Introduction
Colorectal cancer (CRC) is the third most common
cancer worldwide and the second most common cause of cancer-related
death (1). A steady rise of CRC
incidence has been observed as a first common malignancy diagnosed
among men and the third most common among women in Saudi Arabia
(1994-2010) (2,3). During the initial diagnosis,
metastasis occurs in 18% of patients with rectal cancer and in
20-25% of patients with colon cancer (4). Growth and progression of the tumor
during tumor-associated inflammation includes multiple mechanisms
such as anti-apoptosis, abnormal proliferation, angiogenesis, cell
invasion and metastasis (5). As a
key step in enhancing cancer cell invasion and metastasis,
epithelial-mesenchymal transition (EMT) plays an important role in
CRC progression. During the EMT process, suppression of E-cadherin
by the transcription factors Snail and Slug provokes the loss of
the epithelial properties and a higher migration and invasion
capacity of the cancer cells (6).
Thus, the loss of E-cadherin expression and gain of N-cadherin
expression in cancer cells, occasionally called 'the cadherin
switch', have functional significance in cancer progression
(7). EMT is triggered by a
variety of signaling pathways, among which Wnt-β-catenin signaling
pathway has been implicated as one of the many primary inducer
(8). The human mucin family of
proteins includes large proteins with heavy glycosylation
designated MUC1 to MUC21 (9).
Mucin proteins are divided into secretory (MUC2, MUC5AC, MUC5B and
MUC6) and transmembrane forms (MUC1, MUC4, MUC13 and MUC16). The
mucins form a physical barrier that provides protection for
epithelial cells that line the respiratory and gastrointestinal
tracts and form the ductal surfaces of organs such as the liver,
breast, pancreas and kidney (9).
Mucin like 1 (MUCL1), a small glycoprotein also known as small
breast epithelium mucin (SBEM) was first discovered ~2 decades ago
(10). MUCL1 is a breast specific
gene which is highly expressed in breast and salivary glands
(11). An immunohistochemical
study revealed that MUCL1 protein is mostly expressed in breast
cancer (10). Previous studies
have indicated that MUCL1 expression strongly correlates with TNM
staging, higher tumor grade and lymph node metastasis (11-13). Other studies have demonstrated the
importance of MUCL1 detection in breast cancer patients as a
biomarker for tumor progression and metastasis (14,15). Liu et al (16) detected SBEM expression in breast
tumor tissues and blood samples and proposes it as a marker for
predicting hematogenous micrometastasis and neoadjuvant
chemotherapy response in breast cancer. Recently, Li et al
(17) described the role of SBEM
in promoting invasion and metastasis by inducing EMT in breast
cancer cells. To the best of our knowledge, the association between
MUCL1 expression and its function in CRC is still largely unknown.
In the present study, the expression of MUCL1 and functional
significance in human CRC cell lines was investigated. The
expression profile of MUCL1 from TCGA databases was explored in CRC
tumor tissues as compared with adjacent normal tissues. The effect
of silencing MUCL1 was assessed on cell proliferation, EMT,
invasion-migration and drug sensitivity in CRC.
Materials and methods
Cell culture
Human CRC cell lines HT-29, SW480 and SW620 were
purchased from American Type Culture Collection and cultured in
RPMI-1640 media consisting of 10% fetal bovine serum, 100 Unit/ml
penicillin and 2 mM L-glutamine (all from Thermo Fisher Scientific,
Inc.). SW480 were grown in DMEM media (Thermo Fisher Scientific,
Inc.) containing the aforementioned supplements. STR analysis was
performed to confirm new batches of cells. All the cell lines were
checked for mycoplasma contamination.
MUCL1 expression profile data
GEPIA (http://gepia.cancer-pku.cn/) (18) was used for The Cancer Genome Atlas
(TCGA) database and Genotype Tissue Expression (GTEx) projects
provided mRNA expression data from colon adenocarcinoma (COAD) and
rectal adenocarcinoma (READ). The analysis of the MUCL1 gene (NCBI
Entrez Gene ID: 118430) between CRC tumor and normal samples
expression data from the TCGA and GTEx projects was investigated in
COAD (T=275; N=349) and READ (T=92; N=318). UALCAN data - base
(http://ualcan.path.uab.edu/index.html) (19) is based on the TCGA data and is
used for gene expression profiling. In the present study, the
transcription expression profile of MUCL1 for COAD was evaluated
with the default setting in UALCAN. This tool uses Student's t-test
and normalized mRNA level as transcript per million (TPM).
P<0.05 was considered to indicate a statistically significant
difference.
Generation of stable MUCL1 short
interfering (si)RNA cells
HT-29 and SW620 cells (2×105) were
cultured in 2 ml complete media in 6-well plates. After 24 h, when
the cells reached 50-60% confluency, Lipofectamine RNAi/Max reagent
(10 µl; Thermo Fisher Scientific, Inc.), control siRNA (5
µl; 50 pmols); cat. no. SC37007 Santa Cruz Biotechnology,
Inc.) and MUCL1 siRNA (5 µl; 50 pmols); cat. no. SC95777;
Santa Cruz Biotechnology, Inc.) were diluted in Opti-MEM medium
(150 µl; Thermo Fisher Scientific, Inc.). Diluted siRNAs and
Lipofectamine RNAi/Max reagent were mixed together (1:1 ratio) and
kept under the cell culture hood for 30 min at room temperature.
Complex of siRNA and Lipofectamine was added drop-wise to cells and
incubated at 37°C for 6 h. Then 1 ml of complete medium was further
added. The following day, fresh medium was replaced and the mixture
was incubated for 48 h at 37°C. For selection of stably transfected
cells, cells were treated with puromycin (MilliporeSigma; 2
µg/ml) and incubated at 37°C further for 3-5 days. Gradually
fresh media was replaced every 3-4 days. Stable cells were
maintained at 0.5 µg/ml puromycin.
Western blotting
HT-29 control siRNA and HT-29 MUCL1 siRNA clones
(ML1 and 2) as well as SW620 control siRNA and SW620 MUCL1 siRNA
clones (ML1 and 2) were cultured in RPMI-1640 complete medium with
puromycin (0.5 µg/ml). Preparation of total cell lysates was
conducted by harvesting cells followed by PBS washing. The cell
pellets were added with RIPA lysis buffer (Boston Bioproducts,
Inc.) for 15 min at 4°C followed by centrifugation at 17,530 × g
for 15 min at 4°C (20).
Supernatant having the soluble proteins was collected and the
concentration of proteins was determined on Bio-Rad SmartSpec Plus
spectrophotometer using Bradford protein assay reagent (Bio-Rad
Laboratories, Inc.). Equal amount of proteins (10-20 µg)
were loaded on electrophoresis gels (4-20% Mini-Protean TGX precast
gels; Bio-Rad Laboratories, Inc.). The gels were transferred to
0.2-µm PVDF membrane by turbo transfer system (Bio-Rad
Laboratories, Inc.). The membranes with transferred proteins were
blocked in Sea Block blocking buffer (cat. no. 37527; Thermo
Scientific, Inc.) for 1 h at room temperature, followed by washing
twice with PBS containing 0.1% Tween-20 (PBST). Subsequently, the
membranes were incubated overnight at 4°C with the following
primary antibodies: MUCL1 (cat. no. NBP1-92366; 1:500; Novus
Biologicals, LLC), Bcl2 (cat. no. sc-492; 1:1,000), BclxL (cat. no.
sc-56021; 1:1,000), caspase-3 (cat. no. sc-56053; 1:1,000),
E-cadherin (cat. no. sc-8426; 1:1,000), vimentin (cat. no. sc-6260;
1:1,000), Lamin B (cat. no. sc-374015; 1:1,000) and β-actin (cat.
no. sc-47778; 1:2,000) (all from Santa Cruz Biotechnology, Inc.).
Phospho-β-catenin-Ser-552 (cat. no. 9566; 1:1,000) and β-catenin
(cat. no. 8480; 1:1,000) were purchased from Cell Signaling
Technology, Inc. The following day after washing, the blots were
incubated with HRP-conjugated rabbit secondary (cat. no. sc-2357;
1:3,000) and mouse secondary (1:3,000; cat. no. sc-516102) (both
from Santa Cruz Biotechnology, Inc.) antibodies on a shaker for 1 h
at 25°C. Chemiluminescence signal was detected by incubating the
blot membrane with Pierce ECL western blotting substrate (Thermo
Fisher Scientific, Inc.) and detected on a C-DiGit blot scanner
(LI-COR Biosciences). Densitometry was carried out using C-DiGiT
blot scanner software (Image Studio Digits 3.1; LI-COR
Biosciences).
Colony formation assay
Colony formation assay was performed as previously
described (20). HT-29 control
siRNA and HT-29 MUCL1 siRNA clones (ML1 and 2) as well as SW620
control siRNA and MUCL1 siRNA clones (ML1 and 2) were harvested and
centrifuged at 252 × g for 5 min at 4°C. The respective cells were
added into 6-well plates at 500 cells/well containing 2.0 ml
RPMI-1640 media. The media was replaced with fresh medium every 3-4
days. The plates were incubated at 37°C for 10-12 days in a 5%
CO2 incubator. Groups of >50 cells were considered a
colony. Using 4% paraformaldehyde the colonies were fixed for 10
min at room temperature and stained with 0.1% crystal violet for 15
min at room temperature. The quantification of colonies was
performed by using a light inverted microscope (Micros Austria) at
×10 magnification.
Cell proliferation assay
Cell Counting Kit-8 (CCK-8) assay was performed
using CCK-8 reagent (APExBIO Technology LLC). HT-29 control siRNA
and HT-29 MUCL1 siRNA (ML1 and 2) as well as SW620 control siRNA
and SW620 MUCL1 siRNA (ML1 and 2) transfected cells were seeded
into a 96-well plate at 5,000 cells/well. After 24, 48, 72 and 96 h
cells were incubated with CCK-8 reagent (10 µl) at 37°C for
2 h, and the absorbance was measured at 450 nm using a microplate
reader (BioTek Instruments, Inc.). The mean and standard deviation
(SD) values of three independent experiments were collected.
Transwell invasion/migration assay
Migration was analyzed by Transwell chamber assay
using cell culture inserts with a polycarbonate filter (24-wells,
8-µm pore size with polycarbonate membrane; Thermo Fisher
Scientific, Inc.). Invasion assay was performed by using the same
cell culture insert coated with growth factor-reduced Matrigel.
Coating was done with cold Matrigel followed by overnight
incubation at 37°C. HT-29 control siRNA and HT-29 MUCL1 siRNA (ML1
and 2) as well as SW620 control siRNA and SW620 MUCL1 siRNA (ML1
and 2) (1×105/well) were seeded onto Matrigel-coated
inserts in serum free RPMI-1640 media with lower chamber containing
complete RPMI-1640 media. The cells were allowed to invade for 48 h
at 37°C. Remaining cells above the insert membrane were cleared
with a cotton swab. The invasive cells were fixed in 25% methanol
for 15 min at room temperature followed by washing with cold 1X
PBS. The cells were stained with 0.1% crystal violet for 15 min at
room temperature followed by 2-3 times washing with 1X PBS and
air-dried. The invasive cells were counted on the representative
sections using an inverted light microscope (Micros Austria) at ×10
magnification. Counting was performed in five random fields in each
group; the number of invasive cells for each sample represents the
average of triplicate wells over three experiments. Similarly,
migration assay was carried out using normal cell culture
inserts.
Cell fractionation
Cells were harvested and washed with PBS and pellets
were incubated with 500 µl fractionation buffer (20 mM
HEPES, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT and
protease inhibitor) for 15 min on ice. Cell suspension was passed
through 1-ml syringe with 27-gauge needle 10 times and incubated
for 20 min on ice. The mixture was centrifuged 805 × g for 5 min at
4°C. The pellet contained nuclei; supernatant would contain
cytoplasm, membrane and mitochondria. This supernatant was further
centrifuged at 10,000 × g for 30 min at 4°C to obtain the cytosolic
fraction. Nuclear pellet was incubated with 500 µl of
fractionation buffer again for 15 min on ice and passed through a
25-gauge needle 10 times and centrifuged at 805 × g for 10 min at
4°C resulting in the nuclei-containing pellet. This pellet was
incubated with RIPA lysis buffer on ice for 15 min. The suspension
was sonicated briefly and centrifuged at 17,530 × g for 15 min at
4°C. The supernatant was nuclear fraction which was stored in -80°C
until further use.
Flow cytometric analysis of
apoptosis
Apoptosis (early and late) and necrosis were
determined as previously described (20). Briefly, HT-29 control siRNA and
HT-29 MUCL1 siRNA (ML1 and 2) as well as SW620 control siRNA and
SW620 MUCL1 siRNA (ML1 and 2) cells were plated in 6-well plate at
a density of 1×105/well and cultured for 24 h at 37°C.
The cells were treated with different concentrations (50 and 100
µM) of irinotecan (IRI) for 24 h at 37°C. The cells were
harvested along with media by centrifugation at 252 × g at 4°C.
After centrifugation, the cells were washed twice with cold PBS.
Annexin V/Dead cell apoptosis kit (cat. no. V13242; Thermo Fisher
Scientific, Inc.) was used according to the manufacturer's
instructions to detect cell death. The cells were resuspended in
binding buffer followed by addition of Annexin V-FITC (5 µl)
and 1 µl propidium iodide for 15 min in dark at room
temperature. The acquisition and analysis of data were performed
using a flow cytometer (BD FACSCalibur) and CellQuest Pro Ver 6.0
software (both from BD Biosciences).
Statistical analysis
All the experiments were performed in triplicate and
the results were expressed as the mean of three independent
experiments (mean ± standard deviation). Statistical analyses were
performed using GraphPad Prism 7.0 (GraphPad Software, Inc.).
Comparison between the control and multiple groups was performed
using one-way analysis of variance (ANOVA) followed by a Tukey's
post hoc test. In certain analyses, two-way ANOVA was performed
followed by Bonferroni post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
MUCL1 expression profile in CRC
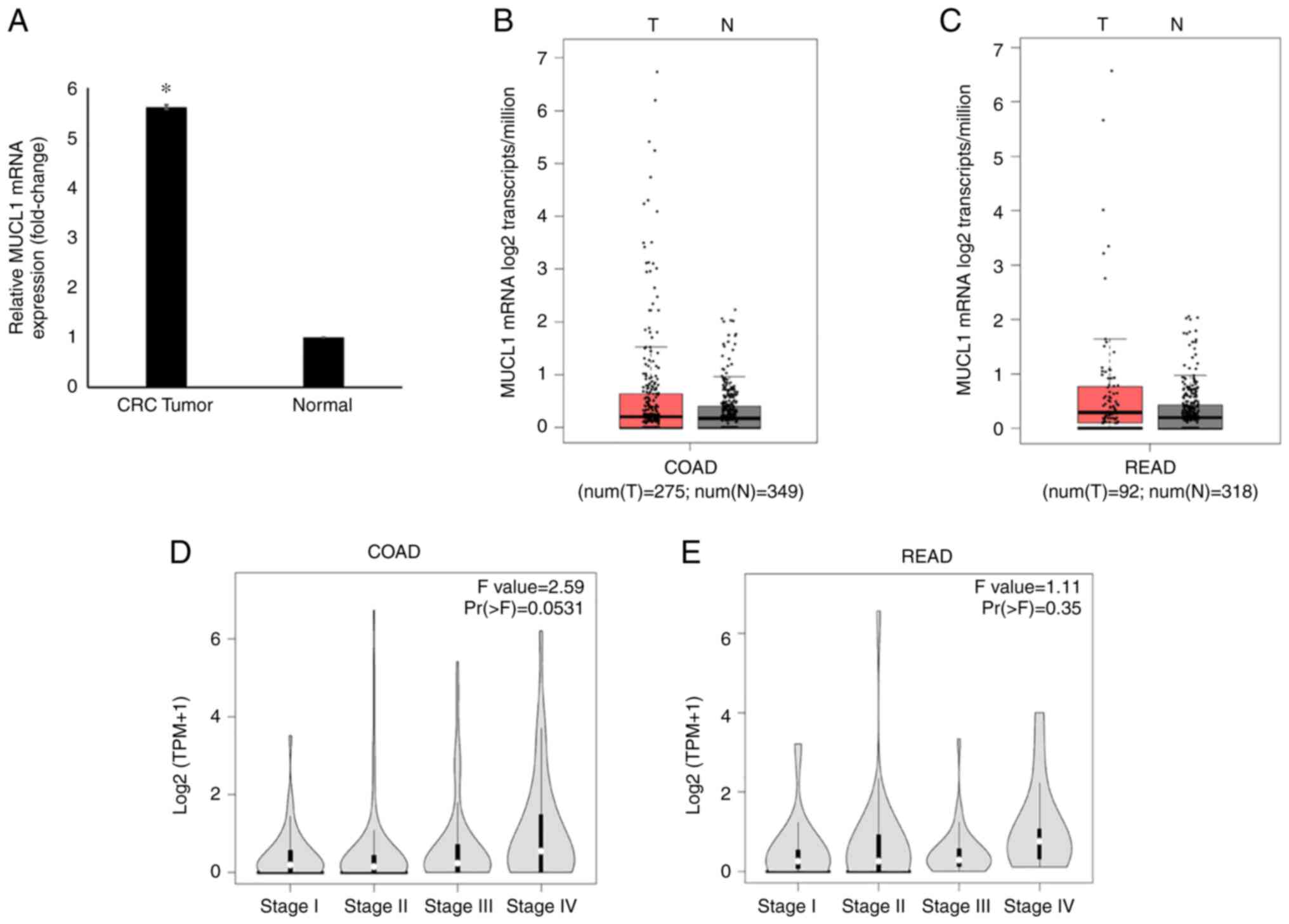
Global mRNA gene expression profiling revealed
significantly increased levels of MUCL1 mRNA in CRC tumor tissues
compared with adjacent normal tissues (Fig. 1A) (21). The MUCL1 gene expression in GEPIA
database was then analyzed. It was identified that MUCL1 expression
was upregulated in COAD (n=275) compared with normal tissue (n=349)
when analyzed in the TCGA/GTEx COAD dataset (Fig. 1B). Similarly, MUCL1 gene
expression was revealed to be higher in READ (n=92) when compared
with normal tissue (n=318) (Fig.
1C). Notably, MUCL1 expression was increased during CRC stage
IV compared with other stages when analyzed in COAD and READ
(Fig. 1D and E).
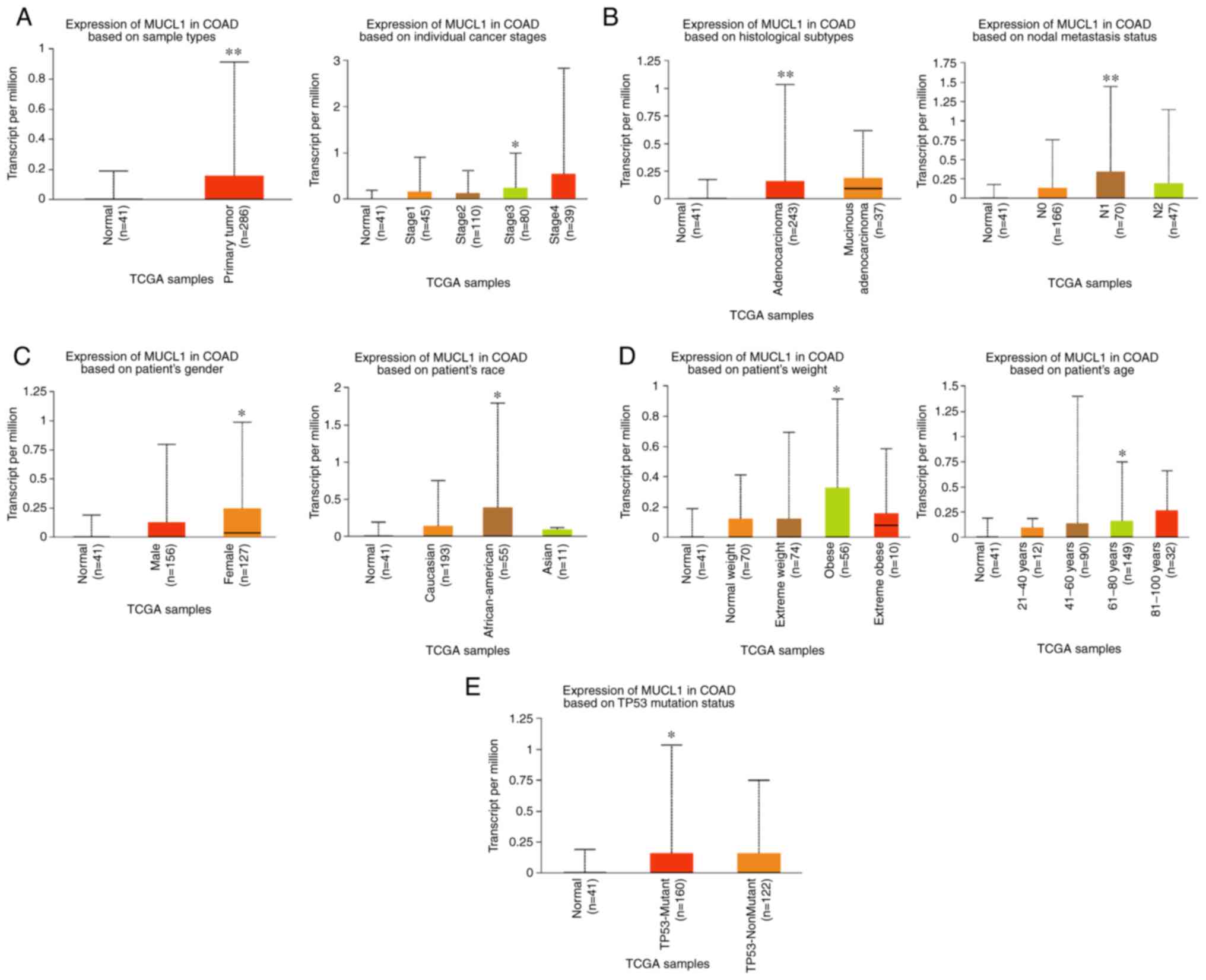
By exploring the UALCAN database, MUCL1 expression
was further analyzed in COAD. The results revealed the
significantly elevated levels of MUCL1 mRNA in primary COAD tumor
(n=286) compared with normal tissue (n=41). Stage-wise analysis of
COAD revealed that MUCL1 expression was significantly higher in
stage III as compared with normal tissue (Fig. 2A). Furthermore, MUCL1 expression
was significantly higher in adenocarcinoma compared with normal
tissue. Expression of MUCL1 was significantly elevated in N1
compared with normal (Fig. 2B).
Additionally, MUCL1 expression was significantly higher in women,
in patients of African-American decent, in obese patients, in 61-80
years old patients and in individuals bearing the TP53-mutant
(Fig. 2C-E). Thus, these findings
demonstrated that MUCL1 gene expression was higher in CRC.
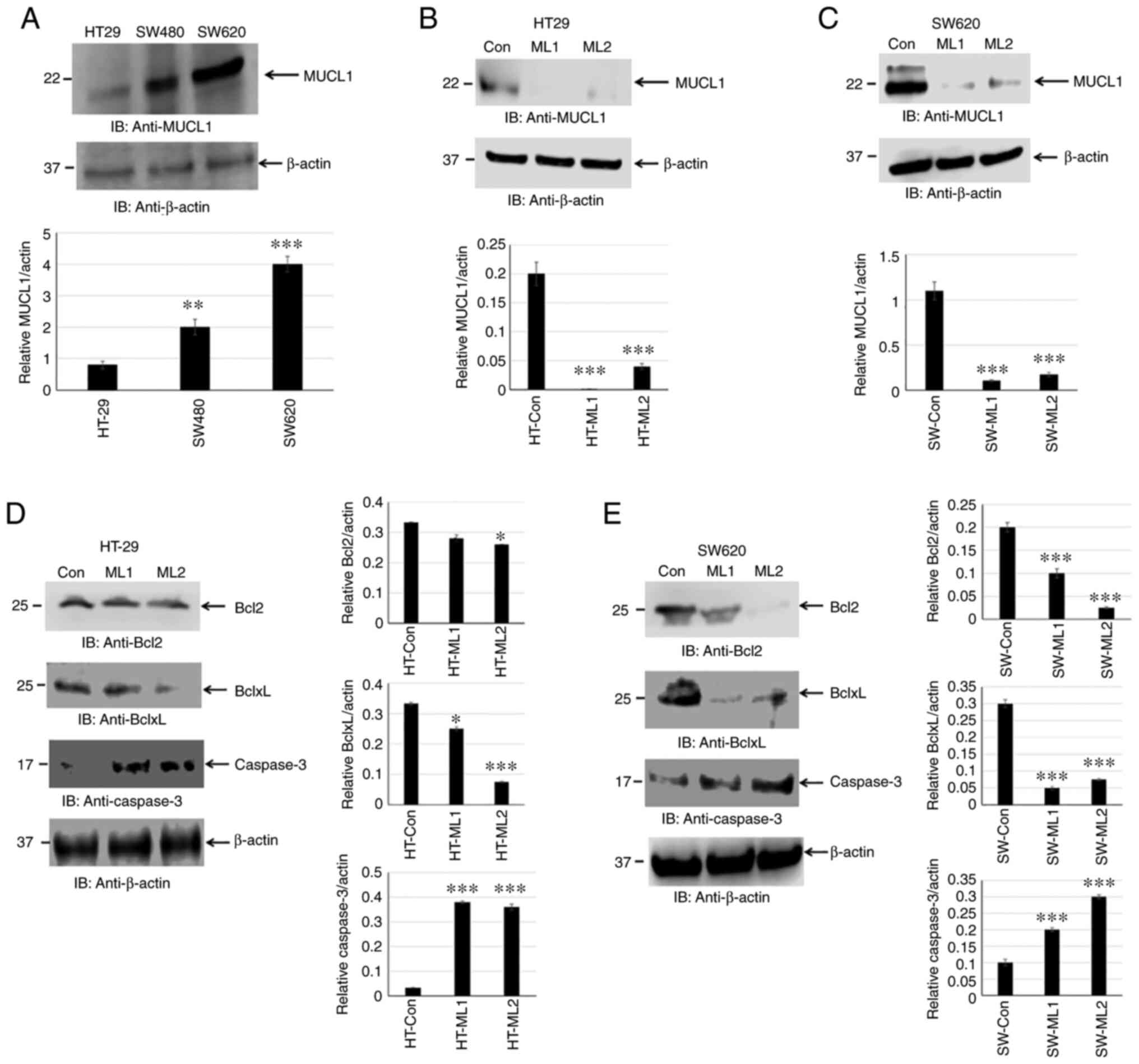
MUCL1 protein expression has not been reported in
human CRC cell lines. The present study attempted to determine
MUCL1 expression by western blotting. MUCL1 was identified to be
expressed in HT-29, SW480 and SW620 cells. MUCL1 expression was low
to moderate in adenocarcinoma cell lines (HT-29 and SW480) with
significantly higher expression observed in the metastatic CRC cell
line SW620 compared with HT-29 (Fig.
3A). This finding indicated that MUCL1 was expressed to a
varied degree in CRC cell lines. To understand the functional
significance of MUCL1 expression in CRC, MUCL1 was silenced in
HT-29 and SW620 cells using siRNA; two clones (designated as ML1
and 2) were selected for further experimentation. MUCL1 gene was
revealed to be inhibited with siRNA in both cell lines as shown by
western blotting (Fig. 3B and
C).
Inhibiting MUCL1 decreases Bcl2 family
protein and activates caspase-3
Cancer cell survival depends on a set of proteins
that regulate cell proliferation and block apoptosis. Bcl2 family
proteins consist of pro-apoptotic (Bax and Bak) and anti-apoptotic
(Bcl2, BclxL and Mcl1) proteins (22). The balance between pro-apoptotic
vs. anti-apoptotic proteins determines the cancer cell fate. MUCL1
silencing was found to significantly inhibit BclxL and, to certain
extent, Bcl2 in HT-29 cells. Both Bcl2 and BclxL were significantly
inhibited in SW620 cells (Fig. 3D and
E). In the apoptotic pathway, cytochrome c release into cytosol
forms a complex with Apaf1 and procaspase-9, which is called
apoptosome and results in the autoactivation of caspase-9 and
activation of downstream caspase-3 (23). It was investigated if there was
any caspase activation by measuring cleaved caspase-3. Indeed,
targeting MUCL1 by siRNA led to the significant increase in the
activation of caspase-3 (Fig. 3D and
E). This result demonstrated that targeting MUCL1 in CRC cells
resulted in the inhibition of Bcl2 and BclxL and further activation
of caspase-3, thereby confirming the significant role of MUCL1 in
controlling the apoptotic pathway.
MUCL1 promotes cell proliferation and
colony formation
To study the oncogenic potential of MUCL1 in CRC,
colony formation assay was performed in HT-29 control siRNA and two
MUCL1 siRNA clones. Inhibition of MUCL1 resulted in a significant
depletion in the number of colonies in both silenced clones
compared with the control (Fig.
4A). Similarly, the colony number was significantly decreased
in MUCL1-silenced SW620 cells compared with control siRNA cells
(Fig. 4B). To understand the
physiological role of MUCL1 in CRC, the cell proliferation was
evaluated at different time points. MUCL1-knockdown resulted in the
significant inhibition of cell proliferation in HT-29 cells
compared with control cells (Fig.
4C). A similar result was observed in the metastatic cell line
SW620 (Fig. 4D). These findings
thus indicated that MUCL1 expression is notable in mediating the
tumorigenesis in CRC.
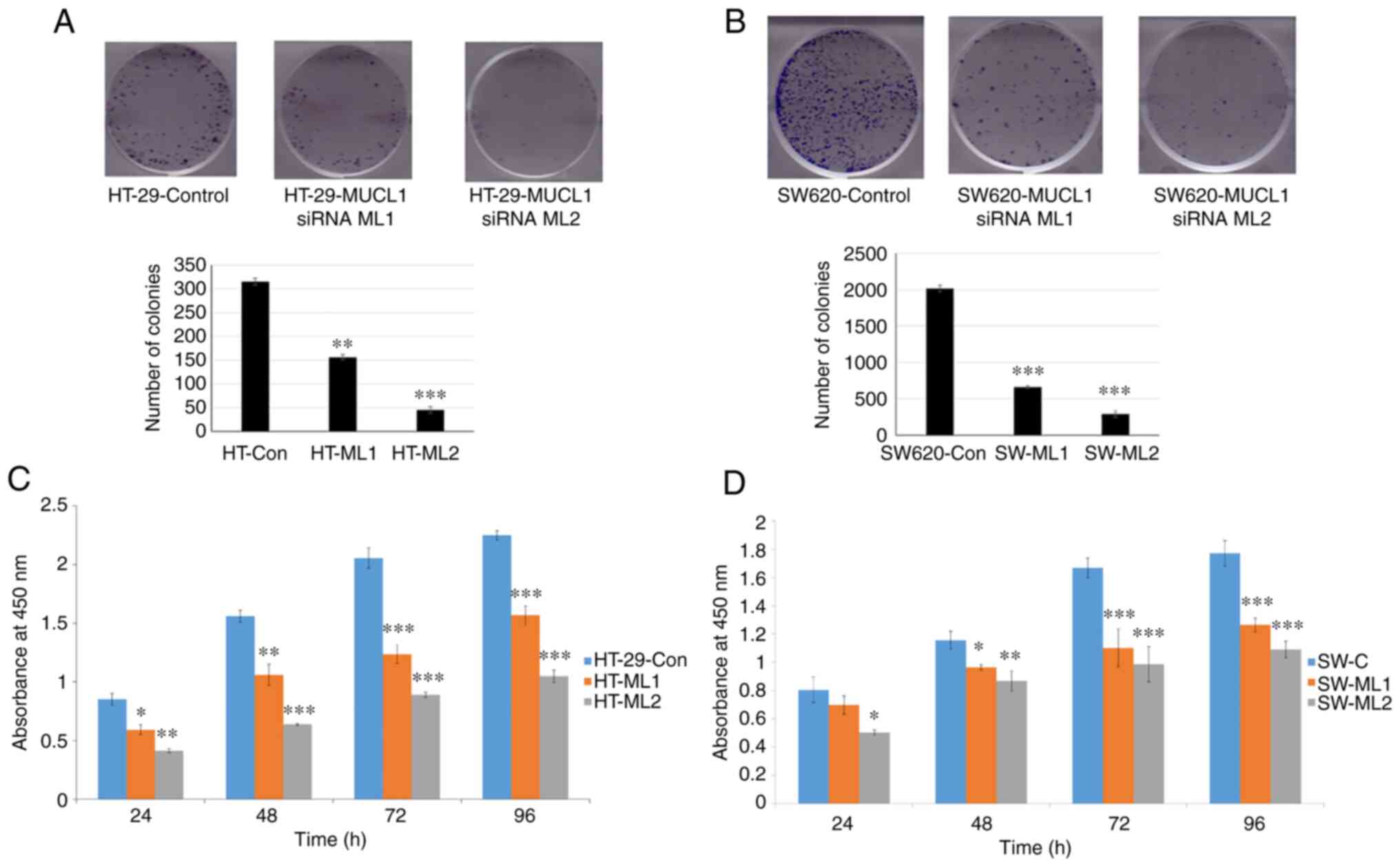 | Figure 4MUCL1 promotes cell proliferation.
(A) HT-29-Control siRNA and HT-29-MUCL1 siRNA clones ML1 and 2 and
(B) SW620-Control siRNA and SW620-MUCL1 siRNA clones ML1 and 2
cells were seeded (500/well) in 6-well plate and incubated at 37°C.
After 10-12 days of incubation crystal violet staining was
performed, colonies were quantified and images were captured by
Bio-Rad gel-doc system. (C) HT-29-Control siRNA and HT-29-MUCL1
siRNA clones ML1 and 2 were seeded at 5,000 cells/well in 96-well
plates and incubated at 37°C. Cell proliferation was determined by
Cell Counting Kit-8 assay on 24, 48, 72 and 96 h. (D) SW620-Control
siRNA and SW620-MUCL1siRNA clone ML1 and 2 cells were seeded at
5,000 cells/well in 96-well plate for incubation at 37°C. Cell
proliferation was measured by CCK-8 assay on 24, 48, 72 and 96 h.
The results are expressed as the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. control. MUCL1, mucin-like 1; si-,
small interfering; Con, control. |
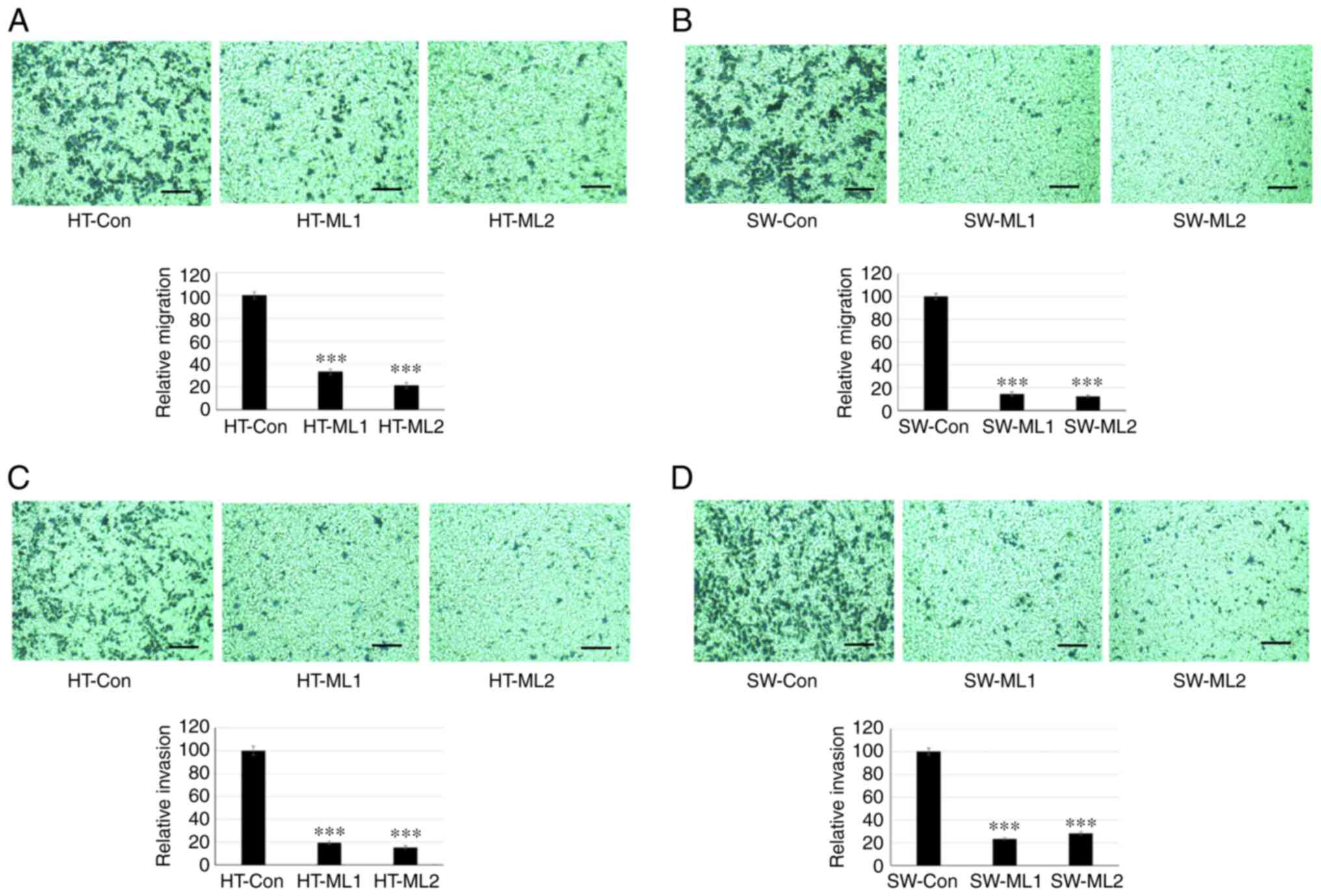
MUCL1 induces migration and invasion
Cancer metastasis is initiated by invasive behavior
of cancer cells and migration process (24). To study the role of MUCL1 in cell
invasion and migration, a Transwell assay was performed. The
results showed that HT-29 control cells migrated to the lower side
of the chambers after 48 h; however silencing MUCL1 significantly
inhibited the cell migration compared with control cells (Fig. 5A). Similarly, silencing MUCL1 in
SW620 cells also inhibited cell migration significantly compared
with control cells (Fig. 5B). The
quantification of migrating cells by crystal violet staining
demonstrated that MUCL1 silencing significantly inhibited the
relative migration ability of HT-29 and SW620 cells by >70 and
>80%, respectively (Fig. 5A and
B). Furthermore, cell invasion was evaluated using
Matrigel-coated inserts. As revealed in Fig. 5C, silencing MUCL1 significantly
inhibited the invasive potential of HT-29 compared with control
cells. SW620 cells with MUCL1 silencing also demonstrated
significant inhibition of cell invasion compared with control cells
(Fig. 5D). Invading cells were
quantified by crystal violet staining and it was revealed that
knockdown of MUCL1 significantly inhibited the relative invasive
ability of HT-29 and SW620 cells by ~80 and 75%, respectively
(Fig. 5C and D). These results
demonstrated that MUCL1 promotes cell migration and invasion in CRC
cells.
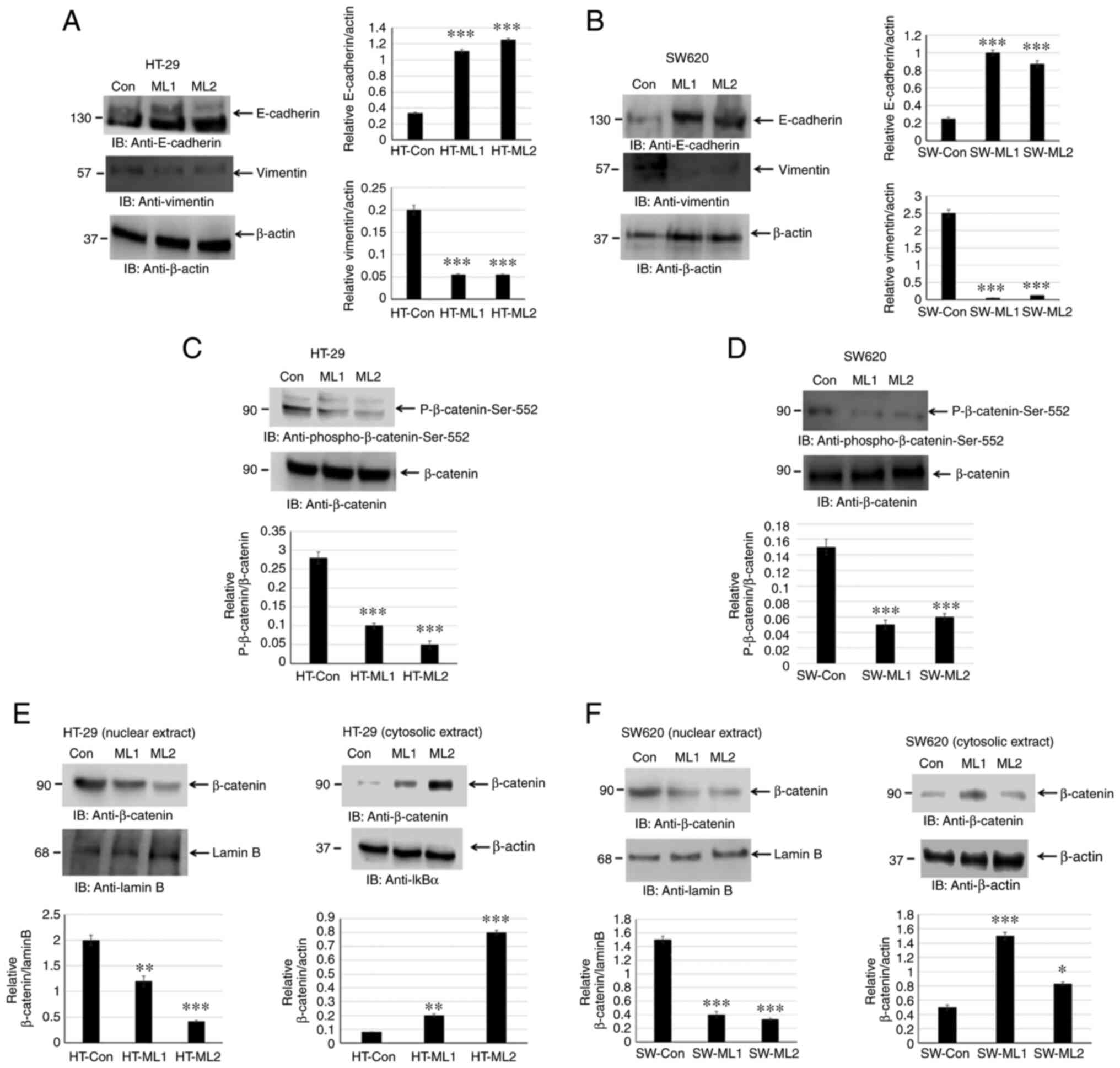
MUCL1 promotes EMT by inducing β-catenin
activation
EMT is associated with an invasive and metastatic
phenotype in CRC. EMT leads to downregulation of E-cadherin and
upregulation of N-cadherin and vimentin by modulating EMT-related
signaling pathways, for instance Wnt/β-catenin, TGF-β and ZEB1
(6-8). The present study investigated if
silencing MUCL1 in CRC leads to alteration in E-cadherin and
vimentin expression levels. Indeed, silencing MUCL1 in HT-29 cells
led to a significant upregulation of E-cadherin and downregulation
of vimentin (Fig. 6A). A similar
result was observed in SW620 MUCL1 siRNA cells (Fig. 6B). These findings thus indicated
that MUCL1 is of importance for EMT process and thereby targeting
MUCL1 may lead to potential therapeutics.
The activation of β-catenin results in EMT and is
directly associated with invasion and metastasis of various cancers
(8). The activation of β-catenin
in HT-29 control siRNA and MUCL1 siRNA cells was analyzed by
measuring β-catenin-Ser552 phosphorylation. Silencing MUCL1 in
HT-29 cells resulted in the significant downregulation of
β-catenin-Ser552 phosphorylation compared with control cells
(Fig. 6C). Similarly, silencing
MUCL1 in SW620 cells resulted in the depletion of phosphorylation
of β-catenin-Ser552 (Fig.
6D).
Translocation of β-catenin from the cytosol into the
nucleus is important for transcriptional activation (25). The present study sought to explore
the localization of β-catenin in nuclear and cytosolic fraction. It
was revealed that β-catenin was maintained in the cytosol during
normal homeostasis; however during transformation and tumor
progression, β-catenin translocated into the nucleus to activate
β-catenin responsive genes. β-catenin was found to be mainly
localized in the nucleus in HT-29 control cells; however inhibiting
MUCL1 resulted in the significant depletion of nuclear β-catenin
and significantly increased its cytosolic level (Fig. 6E). Similarly, in SW620 control
cells, β-catenin was mainly localized in nucleus and less in the
cytosol (Fig. 6F). MUCL1
knockdown resulted in the inhibition of nuclear levels and enhanced
β-catenin levels in the cytoplasm. Thus, these findings
demonstrated that MUCL1 regulates the β-catenin activation and
thereby EMT in CRC cells.
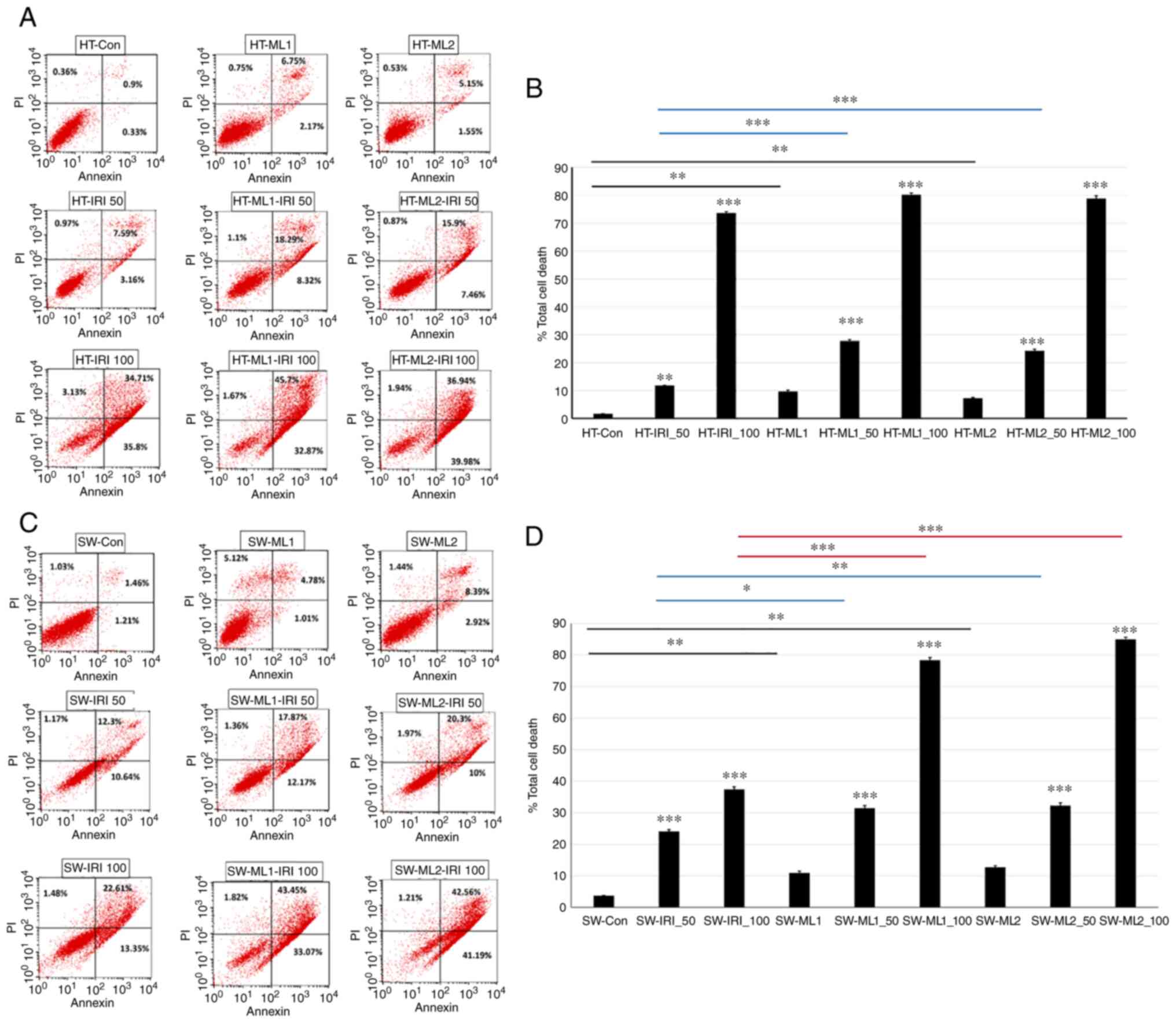
Silencing MUCL1 increases IRI
sensitivity
IRI is an integral part of CRC chemotherapy, but
response rates are not satisfactory and resistance mechanisms
remain unknown. To understand if MUCL1 regulates drug sensitivity
for CRC, HT-29 control and MUCL1 siRNA clones ML1 and 2 were
treated with IRI (50 and 100 µM) for 24 h and analyzed for
total cell death by flow cytometry. As revealed in Fig. 7A and B, HT-29 control cells
treated with 50 and 100 µM of IRI resulted in 11.72 and
73.6% total cell death respectively compared with untreated cells.
Similarly, treatment of HT-ML1 with 50 and 100 µM IRI
resulted in the significant increase in total cell death of 27.71
and 80.24% respectively compared with untreated HT-ML1 cells
(9.67%; Fig. 7B). Similar
increase was observed with HT-ML2 cells. Treatment of HT-ML2 cells
with 50 and 100 µM of IRI resulted in the significant
induction of total cell death by 24.23 and 78.86% compared with
untreated HT-ML2 cells (Fig. 7B).
Untreated HT-ML1 and HT-ML2 cells exhibited significant increase in
9.67 and 7.23% total cell death compared with HT-29 control cells
(1.6%). HT-ML1 and HT-ML2 cells treated with 50 µM of IRI
resulted in 27.71 and 24.23% total cell death which was
significantly higher compared with HT-29-Control siRNA treated
cells (50 IRI) (11.71%). Similarly, treatment of HT-ML1 and HT-ML2
cells with 100 µM IRI also showed mild increase in total
cell death of 80.24 and 78.86% compared with control cells
(HT-IRI_100; 73.6%). SW620 control cells treated with 50 and 100
µM of IRI were found to significantly increase in total cell
death of 24.11 and 37.44% compared with untreated cells (Fig. 7C). Treatment of SW620-ML1 cells
with IRI (50 and 100 µM) exhibited significant increase of
31.4 and 78.34% total cell death compared with untreated SW-ML1
control cells (10.91%). SW-ML2 cells treated with 50 and 100
µM of IRI were found to exhibit significant increase of
32.27 and 84.97% in total cell death compared with 12.75% for
untreated SW-ML2 control cells (Fig.
7D). SW-ML1 and SW-ML2 cells treated with 50 µM IRI
resulted in significant increase of 31.4 and 32.27% in total cell
death compared with SW620 control siRNA cells (treated with 50 IRI)
(24.11%). Treatment of SW-ML1 and SW-ML2 cells with 100 µM
of IRI resulted in significant increase in total cell death of
78.34 and 84.97%, respectively, compared with SW620 control siRNA
cells treated with 100 µM IRI (37.44%). Thus, these findings
demonstrated that silencing MUCL1 in HT-29 and SW620 cells resulted
in increased sensitivity towards IRI.
Discussion
Globally, CRC is the third most common detected
cancer and second leading cause of cancer-related mortality
according to GLOBOCAN 2020 (26).
According to the latest JAMA report, a total of 25% of patients
present with advanced localized disease that eventually develops
into metastasis and 20% of patients with CRC have metastasis at
diagnosis (27). Treatment for
unresectable metastatic CRC involves cytotoxic drugs (5FU,
Oxoplatinum and IRI), antibodies (cetuximab and panitumumab) and
immunotherapy in combination (27). MUCL1 is a small glycoprotein and
belongs to the mucin family of proteins. MUCL1 has been extensively
studied in breast cancer mostly as a diagnostic biomarker for
micrometastasis (10-13). The therapeutic role of MUCL1
targeting in breast cancer has been previously reported (17). Conley et al (28) reported that Her2 drives MUCL1
expression and regulates cell proliferation in breast cancer. MUCL1
mRNA showed high expression in stomach cancer (29). Aziz et al (30) reported that MUCL1 was upregulated
(1.17-fold) in matched-pair CRC tumor. Apart from breast cancer,
MUCL1 function has not been explored for any other cancer, to the
best of our knowledge.
In the present study, it was demonstrated that MUCL1
gene expression was upregulated in CRC-matched adjacent normal
tissues. This finding was confirmed by analyzing the MUCL1 gene
expression in CRC using GEPIA and UALCAN database and it was found
that its expression was significantly higher compared with normal
adjacent tissues. MUCL1 expression has not been reported in human
cancer cell lines with the exception of breast cancer. In the
present study, for the first time, it was revealed that MUCL1 is
highly expressed in human CRC cell lines. HT-29 cells expressed a
low amount of MUCL1 protein. MUCL1 exhibited moderate to high
expression in SW480 and higher expression in SW620 cells. MUCL1
expression varied from low in adenocarcinoma to high in metastatic
CRC cells.
To understand the potential oncogenic role of MUCL1
in CRC, MUCL1 was silenced in HT-29 and SW620 cells and functional
studies were performed. In the present study, MUCL1-silencing
inhibited cell proliferation and colony formation of CRC cell
lines. Knockdown of MUCL1 in CRC cell lines resulted in the
downregulation of antiapoptotic proteins Bcl2 and BclxL, confirming
its role in the regulation of cancer cell survival. Furthermore,
targeting MUCL1 showed activation of caspase-3, which indicated
that it plays an important role in apoptosis.
Dysregulation of the Bcl2 family proteins are of
prime significance for targeting CRC (31). Bcl2 and BclxL proteins are
important regulators of proliferation and apoptosis and have been
implicated in CRC initiation, progression and metastasis (31). Targeting MUCL1 protein provides a
therapeutic opportunity to inhibit the Bcl2-related pathway and
evaluate their potential for CRC treatment. Conley et al
(28) identified that MUCL1
knockdown led to cell cycle arrest by altering the cell cycle
inducers (Cyclin D1 and D3) and inhibitors (p21 and p27) and
proposed that MUCL1 interacts with focal adhesion kinase (FAK);
thereby leading to its activation and further activation of the
JNK-cJun pathway, resulting in modulation of the cell cycle. Cell
invasion and motility play a key role in cancer cell migration to
distant metastasis (6). MUCL1
silencing inhibited the migration ability of CRC cells.
MUCL1-silenced HT-29 and SW620 cells showed inhibition in the
number of invasive cells compared with control siRNA cells, which
exhibited significantly higher invasion ability. Li et al
(17) showed that knockdown of
MUCL1 in MCF7 and MDA-MB-231 inhibited the invasion and
migration.
EMT is a biological process by which epithelial
cells acquire mesenchymal characteristics (32). EMT plays an important role in
embryonic development, tissue fibrosis and tumor progression
(33). During tumor progression,
mesenchymal phenotype with malignant feature associates with
migration, invasion and metastasis (34). The loss of E-cadherin and
upregulation of N-cadherin/vimentin is considered as a key feature
of EMT (7). Therefore, blocking
the EMT process serves as an attractive target for cancer
therapeutics. In the present study, it was found that MUCL1
silencing inhibited EMT phenomenon, as evidenced by increased
E-cadherin and decreased vimentin expression levels. Consistent
with the aforementioned finding, it was previously reported that
knockdown of MUCL1 in breast cancer cells inhibits EMT (17). The mucin proteins, specifically
MUC1, have been shown to play a key role in tumorigenesis by
integrating EMT program (35).
The C-terminus of MUC1 induces EMT by activating NFκB pathway and
ZEB1, a well-known transcription suppressor of EMT in breast and
colon cancer (35-38). The Wnt pathway effector,
β-catenin, plays important roles in proliferation, survival and EMT
of CRC cells. Knockdown of MUCL1 in CRC cell lines resulted in the
inhibition of β-catenin activation, as shown by decreased
phosphorylation of Ser-552. Phosphorylation of β-catenin-Ser 552 by
AKT/PKA induces nuclear accumulation and transcription activation
(39,40). Cellular fractionation of control
and MUCL1 siRNA cells indicated that targeting MUCL1 resulted in
inhibition of nuclear β-catenin and increased its appearance in the
cytosol. This finding confirmed that MUCL1 regulates β-catenin and
thereby EMT.
A previous study has supported the critical role of
β-catenin in regulating EMT (41). The role of mucin in regulating
β-catenin has been extensively studied (9,42).
MUCL1 regulates the β-catenin in CRC. The results of the present
study showed that silencing MUCL1 inhibited phosphorylation of
β-catenin-Ser556 and depleted the nuclear accumulation. In the
absence of Wnt, GSK-3β and CK1 phosphorylate β-catenin (Ser-33,
-37, Thr-41) in the cytosol, leading to interaction with the
destruction complex (APC, GSK3β, CK1, Axin) (25). This destruction complex binds to
the E3 ubiquitin ligase (β-TrCP) through the β-catenin, which
enhances its ubiquitination leading to proteasomal degradation
(25). In cancer cells with Wnt
activation, the Wnt-Frizzled-Axin-LRP-5/6 complex sequesters
cytosolic GSK-3β, thereby blocking the phosphorylation of β-catenin
(25). This leads to accumulation
of non-phosphorylated β-catenin in the cytosol, which translocates
to the nucleus, binds to T cell-specific factor/lymphoid
enhancer-binding factor and co-activators on the promoter to
activate the target genes (c-Myc, cyclin D1 and Cdkn1a) (25). Other oncogenes and modifiers also
block upstream kinases (CK and GSK3β) leading to nuclear
translocation of β-catenin (25).
In the present study, MUCL1 expression increased the translocation
of β-catenin from cytosol into nucleus, and thereby increased
transcriptional activation of its target genes.
MUC1-C has been shown to bind directly to β-catenin
and regulate its transcription (43). MUC1, MUC4 and MUC16 have been
shown to regulate localization and transcriptional activation of
β-catenin (42,43). Similar to these mucins, the
present study demonstrated that MUCL1 influenced the nuclear
localization and stabilization of β-catenin. With known correlation
between MUC1 and the β-catenin pathway (43), MUCL1 may regulate β-catenin
directly or indirectly through upstream kinases. FAK could be one
such kinase that may activate β-catenin by interacting with MUCL1.
There is increasing evidence of a functional crosstalk between the
FAK and Wnt-β-catenin signaling pathways during cancer progression
(44,45). This may serve an interesting area
for future study to elucidate the direct link between
MUCL1-β-catenin.
Finally, the physiological significance of MUCL1 in
increasing sensitivity towards anticancer drug for CRC was
investigated. Tumor resistance is a frequent cause of chemotherapy
failure. New treatments are required to improve survival of CRC
patients specifically in IRI refractory patients. IRI (IRI/CPT-11)
in combination with 5-FU and the modulator leucovorin, has been
approved as first-line chemotherapy for patients with mCRC
(46,47). Knockdown of MUCL1 in CRC cell
lines increased the total cell death in response to IRI compared
with control siRNA cells. Interestingly, it was demonstrated that
IRI (50 µM) showed higher sensitivity in HT-29 cells
compared with 100 µM of IRI. In SW620 cells, both 50 and 100
µM of IRI displayed increased sensitivity in MUCL1-silenced
cells compared with the control. This dose difference in response
could be due to different mutation status in these cell lines and
cell origin. IRI resistance in CRC is attributed to Topo I
expression level, Topo I mutation, NFκB activation and ABC family
of drug transporters (48). Cdk1
inhibition enhanced IRI sensitivity in human CRC cells (HT-29)
(49). A previous study revealed
that low expression of ABCG2 showed increased sensitivity to IRI in
colorectal adenocarcinoma (50).
FGFR3 overexpression alters IRI-induced apoptosis in CRC (51). In consistency with these reports,
the results of the present study supported that MUCL1 is
overexpressed in CRC and that targeting MUCL1 enhanced IRI-mediated
apoptosis. In conclusion, MUCL1: i) acts as a modifier of other
pathways that drive proliferation and colony formation in CRC cell
lines; ii) is necessary for expression of the Bcl2 family protein;
iii) enhances invasion and migration; and iv) induces EMT and
β-catenin activation. Consistent with these results, targeting
MUCL1 enhances IRI sensitivity in CRC. Thus, MUCL1 alters other
pathways that are important for CRC progression, and MUCL1 may act
as a potential target for CRC therapeutics.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
RA and MA conceptualized the idea, designed and
performed the experiments and wrote the manuscript. MAVM, MSEW, NA
and TBT performed experiments. KAK, AZ and OAO analyzed the data.
All authors have read and approved the final version of the
manuscript. RA and MA confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The present study was supported by the Deanship of
Scientific Research, King Saud University, through the Vice
Deanship of Scientific Research Chairs.
Funding
No funding was received.
References
|
1
|
Ahmad R, Singh JK, Wunnava A, Al-Obeed O,
Abdulla M and Srivastava SK: Emerging trends in colorectal cancer:
Dysregulated signaling pathways (Review). Int J Mol Med. 47:142021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alsanea N, Abduljabbar AS, Alhomoud S,
Ashari LH, Hibbert D and Bazarbashi S: Colorectal cancer in Saudi
Arabia: Incidence, survival, demographics and implications for
national policies. Ann Saudi Med. 35:196–202. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bazarbashi S, Al Eid H and Minguet J:
Cancer Incidence in Saudi Arabia: 2012 Data from the Saudi cancer
registry. Asian Pac J Cancer Prev. 18:2437–2444. 2017.PubMed/NCBI
|
|
4
|
Riihimäki M, Hemminki A, Sundquist J and
Hemminki K: Patterns of metastasis in colon and rectal cancer. Sci
Rep. 6:297652016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andersen NN and Jess T: Has the risk of
colorectal cancer in inflammatory bowel disease decreased? World J
Gastroenterol. 19:7561–7568. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu Y, Zheng Y, Dai M, Wu J, Yu B, Zhang H,
Kong W, Wu H and Yu X: Snail2 induced E-cadherin suppression and
metastasis in lung carcinoma facilitated by G9a and HDACs. Cell Adh
Migr. 13:285–292. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaszak I, Witkowska-Piłaszewicz O,
Niewiadomska Z, Dworecka-Kaszak B, Ngosa Toka F and Jurka P: Role
of cadherins in cancer-a review. Int J Mol Sci. 21:76242020.
View Article : Google Scholar
|
|
8
|
Kim WK, Kwon Y, Jang M, Park M, Kim J, Cho
S, Jang DG, Lee WB, Jung SH, Choi HJ, et al: β-catenin activation
down-regulates cell-cell junction-related genes and induces
epithelial-to-mesenchymal transition in colorectal cancers. Sci
Rep. 9:184402019. View Article : Google Scholar
|
|
9
|
Kufe DW: Mucins in cancer: Function,
prognosis and therapy. Nat Rev Cancer. 9:874–885. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Colpitts TL, Billing P, Granados E, Hayden
M, Hodges S, Roberts L, Russell J, Friedman P and Stroupe S:
Identification and immunohistochemical characterization of a
mucin-like glycoprotein expressed in early stage breast carcinoma.
Tumour Biol. 23:263–278. 2002. View Article : Google Scholar
|
|
11
|
Miksicek RJ, Myal Y, Watson PH, Walker C,
Murphy LC and Leygue E: Identification of a novel breast- and
salivary gland-specific, mucin-like gene strongly expressed in
normal and tumor human mammary epithelium. Cancer Res.
62:2736–2740. 2002.PubMed/NCBI
|
|
12
|
Skliris GP, Hubé F, Gheorghiu I, Mutawe
MM, Penner C, Watson PH, Murphy LC, Leygue E and Myal Y: Expression
of small breast epithelial mucin (SBEM) protein in tissue
microarrays (TMAs) of primary invasive breast cancers.
Histopathology. 52:355–369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ayerbes MV, Diaz-Prado S, Ayude D, Campelo
RG, Iglesias P, Haz M, Medina V, Gallegos I and Quindós M: In
silico and in vitro analysis of small breast epithelial mucin as a
marker for bone marrow micrometastasis in breast cancer. Adv Exp
Med Biol. 617:331–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Valladares-Ayerbes M, Iglesias-Diaz P,
Diaz-Prado S, Ayude D, Medina V, Haz M, Reboredo M, Antolín S,
Calvo L and Antón-Aparicio LM: Diagnostic accuracy of small breast
epithelial mucin mRNA as a marker for bone marrow micrometastasis
in breast cancer: A pilot study. J Cancer Res Clin Oncol.
135:1185–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weigelt B, Verduijn P, Bosma AJ, Rutgers
EJ, Peterse HL and van't Veer LJ: Detection of metastases in
sentinel lymph nodes of breast cancer patients by multiple mRNA
markers. Br J Cancer. 90:1531–1537. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu ZZ, Xie XD, Qu SX, Zheng ZD and Wang
YK: Small breast epithelial mucin (SBEM) has the potential to be a
marker for predicting hematogenous micrometastasis and response to
neoadjuvant chemotherapy in breast cancer. Clin Exp Metastasis.
27:251–259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li QH, Liu ZZ, Ge YN, Liu X, Xie XD, Zheng
ZD, Ma YH and Liu B: Small breast epithelial mucin promotes the
invasion and metastasis of breast cancer cells via promoting
epithelial-to-mesenchymal transition. Oncol Rep. 44:509–518. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Khayal K, Vaali-Mohammed MA, Elwatidy
M, Bin Traiki T, Al-Obeed O, Azam M, Khan Z, Abdulla M and Ahmad R:
A novel coordination complex of platinum (PT) induces cell death in
colorectal cancer by altering redox balance and modulating MAPK
pathway. BMC Cancer. 20:6852020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vishnubalaji R, Hamam R, Abdulla MH,
Mohammed MA, Kassem M, Al-Obeed O, Aldahmash A and Alajez NM:
Genome-wide mRNA and miRNA expression profiling reveal multiple
regulatory networks in colorectal cancer. Cell Death Dis.
6:e16142015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hardwick JM and Soane L: Multiple
functions of Bcl2 family proteins. Cold Spring Harb Prospect Biol.
5:a0087222013.
|
|
23
|
Baig S, Seevasant I, Mohamad J, Mukheem A,
Huri HZ and Kamarul T: Potential of apoptotic pathway-targeted
cancer therapeutic research: Where do we stand? Cell Death Dis.
7:e20582016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Sig Transduct Target Ther. 5:282020. View Article : Google Scholar
|
|
25
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Biller LH and Schrag D: Diagnosis and
treatment of metastatic colorectal cancer: A review. JAMA.
325:669–685. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Conley SJ, Bosco EE, Tice DA,
Hollingsworth RE, Herbst R and Xiao Z: HER2 drives Mucin-like 1 to
control proliferation in breast cancer cells. Oncogene.
35:4225–4234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
King RJ, Yu F and Singh PK: Genomic
alterations in mucins across cancers. Oncotarget. 8:67152–67168.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aziz MA, AlOtaibi M, AlAbdulrahman A,
AlDrees M and AlAbdulkarim I: Mucin family genes are downregulated
in colorectal cancer patients. J Carcinogene Mutagene S10:.
009:2014.
|
|
31
|
Ramesh P and Medema JP: BCL-2 family
deregulation in colorectal cancer: Potential for BH3 mimetics in
therapy. Apoptosis. 25:305–320. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rajabi H, Alam M, Takahashi H, Kharbanda
A, Guha M, Ahmad R and Kufe D: MUC1-C oncoprotein activates the
ZEB1/miR-200c regulatory loop and epithelial-mesenchymal
transition. Oncogene. 33:1680–1689. 2014. View Article : Google Scholar
|
|
36
|
Takahashi H, Jin C, Rajabi H, Pitroda S,
Alam M, Ahmad R, Raina D, Hasegawa M, Suzuki Y, Tagde A, et al:
MUC1-C activates the TAK1 inflammatory pathway in colon cancer.
Oncogene. 34:5187–5197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahmad R, Raina D, Trivedi V, Ren J, Rajabi
H, Kharbanda S and Kufe D: MUC1 oncoprotein activates the IkappaB
kinase beta complex and constitutive NF-kappaB signalling. Nat Cell
Biol. 9:1419–1427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ahmad R, Raina D, Joshi MD, Kawano T,
Kharbanda S and Kufe D: MUC1-C oncoprotein functions as a direct
activator of the nuclear factor-kappaB p65 transcription factor.
Cancer Res. 69:7013–7021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fang D, Hawke D, Zheng Y, Xia Y,
Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T and Lu Z:
Phosphorylation of beta-catenin by AKT promotes beta-catenin
transcriptional activity. J Biol Chem. 282:11221–11229. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sánchez-Tilló E, de Barrios O, Siles L,
Cuatrecasas M, Castells A and Postigo A: β-catenin/TCF4 complex
induces the epithelial-to-mesenchymal transition (EMT)-activator
ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA.
108:19204–19209. 2011. View Article : Google Scholar
|
|
42
|
Pai P, Rachagani S, Dhawan P and Batra SK:
Mucins and Wnt/β-catenin signaling in gastrointestinal cancers: An
unholy nexus. Carcinogenesis. 37:223–232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang L, Chen D, Liu D, Yin L, Kharbanda S
and Kufe D: MUC1 oncoprotein blocks glycogen synthase kinase
3beta-mediated phosphorylation and degradation of beta-catenin.
Cancer Res. 65:10413–10422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wörthmüller J and Rüegg C: The crosstalk
between FAK and Wnt signaling pathway in cancer and its therapeutic
implication. Int J Mol Sci. 21:91072020. View Article : Google Scholar
|
|
45
|
Gao C, Chen G, Kuan SF, Zhang DH,
Schlaepfer DD and Hu J: FAK/PYK2 promotes the Wnt/β-catenin pathway
and intestinal tumorigenesis by phosphorylating GSK3β. Elife.
4:e100722015. View Article : Google Scholar
|
|
46
|
Rougier P and Mitry E: Review of the role
of CPT-11 in the treatment of colorectal cancer. Clin Colorectal
Cancer. 1:87–94. 2001. View Article : Google Scholar
|
|
47
|
Vanhoefer U, Harstrick A, Achterrath W,
Cao S, Seeber S and Rustum YM: Irinotecan in the treatment of
colorectal cancer: Clinical overview. J Clin Oncol. 19:1501–1518.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu Y and Villalona-Calero MA: Irinotecan:
Mechanisms of tumor resistance and novel strategies for modulating
its activity. Ann Oncol. 13:1841–1851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Abal M, Bras-Goncalves R, Judde JG, Fsihi
H, De Cremoux P, Louvard D, Magdelenat H, Robine S and Poupon MF:
Enhanced sensitivity to irinotecan by Cdk1 inhibition in the
p53-deficient HT29 human colon cancer cell line. Oncogene.
23:1737–1744. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tuy HD, Shiomi H, Mukaisho KI, Naka S,
Shimizu T, Sonoda H, Mekata E, Endo Y, Kurumi Y, Sugihara H, et al:
ABCG2 expression in colorectal adenocarcinomas may predict
resistance to irinotecan. Oncol Lett. 12:2752–2760. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Erdem ZN, Schwarz S, Drev D, Heinzle C,
Reti A, Heffeter P, Hudec X, Holzmann K, Grasl-Kraupp B, Berger W,
et al: Irinotecan upregulates fibroblast growth factor receptor 3
expression in colorectal cancer cells, which mitigates irinotecan
induced apoptosis. Transl Oncol. 10:332–339. 2017. View Article : Google Scholar : PubMed/NCBI
|