Introduction
Unsuccessful efforts to destroy tumors suggest that
therapies focusing only on cancer cells are usually not sufficient
to eradicate malignant tumors (1). Tumors are surrounded by several
non-cancerous components, including fibroblasts, immunocytes,
adipose cells, blood vessels composed of endothelial cells and
pericytes, extracellular matrix, infiltrated nerves and a variety
of soluble molecules, such as chemokines, interleukins, lactate and
vascular endothelial growth factor (VEGF). These factors have been
associated with tumor cells and form the tumor microenvironment
(TME), which provides an essential foundation to support the
growth, metabolic reprogramming, immunosuppressive abilities,
metastasis, recurrence and treatment resistance of malignant tumors
(2,3). In the past decade, an increasing
number of studies have highlighted the importance of the
interactions between the TME and tumor cells (4-6)
and targeting the tumor stroma has emerged as a novel cancer
treatment paradigm (3,4,7).
However, the molecular mechanisms underlying the interactions
between tumor cells and the TME are complicated and the
interactions between different types of stromal cells in the TME
remain unknown.
Hepatocellular carcinoma (HCC) is one of the most
aggressive malignant tumors and has been associated with a high
mortality rate worldwide (8).
Cancer-associated fibroblasts (CAFs) and tumor-associated
macrophages (TAMs) are the main stromal cells in the TME of HCC
(9). Interactions between CAFs
and TAMs have been reported to promote tumor progression in several
types of cancer, such as prostate cancer (PCa) (4), pancreatic ductal adenocarcinoma
(PDAC) (5), esophageal squamous
cell carcinoma (ESCC) (10),
neuroblastoma (11), intrahepatic
cholangiocarcinoma (IHCC) (12),
breast cancer (13) and oral
squamous cell carcinoma (OSCC) (14). In HCC, activated hepatic stellate
cells (HSCs) can reprogram monocytes to acquire an
immunosuppressive phenotype, thereby altering their paracrine
factor environment to support the development of HCC (15). This suggested that CAFs could
regulate tumor inflammation by modifying the presence and phenotype
of invasive TAMs.
Plasminogen activator inhibitor-1 (PAI-1) is a
serine protease inhibitor encoded by the serpin family E member 1
gene (16). PAI-1 was found to be
upregulated in a variety of different tumors, such as breast
(17), ovarian (18) and colon cancers (19), and possess several pro-tumorigenic
functions, including supporting proliferation and angiogenesis,
inhibiting cell apoptosis and promoting metastasis (17,18,20). Along with cancer cells, several
other components of the TME, including platelets, macrophages and
vascular cells, also secrete PAI-1 (16,21). In HCC, it has been observed that
PAI-1 was upregulated and high PAI-1 protein expression level was
predictive of unfavorable tumor behavior and prognosis (22). However, the biological roles of
PAI-1 in the TME are yet to be fully elucidated.
The present study established an in vitro
model, using the indirect contact coculture method to simulate the
crosstalk between cancer cells, CAFs and TAMs. It was investigated
whether CAFs could induce TAMs to adapt their polarization into a
special phenotype and whether they could mediate their paracrine
signaling to promote the malignant behavior of HCC. Furthermore,
important factors involved in this process were investigated to
provide evidence that inhibiting the crosstalk in the TME, by
targeting these factors, may be beneficial for improving the
prognosis of patients with HCC.
Materials and methods
Cell culturing and reagents
The human HCC cell line, Huh-7 (RIKEN BioResource
Research Centre) and the human HSC line, Lx-2 (Merck KGaA) were
cultured in DMEM, supplemented with 10% FBS and a 1%
penicillin-streptomycin solution (all from Thermo Fisher
Scientific, Inc.). The human monocyte cell line, THP-1, was
obtained from the Japanese Collection of Research Bioresources Cell
Bank and was cultured in RPMI-1640 medium, supplemented with 10%
FBS and a 1% penicillin-streptomycin solution (all from Thermo
Fisher Scientific, Inc.). All experiments were performed with
mycoplasma-free cells. The cells were cultured at 37°C in a
humidified incubator with 5% CO2 and were selected for
experiments in the logarithmic growth phase.
To induce the M0 macrophages (M0), the THP-1 cells
were treated with 150 nM phorbol-12-myristate-13-acetate (PMA;
Sigma-Aldrich; Merck KGaA) for 48 h at 37°C. The human C-X-C motif
chemokine 12 (CXCL12) antibody (300 µg/ml; cat. no.
AF-310-NA), normal human IgG control (300 µg/ml; cat. no.
1-001-A) and recombinant human PAI-1 (rPAI-1; 100 ng/ml; cat. no.
1786-PI) were purchased from R&D Systems, Inc. Tiplaxtinin
(PAI-039; a selective PAI-1 inhibitor; cat. no. s7922) was
purchased from Selleck Chemicals.
Preparation of conditioned medium
(CM)
The Huh-7 and Lx-2 cells, and M0 were cultured to
80% confluency, washed twice with PBS, then incubated with DMEM
containing 1% FBS for 48 h at 37°C. The supernatant was collected,
centrifuged (500 × g; 20 min) at room temperature and filtered
using a 0.2-µm filter to remove the cell debris. The CM was
stored at −80°C, and repeated freeze-thaw cycles were avoided. For
treating the indicated cells, the CM was added to the complete
growth medium at a ratio of 1:2, and the cells were stimulated for
48 h at 37°C. The Lx-2 cell line was treated with Huh-7-derived CM
to obtain CAF(Ca), while M0 was treated with Huh-7-derived CM to
obtain TAM(Ca). Next, M0 was treated with CAF(Ca)-derived CM and
TAM(Ca)-derived CM to obtain TAM(CAF) and TAM(TAM), respectively
(Fig. 1A). Then, the CM from M0,
TAM(Ca), TAM(CAF) were further collected in the same manner as
aforementioned and was added to the complete growth medium at a
ratio of 1:2 for the next experiment.
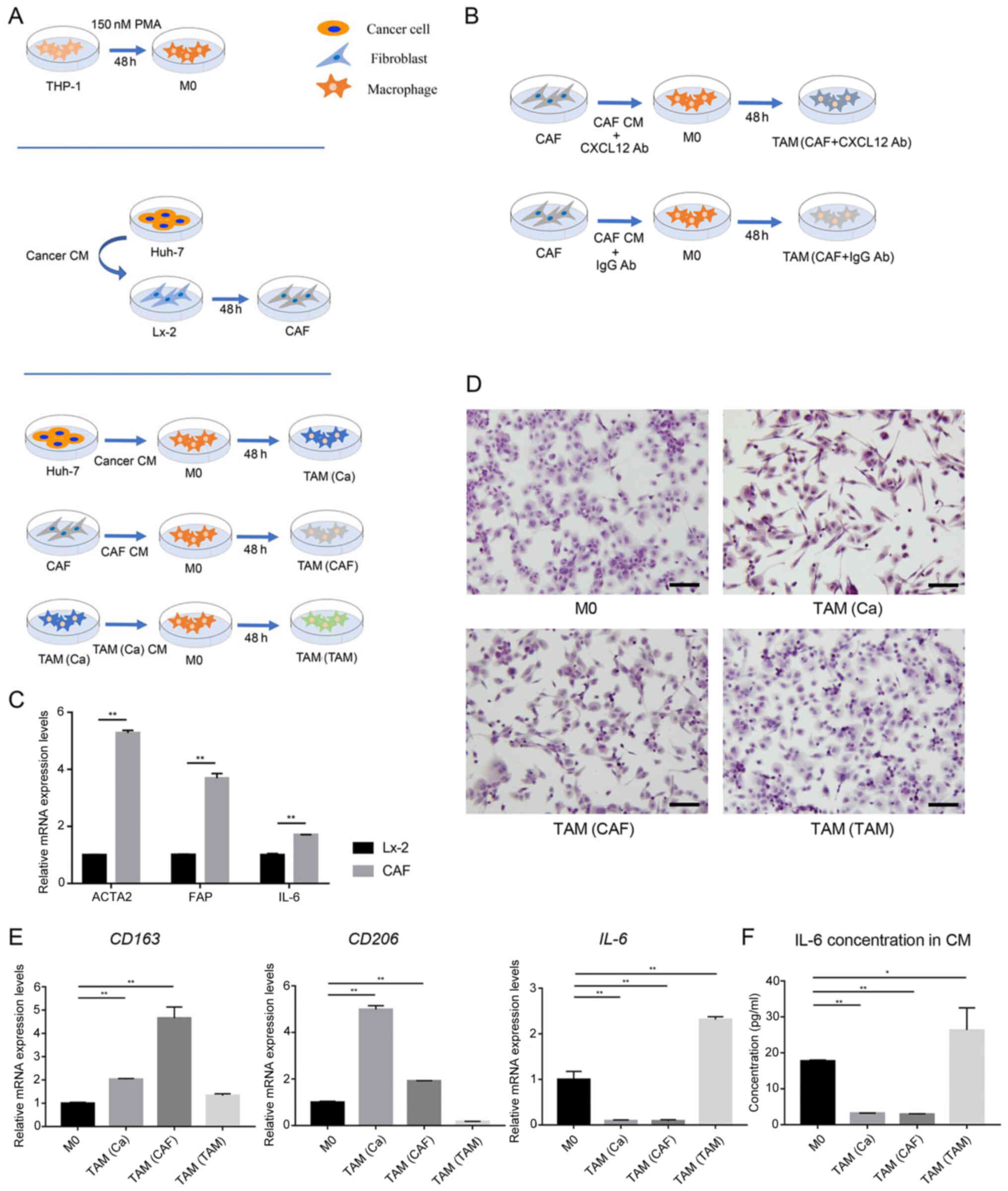 | Figure 1Cancer cells and CAFs induce M2
polarization in human macrophages. (A) Schematic representation of
the generation of M0, CAFs and the three types of TAMs. (B)
Schematic representation of anti-CXCL12 treatment in CAF-induced
TAMs. (C) After treatment with CM from the Huh-7 cells, the gene
expression levels of the CAFs markers were detected in the Lx-2
cells using RT-qPCR analysis. (D) Typical morphological changes in
the M0 macrophages and different types of TAMs are shown following
staining with H&E. Scale bar, 100 µm. (E) Gene
expression levels of M2-polarized macrophage markers were detected
in M0 macrophages and different types of TAMs using RT-qPCR
analysis. (F) Secretion of IL-6 in the CM of M0 and different types
of TAMs. *P<0.05;**P<0.01. CAF,
cancer-associated fibroblast; CM, conditioned medium; RT-qPCR,
reverse transcription-quantitative PCR; TAM, tumor-associated
macrophage; PMA, phorbol-12-myristate-13-acetate; M0, macrophages;
FAP, fibroblast activation protein; ACTA2, actin α-2; CXCL12, C-X-C
motif chemokine ligand 12; Ab, antibody. |
For the PAI-1 inhibition experiment, 20 µM
tiplaxtinin was added into normal DMEM, M0-CM, TAM(Ca)-CM and
TAM(CAF)-CM. To determine the direct effects of PAI-1, 100 ng/ml
rPAI-1 was added into normal DMEM (control). These CM were used to
perform the cell proliferation, colony formation, wound healing,
migration and invasion assays.
For the CXLC12 neutralization experiment, 300
µg/ml CXCL12 antibody or IgG control was added into the
CAF-CM, which was subsequently used to stimulate M0 to obtain
TAM(CAF + CXCL12 Ab) and TAM(CAF + IgG Ab), respectively (Fig. 1B).
Morphology analysis
For H&E staining, M0, TAM(Ca), TAM(CAF) and
TAM(TAM) were seeded in the Nunc Lab-Tek II Chamber Slide system
(cat. no. 154526PK; Thermo Fisher Scientific, Inc.). The subsequent
steps were all performed at room temperature. The slides were
gently washed twice with PBS and fixed with 90% ethanol for 15 min.
Then, the slides were stained with hematoxylin (Muto Pure Chemicals
Co., Ltd.) for 10 min, washed under running water for 5 min and
stained with eosin Y (FUJIFILM Wako Pure Chemical Corporation) for
5 min. After washing under running water for 5 min to remove the
excess dye, the slides were sealed with a coverslip and neutral
resin. Differences between each group were compared under a light
microscope (magnification, ×200).
Cell proliferation assay
The tumor cells were seeded at a density of
2×104 cells/well into 96-well plates in complete medium
and cultured in an incubator at 37°C. After adhesion, the cells
were washed with PBS and cultured with the indicated CM at 37°C and
the proliferative state of the cells was observed every 24 h
between days 1 and 4. The Cell Counting Kit-8 (CCK-8) solution
(Dojindo Molecular Technologies, Inc.) was added to the medium, as
a 10% volume, and cell proliferation was analyzed by measuring the
absorbance values at 450 nm with a microplate reader (SpectraMax
i3; Molecular Devices, LLC) after 2 h, according to the
manufacturer's instructions.
Colony formation assay
The cancer cells were seeded at a density of 200
cells/well in 6-well plates containing culture medium, then the
plates were placed in an incubator at 37°C. After 3 days, the
growth medium was removed and the indicated CM was added in each
group. The cells were cultured for 14 days and the medium was
changed every 5 days. At the endpoint, 4% paraformaldehyde and 0.1%
crystal violet was used to fix and stain the cells for 20 min at
room temperature, respectively. After washing with PBS, images of
the cell colonies from the different groups were captured using a
light microscope (magnification, ×40; BX43F; Olympus Corporation)
and counted. The colony was only counted if it contained >50
cells.
Wound healing assay
The cancer cells were seeded, at a density of
5×105 cells/well into 6-well plates, and once they
formed a confluent monolayer at 90%, a 200-µl pipette tip
was used to scratch a wound through the entire center of the well.
After washing with PBS, the cells were cultured with indicated CM
in the absence of FBS in each group for 12 h at 37°C. The areas of
the wounds were observed, and images were captured using a light
microscope (magnification, ×40; DP22-CU; Olympus Corporation) at 0
h and 12 h after scratching. The cell migration rates were
calculated using ImageJ v1.46r software (National Institutes of
Health) and using the following equation: Relative migration
rate=[width (0 h)−width (12 h)]/width (0 h) ×100%.
Migration and invasion assays
A 24-well Transwell system. with 8.0-µm pores
(Corning, Inc.) was used for the migration and invasion assays.
Serum-starved cancer cells were resuspended, adjusted to a
concentration of 2×105/ml, then seeded into the upper
chamber, with 100-µl cell suspension per well. Following
cell attachment, the culture medium was changed to indicated CM in
each group. The FBS concentration was 5% in the upper chambers and
10% in the lower chambers. After incubation for 24 h for migration
assay and 36 h for invasion assay at 37°C, the upper chambers were
fixed with 100% methanol for 20 min and stained with 0.1% crystal
violet for 20 min, both at room temperature, following which the
non-migrated cells on the upper chambers were removed using a
cotton swab. The number of remaining cells was calculated and
quantified in 5 random fields of view under a light microscopic
(magnification, ×100). For the invasion assays, the upper chambers
of the Transwell system were precoated with Matrigel (Corning,
Inc.) overnight at 37°C.
Cytokine array
A Human XL Cytokine Array kit (cat. no. ARY022B;
R&D Systems, Inc.) was used to detect molecules in the CM from
the Huh-7, Lx-2, CAF(Ca), M0, TAM(Ca) and TAM(CAF) cells, according
to the manufacturer's instructions. Briefly, after blocking with
Array Buffer 6 (2 ml for one membrane) for 1 h at room temperature,
the membrane containing 105 different capture antibodies was
incubated overnight with 400 µl indicated CM at 4°C in each
group. The following day, the membranes were washed with 1X Wash
Buffer (diluted 1:25 with distilled water from Wash Buffer
Concentrate) and incubated with the detection antibody cocktail and
streptavidin-HRP in sequence. Finally, the membranes were treated
with a chemiluminescent detection reagent for 1 min at room
temperature and an Amersham Imager 600 (Cytiva) was used to detect
the signal intensities on each membrane. Each pair of positive dots
represented the signals of highly expressed molecules and the
luminescence intensity was quantified using ImageJ v1.46r software
(National Institutes of Health). The full list of all the
antibodies is available in the product datasheet.
ELISA
The Human IL-6 Quantikine ELISA kit (cat. no.
D6050), Human Serpin E1/PAI-1 Quantikine ELISA kit (cat. no.
DSE100) and Human CXCL12/stromal cell-derived factor 1α Quantikine
ELISA kit (cat. no. DSA00) were purchased from R&D Systems
Inc., to detect the concentrations of IL-6, PAI-1 and CXCL12 in the
CM, according to the manufacturer's instructions. The absorbance at
450 nm was measured using a microplate reader and 540 nm was set as
the reference wavelength.
Reverse transcription-quantitative PCR
(RT-qPCR)
The total RNA, in each sample, was extracted using a
RNeasy Mini kit (Qiagen GmbH), in accordance with the
manufacturer's instructions, and a spectrophotometer (NanoDrop™
2000; Thermo Fisher Scientific, Inc.) was used to measure and
calculate the RNA concentration of the samples. Subsequently, 2.5
µg RNA was reverse transcribed into cDNA, in a total 50
µl reaction system, using a High-Capacity cDNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Then, TaqMan
qPCR was performed using a StepOnePlus Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: Initial denaturation
at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C
for 30 sec, annealing at 58°C for 30 sec and extension at 72°C for
45 sec; and a final extension at 72°C for 10 min. The following
TaqMan gene expression assays were used: ACTA2 (assay ID,
Hs00426835_g1), CD163 (assay ID, Hs00174705_m1), CD206 (assay ID,
Hs00267207_ m1), E-cadherin (assay ID, Hs00170423_m1), N-cadherin
(assay ID, Hs00169953_m1), CXCL12 (assay ID, Hs00930455_ m1), CXCR4
(assay ID, Hs00976734_m1), FAP (assay ID, Hs00990791_m1), IL-6
(assay ID, Hs00985639_m1), PAI-1 (assay ID, Hs01126606_m1) (all
from Thermo Fisher Scientific, Inc.). GAPDH (assay ID, 4326317E;
Thermo Fisher Scientific, Inc.) was used as the internal control to
normalize the raw data. Data analysis was performed using the
2−∆∆Cq method for relative quantification (23) and the results are presented as the
fold changes of the relative mRNA expression for each experimental
group compared with that in the control group.
Western blot analysis
Total protein was extracted from the cell lines
using freshly prepared RIPA lysis buffer (Thermo Fisher Scientific,
Inc.), containing a protease inhibitor cocktail (Sigma-Aldrich;
Merck KGaA) and a PhosSTOP phosphatase inhibitor cocktail (Roche
Diagnostics). A BCA kit (Thermo Fisher Scientific Inc.) was used to
measure the concentration of the protein. Equal amounts of protein
(20 µg) were separated on 12% (for Snail and Twist1) or 10%
(for other proteins) SDS-PAGE and transferred onto PVDF membranes
(Bio-Rad Laboratories, Inc.). To evaluate protein expression, the
blots were blocked with 5% skimmed milk for 1 h at room temperature
and incubated overnight at 4°C with the following primary
antibodies: Anti-PAI-1 (1:1,000; cat. no. 11907; Cell Signaling
Technology, Inc.), anti-E-cadherin (1:1,000; cat. no. ab1416;
Abcam), anti-N-cadherin (1:1,000; cat. no. 13116; Cell Signaling
Technology, Inc.), anti-Snail family transcriptional repressor 1
(Snail; 1:1,000; cat. no. ab85936; Abcam), anti-Twist family bHLH
transcription factor 1 (Twist1; 1:1,000; cat. no. SC-15393, Cosmo
Bio Co., Ltd.) and anti-β-actin (1:2,000; cat. no. 4970; Cell
Signaling Technology, Inc.). Then, anti-rabbit IgG, HRP-linked
(1:2,000; cat. no. 7074; Cell Signaling Technology, Inc.) and
anti-mouse IgG, HRP-linked (1:2,000; cat. no. 7076; Cell Signaling
Technology, Inc.) were used as secondary antibodies, according to
the species of the primary antibodies, for 1 h at room temperature.
The proteins were detected with ECL reagents (Cytiva).
Statistical analysis
All the data are presented as the mean ± SD.
Statistical analysis and construction of the graphs was performed
using GraphPad Prism v7.0 software (GraphPad Software, Inc.) and
ImageJ v1.46r software (National Institutes of Health). Comparisons
between two groups were evaluated using an unpaired Student's
t-test or a Mann-Whitney U test. Differences among multiple groups
were analyzed using one-way ANOVA followed by Tukey's post hoc
test. All the experiments were repeated ≥3 times. P<0.05
(two-sided) was considered to indicate a statistically significant
difference.
Results
Cancer cells and CAFs induce M2
polarization in human macrophages
The THP-1 cells were treated with PMA for 48 h to
induce their differentiation into M0 (Fig. 1A), as previously reported
(24). CM from the HCC cells was
used to stimulate the human HSC line, Lx-2 for 48 h. After
activation by the cancer cells, the CAF markers, actin α-2
(ACTA2), fibroblast activation protein (FAP) and
IL-6 were markedly upregulated in the Lx-2 cells, indicating
that the HSCs transdifferentiated into CAFs (Fig. 1C).
To mimic the crosstalk between tumor cells, CAFs and
TAMs in the TME, the THP-1-induced M0 were treated with cancer
cell-derived CM, CAF-derived CM and cancer cell-pretreated M0
macrophage-derived CM to obtain TAM(Ca), TAM(CAF) and TAM(TAM),
respectively (Fig. 1A).
Macrophage polarization is characterized by distinct morphological
features (25). Using H&E
staining, it was identified that TAM(Ca) and TAM(CAF) exhibited an
elongated morphology, which was similar to that of M2-polarized
macrophages. However, TAM(TAM) were predominantly round and more
closely resembled M1-polarized macrophages (Fig. 1D).
Subsequently, the mRNA expression levels and the
concentration of M2 polarization-related markers were measured
using RT-qPCR and ELISA, respectively. TAM(Ca) and TAM(CAF)
exhibited increased mRNA expression levels of CD163 and
CD206. In addition, the concentration of the M1
polarization-related marker, IL-6 was decreased compared with that
in the M0, confirming the M2 polarization of the macrophages.
However, TAM(TAM) failed to express the M2 polarization-related
markers and IL-6 was upregulated at both the gene and protein
levels (Fig. 1E and F).
Therefore, the current study focused on TAM(Ca) and TAM(CAF) in the
subsequent experiments.
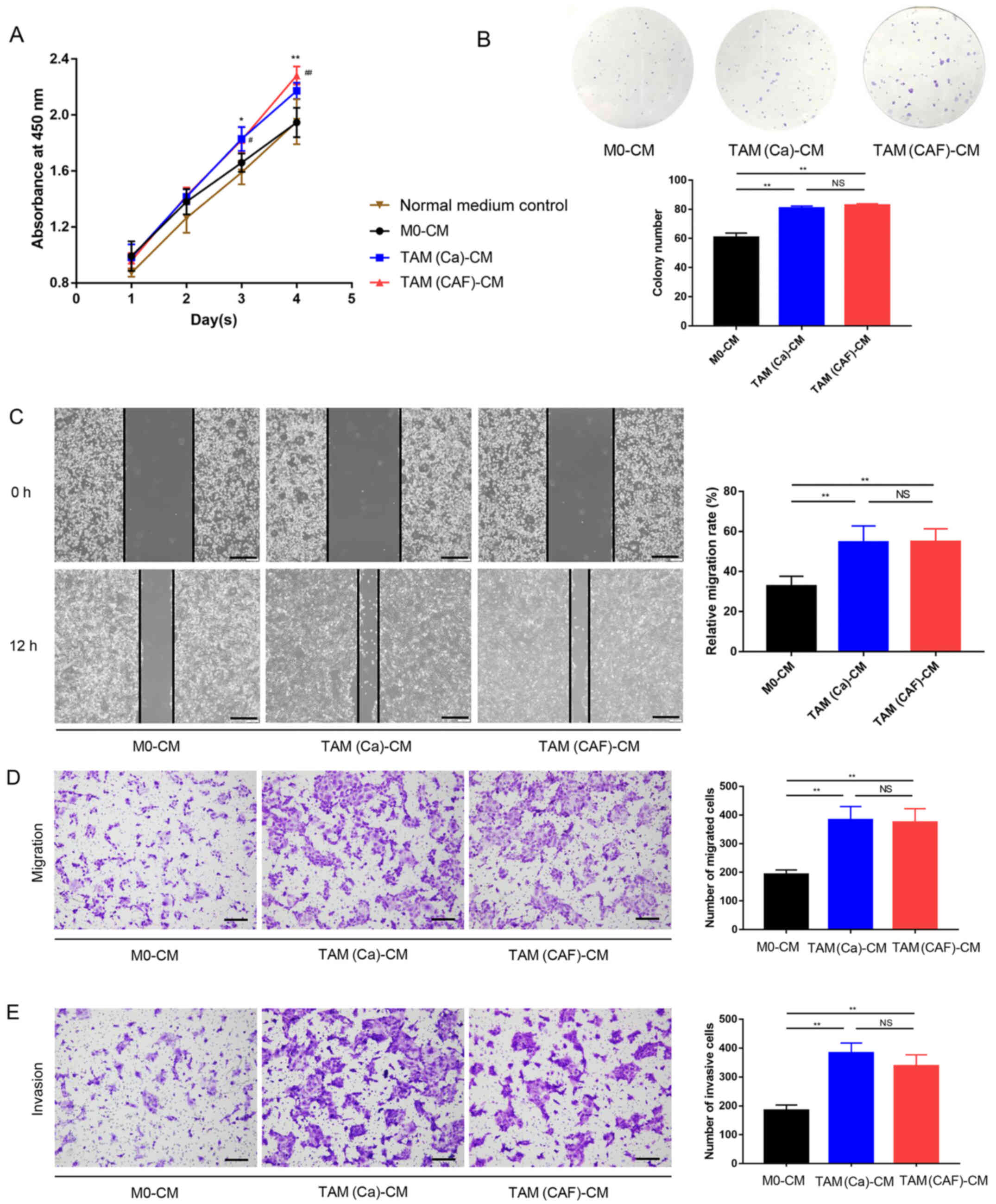
TAM(Ca) and TAM(CAF) promote HCC cell
malignant behavior in vitro
To investigate the effects of different types of
TAMs on the HCC cells, Huh-7 was treated with CM collected from M0,
TAM(Ca) and TAM(CAF), and normal culture medium was used as
control. The CCK-8 assay results demonstrated that after 4 days of
culture, the M0 did not increase the proliferation of the Huh-7
cells compared with that in the control group, while the TAM(Ca)
and TAM(CAF) groups significantly promoted the proliferation of the
Huh-7 cells compared with that in the M0 and normal medium control
groups. However, there was no significant difference between the
TAM(Ca) and TAM(CAF) groups (Fig.
2A). Similar results were obtained from the colony formation
assays, which revealed that the TAM(Ca) and TAM(CAF) experimental
groups significantly promoted the colony formation of Huh-7 cells
compared with that in the M0 group (Fig. 2B). Furthermore, compared with that
in the M0, TAM(Ca) and TAM(CAF) significantly promoted the
migration of the Huh-7 cells, as detected in the wound healing
(Fig. 2C) and Transwell (Fig. 2D) assays. The Matrigel assay
results identified that the invasive ability of the Huh-7 cells was
also enhanced by the two types of TAMs (Fig. 2E). Collectively, these results
suggested that both TAM(Ca) and TAM(CAF) promoted HCC cell
proliferation, migration and invasion in vitro.
PAI-1 expression is upregulated in
CAF-induced TAMs compared with that in the cancer cell-induced
TAMs
To further investigate the interactions between CAFs
and TAMs, the factors secreted by TAM(Ca) and TAM(CAF) were
compared. According to the results of the cytokine array, several
factors, such as PAI-1, IL-8, platelet derived growth factor-AA
(PDGF-AA) and T-cell immunoglobulin mucin 3 (TIM-3) were
upregulated in the CM from the M0 after stimulation with the CM
from cancer cells and CAFs (Fig.
3A). In addition, a total of 7 factors with notable differences
in secretion between the TAM(Ca) and TAM(CAF) were selected and
quantified (Fig. 3B). Among
these, PAI-1 exhibited the highest fold change. The western blot
analysis and ELISA results demonstrated that TAM(CAF) expressed
higher protein expression levels and released higher concentrations
of PAI-1, respectively, compared with that in the TAM(Ca) and M0
(Fig. 3C and D). The Huh-7 cells
also secreted PAI-1; however, the concentration of PAI-1 in the CM
of TAM(CAF) was significantly higher compared with that in the
Huh-7 cells (Fig. 3D).
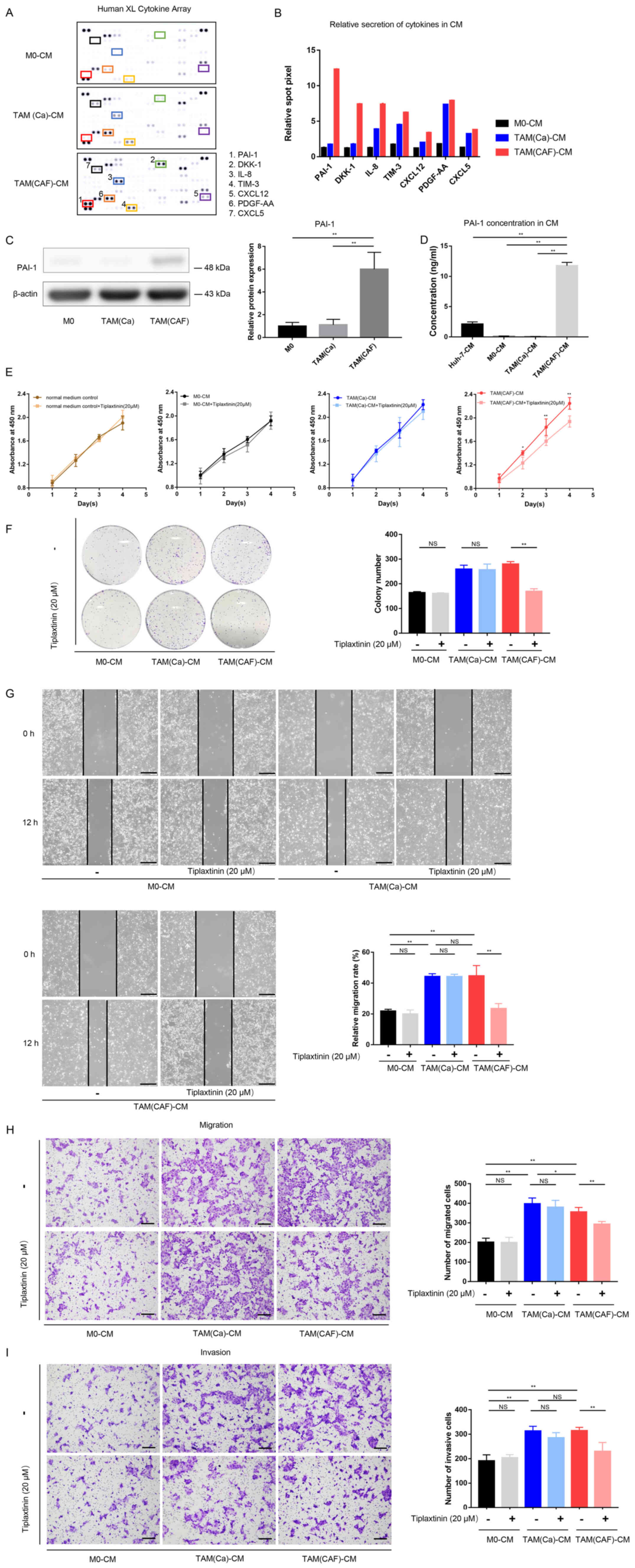 | Figure 3PAI-1 is the key factor secreted by
TAMs following CAF-CM stimulation and promotes tumor malignant
behavior in vitro. (A) Original image and (B) spot pixel
value from a cytokine array revealed the profiles of paracrine
factors in the M0-CM, TAM (Ca)-CM and TAM(CAF)-CM. (C) Western blot
and (D) ELISA results indicated that PAI-1 was upregulated in the
TAM(CAF) group compared with that in the M0 and TAM(Ca) groups. (E)
Cell Counting Kit-8 and (F) colony formation assays showed that the
inhibition of PAI-1 in TAM(CAF)-CM suppressed the enhanced
proliferation of Huh-7 cells. (G) Wound healing (scale bar, 400
µm) and (H) Transwell and (I) Matrigel (scale bar, 200
µm) assays indicated that the inhibition of PAI-1 in the
TAM(CAF)-CM group suppressed the enhanced migration and invasion of
the Huh-7 cells. *P<0.05; **P<0.01.
CAF, cancer-associated fibroblast; CM, conditioned medium; CXCL12,
C-X-C motif chemokine ligand 12; CXCL5, C-X-C motif chemokine
ligand 5; PAI-1, plasminogen activator inhibitor-1; PDGF-AA,
platelet derived growth factor-AA; TAM, tumor-associated
macrophage; TIM-3, T-cell immunoglobulin mucin 3; M0, macrophages;
NS, not significant. |
Accumulating evidence has suggested that PAI-1
contributed to tumor cell proliferation, migration, invasion, drug
resistance and epithelial-mesenchymal transition (EMT), and that
high PAI-1 protein expression was associated with unfavorable
biological behavior and patient prognosis in HCC (19,21,22). Therefore, the present study
selected PAI-1 for subsequent functional analysis and investigated
the roles of PAI-1 in TAM(CAF).
Inhibition of PAI-1 suppresses the
tumor-promoting effects of TAM(CAF) in vitro
To evaluate the functions of PAI-1 in the tumors
cells, the functional activity of PAI-1 in the CM of TAM(CAF) was
inhibited using tiplaxtinin, a small-molecule specific inhibitor of
PAI-1 (26). First, the effect of
tiplaxtinin on the proliferation of the Huh-7 cell line was
determined. As the data showed, tiplaxtinin at 20 µM had no
effect on proliferation (Fig.
3E). The data from the CCK-8 and colony formation assays
demonstrated that the proliferation-promoting effect of TAM(CAF),
but not TAM(Ca) or M0, was impaired following the inhibition of
PAI-1 (Fig. 3E and F). These
results indicated that the decreased proliferation was due to the
inhibition of PAI-1 function rather than the direct cytotoxic
effects of tiplaxtinin. In addition, following the inhibition of
PAI-1, the migratory and invasive abilities of the HCC cells were
significantly decreased in the TAM(CAF) group compared with that in
the TAM(CAF) group without tiplaxtinin (Fig. 3G-I). It was also found that
treatment with rPAI-1 directly increased the malignancy of the HCC
cells (Fig. 4A-D).
Mechanistically, the downregulation of E-cadherin and upregulation
of N-cadherin, Snail and Twist1 revealed that TAM(Ca) and TAM(CAF)
promoted EMT in the cancer cells compared with that in the M0
group, while PAI-1 inhibition diminished this effect in the
TAM(CAF) group (Fig. 4E and F).
Taken together, these results suggested that TAM(CAF)-secreted
PAI-1 contributed to the malignant behavior of the HCC cells by
mediating EMT.
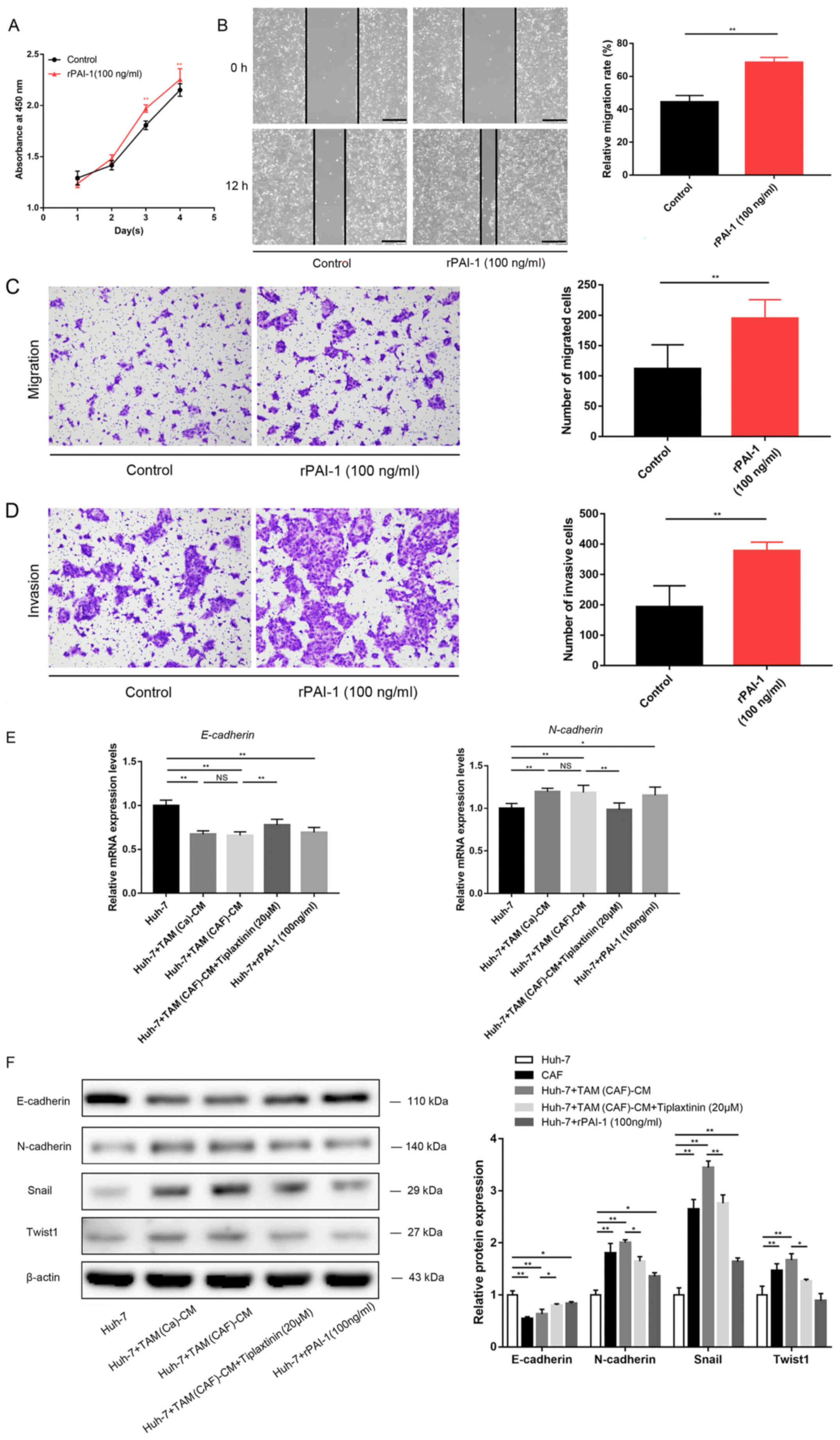 | Figure 4PAI-1 promotes HCC malignant behavior
by mediating EMT. (A) Cell Counting Kit-8, (B) wound healing (scale
bar, 400 µm), (C) Transwell and (D) Matrigel (scale bar, 200
µm) assays demonstrated the direct effects of PAI-1 on tumor
cell malignant behavior. (E) EMT-related gene expression was
detected using reverse transcription-quantitative PCR in the Huh-7
cells. (F) Western blot analysis revealed the protein expression
levels of E-cadherin, N-cadherin, Snail and Twist1 in the Huh-7
cells under the specified treatment conditions.
*P<0.05;**P<0.01. CAF,
cancer-associated fibroblast; CM, conditioned medium; PAI-1,
plasminogen activator inhibitor-1; Snail, snail family
transcriptional repressor 1; TAM, tumor-associated macrophage;
Twist1, twist family bHLH transcription factor 1; NS, not
significant. |
CAF-derived CXCL12 contributes to the
secretion of PAI-1 in TAM(CAF)
As the aforementioned results suggested a
significant role of PAI-1 in tumor-promoting processes, the
mechanisms underlying the specific secretion of PAI-1 in TAM(CAF)
was further investigated. The differences in the factors between
the CM from the Huh-7 cells and the CAFs were compared (Fig. 5A). The results of the cytokine
array indicated that the levels of CXCL12 and C-C motif chemokine
ligand 5 were markedly higher in the CM from the CAFs compared with
that in the CM from the cancer cells (Fig. 5B). As CXCL12 is known to promote
the transcription and secretion of PAI-1 (27), CXCL12 was selected for subsequent
analysis.
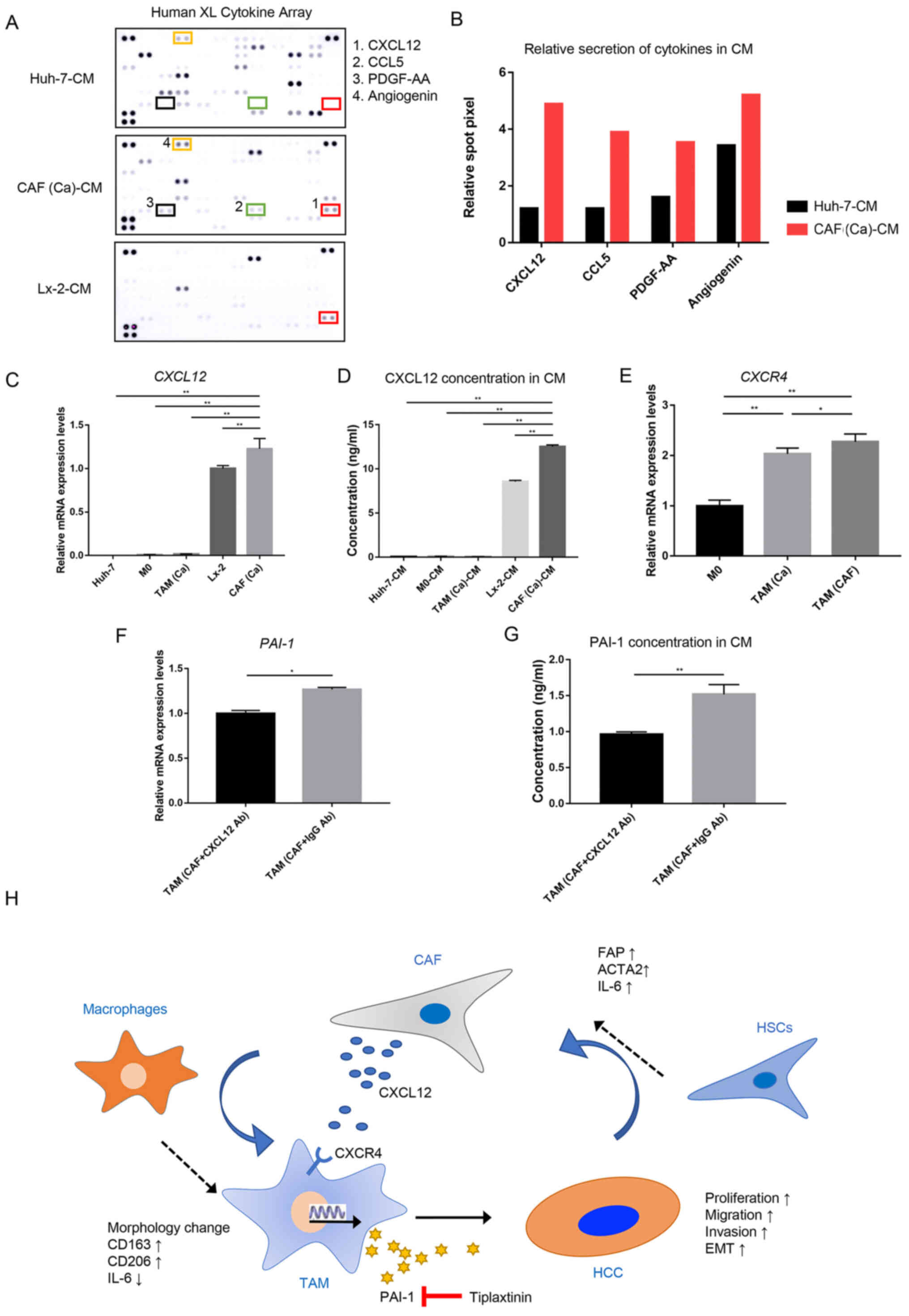 | Figure 5CAF-derived CXCL12 induces the
secretion of PAI-1 in TAM(CAF). (A) Original image and (B) spot
pixel value from a cytokine array to identify the different
patterns of molecules in cancer-CM and CAFs-CM. CXCL12 gene
expression and its secretion were increased in CAFs compared with
that in the Huh-7 and Lx-2 cells following (C) RT-qPCR and (D)
ELISA. (E) CXCR4 gene expression in M0, TAM(Ca) and TAM(CAF)
was analyzed using RT-qPCR. (F) RT-qPCR and (G) ELISA results
demonstrated that the gene expression level and secretion of PAI-1
were decreased in TAM(CAF) after CXCL12 neutralization in CAF-CM,
respectively. (H) Schematic representation of the proposed
interactions between HCC, TAMs and CAFs. *P<0.05;
**P<0.01. CAF, cancer-associated fibroblast; CM,
conditioned medium; CCL5, C-C motif chemokine ligand 5; CXCL12,
C-X-C motif chemokine ligand 12; CXCR4, C-X-C Motif chemokine
receptor 4; HCC, hepatocellular carcinoma; HSCs, hepatic stellate
cells; PAI-1, plasminogen activator inhibitor-1; PDGF-AA, platelet
derived growth factor-AA; RT-qPCR, reverse
transcription-quantitative PCR; TAM, tumor-associated macrophage;
Ab, antibody. |
The RT-qPCR and ELISA results demonstrated that the
CAFs had higher mRNA expression level and secreted more CXCL12
compared with that in the normal fibroblasts, and these were the
primary source of CXCL12 in the TME (Fig. 5C and D). Next, the mRNA expression
level of C-X-C motif chemokine receptor 4 (CXCR4), which is
the receptor of CXCL12 (28), was
measured in the M0, TAM(Ca) and TAM(CAF). Consistent with the
current hypothesis, it was found that TAM(CAF) overexpressed the
CXCR4 gene (Fig. 5E).
Furthermore, CXCL12 neutralization with a CXCL12 antibody in the CM
of CAFs reduced the mRNA expression level and the secretion of
PAI-1 in the TAM(CAF) (Figs. 1B
and 5F and G), suggesting that
CXCL12 could be an inducer of PAI-1 secretion in TAM(CAF).
Collectively, these results indicated that the HCC cells induced
the transformation of HSCs into CAFs and upregulated the expression
level of CXCL12. Through various interactions, CAFs interacted with
the macrophages to induce their M2 polarization and stimulated the
secretion of PAI-1 via CXCL12. In turn, PAI-1 produced by TAM(CAF)
enhanced the malignant behavior of HCC cells (Fig. 5H).
Discussion
The present study demonstrated that the HCC cells
induced the differentiation of HSCs into CAFs and upregulated the
mRNA expression level of CXCL12. CAF-induced macrophages exhibited
a special M2 polarization phenotype and secreted PAI-1 to promote
the malignant behavior of HCC cells in vitro. The current
study also provided evidence that inhibiting the crosstalk in the
TME by targeting these factors may be beneficial for HCC
treatment.
CAFs are a heterogeneous population; however, HSCs
with high ACTA2 and FAP mRNA expression levels
activated by adjacent tumor cells are considered to be the main
source of CAFs in HCC (29). In
the TME, infiltrated monocytes or resident macrophages are
recruited by several factors, such as CCL5 and TGF-β, and form a
diverse spectrum of TAMs, which are the main regulators of
tumor-related inflammation (30,31). TAMs are mainly divided into two
different polarization types. Classically activated
anti-tumorigenic M1 macrophages have pro-inflammatory
characteristics and secrete IL-1 and IL-6, while alternatively
activated M2 macrophages exhibit anti-inflammatory and
pro-tumorigenic functions and mainly express CD163 and
CD206 (32).
The present results demonstrated that both cancer
cells and CAFs could induce the polarization of monocyte-derived
macrophages into M2-polarized macrophages, which promoted the
malignant characteristics of the HCC cells. Furthermore, it was
identified that IL-6 concentration and expression level was
significantly increased in the CAFs compared with that in the HSCs.
Previous studies have reported that IL-6 could modulate the M2
macrophage polarization, and this may be a possible mechanism for
CAF-CM to induce the change from the M0 macrophage phenotype into
TAM (33,34). Furthermore, previous reports have
revealed that, in addition to cancer cells, CAFs also induced the
chemotaxis of monocytes to localize to the tumor cells via
paracrine actions and induced the M2 polarization phenotype in HCC
(15), PCa (35), PDAC (5), IHCC (12), breast cancer (13), OSCC (36) and ESCC (10). Similar to CAFs, macrophages
display abnormal transcriptomic characteristics and functions under
specific stimuli from tumor cells or stromal cells (37). Furthermore, heterogeneous TAMs
secrete a variety of factors, such as IL-8, CCL5 and VEGF, which
exert various effects on cancer cells and other stromal cells,
resulting in enhanced cancer cell proliferation, stemness,
invasion, drug resistance and immune escape (6,32,38). The present results identified that
TAM(CAF) exhibited significantly increased expression levels and
secretion of a variety of factors, including PAI-1.
The paradoxical tumor-promoting abilities of PAI-1
in cancer have been a topic of increased interest. It is currently
considered that the complex functional domains and various sources
of PAI-1 contribute to its multiple roles in promoting cancer
progression (21). While cancer
cells also secrete PAI-1, the present results suggested that only
TAMs treated with CAF-derived CM exhibited a significant increase
in PAI-1 protein expression and secretion. However, the intensity
of the expression levels of PAI-1 from western blot analysis was
not as high as that in the cytokine array. We hypothesized that as
a secretory protein, PAI-1 could be secreted and enriched in the
supernatant, which may lead to a higher level of PAI-1 in the
supernatant.
To elucidate the mechanism in which the CAFs
specifically increased PAI-1 secretion in the TAMs, the current
study examined the differences in factors in the CM between the
cancer cells and CAFs. It was found that CAFs secreted high amounts
of CXCL12, whereas cancer cells lacked this ability. Furthermore,
CXCL12 mRNA expression level was increased during the
transformation of the HSCs into CAFs stimulated by cancer cells,
which was consistent with findings from previous studies (39,40). In addition to the main regulators
of PAI-1, such as TNF-α and TGF-β (16), CXCL12 was also found to stimulate
PAI-1 expression (27). CXCL12
achieved its chemotactic effects by interacting with CXCR4. Oh
et al (27) reported that
glioma cells expressing high protein levels of CXCR4 could
upregulate PAI-1 mRNA and protein expression levels under the
stimulation of CXCL12, and identified that the activation of Gi
protein α subunit and the ERK/MAPK signaling pathways were
necessary for this process. The current results demonstrated that
CXCR4 mRNA expression level was upregulated in the TAM(CAF),
and that the neutralization of CXCL12 downregulated both the mRNA
expression level and the secretion of PAI-1. Taken together, it was
suggested that CXCL12 interacted with CXCR4 to promote PAI-1
transcription.
The effects of PAI-1 on tumor progression are well
documented. Several reports have shown that PAI-1 directly
stimulated the proliferation of tumor cells by regulating cell
cycle progression (41,42), and that it exerted protective
effects by inhibiting Fas cell surface death receptor (Fas)/Fas
ligand-mediated apoptosis (18)
and inactivating caspase-3 (43).
In breast cancer, PAI-1 facilitated cancer-associated
adipocyte-mediated collagen remodeling and further promoted cancer
metastasis by activating the PI3K/AKT/forkhead box P1 signaling
pathway (44). Liu et al
(45) reported that PAItrap3, a
specific inhibitor of PAI-1, reduced the migratory and invasive
abilities in breast and cervical cancer cells by inhibiting the
interactions between the F-actin complex and integrins.
EMT is a process involving changes in the epithelial
characteristics of cells to enhance their migratory ability
(46). A decrease in E-cadherin
and increase in N-cadherin mRNA and protein expression levels are
typical changes in EMT (47). Our
previous study revealed that lactate secreted by HCC and PDAC cells
stimulated the M2 phenotypic transformation of macrophages and
enhanced VEGF expression by nuclear factor erythroid 2-related
factor 2 (NRF2) activation, and that M2 macrophages triggered NRF2
signaling activation in cancer cells via paracrine VEGF, thereby
promoting EMT (6). Several
previous studies have shown that PAI-1 contributed to cell invasion
and migration by mediating EMT in colorectal cancer (19), gastric adenocarcinoma (48) and non-small cell lung cancer
(49). Furthermore, the present
results demonstrated that E-cadherin protein expression level was
downregulated, while N-cadherin, Snail and Twist1 protein
expression levels were upregulated in the Huh-7 treated with
TAM(CAF), and these effects were reversed by the inhibition of
PAI-1. Collectively, the current findings suggested that PAI-1
secreted from TAM(CAF) was associated with EMT in HCC cell
lines.
As a selective inhibitor of PAI-1, with an
IC50 of 2.7 µM (50), 20 µM tiplaxtinin was used
to inactivate the PAI-1 in the CM in the present study. Additional
experiments were also performed to confirm that tiplaxtinin, at
this concentration, did not affect the proliferation of Huh-7. To
date, PAI-1 inhibitors have been used in animal models of cancer
and have shown effective anti-tumor activity. For example,
tiplaxtinin has been reported to decrease tumor cell proliferation
and vascularization in mouse models of bladder cancer and cervical
cancer (51). Furthermore, SK-216
exerted an anti-tumor effect by reducing tumor size and inhibiting
angiogenesis and metastasis in mice with lung carcinoma and
melanoma (52). However, the
short effective half-life of these inhibitors in the body has
limited their clinical use, as sufficient concentrations are often
difficult to achieve, particularly in the tumor area, during
long-term cancer treatment (19).
Therefore, anti-PAI-1 therapy in cancer treatment remains a
promising but challenging strategy, and the development of more
effective and safe inhibitors is required.
There are certain limitations to the present study.
The mechanism of TAM polarization and the status of TAMs under
immunosuppressive conditions in the TME require further
investigation. Recent research has shown that the protein
expression levels of PAI-1 and ACTA2 are associated with each
other. For example, in vitro inhibition of PAI-1
downregulated the positive expression level of ACTA2 in CAFs and
promoted CAF apoptosis (53).
PAI-1 also increased monocyte/macrophage recruitment and M2
polarization (54). These
findings suggest that the effects of PAI-1 on tumor stromal cells
require further consideration.
The present study simulated the crosstalk among
cancer cells, CAFs and TAMs in vitro, and to the best of our
knowledge, reported for the first time that CAFs promoted tumor
progression by inducing a special TAM phenotype. CAFs supported
tumors by stimulating the M2 phenotypic transformation of
macrophages and the secretion of PAI-1. PAI-1 promoted the
proliferation and invasion of HCC cells by mediating EMT, and
CXCL12 produced by CAFs contributed to the secretion of PAI-1 in
TAMs. The current results identified CAFs as a source of soluble
factors, which may regulate the phenotype of other stromal cells.
Furthermore, TAMs directly promoted tumor progression via paracrine
factors. Therefore, inhibiting the crosstalk between stromal cells
may be a promising method for cancer treatment in HCC. Further
research should be conducted to fully elucidate the complex
functions of CAFs and TAMs in HCC to improve treatment for patients
with HCC.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The cytokine datasets generated and analyzed during the
current study are available in the Figshare repository (https://doi.org/10.6084/m9.figshare.14386388.v1).
Authors' contributions
SC, KT, YM and MS designed the experiments. SC, YS
and MN performed the experiments and collected the data. SY and TI
analyzed and interpreted the data. SC drafted the manuscript. YM
and MS revised the paper critically for important intellectual
content. SC and YM confirmed the authenticity of all the raw data.
SC and YM agree to be accountable for all aspects of the work in
ensuring that questions related to the accuracy or integrity of any
part of the work are appropriately investigated and resolved. All
the authors have read and approved the final version of the
manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was partly supported by the Research Program on
Hepatitis from Japanese Foundation for Multidisciplinary Treatment
of Cancer and the Japan Agency for Medical Research and Development
(grant nos. JP19fk0210048 and JP20fk0210048), Grant-in-Aid for
Scientific Research (grant nos. 20K08957 and 18K02871) and Taiho
Pharmaceutical Co., Ltd.
Abbreviations:
|
ACTA2
|
actin α-2
|
|
CAFs
|
cancer-associated fibroblasts
|
|
CCK-8
|
Cell Counting Kit-8
|
|
CM
|
conditioned medium
|
|
CXCL12
|
C-X-C motif chemokine ligand 12
|
|
CXCR4
|
C-X-C motif chemokine receptor 4
|
|
EMT
|
epithelial-mesenchymal transition
|
|
ESCC
|
esophageal squamous cell carcinoma
|
|
FAP
|
fibroblast activation protein
|
|
HCC
|
hepatocellular carcinoma
|
|
HSCs
|
hepatic stellate cells
|
|
IHCC
|
intrahepatic cholangiocarcinoma
|
|
NRF2
|
nuclear factor erythroid 2-related
factor 2
|
|
OSCC
|
oral squamous cell carcinoma
|
|
PAI-1
|
plasminogen activator inhibitor-1
|
|
PCa
|
prostate cancer
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
PMA
|
phorbol-12-myristate-13-acetate
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
TAMs
|
tumor-associated macrophages
|
|
TME
|
tumor microenvironment
|
|
VEGF
|
vascular endothelial growth factor
|
References
|
1
|
Metcalf KJ, Alazzeh A, Werb Z and Weaver
VM: Leveraging microenvironmental synthetic lethalities to treat
cancer. J Clin Invest. 131:e1437652021. View Article : Google Scholar
|
|
2
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bejarano L, Jordāo MJ and Joyce JA:
Therapeutic targeting of the tumor microenvironment. Cancer Discov.
11:933–959. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Comito G, Giannoni E, Segura CP,
Barcellos-de-Souza P, Raspollini MR, Baroni G, Lanciotti M, Serni S
and Chiarugi P: Cancer-associated fibroblasts and M2-polarized
macrophages synergize during prostate carcinoma progression.
Oncogene. 33:2423–2431. 2014. View Article : Google Scholar
|
|
5
|
Andersson P, Yang Y, Hosaka K, Zhang Y,
Fischer C, Braun H, Liu S, Yu G, Liu S, Beyaert R, et al: Molecular
mechanisms of IL-33-mediated stromal interactions in cancer
metastasis. JCI Insight. 3:e1223752018. View Article : Google Scholar
|
|
6
|
Feng R, Morine Y, Ikemoto T, Imura S,
Iwahashi S, Saito Y and Shimada M: Nrf2 activation drive
macrophages polarization and cancer cell epithelial-mesenchymal
transition during interaction. Cell Commun Signal. 16:542018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang JI, Kim DH, Sung KW, Shim SM,
Cha-Molstad H, Soung NK, Lee KH, Hwang J, Lee HG, Kwon YT and Kim
BY: p62-Induced cancer-associated fibroblast activation via the
nrf2-atf6 pathway promotes lung tumorigenesis. Cancers (Basel).
13:8642021. View Article : Google Scholar
|
|
8
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu Q, Zhou LY, Lv DD, Zhu X and Tang H:
Exosome-mediated communication in the tumor microenvironment
contributes to hepatocellular carcinoma development and
progression. J Hematol Oncol. 12:532019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Higashino N, Koma YI, Hosono M, Takase N,
Okamoto M, Kodaira H, Nishio M, Shigeoka M, Kakeji Y and Yokozaki
H: Fibroblast activation protein-positive fibroblasts promote tumor
progression through secretion of CCL2 and interleukin-6 in
esophageal squamous cell carcinoma. Lab Invest. 99:777–792. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hashimoto O, Yoshida M, Koma YI, Yanai T,
Hasegawa D, Kosaka Y, Nishimura N and Yokozaki H: Collaboration of
cancer-associated fibroblasts and tumour-associated macrophages for
neuroblastoma development. J Pathol. 240:211–223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W,
Dang Y, Chu Y, Fan J and He R: Fap promotes immunosuppression by
cancer-associated fibroblasts in the tumor microenvironment via
STAT3-CCL2 signaling. Cancer Res. 76:4124–4135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Allaoui R, Bergenfelz C, Mohlin S,
Hagerling C, Salari K, Werb Z, Anderson RL, Ethier SP, Jirström K,
Påhlman S, et al: Cancer-associated fibroblast-secreted CXCL16
attracts monocytes to promote stroma activation in triple-negative
breast cancers. Nat Commun. 7:130502016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim
JH, Soh J, Kim HS, Lee H, Kim J, et al: Cancer-stimulated cafs
enhance monocyte differentiation and protumoral tam activation via
il6 and GM-CSF secretion. Clin Cancer Res. 24:5407–5421. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ji J, Eggert T, Budhu A, Forgues M, Takai
A, Dang H, Ye Q, Lee JS, Kim JH, Greten TF and Wang XW: Hepatic
stellate cell and monocyte interaction contributes to poor
prognosis in hepatocellular carcinoma. Hepatology. 62:481–495.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kubala MH and DeClerck YA: The plasminogen
activator inhibitor-1 paradox in cancer: A mechanistic
understanding. Cancer Metastasis Rev. 38:483–492. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li SJ, Wei XH, Zhan XM, He JY, Zeng YQ,
Tian XM, Yuan ST and Sun L: Adipocyte-derived leptin promotes
PAI-1-mediated breast cancer metastasis in a STAT3/miR-34a
dependent manner. Cancers (Basel). 12:38642020. View Article : Google Scholar
|
|
18
|
Mashiko S, Kitatani K, Toyoshima M,
Ichimura A, Dan T, Usui T, Ishibashi M, Shigeta S, Nagase S, Miyata
T and Yaegashi N: Inhibition of plasminogen activator inhibitor-1
is a potential therapeutic strategy in ovarian cancer. Cancer Biol
Ther. 16:253–260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Muñoz-Galván S, Rivero M, Peinado-Serrano
J, Martinez-Pérez J, Fernández-Fernández MC, Ortiz MJ,
García-Heredia JM and Carnero A: PAI1 is a marker of bad prognosis
in rectal cancer but predicts a better response to treatment with
PIM inhibitor AZD1208. Cells. 9:10712020. View Article : Google Scholar :
|
|
20
|
Fang H, Placencio VR and DeClerck YA:
Protumorigenic activity of plasminogen activator inhibitor-1
through an antiapoptotic function. J Natl Cancer Inst.
104:1470–1484. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Placencio VR and DeClerck YA: Plasminogen
activator inhibitor-1 in cancer: Rationale and insight for future
therapeutic testing. Cancer Res. 75:2969–2974. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin Y, Liang ZY, Zhou WX and Zhou L:
Expression, clinicopathologic and prognostic significance of
plasminogen activator inhibitor 1 in hepatocellular carcinoma.
Cancer Biomark. 27:285–293. 2020. View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei
T, Yang JQ, Tang J, Wang J, Chen Y, et al: Hypoxia-inducible
factor-1α/interleukin-1β signaling enhances hepatoma
epithelial-mesenchymal transition through macrophages in a
hypoxic-inflammatory microenvironment. Hepatology. 67:1872–1889.
2018. View Article : Google Scholar
|
|
25
|
Dahlem C, Siow WX, Lopatniuk M, Tse WK,
Kessler SM, Kirsch SH, Hoppstädter J, Vollmar AM, Müller R,
Luzhetskyy A, et al: Thioholgamide A, a new anti-proliferative
anti-tumor agent, modulates macrophage polarization and metabolism.
Cancers (Basel). 12:12882020. View Article : Google Scholar
|
|
26
|
Che Y, Wang JN, Li Y, Lu ZL, Huang JB, Sun
SG, Mao SS, Lei YY, Zang RC, Sun N and He J: Cisplatin-activated
PAI-1 secretion in the cancer-associated fibroblasts with paracrine
effects promoting esophageal squamous cell carcinoma progression
and causing chemoresistance. Cell Death Dis. 9:7592018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oh JW, Olman M and Benveniste EN:
CXCL12-mediated induction of plasminogen activator inhibitor-1
expression in human CXCR4 positive astroglioma cells. Biol Pharm
Bull. 32:573–577. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Phillips RJ, Burdick MD, Lutz M, Belperio
JA, Keane MP and Strieter RM: The stromal derived
factor-1/CXCL12-CXC chemokine receptor 4 biological axis in
non-small cell lung cancer metastases. Am J Respir Crit Care Med.
167:1676–1686. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yin ZL, Dong CY, Jiang KQ, Xu Z, Li R, Guo
K, Shao SJ and Wang L: Heterogeneity of cancer-associated
fibroblasts and roles in the progression, prognosis, and therapy of
hepatocellular carcinoma. J Hematol Oncol. 12:1012019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pham K, Huynh D, Le L, Delitto D, Yang L,
Huang J, Kang Y, Steinberg MB, Li J, Zhang L, et al: E-cigarette
promotes breast carcinoma progression and lung metastasis:
Macrophage-tumor cells crosstalk and the role of CCL5 and VCAM-1.
Cancer Lett. 491:132–145. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan W, Liu X, Ma H, Zhang H, Song X, Gao
L, Liang X and Ma C: Tim-3 fosters HCC development by enhancing
TGF-β-mediated alternative activation of macrophages. Gut.
64:1593–1604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Franklin RA, Liao W, Sarkar A, Kim MV,
Bivona MR, Liu K, Pamer EG and Li MO: The cellular and molecular
origin of tumor-associated macrophages. Science. 344:921–925. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yin Z, Ma TT, Lin Y, Lu X, Zhang CZ, Chen
S and Jian ZX: IL-6/STAT3 pathway intermediates M1/M2 macrophage
polarization during the development of hepatocellular carcinoma. J
Cell Biochem. 119:9419–9432. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Q, He Z, Huang M, Liu T, Wang Y, Xu
H, Duan H, Ma P, Zhang L, Zamvil SS, et al: Vascular niche IL-6
induces alternative macrophage activation in glioblastoma through
HIF-2α. Nat Commun. 9:5592018. View Article : Google Scholar
|
|
35
|
Augsten M, Sjöberg E, Frings O, Vorrink
SU, Frijhoff J, Olsson E, Borg A and Östman A: Cancer-associated
fibroblasts expressing CXCL14 rely upon NOS1-derived nitric oxide
signaling for their tumor-supporting properties. Cancer Res.
74:2999–3010. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li X, Bu WH, Meng L, Liu XC, Wang SS,
Jiang LM, Ren MS, Fan Y and Sun HC: CXCL12/CXCR4 pathway
orchestrates CSC-like properties by CAF recruited tumor associated
macrophage in OSCC. Exp Cell Res. 378:131–138. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Murray PJ: Macrophage polarization. Annu
Rev Physiol. 79:541–566. 2017. View Article : Google Scholar
|
|
38
|
Huang CP, Liu LX and Shyr CR:
Tumor-associated macrophages facilitate bladder cancer progression
by increasing cell growth, migration, invasion and cytokine
expression. Anticancer Res. 40:2715–2724. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Feig C, Jones JO, Kraman M, Wells RJ,
Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL,
et al: Targeting CXCL12 from FAP-expressing carcinoma-associated
fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic
cancer. Proc Natl Acad Sci U S A. 110:20212–20217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tan HX, Gong WZ, Zhou K, Xiao ZG, Hou FT,
Huang T, Zhang L, Dong HY, Zhang WL, Liu Y and Huang ZC:
CXCR4/TGF-β1 mediated hepatic stellate cells differentiation into
carcinoma-associated fibroblasts and promoted liver metastasis of
colon cancer. Cancer Biol Ther. 21:258–268. 2020. View Article : Google Scholar
|
|
41
|
Giacoia EG, Miyake M, Lawton A, Goodison S
and Rosser CJ: PAI-1 leads to G1-phase cell-cycle progression
through cyclin D3/cdk4/6 upregulation. Mol Cancer Res. 12:322–334.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu SL, Wu XS, Li FN, Yao WY, Wu ZY, Dong
P, Wang XF and Gong W: ERRα promotes pancreatic cancer progression
by enhancing the transcription of PAI1 and activating the MEK/ERK
pathway. Am J Cancer Res. 10:3622–3643. 2020.
|
|
43
|
Chen Y, Kelm RJ Jr, Budd RC, Sobel BE and
Schneider DJ: Inhibition of apoptosis and caspase-3 in vascular
smooth muscle cells by plasminogen activator inhibitor type-1. J
Cell Biochem. 92:178–188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wei X, Li S, He J, Du H, Liu Y, Yu W, Hu
H, Han L, Wang C, Li H, et al: Tumor-secreted PAI-1 promotes breast
cancer metastasis via the induction of adipocyte-derived collagen
remodeling. Cell Commun Signal. 17:582019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu J, Chen Z, Huang M, Tang S, Wang Q, Hu
P, Gupta P, Ashby CR Jr, Chen ZS and Zhang L: Plasminogen activator
inhibitor (PAI) trap3, an exocellular peptide inhibitor of PAI-1,
attenuates the rearrangement of F-actin and migration of cancer
cells. Exp Cell Res. 391:1119872020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen S, Kang X, Liu G, Zhang B, Hu X and
Feng Y: α7-Nicotinic acetylcholine receptor promotes
cholangiocarcinoma progression and epithelial-mesenchymal
transition process. Dig Dis Sci. 64:2843–2853. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pal M, Bhattacharya S, Kalyan G and Hazra
S: Cadherin profiling for therapeutic interventions in epithelial
mesenchymal transition (EMT) and tumorigenesis. Exp Cell Res.
368:137–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang JD, Ma L and Zhu Z: SERPINE1 as a
cancer-promoting gene in gastric adenocarcinoma: Facilitates tumour
cell proliferation, migration, and invasion by regulating EMT. J
Chemother. 31:408–418. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lin X, Lin BW, Chen XL, Zhang BL, Xiao XJ,
Shi JS, Lin JD and Chen X: PAI-1/PIAS3/Stat3/miR-34a forms a
positive feedback loop to promote EMT-mediated metastasis through
Stat3 signaling in non-small cell lung cancer. Biochem Biophys Res
Commun. 493:1464–1470. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hennan JK, Morgan GA, Swillo RE, Antrilli
TM, Mugford C, Vlasuk GP, Gardell SJ and Crandall DL: Effect of
tiplaxtinin (PAI-039), an orally bioavailable PAI-1 antagonist, in
a rat model of thrombosis. J Thromb Haemost. 6:1558–1564.
2008.PubMed/NCBI
|
|
51
|
Gomes-Giacoia E, Miyake M, Goodison S and
Rosser CJ: Targeting plasminogen activator inhibitor-1 inhibits
angiogenesis and tumor growth in a human cancer xenograft model.
Mol Cancer Ther. 12:2697–2708. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Masuda T, Hattori N, Senoo T, Akita S,
Ishikawa N, Fujitaka K, Haruta Y, Murai H and Kohno N: SK-216, an
inhibitor of plasminogen activator inhibitor-1, limits tumor
progression and angiogenesis. Mol Cancer Ther. 12:2378–2388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Masuda T, Nakashima T, Namba M, Yamaguchi
K, Sakamoto S, Horimasu Y, Miyamoto S, Iwamoto H, Fujitaka K,
Miyata Y, et al: Inhibition of PAI-1 limits chemotherapy resistance
in lung cancer through suppressing myofibroblast characteristics of
cancer-associated fibroblasts. J Cell Mol Med. 23:2984–2994. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kubala MH, Punj V, Placencio-Hickok VR,
Fang H, Fernandez GE, Sposto R and DeClerck YA: Plasminogen
activator inhibitor-1 promotes the recruitment and polarization of
macrophages in cancer. Cell Rep. 25:2177–2191. 2018. View Article : Google Scholar : PubMed/NCBI
|