identified that Beclin was significantly phosphorylated
(Fig. 5A). Next, HCT116 cells were
stained with LC3 antibody and visualized via immunofluorescence.
Representative images indicated a weak staining in the untreated
cells, while a moderate to strong intensity of LC3 fluorescence was
observed in W922 treated groups (Fig.
5B).
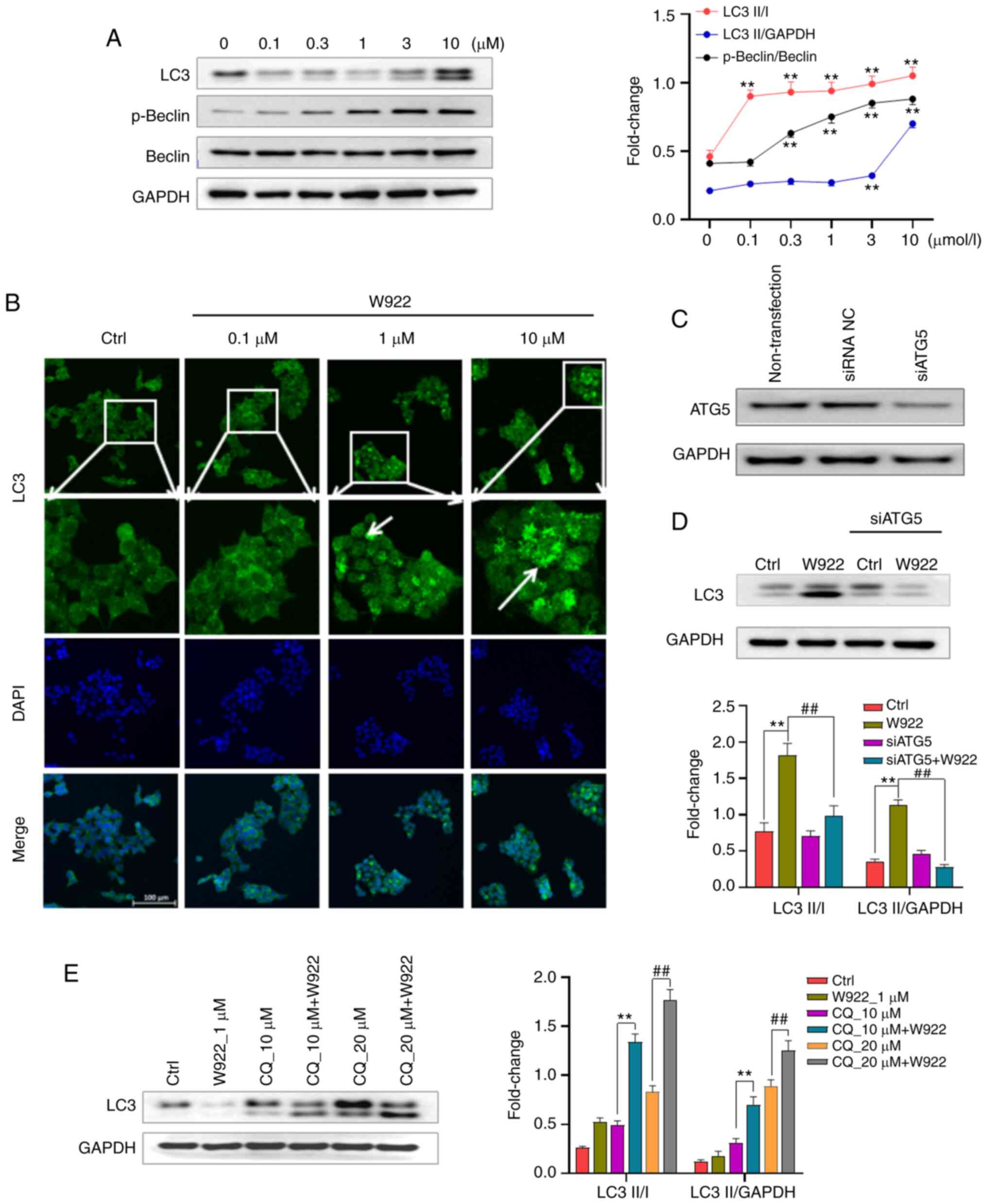 | Figure 5W922 induces autophagy in HCT116
cells. (A) HCT116 cells were treated with desired concentrations of
W922 for 24 h. Collected cells were lysed and subsequently the
lysates were immunoblotted with anti-LC3, anti-p-Beclin or
anti-Beclin antibodies (n=3; one-way ANOVA followed by Dunnett's
test). **P<0.01 vs. 0 µM treatment. (B) HCT116
cells were treated with W922 for 24 h, fixed and stained with
anti-LC3 antibody (green) and with DAPI (blue), magnification, ×20.
Quantification on the right showed LC3 fluorescence determined by
ImageJ (n=3). **P<0.01 vs. Ctrl. (C) Transfection
efficacy detection. HCT116 cells were transfected with siATG5 or
siRNA NC, and the expression of ATG5 was detected. (D) HCT116 cells
were successfully transfected with siATG5 and the expression of LC3
after W922 treatment was measured via western blotting (n=3;
one-way ANOVA followed by Dunnett's test). **P<0.01
vs. Ctrl; ##P<0.01 vs. W922 group. (E) Autophagic
flux was examined via co-treatment of CQ, an autophagosome-lysosome
fusion inhibitor. HCT116 cells were treated with W922 and/or CQ for
24 h and the expression of LC3 was determined via western blotting
(n=3; unpaired Student's t-test). **P<0.01 vs. 10
µM CQ treatment; ##P<0.01 vs. 20 µM CQ
treatment. CQ, chloroquine; ATG5, autophagy related 5; Ctrl,
control; siRNA, small interfering RNA. |
To further investigate the autophagy induction by
W922, the autophagy signaling was blocked via silencing ATG5. The
results suggested that the conversion of LC3-I to II induced by
W922 was inhibited in ATG5-silenced cells (Fig. 5C).
It remains difficult to distinguish autophagy
inducers and inhibitors as both could increase the expression of
LC3-II level (34). Therefore, in
order to further investigate the role of W922 on autophagy, the
impact on autophagic flux was evaluated via co-treatment with the
autophagosome-lysosome fusion inhibitor, CQ. An enhanced LC3II/I
ratio was observed in cells treated with W922 combined with CQ
compared with W922 treatment alone (Fig. 5D and E). These results suggested
that W922 functioned as an autophagy inducer in HCT116 cells.
Autophagy contributes to W922-induced
cell cycle arrest
An autophagy inhibitor, CQ, was used to investigate
whether W922-induced autophagy was involved in cell cycle arrest.
As aforementioned, W922 treatment resulted in cell cycle arrest in
the G0-G1 phase; however, such phenomenon was
impaired by CQ co-treatment. CQ treatment alone did not demonstrate
a significant impact on the cell cycle distribution of HCT116
cells, but CQ co-treatment significantly decreased >20% of
G0-G1 phase proportion in W922-treatment
cells (Fig. 6A).
Next, the alterations of proteomics induced by CQ
were detected via MS (Fig. 6B).
Top 20 up- and downregulated proteins were sorted and classified
using STRING analysis tool (Fig.
6D). CQ co-treatment led to a suppression in cellular metabolic
process and oxidation-reduction process (Fig. 6C). Moreover, CQ enhanced the
apoptosis of W922-treated cells. The data collected from flow
cytometry assay also confirmed this result. Co-treatment of W922
with CQ led to ~30% apoptotic cells out of a total cellular
population (Fig. 6E). These
findings provide potential evidence for the combined treatment of
W922 and CQ in CRC therapy.
Discussion
The present study demonstrated that the novel PAM
pathway inhibitor, W922, inhibited tumor formation by causing cell
cycle arrest. As aforementioned, W922 also induced autophagy in
HCT116 cells. It was found that W922-induced autophagy contributed
to W922-induced cell cycle arrest, since inhibition of autophagy by
CQ impaired the cell cycle arrest. PAM is one of the pivotal
transduction pathways regulating cellular basic activities
(25,26). Thus, targeting this pathway may
contribute to the tumor growth suppression. Our previous study
developed a series of inhibitors against this cascade, and among
which the most effective compound named W922 was selected.
According to the previous enzymatic results, W922 was found to
exhibit the strongest enzymatic inhibition against the PI3Kα enzyme
compared with other synthesized compounds (21). In the current study, it was
verified that W922 has robust ability to dephosphorylate PAM
signaling proteins.
Inhibition of PI3K/Akt signaling has been suggested
as potential therapeutic strategy for CRC. For example, previous
studies have reported the association of the PI3K/Akt pathway with
colon tumorigenesis (27,28). Moreover, the PAM pathway is well
known as an important regulatory pathway of autophagy. PI3K/Akt
regulates autophagy mainly via modulation of mTOR activity, and
thus decreased mTOR activity by inhibitors could trigger autophagy
(14). Autophagy is an
evolutionarily highly conserved catabolic pathway, and the capacity
of autophagy to maintain cellular metabolism improves the survival
of cells (17). The autophagy
pathway has a broad implication in numerous physiological and
pathological processes, including carcinogenesis (29). Although upregulation and
downregulation of autophagy have both been revealed to serve
important roles in carcinogenesis, in general, autophagy is
identified as a pro-survival mechanism in cancer cells by removing
damaged organelles and recycling nutrients in response to
chemotherapy stress (30).
Therefore, autophagy is considered to be associated with drug
resistance as it may protect cells from the cytotoxicity of
chemotherapy drugs.
The present results suggested that W922 caused cell
cycle arrest at the G0-G1 phase as a single
agent; however, only slight apoptosis was observed after W922
treatment. The cell cycle represents a series of tightly integrated
events that allow cells to proliferate (31). Therefore, cell cycle arrest
contributes to the anti-tumor effect of W922. In response to
stressful microenvironment, cells are able to arrest the cell
cycle, which helps regulate cell proliferation and prevents the
expansion of potential harmful cell populations (31,32).
Cancer frequently represents a dysregulation of the cell cycle, and
the cell cycle can be arrested under stress to protect the cells
from DNA damage (33). Moreover,
autophagy is induced in response to a variety of stress conditions,
where it serves an essential role in protecting cellular viability
(17). While the correlation
between autophagy and cell cycle arrest has been widely studied,
several signaling pathways exhibit opposite effects on the
regulation of autophagy and cell cycle, and increasing evidence
suggests that the opposite regulation may be rational and
coordinated, since there is an interaction between the two
processes (34,38,39).
The present study observed that W922 treatment led
to cell cycle arrest and the upregulation of LC3-II. However, both
autophagy inducers and inhibitors can cause an increase of LC3-II
expression level (34). According
to the guidelines for the use and interpretation of assays for
monitoring autophagy, it is necessary to block lysosomal
degradation of the protein to truly measure in vivo
autophagic flux using LC3-II as a biomarker (40). Thus, co-treatment of CQ is
necessary to examine whether W922 is an autophagy inducer or
inhibitor. CQ is an autophagy inhibitor that can inhibit
autophagosome degradation by blocking autophagic flux (34). If the LC3-II expression in the
combination treatment is higher compared with that in W922
treatment alone, then W922 is an autophagy inducer. An enhanced
LC3II/I ratio observed in combination treatment demonstrated the
autophagy induction characteristic of W922. The cell cycle arrest
induced by W922 was impaired when cells were co-treated with the
autophagy inhibitor CQ, suggesting that autophagy may facilitate
W922-induced cell cycle arrest. Therefore, the cell cycle arrest
induced by W922 may be a protective action caused by the induction
of autophagy.
Increasing preclinical evidence suggests that
targeting autophagy can improve the efficacy of numerous cancer
treatments (35). While various
compounds have been developed to block the different stages of
autophagy, the only clinically approved autophagy inhibitor is
hydroxychloroquine (HCQ) (36).
Preclinical trials have reported that HCQ can enhance tumor
shrinkage either by single or combination treatment (36,37).
Thus, there are multiple ongoing clinical trials combining HCQ with
other chemo-drugs in cancer therapy. HCQ enhances the effect of
temsirolimus on cytotoxicity and promotes apoptosis in renal cell
carcinoma lines (37). The
combination of HCQ and tamoxifen exhibits improved therapeutic
efficacy compared with monotherapy in estrogen receptor-positive
breast cancer cell lines (36).
These findings suggest that combination therapy of a PAM signaling
inhibitor with an autophagy inhibitor could be a novel method for
cancer treatment. Therefore, the present study detected the
combination therapy of W922 with autophagy blockers in
vitro. The results demonstrated that CQ enhanced the inhibitory
effects of W922 on cells, indicating that W922 and autophagy
blockers promote mutual sensitization. These results suggested that
W922 co-treatment with autophagy blockers may be a novel
therapeutic strategy for CRC.
In conclusion, the present study first examine the
function of a novel PAM inhibitor, W922, in the regulation of the
proliferation in HCT116 CRC cells. W922 exerted its anti-tumor
effects as a single agent mainly via the induction of cell cycle
arrest. W922 also induced autophagy in HCT116 cells, and W922-
induced cell cycle arrest was impaired when the cells were
pre-treated with CQ, suggesting that autophagy may contribute to
W922-induced cell cycle arrest. However, the aforementioned results
were only obtained from in vitro experiments, and the same
measurements should be collected in vivo to further verify
the mechanism of the anti-cancer effects of W922. The current study
demonstrated that simultaneously treating the cells with CQ led to
an increase in the number of apoptotic cells. To verify the
enhanced effect of W922 and autophagy inhibitor co-treatment, the
same experiment should be performed in vivo in future
studies. Collectively, the present results support the therapeutic
potential of W922 in the treatment of CRC.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81670001) and Natural
Science Basic Research Project of Shaanxi Province (grant no.
2018JQ8042).
Availability of data and materials
Data in this study are availabe from the
corresponding author upon reasonable request.
Authors' contributions
JW performed most of the experiments, analyzed the
data and was responsible for writing the manuscript. DL helped with
the in vivo experiments and cell immunoflorescence assay. XPZ
conducted the mass spectrometry technique. CFH participated in
western blotting assay. LC participated in data analysis and
manuscript revision. SQZ synthesized W922. XX analyzed data. YXC
and XX provided the funding. SJL and YXC designed the experiment,
revised the manuscript and gave final approval of the version to be
published. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with the National Guidelines for Experimental Animal Welfare and
with approval of the Ethics Committee of Xi'an Jiaotong
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brody H: Colorectal cancer. Nature.
521:S12015. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stein A, Atanackovic D and Bokemeyer C:
Current standards and new trends in the primary treatment of
colorectal cancer. Eur J Cancer. 47(Suppl 3): S312–S314. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
5
|
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P,
van Buren G II, Samuel S, Kim MP, Lim SJ and Ellis LM:
Chemoresistant colorectal cancer cells, the cancer stem cell
phenotype, and increased sensitivity to insulin-like growth
factor-I receptor inhibition. Cancer Res. 69:1951–1957. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O'Hara MH, Hamilton SR and O'Dwyer PJ:
Molecular triage trials in colorectal cancer. Cancer J. 22:218–222.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bendell JC, Varghese AM, Hyman DM, Bauer
TM, Pant S, Callies S, Lin J, Martinez R, Wickremsinhe E, Fink A,
et al: A First-in-human phase 1 study of LY3023414, an oral
PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin
Cancer Res. 24:3253–3262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lech G, Slotwinski R, Slodkowski M and
Krasnodebski IW: Colorectal cancer tumour markers and biomarkers:
Recent therapeutic advances. World J Gastroenterol. 22:1745–1755.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar
|
|
11
|
Dienstmann R, Rodon J, Serra V and
Tabernero J: Picking the point of inhibition: A comparative review
of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 13:1021–1031.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hart S, Novotny-Diermayr V, Goh KC,
Williams M, Tan YC, Ong LC, Cheong A, Ng BK, Amalini C, Madan B, et
al: VS-5584, a novel and highly selective PI3K/mTOR kinase
inhibitor for the treatment of cancer. Mol Cancer Ther. 12:151–161.
2013. View Article : Google Scholar :
|
|
13
|
Kolev VN, Wright QG, Vidal CM, Ring JE,
Shapiro IM, Ricono J, Weaver DT, Padval MV, Pachter JA and Xu Q:
PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem
cells. Cancer Res. 75:446–455. 2015. View Article : Google Scholar
|
|
14
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Noda T and Ohsumi Y: Tor, a
phosphatidylinositol kinase homologue, controls autophagy in yeast.
J Biol Chem. 273:3963–3966. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar :
|
|
17
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poillet-Perez L and White E: Role of tumor
and host autophagy in cancer metabolism. Genes Dev. 33:610–619.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM
and Lotze MT: Cell-mediated autophagy promotes cancer cell
survival. Cancer Res. 72:2970–2979. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsujimoto Y and Shimizu S: Another way to
die: Autophagic programmed cell death. Cell Death Differ. 12(Suppl
2): S1528–S1534. 2005. View Article : Google Scholar
|
|
21
|
Wang HY, Shen Y, Zhang H, Hei YY, Zhao HY,
Xin M, Lu SM and Zhang SQ: Discovery of 2-(aminopyrimidin-5-yl)-
4-(morpholin-4-yl)-6-substituted triazine as PI3K and BRAF dual
inhibitor. Future Med Chem. 10:2445–2455. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singla P, Luxami V and Paul K: Triazine as
a promising scaffold for its versatile biological behavior. Eur J
Med Chem. 102:39–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McDonald WH and Yates JR III: Shotgun
proteomics: Integrating technologies to answer biological
questions. Curr Opin Mol Ther. 5:302–309. 2003.PubMed/NCBI
|
|
24
|
Silva MN, Leite JS, Mello MF, Silva KV,
Corgozinho KB, de Souza HJ, Cunha SC and Ferreira AM: Histologic
evaluation of Ki-67 and cleaved caspase-3 expression in feline
mammary carcinoma. J Feline Med Surg. 19:440–445. 2017. View Article : Google Scholar
|
|
25
|
Pandurangan AK: Potential targets for
prevention of colorectal cancer: A focus on PI3K/Akt/mTOR and Wnt
pathways. Asian Pac J Cancer Prev. 14:2201–2205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fang JY and Richardson BC: The MAPK
signalling pathways and colorectal cancer. Lancet Oncol. 6:322–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Luo Q and Chen D, Fan X, Fu X, Ma T and
Chen D: KRAS and PIK3CA bi-mutations predict a poor prognosis in
colorectal cancer patients: A single-site report. Transl Oncol.
13:1008742020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J, Zhang Y, Qu J, Xu L, Hou K, Zhang
J, Qu X and Liu Y: β-Elemene-induced autophagy protects human
gastric cancer cells from undergoing apoptosis. BMC Cancer.
11:1832011. View Article : Google Scholar
|
|
31
|
Grana X and Reddy EP: Cell cycle control
in mammalian cells: Role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
32
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in cancer
therapy. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zheng K, He Z, Kitazato K and Wang Y:
Selective autophagy regulates cell cycle in cancer therapy.
Theranostics. 9:104–125. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cook KL, Warri A, Soto-Pantoja DR, Clarke
PA, Cruz MI, Zwart A and Clarke R: Hydroxychloroquine inhibits
autophagy to potentiate antiestrogen responsiveness in ER+ breast
cancer. Clin Cancer Res. 20:3222–3232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee HO, Mustafa A, Hudes GR and Kruger WD:
Hydroxychloroquine destabilizes phospho-S6 in human renal carcinoma
cells. PLoS One. 10:e1314642015.
|
|
38
|
Neufeld TP: Autophagy and cell growth-the
yin and yang of nutrient responses. J Cell Sci. 125:2359–2368.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mathiassen SG, De Zio D and Cecconi F:
Autophagy and the cell cycle: A complex landscape. Front Oncol.
7:512017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|