Introduction
Melanoma is the most aggressive and debilitating
form of skin cancer, as it progresses quickly and readily
metastasizes (1). Moreover, the
survival rate of patients with melanoma is markedly decreased as
the condition progresses to more advanced stages, as the five-year
survival rate in patients with localized tumors is >98%, but
this survival rate decreases to <27% once tumors metastasize
(2). Thus, prevention and early
detection of melanoma are key for optimal patient outcomes. Within
the last decade, the development of novel treatment options have
significantly improved the survival rate of patients with early to
mid-stage melanoma (3–5). Smaller, yet significant improvements
were also seen in patients with advanced-stage disease; however,
the overall survival rate remains low in this group (4,5).
Therefore, due to the increased incidence of new cases diagnosed
each year, the ongoing levels of mortality due to melanoma in both
men and women, and the continued difficulty to treat patients with
treat late-stage melanoma, further development of novel treatment
options is required (2).
Disadvantages of current treatment options and the
emergence of drug resistance have contributed to poor outcomes in
patients with cancer (6,7). One current treatment option for
melanoma is conventional chemotherapy. However, it is not a
first-line treatment option and can cause debilitating adverse
effects, such as hepatotoxicity and myelosuppression (8). Both immunotherapy and targeted therapy
have improved treatment outcomes with greater efficacy and
tolerability in patients with melanoma, when compared with
conventional chemotherapy (5,9,10).
However, limitations include a lack of response by numerous
patients with melanoma, the inability to consistently treat
patients with late-stage melanoma, and the cost of immunotherapy
and targeted therapy treatment regimens, particularly if
combination therapies are utilized (11–13).
Moreover, levels of drug resistance contribute to poor treatment
outcomes, as patients fail to adequately respond to the current
available treatment options. This is due, in part, to cellular
mechanisms that allow the cancer cell to overcome or avoid the
anti-tumor effects of treatment. Such mechanisms include the
evasion of cell death due to the ability to inhibit pro-apoptotic
signaling, or the decreased expression of pro-cell death mediators,
such as death receptors and apoptotic protease activating factor-1
(14,15). Another form of resistance in cancer
cells is known as multidrug resistance (MDR). This involves the
expression of drug-resistance proteins, which allows cancer cells
to avoid exposure to cancer drugs (6,16–18).
Three well-known drug resistance proteins are
phosphorylated-glycoprotein 1 (MDR1) (19), multidrug resistance-associated
protein 1 (MRP1) (20) and lung
resistance-related protein (LRP) (21). These enable MDR in cancer cells,
where they serve as specialized efflux pumps that expel
chemotherapeutic drugs from cancer cells and thereby allow tumors
to avoid the associated toxic effects (22). By doing so, MDR proteins promote the
survival of cancer cells following treatment. Thus, it is paramount
that novel and efficient treatment options are made available for
patients with melanoma, including those that are more economical
and accessible, and those that can overcome drug resistance.
The transient receptor potential (TRP) channels
represent a superfamily of non-specific ion channels with numerous
functions in healthy cells. They reside on the plasma membrane and
through their ability to serve as ionophores, TRP channels exhibit
numerous functions, including facilitation of vision and taste
(23,24), detection of heat and pain sensations
(24), enabling contraction of
smooth muscle and migration of squamous cells (25,26),
and the facilitation of cell death (27). Several types of TRP channels have
been studied to determine their potential roles in cancer. One such
channel is the second member of the TRP melastatin sub-family, TRP
melastatin-2 (TRPM2) channel. TRPM2 is a calcium-permeable
non-selective ion channel that is activated in response to
oxidative stress (28). Activation
of TRPM2 channels promotes a number of processes, including
microglial activation (29), immune
cell activation (30) and cell
death (27). It has previously been
reported that antagonism of TRPM2 channels in healthy cells was
beneficial following oxidative injury, leading to increased cell
survival (31,32). However, a paradoxical effect was
observed in several cancer types, where TRPM2 antagonism led to
decreased proliferation and increased cell death. These anti-tumor
effects were observed in numerous cancers, such as breast, prostate
and oral cancer (33–36).
The purpose of the present study was to determine
the effects of TRPM2 antagonism in primary human malignant melanoma
cells by analyzing the effects on proliferation and cell death.
Moreover, the present study aimed to further characterize TRPM2
channels in these cells, in order to identify the potential
differential role of TRPM2 channels in melanoma cells.
Materials and methods
Chemicals
(2E)-N-(2,3-Dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide
(AMG 9810), was prepared as a 1 mM stock solution in dimethyl
sulfoxide (DMSO) for each experiment, and was purchased from Tocris
Bioscience.
N-(3-Aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide
hydrochloride (AMTB; 5 mM stock solution in water) was purchased
from Santa Cruz Biotechnology, Inc. Clotrimazole (25 mM in DMSO)
and temozolomide (TMZ; 10 mM in DMSO) were purchased from
Sigma-Aldrich; Merck KGaA. ApoScreen Propidium Iodide (PI) and
ApoScreen annexin V-fluorescein isothiocyanate (FITC) solutions
were purchased from SouthernBiotech.
Cell lines and cell culture
reagents
The primary human malignant melanoma cell line,
SK-Mel-23, was obtained from the Memorial Sloan-Kettering Cancer
Center. This cell line was previously established from a metastatic
lymph node, the tissue of origin was a rectal tumor, and the
genotype profile is wild-type, with respect to the GTPase N-RAS
(N-RAS) and serine/threonine-protein kinase B-RAF (B-RAF) genes
(37). The primary human malignant
melanoma cell lines, UKRV-Mel-4 and UKRV-Mel-5 were both obtained
from the European Searchable Tumor Line Database, directed by Dr
Graham Pawalec at the University of Tübingen (Germany) (38). The UKRV-Mel-4 cell line was
previously established from a liver metastasis and is wild-type for
both N-RAS and B-RAF (39,40). The UKRV-Mel-5 cell line was
previously established from a subcutaneous lesion and is also
wild-type for N-RAS and B-RAF (40). All three melanoma cell lines were
cultured in RPMI 1640 medium (GenDEPOT, LLC), supplemented with 10%
fetal bovine serum (FBS; characterized; HyClone; Cytiva), 2 mM
L-glutamine (Corning Inc.) and 100 U/ml penicillin/streptomycin
(HyClone; Cytiva) at 37°C in 5% CO2. The human
keratinocyte cell line, HaCat, was purchased from AddexBio
Technologies (cat. no. T0020001). This cell line was authenticated
by the manufacturer using DNA STR profiling and the full statement
of authentication can be accessed online (https://www.addexbio.com/productdetail?pid=117).
The human embryonic kidney cell line, 293T, was purchased from the
American Type Culture Collection (cat. no. CRL-3216). Both the
HaCat and 293T cell lines were cultured in DMEM medium (GenDEPOT,
LLC) supplemented with 10% FBS and 2 mM L-glutamine at 37°C in 5%
CO2.
Reagents
OptiMEM reduced serum medium and
Lipofectamine® 2000 reagent were purchased from
Invitrogen (Thermo Fisher Scientific, Inc.). Protease inhibitor
cocktail tablets (Complete Mini, EDTA-free) were purchased from
Roche Diagnostics, GmbH. Primary antibodies utilized were
polyclonal rabbit anti-human TRPM2 (cat. no. A300-414A; Bethyl
Laboratories, Inc.), polyclonal rabbit anti-human β-actin (cat. no.
600-401-886; Rockland Immunochemicals Inc.), monoclonal mouse
anti-human β-actin, clone AC-74 (cat. no. A5316; Sigma-Aldrich;
Merck KGaA), polyclonal rabbit anti-human manganese superoxide
dismutase (MnSOD; cat. no. 06–984; MilliporeSigma), monoclonal
mouse anti-human lamin B2, clone LN43 (cat. no. MA1-06104; Thermo
Fisher Scientific, Inc.), polyclonal rabbit anti-human MDR1 (cat.
no. 13342S; Cell Signaling Technology, Inc.), polyclonal rabbit
anti-human MRP1 (cat. no. 14685S; Cell Signaling Technology, Inc.)
and monoclonal mouse anti-human LRP, clone 1014 (cat. no. sc-23916;
Santa Cruz Biotechnology, Inc). Two secondary antibodies,
peroxidase-AffiniPure goat anti-rabbit IgG, Fc fragment specific
(cat. no. 111-035-008) and peroxidase-AffiniPure rabbit anti-mouse
IgG, Fc (γ) fragment specific (cat. no. 315-035-008) were purchased
from Jackson ImmunoResearch Laboratories, Inc.
Cell culture
All cells were grown and maintained in RPMI 1640 or
DMEM supplemented with 10% FBS, L-glutamine and 100 U/ml
penicillin/100 µg/ml streptomycin, and incubated at 37°C in 5%
CO2 for further experiments, as previously described.
For transfection experiments, no antibiotics were utilized. Every 2
days in culture, cells were washed once with PBS (pH 7.2) and
cultured in fresh growth medium. Cells were subcultured or
harvested using 0.25% trypsin/0.02% EDTA (Quality Biological,
Inc.).
Cell proliferation analyses
Cells were seeded in triplicate in 24-well cell
culture plates in growth medium at a minimum seeding density of
1×105 cells/ml. Cells were incubated at 37°C overnight
and subsequently treated with 5–50 µM clotrimazole, 1 µM AMG 9810
or 5 µM AMTB. Growth medium and treatments were replaced every 48
h. At time points 0, 1, 2, 3 and 6 days, growth medium was removed
and cells were harvested using trypsinization. Cell numbers were
then determined using a hemocytometer. All counts were performed in
triplicate and all experiments were performed at least twice with
similar results.
Cell death assays
For the quantification of cell death using flow
cytometry, cells were treated with clotrimazole or TRPM2
small-interfering (si)RNA, harvested by trypsinization, washed with
PBS and resuspended in annexin-binding buffer (10 mM HEPES; pH 7.4;
140 mM NaCl; 2.5 mM CaCl2) to ~1×106
cells/ml. For each 100 µl of cell suspension, 4 µl ApoScreen
annexin V-FITC conjugate solution and 2 µl 100 µg/ml ApoScreen
propidium iodide (PI) solution was added. Cells were subsequently
maintained at room temperature for 15 min in the dark. Following
this, 0.4 ml annexin-binding buffer (SouthernBiotech) was added and
cell death was measured by quantifying the percentage of cells that
exhibited annexin V-FITC (FL1 channel) and PI (FL3) fluorescence
using a flow cytometer (BD Accuri model C6; BD Biosciences). Flow
cytometry analysis was performed using BD Accuri C6 software
(version 1.0.264.2; BD Biosciences). Total cell death was
quantified by adding the percentage of cells detected in the upper
left (PI), upper right (PI + annexin V-FITC) and lower right
(annexin V-FITC) quadrants in the flow cytometry dot plots. A
minimum of 1×105 cells was analyzed for each cell death
determination and all experiments were performed in triplicate.
For quantification of cell death using a WST-1
assay, cells were seeded in 96-well cell culture plates at a
density of 50,000 cells/ml and incubated at 37°C. The following
day, cells were treated with 5–50 µM clotrimazole for 2 days.
Following treatment, cell death was determined using the CytoScan
WST-1 Cell Cytotoxicity Assay (Geno Technology, Inc.). This is a
colorimetric assay based upon the reduction of WST-1, a tetrazolium
salt, by cellular dehydrogenases (41). The product, formazan, is detected at
440 nm and any decreased dehydrogenase activity is directly
associated with the amount of cell death following treatment. The
assay was initiated by the addition of 10 µl WST-1/CEC assay dye to
each well and the reaction was carried out for 30 min at 30°C.
Detection was performed using a BioTek Synergy HT microplate reader
using Gen5 software (version 1.05.11). All data points were assayed
in triplicate and all experiments were performed twice with similar
results.
Transfection
The silencing of TRPM2 using transfection was
performed as previously described, using siRNA specific to
nucleotides 4574–4594 (5′-AUAGAUCAGGAACUCCGUCUC-3′) of human TRPM2
(33). This RNA oligo was purchased
as duplexed RNA (Integrated DNA Technologies, Inc.), resuspended in
RNase-free water at a final concentration of 40 µM, and stored at
−20°C. The following non-targeting oligo primers: Forward,
5′AGACAGAAGACAGAUAGGCtt-3′ (sense) and reverse, 5′
GCCUAUCUGUCUUCUGUCUtt-3′ (antisense) were used for all negative
controls (Integrated DNA Technologies, Inc.). For transfection,
cells were plated in 24-well plates containing 0.5 ml medium per
well without antibiotics. Transfections were performed when cells
reached 40–60% confluence. The transfection protocol was initiated
by adding duplex siRNA to 50 µl OptiMEM, and adding 1 µl
Lipofectamine® 2000 to 50 µl OptiMEM in a separate tube.
The two solutions were mixed and maintained at room temperature for
20 min. The final concentration was 100 nM for both TRPM2 siRNA and
the corresponding negative control. The mixtures were added to each
well in drops, mixed and the cells were cultured for an additional
48 h at 37°C. Western blotting and cell death analyses were
subsequently performed 48 h after transfections.
Whole cell lysate extraction
Transfected cells were harvested using
trypsinization, centrifuged at 112 × g for 5 min at 4°C, and washed
once with 0.2 ml ice-cold PBS. The pellet was resuspended in 0.2 ml
lysis buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM
EDTA, 1 mM EGTA, 1% NP-40 and protease inhibitors. Suspensions were
placed on ice for 30 min and vortexed every 10 min. The cell
lysates were cleared by centrifugation at 16,000 × g for 10 min at
4°C and the protein concentration for all sample lysates was
immediately obtained using the Pierce BCA Protein Assay kit (Thermo
Fisher Scientific, Inc.). Subsequently, 10X SDS-PAGE sample buffer
[500 mM Tris-HCl (pH 6.8), 10% SDS, 25% glycerol, 0.05% bromophenol
blue and 100 mM dithiothreitol] was added to the supernatants to
reach a final concentration of 1X (50 mM Tris-HCl, pH, 6.8, 1% SDS,
2.5% glycerol, 0.005% bromophenol blue and 10 mM dithiothreitol).
Samples were then heated for 2 min at 95°C.
Subcellular fractionations
Cells were grown in 60-mm tissue culture dishes at
37°C for 48 h (~2×106 cells/dish) and harvested by
scraping in ice-cold PBS. Cell suspensions were centrifuged 200 × g
for 5 min at 4°C and the resulting cell pellets were fractionated
using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo
Fisher Scientific, Inc.). Briefly, the cell pellets were washed
with 1 ml ice-cold PBS and centrifuged at 500 × g for 3 min at 4°C.
The supernatant was removed, and the pellet was resuspended in 0.2
ml ice-cold CER I solution (a kit component) containing protease
inhibitors. The cell suspension was vortexed for 15 sec, placed on
ice for 10 min and 11 µl CER II solution (a kit component) was
added. The suspension was vortexed for a further 5 sec, placed on
ice for 1 min and subsequently vortexed again. The resulting
extract was centrifuged at 16,000 × g for 5 min at 4°C and the
supernatant (cytoplasmic fraction) was removed. The pellet was
washed with PBS and centrifuged at 16,000 × g for 5 min at 4°C then
resuspended in 0.1 ml of ice-cold NER solution (a kit component).
The suspension was vortexed and placed on ice for 10 min, and the
vortexing/ice procedure was repeated three times for a total of 40
min. The resulting extract was centrifuged at 16,000 × g at 4°C for
10 min and the supernatant (nuclear fraction) was removed. Protein
concentrations for cytoplasmic and nuclear fractions were obtained
using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.). Subsequently, 10X SDS-PAGE sample buffer [500 mM Tris-HCl
(pH 6.8), 10% SDS, 25% glycerol, 0.05% bromophenol blue and 100 mM
dithiothreitol] was added to each cytoplasmic and nuclear fraction
to reach a final concentration of 1X [50 mM Tris-HCl (pH 6.8), 1%
SDS, 2.5% glycerol, 0.005% bromophenol blue and 10 mM
dithiothreitol]. Samples were then heated for 2 min at 95°C.
Western blotting
For each western blot analysis, total proteins were
originally extracted using either: i) Lysis buffer containing 25 mM
Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40 and
protease inhibitors (for RNAi and MDR protein extracts), ii) CER
I/CER II solution (Thermo Fisher Scientific, Inc.; for cytoplasmic
fractionation extracts) or iii) NER solution (Thermo Fisher
Scientific, Inc.; for nuclear fractionation extracts. Total
proteins were subsequently added to 10X SDS sample buffer [500 mM
Tris-HCl (pH 6.8), 10% SDS, 25% glycerol, 0.05% bromophenol blue
and 100 mM dithiothreitol] to reach a final concentration of 1X [50
mM Tris-HCl (pH 6.8), 1% SDS, 2.5% glycerol, 0.005% bromophenol
blue and 10 mM dithiothreitol]. Protein concentrations for all
sample lysates were obtained using the Pierce BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). Following protein determination,
40 µg of whole cell lysates or cell fractionations were loaded into
each lane on a 7.5% SDS-PAGE gel. Following separation, the
proteins were transferred to 0.45-µm nitrocellulose (Protran BA 85;
GE Healthcare Bio-Sciences) by semi-dry transfer at 25V for 30 min
using a Trans-blot SD apparatus (BioRad Laboratories, Inc.).
Membranes were blocked with PBS containing 0.05% Tween-20 (PBST)
and 5% milk at room temperature for 1 h, then incubated with the
following primary antibodies: Anti-TRPM2 (1:1,000), polylconal
β-actin (1:2,000), monoclonal β-actin (loading control for RNAi
experiments; 1:5,000), anti-MnSOD (1:1,000), anti-lamin B2
(1:1,000), anti-MDR1 (1:1,000), anti-MRP1 (1:1,000) and anti-LRP
(1:1,000) in PBST + 5% milk overnight at 4°C with shaking.
Membranes were subsequently washed with PBST three times and
incubated with anti-rabbit (1:5,000) or anti-mouse (1:5,000)
secondary antibodies for 1 h at room temperature. The membranes
were washed as previously described, and chemiluminescence was
induced using the SuperSignal West Pico Chemiluminescent Substrate
(Thermo Fisher Scientific, Inc.) for immunodetection of β-actin,
MnSOD, lamin B2, MDR1 and LRP. SuperSignal West Femto
Chemiluminescent Substrate (Thermo Fisher Scientific, Inc.) was
utilized for immunodetection of TRPM2 and MRP1. Densitometric
analysis was completed using Adobe Photoshop CSS Extended (version
12.1×64; Adobe Systems, Inc.). Chemiluminescent detection was
achieved using a ChemiDoc XRS gel imaging system (Bio-Rad
Laboratories, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
For gene expression analyses using RT-qPCR, relative
quantification using the comparative 2−ΔΔCq method was
utilized (42). Total RNA for each
cell line was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and cDNA was obtained
using the High Capacity cDNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Briefly, 10 µl total
RNA was added to 10 µl reaction mix containing primers, dNTPs and
MultiScribe Reverse Transcriptase (all included in the kit). The
reaction was carried out in four steps: 25°C for 10 min, 37°C for
120 min, 85°C for 5 min followed maintenance at 4°C. For qPCR, each
reaction mix (20 µl total) contained 10 ng cDNA, 10 µl PowerUp SYBR
Green Master mix (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and 5 pmol of the following primer sets: MDR1 forward,
5′-TTGCTGCTTACATTCAGGTTTCA-3′ and reverse,
5′-AGCCTATCTCCTGTCGCATTA-3′; MRP1 forward,
5′-AGAACCTCAGTGTCGGGCAGCG-3′ and reverse,
5′-TCGCATCTCTGTCTCTCCTGGG-3′; LRP forward,
5′-CCTCGAGATCCATTGTGCTGG-3′ and reverse,
5′-CACAGGGTTGGCCACTGTGGA-3′. β-actin was amplified as an internal
control using the following primer set, as previously reported
(43): Forward,
5′-CACCAACTGGGACGACAT-3′; and reverse, 5′-ACAGCCTGGATAGCAACG-3′.
Following initial denaturation at 94°C for 2 min, qPCR
amplification was conducted using 40 cycles of 94°C for 30 sec,
57°C for 15 sec and 72°C for 30 sec in a Bio-Rad DNAEngine thermal
cycler with Chromo4 Continuous Fluorescence Detector (Bio-Rad
Laboratories, Inc.). Gene expression was analyzed using MJ Opticon
Monitor Analysis software, version 3.1.32 (Bio-Rad Laboratories,
Inc.). Each reaction was repeated in quadruplicate for each
experiment. A total of four separate qPCR quantifications were
completed for each gene analysis.
Statistical analysis
All error bars for cell death and growth
quantifications represented the standard error of the mean (SEM).
Statistical analyses were carried out using one-way ANOVA followed
by Dunnett's post-hoc test. P<0.05 was considered to indicate a
statistically significant difference. Statistical analyses were
completed using Microsoft Excel, version 14.1.0. All experiments
were performed a minimum of three times and similar results were
obtained.
Results
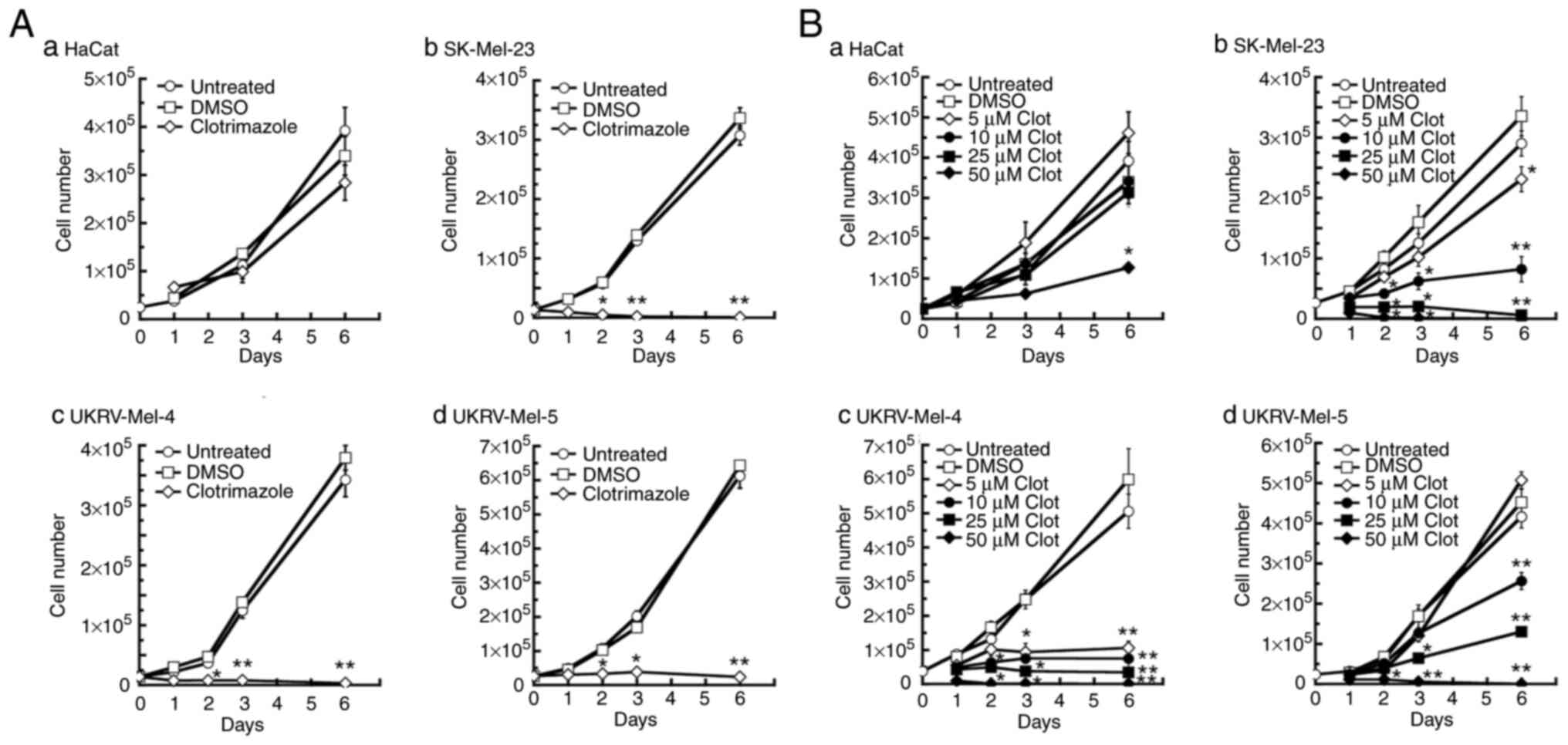
Clotrimazole treatment causes
decreased proliferation in human malignant melanoma cells
To determine the effects of TRPM2 inhibition on the
proliferation of human melanoma cells, three primary human
malignant melanoma cell lines were treated with the TRPM2
inhibitor, clotrimazole (44).
Treatment with 30 µM clotrimazole led to significant decreases in
proliferation after 2 days in all melanoma lines used, compared
with the untreated and DMSO-treated controls (Fig. 1A-b-d). Further cell proliferation
was not observed in the SK-Mel-23 and UKRV-Mel-4 melanoma cell
lines after 2 days of treatment, compared with the positive growth
observed in the same melanoma cell lines that were either untreated
or treated with DMSO (Fig. 1A-b and
-c). In the UKRV-Mel-5 melanoma line, cell proliferation was
decreased by ~84% by day 3 of clotrimazole treatment (Fig. 1A-d); however, no further cell
proliferation was observed subsequently. No significant decrease in
cell proliferation was observed in human keratinocytes (HaCat
cells) during 1–6 days of treatment with clotrimazole (Fig. 1A-a). These results demonstrated that
clotrimazole treatment led to a notable decrease in cell
proliferation in human malignant melanoma cells compared with HaCat
cells.
Subsequently, the dose-dependent response of human
melanoma proliferation to clotrimazole was investigated by treating
each cell line with 5–50 µM clotrimazole. Significant decreases in
proliferation were observed in SK-Mel-23 and UKRV-Mel-4 cells
following each dose of clotrimazole after 3 days of treatment
(Fig. 1B-b and A-c). In the
SK-Mel-23 cell line, in contrast with the DMSO-treated negative
control groups, a 30% reduction in cell proliferation was observed
following treatment with 5 µM clotrimazole after 6 days of
treatment, a 75% reduction was observed after 10 µM treatment, and
cell proliferation was completely inhibited following treatment
with 25 µM clotrimazole (Fig.
1B-b). In the UKRV-Mel-4 line, notable decreases in cell
proliferation were observed at all doses by day 3 of treatment,
compared with the untreated and DMSO-treated controls (Fig. 1B-c). By day 6, cell proliferation
was decreased 85% following treatment with 5 µM clotrimazole, 90%
following treatment with 10 µM clotrimazole, and >98% following
treatment with 25 µM clotrimazole, compared with the DMSO-treated
negative control groups (Fig.
1B-c). In the UKRV-Mel-5 line, a significant decrease in cell
proliferation was observed after 3 days of treatment with 25 or 50
µM clotrimazole, compared with the untreated and DMSO-treated
groups after 3 days (Fig. 1B-d). By
day 6 of treatment, cell proliferation was reduced by ~50%
following treatment with 10 µM clotrimazole and >80% following
treatment with a dose of 25 µM, compared with the cell growth
observed in the corresponding DMSO-treated cells at day 6 (Fig. 1B-d). In all melanoma cell lines,
cell proliferation was not observed following treatment with 50 µM
clotrimazole, compared with the maximal levels of cell growth
observed in untreated and DMSO-treated cells. In HaCat cells, no
significant effect on cell proliferation was observed following
treatment with 5–25 µM clotrimazole for 1–6 days. However, at the
highest dose of clotrimazole (50 µM), significant effects on cell
proliferation were observed at days 3 and 6, with a ~60% decrease
in cell proliferation by day 6. These results demonstrated that
clotrimazole treatment led to dose-dependent decreases in cell
proliferation in human malignant melanoma cells, with no
significant effect on normal human keratinocytes within the 5–25 µM
dose range.
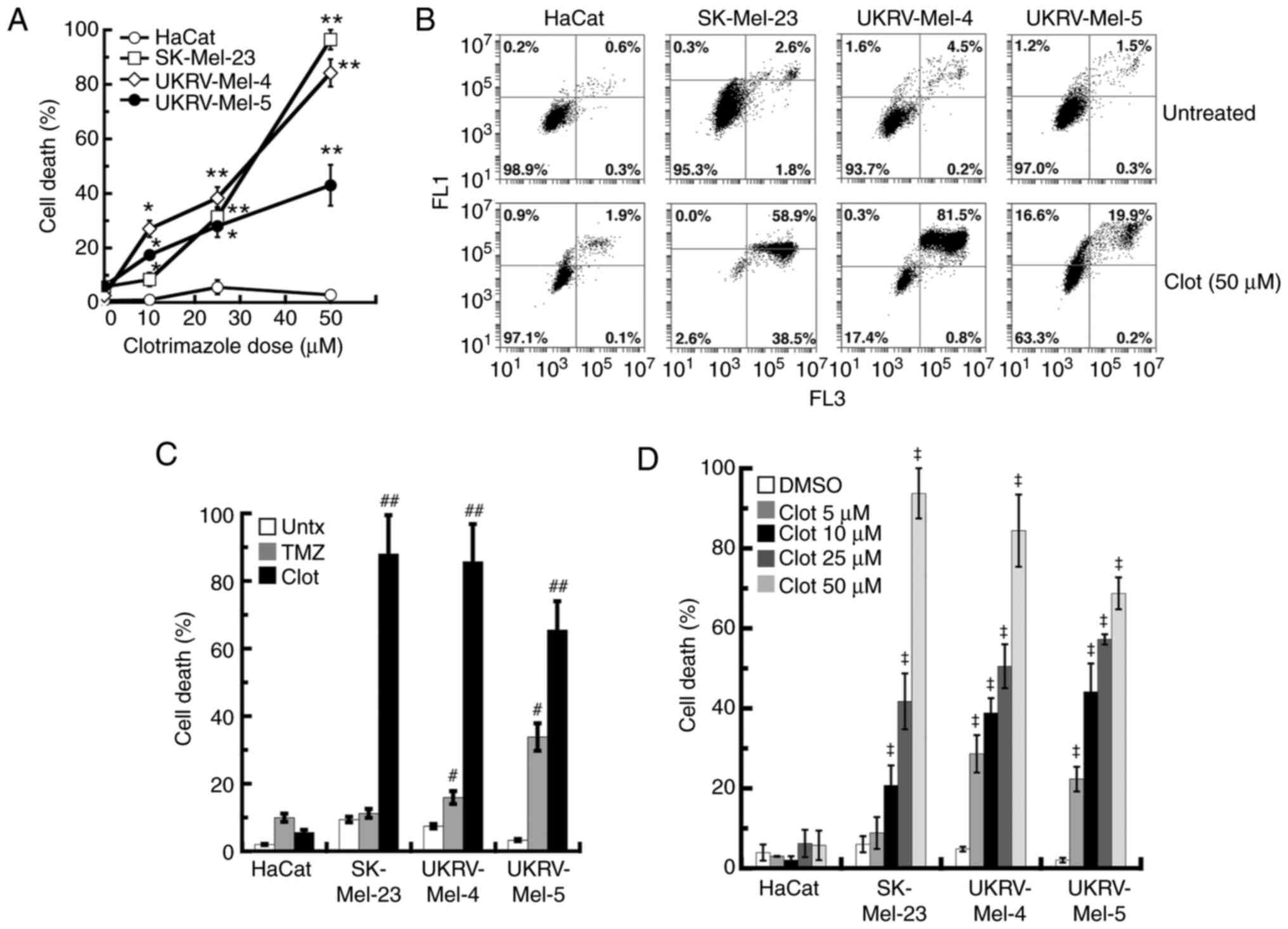
Clotrimazole causes increases in cell
death in human primary malignant melanoma cells in a dose-dependent
manner
To determine if the effects of clotrimazole on the
primary human melanoma cell lines involved the induction of cell
death, levels of cell death were determined in each cell line
following 2 days treatment with 10–50 µM clotrimazole using flow
cytometry. The results of the present study demonstrated that
clotrimazole treatment led to dose-dependent increases in cell
death in each human melanoma line, when compared with similar
treatments in noncancerous keratinocytes at each dose (Fig. 2A). While statistically significant
levels of cell death were observed in each human melanoma line
following 10 µM treatment, 25 µM treatment led to increased levels
of cell death (28% for UKRV-Mel-5, 31% for SK-Mel-23 and 38% for
UKRV-Mel-4). Among all cell lines analyzed, the highest levels of
cell death were observed following 50 µM treatment of clotrimazole,
where the levels of cell death were ~95% for the SK-Mel-23 line and
82% for the UKRV-Mel-4 line. Cell death in the UKRV-Mel-5 line was
reduced compared with the SK-Mel-23 and UKRV-Mel-4 cell lines,
following 50 µM treatment of clotrimazole (42%), but significantly
increased compared with 25 µM treatment. Analysis of cell death
following treatment with clotrimazole in each cell line indicated
that the primary form of cell death induced was most likely
apoptosis, due to dual PI and annexin V-FITC fluorescence detection
in most of the human melanoma cells (Fig. 2B). No significant levels of cell
death were observed in HaCat cells following 10–50 µM clotrimazole
treatment (Figs. 2A and B). These
results demonstrated that clotrimazole caused dose-dependent
increases in cell death in primary human malignant melanoma
cells.
Each human melanoma cell line was further
investigated by comparing the levels of cell death induced by
clotrimazole with the levels induced by TMZ, a conventional
chemotherapeutic treatment option previously utilized in patients
with melanoma (8). Treatment with
10 µM TMZ led to significant increases in cell death 3 days after
treatment in the UKRV-Mel-4 and UKRV-Mel-5 melanoma lines, compared
with untreated cells in each group (Fig. 2C). The greater response was observed
in the latter cell line, where 33% cell death was determined.
However, when compared with 10 µM TMZ treatment, 3 days of 50 µM
clotrimazole treatment caused increased levels of cell death in all
melanoma lines, with the highest levels observed in the SK-Mel-23
line (88%) and UKRV-Mel-4 line (85%; Fig. 2C). While TMZ achieved an ~10% level
of cell death in HaCat cells, cell death following clotrimazole
treatment was markedly lower (4%; Fig.
2C). These results demonstrated that treatment with
clotrimazole leads to increased levels of cell death in human
melanoma cells compared with treatment with TMZ.
Levels of cell death induced by clotrimazole
treatment were also investigated in the primary human melanoma
lines using an alternative cell death assay. Following 2 days of
treatment with clotrimazole, significant levels of cell death were
observed using a WST-1 assay in SK-Mel-23 cells, where 25 µM
treatment caused 42% cell death and 50 µM caused 94% cell death
(Fig. 2D). This was comparable to
the levels of cell death detected using flow cytometry (31%
following 25 µM treatment, 95% following 50 µM treatment; Fig. 2A). In UKRV-Mel-4 cells, results of
the WST-1 assay demonstrated 49% cell death after 25 µM treatment
and 84% after 50 µM treatment (Fig.
2D). This was comparable to the levels observed using flow
cytometry (38% after 25 µM treatment, 83% after 50 µM treatment;
Fig. 2A). However, in UKRV-Mel-5
cells, the levels of cell death levels determined using the WST-1
assay were significantly increased, compared with those quantified
using flow cytometry. More specifically, while cell death
quantified using flow cytometry in these cells was 28% after 25 µM
treatment and 42% after 50 µM treatment (Fig. 2A), results of the WST-1 assay
demonstrated 57% cell death after 25 µM and 68% after 50 µM
treatment (Fig. 2D). Minimal levels
of cell death were observed following treatment with all doses of
clotrimazole in HaCat cells, when compared with the levels of cell
death observed in untreated HaCat cells (Fig. 2D). Collectively, these results
demonstrated that clotrimazole treatment led to significant levels
of cell death in human melanoma cells. Furthermore, the increased
levels of UKRV-Mel-5 cell death detected using the WST-1 assay
compared with the levels quantified using flow cytometry indicated
that an additional cell death pathway, one that was not detected
using flow cytometry, may have been induced.
Treatment with an antagonist of the
TRP vanilloid-1 (TRPV1) or TRP melastatin-8 (TRPM8) channel
exhibits minimal effects on the proliferation of human malignant
melanoma cells
To determine whether effects similar to clotrimazole
treatment may be induced by the antagonism of other TRP channels
involved in skin function or skin cancer development, the human
melanoma cell lines were treated with an antagonist of the TRPV1
channel. The TRPV1 channel is highly expressed in skin (45), and it was found to be overexpressed
in several cancers, including squamous cell carcinoma and
glioblastoma (46,47). In the present study, AMG 9810, a
competitive and selective TRPV1 channel antagonist was used.
Following treatment with 1 µM AMG 9810, which is higher than the
IC50 value of 25 nM (48), no significant effect was observed in
UKRV-Mel-4 or UKRV-Mel-5 cells up to 4 days of treatment (Fig. 3C and D). A total of 6 days of
treatment led to a 25% reduction in cell proliferation in
UKRV-Mel-4 cells (Fig. 3C), but no
significant effect in UKRV-Mel-5 cells (Fig. 3D). In SK-Mel-23 cells, AMG 9810
exerted no significant effect on cell proliferation after 2 days of
treatment, but exerted a 34% reduction in cell proliferation after
4 days of treatment (Fig. 3B).
However, no significant effect was observed following 6 days of
treatment. In human keratinocytes, AMG 9810 treatment for up to six
days had no significant effect on cell proliferation (Fig. 3A). Collectively, these results
indicated that AMG 9810, and thus, antagonism of TRPV1 channels,
caused minimal overall effects on cell proliferation in the human
malignant melanoma cell lines, as these effects were similar to the
minimal effects observed on growth in each cell line after DMSO
treatment. However, the reduction in proliferation of SK-Mel-23
cells after 3 days of treatment suggested a potential role of TRPV1
in these cells.
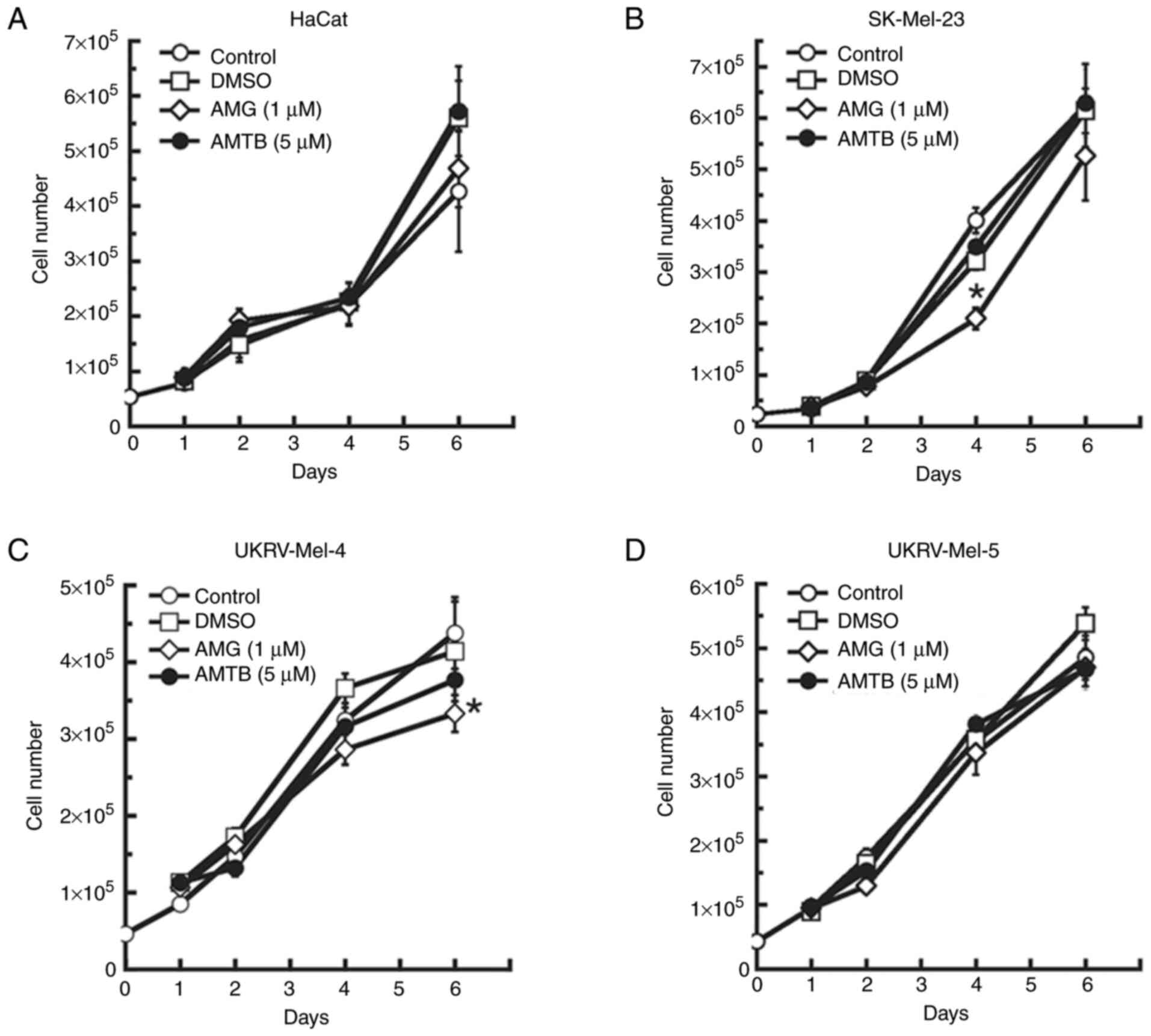 | Figure 3.Effects of TRPV1 and TRPM8
antagonists on proliferation of human malignant melanoma cells.
Treatment of (A) non-cancerous human keratinocytes (HaCat cells)
and the primary malignant melanoma cell lines (B) SK-Mel-23, (C)
UKRV-Mel-4 and (D) UKRV-Mel-5 for 1–6 days with 1 µM of the TRPV1
inhibitor, AMG 9810 or 5 µM of the TRPM8 inhibitor, AMTB. All cell
counts were performed in triplicate and all experiments were
performed at least three times with similar results. *P<0.05 vs.
DMSO-treated; error bars represent the standard error of the mean.
TRP, transient receptor potential; TRPM8, TRP melastatin-8; TRPV1,
TRP vanilloid-1; AMG 9810/AMG,
(2E)-N-(2,3-Dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide;
AMTB, N-(3-Aminopropyl)-2-[(3-methylpheny
l)methoxy]-N-(2-thienylmethyl)benzamide hydrochloride. |
Subsequently, due to the involvement of the TRPM8
channel in skin cancer development (49,50),
AMTB, a competitive and selective TRPM8 antagonist was used in the
present study. Treatment of the human melanoma cell lines with a
dose of 5 µM AMTB, which is higher than the IC50 of 0.6
µM (51), resulted in no
significant effect on cell proliferation in any of the cell lines
following 1–6 days of treatment as compared to untreated and
DMSO-treated negative control groups (Fig. 3A-D). These results indicated that
AMTB, and thus, antagonism of TRPM8 channels, did not adversely
affect proliferation in the human malignant melanoma cell
lines.
Knockdown of TRPM2 leads to increased
death in primary human malignant melanoma cells
To determine whether the effects of clotrimazole
were caused by the antagonism of TRPM2, the effects of TRPM2
knockdown on the levels of cell death were determined. In all cell
lines, TRPM2 siRNA was used to knockdown the expression levels of
TRPM2, as previously described using breast cancer cells (33). TRPM2 expression levels were
decreased in all cell lines, determined using western blot analysis
(Fig. 4A-a, -c, -e and -g) and
quantification of TRPM2 knockdown was determined using densitometry
analysis as outlined in the Material and Methods section (Fig. 4A-b, -d, -f and -h). In SK-Mel-23
cells, knockdown of TRPM2 expression levels by 80% (Fig. 4A-c and -d) led to ~38% cell death
(Fig. 4B-b). TRPM2 knockdown of
>70% in UKRV-Mel-4 cells (Fig. 4A-e
and -f) led to similar levels of cell death (39%; Fig. 4B-c). For UKRV-Mel-5 cells, TRPM2
knockdown by 90% (Fig. 4A-g and -h)
caused ~23% cell death (Fig. 4B-d).
Minimal effects on cell death were observed in human keratinocytes,
as 61% TRPM2 knockdown in HaCat cells (Fig. 4A-a and b) led to only 12% cell death
(Fig. 4B-a). These results
indicated that TRPM2 knockdown led to increased levels of cell
death in human malignant melanoma cells, compared with the minimal
levels of cell death observed in the cells transfected with
non-silencing siRNA. Results of the present study also suggested
that the specific inhibition of TRPM2 function in these cells may
be a primary mechanism underlying the anti-tumor effects caused by
treatment with clotrimazole.
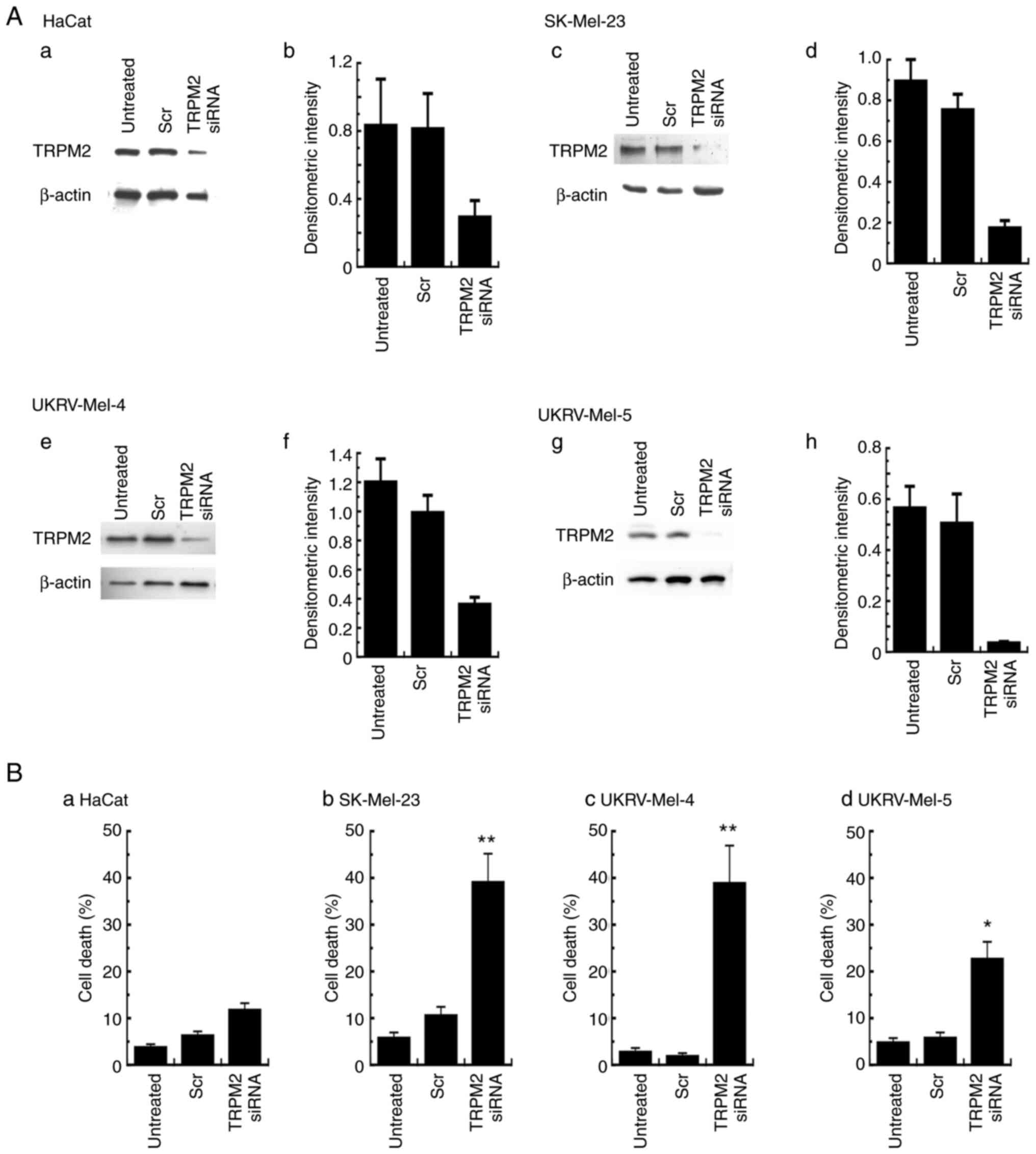 | Figure 4.Cell death levels in human malignant
melanoma cells following TRPM2 silencing. (A) TRPM2 protein
expression levels were determined using western blot analysis in
(a) non-cancerous human keratinocytes (HaCat) and primary malignant
melanoma cell lines, (c) SK-Mel-23, (e) UKRV-Mel-4 and (g)
UKRV-Mel-5 following TRPM2 silencing for 48 h. Quantification of
transfection was performed using densitometric analysis of TRPM2
protein expression levels in (b) non-cancerous human keratinocytes
(HaCat) and primary malignant melanoma cell lines, (d) SK-Mel-23,
(f) UKRV-Mel-4 and (h) UKRV-Mel-5. β-actin was used as the internal
control. Densitometry values represent TRPM2/β-actin ratios.
Untreated cells and cells treated with non-silencing siRNA were
used as negative controls. (B) Cell death levels analyzed in (a)
HaCat cells and primary malignant melanoma cell lines, (b)
SK-Mel-23, (c) UKRV-Mel-4 and (d) UKRV-Mel-5, 48 h after
transfection with TRPM2 siRNA. All cell death analyses were run in
duplicate and all experiments were performed at least three times
with similar results. *P<0.05 and **P<0.01 vs. non-silencing
control transfections; error bars represent the standard error of
the mean. TRPM2, transient receptor potential melastatin-2; siRNA,
small-interfering RNA; Scr, non-silencing siRNA. |
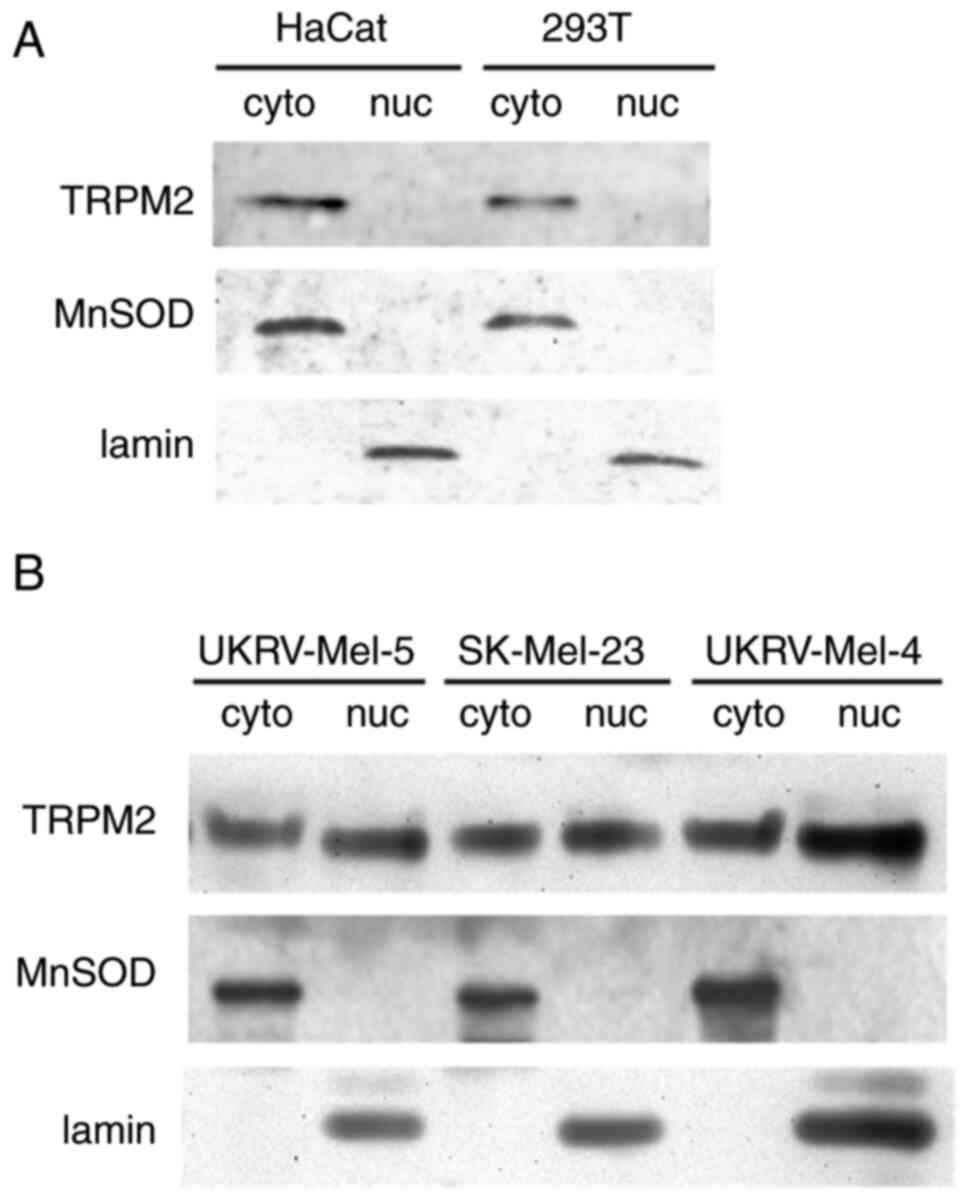
TRPM2 exhibits nuclear localization in
primary human malignant melanoma cells
To further characterize TRPM2 in human malignant
melanoma cells, its cellular localization was investigated.
Subcellular fractionation of each cell line was carried out to form
both nuclear and cytoplasmic fractions. Subsequent western blot
analysis determined that TRPM2, normally localized to the plasma
membrane in healthy cells, exhibited a nuclear localization in all
human malignant melanoma cell lines investigated (Fig. 5B). In comparison, this was in
contrast to the results of the Western blot analysis of the normal
human keratinocyte cell line, where after fractionation, TRPM2
localization was exclusively observed in the cytoplasmic fraction
and not observed in the nuclear fraction (Fig. 5A). Furthermore, additional
comparison via analysis of an additional non-cancerous cell line,
the human embryonic kidney cell line (293T), also revealed TRPM2
localization exclusively to the cytoplasmic fraction (Fig. 5A). These results confirmed that
TRPM2 exhibits a cytoplasmic localization in noncancerous cells
(HaCat and 293T), but in human malignant melanoma cells, TRPM2
exhibits a unique nuclear localization. This highlighted the
potential alternative role of TRPM2 channels in melanoma cells.
Characterization of MDR protein
expression levels in primary human malignant melanoma cells
Numerous patients with melanoma exhibit MDR; thus,
the expression levels of three MDR proteins were investigate in all
cell lines used in the present study. For each human primary
malignant melanoma cell line used, expression levels of MDR1, MRP1
and LRP were analyzed using western blot analyses. Results of the
present study demonstrated that MDR1 expression was not observed in
the normal human keratinocyte cell line and the three human
malignant melanoma lines (Fig.
6A-a); however, results of the RT-qPCR analysis revealed the
reduced, yet significant, expression level of MDR1 mRNA in the
SK-Mel-23 and UKRV-Mel-4 cell lines when compared with the
expression levels of the other MDR genes (Fig. 6B). All human malignant melanoma cell
lines, as well as the human keratinocyte cell line, exhibited
expression of MRP1 (Fig. 6A-b and
B). Moreover, each human melanoma cell line exhibited
significant levels of LRP expression via western blot analysis, but
no significant expression levels were observed in human
keratinocytes (Fig. 6A-c). Notably,
results of the RT-qPCR analysis demonstrated that high levels of
LRP mRNA were present in each human malignant melanoma cell line,
with the highest level observed in the UKRV-Mel-4 line (Fig. 6B). These results further verified
the expression of MRP1 and/or LRP in each human malignant melanoma
cell line investigated. These findings are suggestive of the
ability of TRPM2 antagonism to successfully treat melanoma tumors
that exhibit MDR.
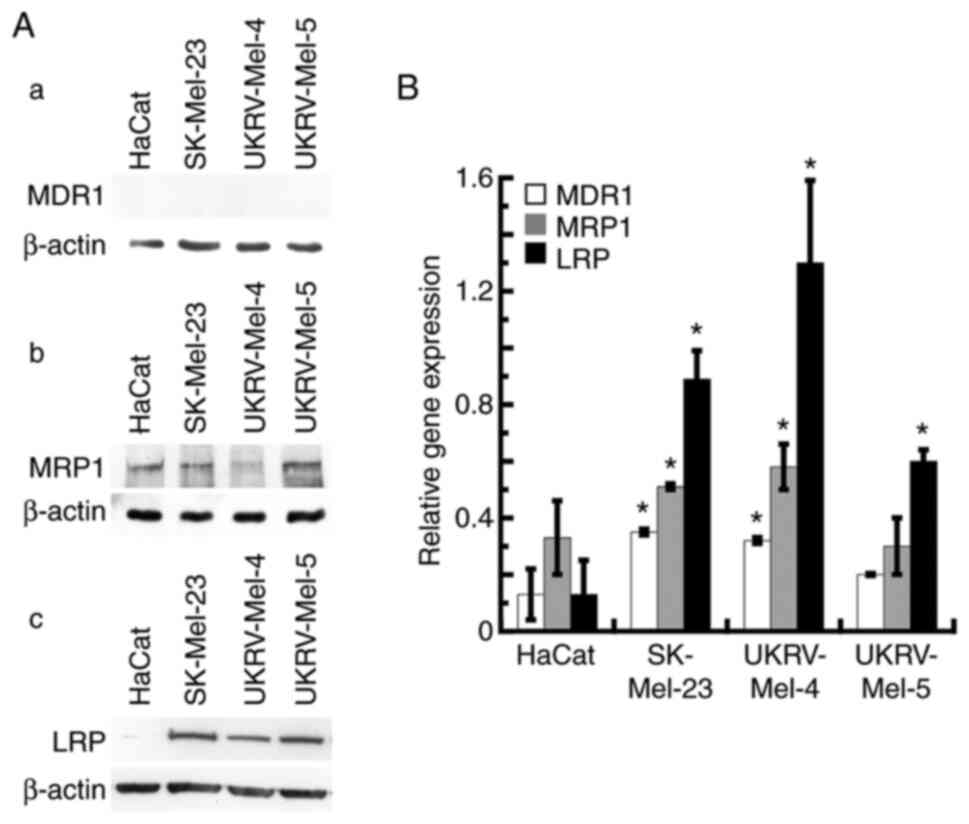 | Figure 6.Expression levels of MDR genes in
human malignant melanoma cells. (A) Non-cancerous human
keratinocytes (HaCat) and the human malignant melanoma cells
SK-Mel-23, UKRV-Mel-4 and UKRV-Mel-5 were analyzed using western
blot analysis, for the detection of MDR genes, (a) MDR1, (b) MRP1
and (c) LRP. β-actin was used as the internal control. Western blot
analysis was performed at least three times with similar results.
(B) HaCat cells and human malignant melanoma cells (SK-Mel-23,
UKRV-Mel-4 and UKRV-Mel-5) were analyzed using RT-qPCR to detect
the gene expression levels of MDR1, MRP1 and LRP. All RT-qPCR
assays were run in triplicate. Values represent four separate
RT-qPCR experiments for each gene in each cell line, for a minimum
of 16 RT-qPCR determinations. *P<0.05 vs. HaCat; error bars
represent the standard error of the mean. RT-qPCR, reverse
transcription-quantitative PCR; MDR, multidrug resistance; MRP1,
multidrug resistance-associated protein 1; MDR1,
phosphorylated-glycoprotein 1; LRP, lung resistance-related
protein. |
Discussion
Results of the present study demonstrated that
clotrimazole may decrease the levels of proliferation and induce
cell death in human malignant melanoma cells. Due to the minimal
deleterious effects observed in healthy human keratinocytes (HaCat
cells), there may be a high level of specificity of clotrimazole in
melanoma cells. Clotrimazole is a well-established drug that has
been used for decades as an imidazole antifungal agent (52). Its use as a fungicidal is based upon
attaining blood levels of clotrimazole up to 20 µg/ml in humans,
and its normal oral dose is 10 mg up to five times daily (53). It is also currently utilized in
chemotherapy regimens for prophylaxis of oral candidiasis at a dose
of 10 mg three times daily (54).
The doses utilized in the treatment of human melanoma cells in the
present study ranged from 1.7–17 µg/ml. Thus, the doses utilized in
the present study are comparable to those currently in use in
chemotherapy regimens, and the in vitro levels utilized in
the present study are comparable with the desired therapeutic blood
levels of clotrimazole in treating oral mycoses. Moreover,
therapeutic doses of clotrimazole in the potential treatment of
melanoma may be obtainable.
As clotrimazole has previously been used, the
potential side effects and toxicity profiles of clotrimazole are
well-established, and overall, its use is well-tolerated (52). While most adverse effects
experienced following clotrimazole administration occur after oral
use, clotrimazole is still administered both orally and topically
(52,55). Topical administration may be more
advantageous in patients with melanoma, due to the ability to
minimize adverse effects and the potential to readily treat primary
tumors in the skin. Notably, an additional advantage is that
clotrimazole is an economical drug that is available for purchase
over-the-counter (52). Thus, the
potential treatment regimens in patients with melanoma may cost
significantly less than the cost of conventional chemotherapy,
immunotherapy or targeted therapy. In the present study, TRPM2 was
characterized as a potential target in the treatment of melanoma.
Moreover, in addition to the benefits of utilizing clotrimazole to
target TRPM2 for its anti-tumor effects, this potential treatment
option may also be widely accessible, affordable and safe.
Clotrimazole is an antagonist of TRPM2 channels
(44). As it may also exhibit
interactions with other targets or cellular processes, inhibitors
of other types of TRP channels were used in the present study, to
determine if similar effects were induced. The lack of similar
effects following this treatment demonstrated that antagonism of
two other types of TRP channels was not the source of the
anti-cancer effects. While the results of the present study do not
demonstrate any favorable effects in targeting these TRP channels
in the human metastatic melanoma cell lines utilized, they do
remain potential targets in melanoma. In A375 human melanoma cells,
modulation of TRPV1 or TRPM8 channels led to increased cell death
(56,57). Moreover, A375 cells harbor the BRAF
V600E mutation (58), while the
melanoma cell lines utilized in the present study were wild-type,
and thus, do not exhibit this genotype. Collectively, the results
indicated that successfully targeting TRP channels in melanoma may
be dependent on the activation or inhibition of their ionophore
function, or the presence of absence of mutations, where specific
TRP channels can be targeted in different melanoma tumors based on
their genotype.
To determine whether these effects were
TRPM2-specific, gene silencing was carried out in the present
study. Knockdown of TRPM2 led to notable levels of cell death in
each human melanoma cell line investigated. These results indicated
that the effects observed following clotrimazole treatment in human
melanoma cells were primarily mediated by TRPM2 antagonism.
However, other mechanisms may play a role, including the ability of
clotrimazole to inhibit hexokinase and decrease overall glycolytic
energy metabolism in melanoma cells (59,60).
Nevertheless, the results of the present study and others
previously reported (59,60) demonstrated a high level of
specificity for clotrimazole in inhibiting proliferation and
inducing cell death in cancer cells. Therefore, clotrimazole may
act as a novel potential treatment option for patients with
melanoma, primarily via TRPM2 antagonism, but also in addition to
other contributing mechanisms.
TRPM2 channels have been identified as novel targets
in several other types of cancer. These include breast cancer
(33,34), head and neck cancer (36), acute myeloid leukemia (61), prostate cancer (35) and neuroblastoma (62). The anti-cancer effects of
clotrimazole in melanoma were previously reported by Benzaquen
et al, and the results of this study demonstrated that
clotrimazole decreased Ca2+ gating (63). Subsequent studies also determined
that clotrimazole functioned to decrease hexokinase and overall
glycolytic activity in melanoma cells (59,60).
As Hill et al (44)
demonstrated that clotrimazole antagonized TRPM2 channels, the
present study aimed to determine whether TRPM2 antagonism was the
primary mechanism through which clotrimazole exerts its effects in
human melanoma cells. TRPM2 silencing in the present study
highlighted TRPM2 antagonism as a major contributor to the
anti-cancer effects observed in human melanoma cells. However,
results of the present study also demonstrated that TRPM2 played a
unique role in melanoma due to its observed nuclear localization.
Thus, the specificity of TRPM2 antagonism may be due to the
prevention of its nuclear role in melanoma, and not its role as an
ion channel at the plasma membrane. A similar nuclear localization
of TRPM2 in other cancer cells, including breast (33), prostate (35) and oral cancer cells (36) indicated that a similar level of
specificity may occur following treatment with TRPM2
antagonism.
Results of the present study also demonstrated that
human melanoma cell lines express MDR proteins. MDR is present in
melanoma and often involves overexpression of one or more of the
following drug resistance genes: MRP1, MDR1 and LRP (6,16,17).
These enable MDR in cancer cells, where they remove
chemotherapeutic drugs from the cell; thus, promoting survival
following treatment due to the lack of active drugs at the tumor
site. A previous study focused on analyzing the expression of MDR1
in melanoma cells, with the authors hypothesizing that additional
MDR genes may exhibit clinical relevance in melanoma (64). The present study further
characterized the expression of additional MDR genes in both
melanoma and non-cancerous cells. Results of the present study
demonstrated that treatment with clotrimazole led to increased cell
death and decreased proliferation in primary human malignant
melanoma cells that express both MRP1 and LRP proteins. The ability
to successfully treat human melanoma cells that exhibit MDR
enhances the therapeutic potential of TRPM2 antagonism, as the
presence of MDR can lead to poor treatment outcomes in patients
with melanoma (16,17). However, further studies using
numerous human melanoma cell lines that exhibit MDR are required in
order to definitively determine whether TRPM2 antagonism can be
used to treat drug-resistant melanoma.
In conclusion, results of the present study
demonstrated that TRPM2 antagonism led to decreased proliferation,
as well as increased cell death, in human malignant melanoma cells
exhibiting MDR. In addition, TRPM2 in these melanoma cells
exhibited notable nuclear localization. Results of the present
study further indicated that TRPM2 may act as a novel potential
target in the treatment of melanoma. Based on the results of the
present study, future studies should focus on elucidating the
potential alternative role of TRPM2 in the nuclei of melanoma
cells, and determining the overall susceptibility of melanoma cells
that express MDR1, MRP1 or LRP to TRPM2 antagonism. Moreover, as
the melanoma cell lines used in the present study were wild-type
cell lines, future studies should also investigate the
susceptibility of alternative melanoma cell lines to TRPM2
antagonism, that are also unresponsive to current treatment
options, including those that exhibit N-RAS Q61R or B-RAF V600E
mutations.
Acknowledgements
We would like to thank Beatrice Yin, MA (yinb@mskcc.org) at the laboratory of Dr
Jedd Wolchok at the Memorial Sloan-Kettering Cancer Center for
providing the SK-Mel-23 melanoma cell line.
Funding
The present research was supported in part by the Bower, Bennet
and Bennet Endowed Chair Research Award obtained from Ohio Northern
University (ONU), and the ONU Summer Research Stipend program.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SGM performed proliferation assays, cell death
assays, assisted in western blot analyses and prepared the
manuscript. LRJ performed RT-qPCR experiments and assisted in
manuscript preparation. CMT performed proliferation assays, cell
death assays, transfection and assisted in manuscript preparation.
SDB performed proliferation assays, cell death assays and
transfection. DPP performed proliferation and cell death assays.
ATD performed RT-qPCR experiments. DWK directed all experiments,
performed proliferation assays, cell death assays, western blot
analysis, RT-qPCR and manuscript preparation. SGM, LRJ, and DWK
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AMG 9810
|
(2E)-N-(2,3-Dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide
|
|
AMTB
|
N-(3-Aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide
hydrochloride
|
|
MDR
|
multidrug resistance
|
|
MnSOD
|
manganese superoxide dismutase
|
|
TMZ
|
temozolomide
|
|
TRP
|
transient receptor potential
|
|
TRPM2
|
transient receptor potential
melastatin-2
|
References
|
1
|
Meier F, Will S, Ellwanger U,
Schlagenhauff B, Schittek B, Rassner G and Garbe C: Metastatic
pathways and time courses in the orderly progression of cutaneous
melanoma. Br J Dermatol. 147:62–70. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer Statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Larkin J, Chiarion-Sileni V, Gonzalez R,
Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J,
Dummer R, et al: Five-year survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 381:1535–1546. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hamid O, Robert C, Daud A, Hodi FS, Hwu
WJ, Kefford R, Wolchok JD, Hersey P, Joseph R, Weber JS, et al:
Five-year survival outcomes for patients with advanced melanoma
treated with pembrolizumab in KEYNOTE-001. Ann Oncol. 30:582–588.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wolchok JD, Kluger H, Callahan MK, Postow
MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K,
et al: Nivolumab plus ipilimumab in advanced melanoma. N Engl J
Med. 369:122–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schadendorf D, Makki A, Stahr C, van Dyck
A, Wanner R, Scheffer GL, Flens MJ, Scheper R and Henz BM: Membrane
transport proteins associated with drug resistance expressed in
human melanoma. Am J Pathol. 147:1545–1552. 1995.PubMed/NCBI
|
|
7
|
Vinay DS, Ryan EP, Pawelec G, Talib WH,
Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, et
al: Immune evasion in cancer: Mechanistic basis and therapeutic
strategies. Semin Cancer Biol. 35 Suppl:S185–S198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Middleton MR, Grob JJ, Aaronson N,
Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates
A, et al: Randomized phase III study of temozolomide versus
dacarbazine in the treatment of patients with advanced metastatic
malignant melanoma. J Clin Oncol. 18:158–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sosman JA, Kim KB, Schuchter L, Gonzalez
R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ,
Flaherty KT, et al: Survival in BRAF V600-mutant advanced melanoma
treated with vemurafenib. N Engl J Med. 366:707–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klink AJ, Chmielowski B, Feinberg B, Ahsan
S, Nero D and Liu FX: Health care resource utilization and costs in
first-line treatments for patients with metastatic melanoma in the
United States. J Manag Care Spec Pharm. 25:869–877. 2019.PubMed/NCBI
|
|
12
|
Kandel M, Allayous C, Dalle S, Mortier L,
Dalac S, Dutriaux C, Leccia MT, Guillot B, Saiag P, Lacour JP, et
al: Update of survival and cost of metastatic melanoma with new
drugs: Estimations from the MelBase cohort. Eur J Cancer.
105:33–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ottaviano M, De Placido S and Ascierto PA:
Recent success and limitations of immune checkpoint inhibitors for
cancer: A lesson from melanoma. Virchows Arch. 474:421–432. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Helmbach H, Kern MA, Rossmann E, Renz K,
Kissel C, Gschwendt B and Schadendorf D: Drug resistance towards
etoposide and cisplatin in human melanoma cells is associated with
drug-dependent apoptosis deficiency. J Invest Dermatol.
118:923–932. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Soengas MS and Lowe SW: Apoptosis and
melanoma chemoresistance. Oncogene. 22:3138–3151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen KG, Valencia JC, Gillet JP, Hearing
VJ and Gottesman MM: Involvement of ABC transporters in
melanogenesis and the development of multidrug resistance of
melanoma. Pigment Cell Melanoma Res. 22:740–749. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ichihashi N and Kitajima Y: Chemotherapy
induces or increases expression of multidrug resistance-associated
protein in malignant melanoma cells. Br J Dermatol. 144:745–750.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Knorr C, Pelz JO, Gohl J, Hohenberger W
and Meyer T: Expression of chemoresistance-related genes and heat
shock protein 72 in hyperthermic isolated limb perfusion of
malignant melanoma: An experimental study. J Oncol.
2010:1387582010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
van Helvoort A, Smith AJ, Sprong H,
Fritzsche I, Schinkel AH, Borst P and van Meer G: MDR1
P-glycoprotein is a lipid translocase of broad specificity, while
MDR3 P-glycoprotein specifically translocates phosphatidylcholine.
Cell. 87:507–517. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnson ZL and Chen J: Structural basis of
substrate recognition by the multidrug resistance protein MRP1.
Cell. 168:1075–1085. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Izquierdo MA, Scheffer GL, Flens MJ,
Shoemaker RH, Rome LH and Scheper RJ: Relationship of LRP-human
major vault protein to in vitro and clinical resistance to
anticancer drugs. Cytotechnology. 19:191–197. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Robey RW, Pluchino KM, Hall MD, Fojo AT,
Bates SE and Gottesman MM: Revisiting the role of ABC transporters
in multidrug-resistant cancer. Nat Rev Cancer. 18:452–464. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chyb S, Raghu P and Hardie RC:
Polyunsaturated fatty acids activate the Drosophila light-sensitive
channels TRP and TRPL. Nature. 397:255–259. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aroke EN, Powell-Roach KL, Jaime-Lara RB,
Tesfaye M, Roy A, Jackson P and Joseph PV: Taste the pain: The role
of TRP channels in pain and taste perception. Int J Mol Sci.
21:59292020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bergdahl A, Gomez MF, Wihlborg AK, Erlinge
D, Eyjolfson A, Xu SZ, Beech DJ, Dreja K and Hellstrand P:
Plasticity of TRPC expression in arterial smooth muscle:
Correlation with store-operated Ca2+ entry. Am J Physiol Cell
Physiol. 288:C872–C880. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Okamoto Y, Ohkubo T, Ikebe T and Yamazaki
J: Blockade of TRPM8 activity reduces the invasion potential of
oral squamous carcinoma cell lines. Int J Oncol. 40:1431–1440.
2012.PubMed/NCBI
|
|
27
|
Zhang W, Hirschler-Laszkiewicz I, Tong Q,
Conrad K, Sun SC, Penn L, Barber DL, Stahl R, Carey DJ, Cheung JY
and Miller BA: TRPM2 is an ion channel that modulates hematopoietic
cell death through activation of caspases and PARP cleavage. Am J
Physiol Cell Physiol. 290:C1146–C1159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perraud AL, Takanishi CL, Shen B, Kang S,
Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, et al:
Accumulation of free ADP-ribose from mitochondria mediates
oxidative stress-induced gating of TRPM2 cation channels. J Biol
Chem. 280:6138–6148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeong H, Kim YH, Lee Y, Jung SJ and Oh SB:
TRPM2 contributes to LPC-induced intracellular Ca2+
influx and microglial activation. Biochem Biophys Res Commun.
485:301–306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Melzer N, Hicking G, Gobel K and Wiendl H:
TRPM2 cation channels modulate T cell effector functions and
contribute to autoimmune CNS inflammation. PLoS One. 7:e476172012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hara Y, Wakamori M, Ishii M, Maeno E,
Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, et al:
LTRPC2 Ca2+-permeable channel activated by changes in
redox status confers susceptibility to cell death. Mol Cell.
9:163–173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fonfria E, Marshall IC, Benham CD,
Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD and McNulty S:
TRPM2 channel opening in response to oxidative stress is dependent
on activation of poly(ADP-ribose) polymerase. Br J Pharmacol.
143:186–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hopkins MM, Feng X, Liu M, Parker LP and
Koh DW: Inhibition of the transient receptor potential melastatin-2
channel causes increased DNA damage and decreased proliferation in
breast adenocarcinoma cells. Int J Oncol. 46:2267–2276. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koh DW, Powell DP, Blake SD, Hoffman JL,
Hopkins MM and Feng X: Enhanced cytotoxicity in triple-negative and
estrogen receptor positive breast adenocarcinoma cells due to
inhibition of the transient receptor potential melastatin-2
channel. Oncol Rep. 34:1589–1598. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeng X, Sikka SC, Huang L, Sun C, Xu C,
Jia D, Abdel-Mageed AB, Pottle JE, Taylor JT and Li M: Novel role
for the transient receptor potential channel TRPM2 in prostate
cancer cell proliferation. Prostate Cancer Prostatic Dis.
13:195–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao LY, Xu WL, Xu ZQ, Qi C, Li Y, Cheng
J, Liu LK, Wu YN, Gao J and Ye JH: The overexpressed functional
transient receptor potential channel TRPM2 in oral squamous cell
carcinoma. Sci Rep. 6:384712016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shiku H, Takahashi T and Oettgen HF: Cell
surface antigens of human malignant melanoma. II. Serological
typing with immune adherence assays and definition of two new
surface antigents. J Exp Med. 144:873–881. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pawelec G and Marsh SG: ESTDAB: A
collection of immunologically characterised melanoma cell lines and
searchable databank. Cancer Immunol Immunother. 55:623–627. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schadendorf D, Fichtner I, Makki A,
Alijagic S, Küpper M, Mrowietz U and Henz BM: Metastatic potential
of human melanoma cells in nude mice-characterization of phenotype,
cytokine secretion and tumour-associated proteins. Br J Cancer.
74:194–199. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bloethner S, Chen B, Hemminki K,
Müller-Berghaus J, Ugurel S, Schadendorf D and Kumar R: Effect of
common B-RAF and N-RAS mutations on global gene expression in
melanoma cell lines. Carcinogenesis. 26:1224–1232. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Berridge MV, Herst PM and Tan AS:
Tetrazolium dyes as tools in cell biology: New insights into their
cellular reduction. Biotechnol Annu Rev. 11:127–152. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiang X, Deng Z, Zhuang X, Ju S, Mu J,
Jiang H, Zhang L, Yan J, Miller D and Zhang HG: Grhl2 determines
the epithelial phenotype of breast cancers and promotes tumor
progression. PLoS One. 7:e507812012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hill K, McNulty S and Randall AD:
Inhibition of TRPM2 channels by the antifungal agents clotrimazole
and econazole. Naunyn Schmiedebergs Arch Pharmacol. 370:227–237.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Denda M, Fuziwara S, Inoue K, Denda S,
Akamatsu H, Tomitaka A and Matsunaga K: Immunoreactivity of VR1 on
epidermal keratinocyte of human skin. Biochem Biophys Res Commun.
285:1250–1252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Marincsak R, Toth BI, Czifra G, Márton I,
Rédl P, Tar I, Tóth L, Kovács L and Bíró T: Increased expression of
TRPV1 in squamous cell carcinoma of the human tongue. Oral Dis.
15:328–335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Alptekin M, Eroglu S, Tutar E, Sencan S,
Geyik MA, Ulasli M, Demiryurek AT and Camci C: Gene expressions of
TRP channels in glioblastoma multiforme and relation with survival.
Tumour Biol. 36:9209–9213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang
TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, et al: AMG 9810
[(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4]
dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1)
antagonist with antihyperalgesic properties. J Pharmacol Exp Ther.
313:474–484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Oh ST, Yang KJ, Kim YH, Bae JM, Park HJ,
Kim JW and Park YM: Increased immunoreactivity for TRPM8 in
cutaneous squamous cell carcinoma. J Cutan Pathol. 45:970–972.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hemida AS, Hammam MA, Heriz NAEM and
Shehata WA: Expression of transient receptor potential channel of
melastatin number 8 (TRPM8) in non-melanoma skin cancer: A clinical
and immunohistochemical study. J Immunoassay Immunochem.
42:620–632. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lashinger ES, Steiginga MS, Hieble JP,
Leon LA, Gardner SD, Nagilla R, Davenport EA, Hoffman BE, Laping NJ
and Su X: AMTB, a TRPM8 channel blocker: Evidence in rats for
activity in overactive bladder and painful bladder syndrome. Am J
Physiol Renal Physiol. 295:F803–F810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Crowley PD and Gallagher HC: Clotrimazole
as a pharmaceutical: Past, present and future. J Appl Microbiol.
117:611–617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
PubChem [Internet]. Bethesda (MD), .
National Library of Medicine (US), National Center for
Biotechnology Information; 2004. PubChem Compound Summary for CID
2812, Clotrimazole. Available from. https://pubchem.ncbi.nlm.nih.gov/compound/Clotrimazole
|
|
54
|
Cuttner J, Troy KM, Funaro L, Brenden R
and Bottone EJ: Clotrimazole treatment for prevention of oral
candidiasis in patients with acute leukemia undergoing
chemotherapy. Results of a double-blind study. Am J Med.
81:771–774. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Taudorf EH, Jemec GBE, Hay RJ and Saunte
DML: Cutaneous candidiasis-an evidence-based review of topical and
systemic treatments to inform clinical practice. J Eur Acad
Dermatol Venereol. 33:1863–1873. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zheng J, Liu F, Du S, Li M, Wu T, Tan X
and Cheng W: Mechanism for regulation of melanoma cell death via
activation of thermo-TRPV4 and TRPV2. J Oncol. 2019:73628752019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yamamura H, Ugawa S, Ueda T, Morita A and
Shimada S: TRPM8 activation suppresses cellular viability in human
melanoma. Am J Physiol Cell Physiol. 295:C296–C301. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dratkiewicz E, Simiczyjew A,
Pietraszek-Gremplewicz K, Mazurkiewicz J and Nowak D:
Characterization of melanoma cell lines resistant to vemurafenib
and evaluation of their responsiveness to EGFR- and MET-inhibitor
treatment. Int J Mol Sci. 21:1132019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Penso J and Beitner R: Clotrimazole and
bifonazole detach hexokinase from mitochondria of melanoma cells.
Eur J Pharmacol. 342:113–117. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zancan P, Rosas AO, Marcondes MC,
Marinho-Carvalho MM and Sola-Penna M: Clotrimazole inhibits and
modulates heterologous association of the key glycolytic enzyme
6-phosphofructo-1-kinase. Biochem Pharmacol. 73:1520–1527. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen SJ, Bao L, Keefer K, Shanmughapriya
S, Chen L, Lee J, Wang J, Zhang XQ, Hirschler-Laszkiewicz I, Merali
S, et al: Transient receptor potential ion channel TRPM2 promotes
AML proliferation and survival through modulation of mitochondrial
function, ROS, and autophagy. Cell Death Dis. 11:2472020.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hirschler-Laszkiewicz I, Chen SJ, Bao L,
Wang J, Zhang XQ, Shanmughapriya S, Keefer K, Madesh M, Cheung JY
and Miller BA: The human ion channel TRPM2 modulates neuroblastoma
cell survival and mitochondrial function through Pyk2, CREB, and
MCU activation. Am J Physiol Cell Physiol. 315:C571–C586. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Benzaquen LR, Brugnara C, Byers HR,
Gatton-Celli S and Halperin JA: Clotrimazole inhibits cell
proliferation in vitro and in vivo. Nat Med. 1:534–540. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Schadendorf D, Herfordt R and Czarnetzki
BM: P-glycoprotein expression in primary and metastatic malignant
melanoma. Br J Dermatol. 132:551–555. 1995. View Article : Google Scholar : PubMed/NCBI
|