Introduction
Chronic myeloid leukemia (CML), which is a malignant
myeloproliferative disorder caused by hematopoietic stem cells
(HSCs), is a clonal disorder of HSCs (1,2).
Various treatments are available for patients with CML, including
tyrosine kinase inhibitors (TKIs), HSC transplantation and
chemotherapy (3). Imatinib (IM)
is a targeted agent after clinical experiments that has become an
established treatment for CML, and was first approved as a
first-line drug in 2003 (4,5).
IM is a TKI that has revolutionized the treatment of CML, and a
large amount of clinical data have shown that this drug can induce
complete remission of CML and change the clinical course (6,7).
However, finding an effective treatment for IM-resistant patients
remains challenging despite the progress and success of IM in CML
treatment. Drug resistance is undoubtedly emerging as a major
problem in the treatment of CML. Thus, finding ways and mechanisms
to tackle tumor drug resistance is urgent. In the present study,
the K562 CML cell line and its IM-resistant cell line (KG) were
used to investigate the mechanism underlying CML resistance to IM
and to identify potential strategies to reverse resistance.
Tribbles pseudokinase 2 (TRIB2) is a member of the
tribbles family (8). TRIB2
includes an N-terminal domain, a C-terminal domain and a central
pseudokinase domain. Under the absence of kinase activity, it can
regulate different signaling pathways in basic biological processes
and pathology. For instance, TRIB2 blocks cellular senescence
through AP4/p21 signaling and can also regulate the differentiation
of myeloid progenitors transduced with mll-tet1 (9,10).
TRIB2 plays an important role in regulating tumor cell
proliferation, apoptosis and drug resistance (11,12). TRIB2 acts as a tumor promoting
factor and plays a promoting role in tumorigenesis and development.
Link et al (13) have
reported that TRIB2 suppresses FOXO and acts as an oncogenic
protein in melanoma. TRIB2 also plays a tremendous role in tumor
cell drug resistance. Wang et al (14) found that combined elevation of
TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to
temozolomide in glioblastoma. Meanwhile, Hill et al
(15) reported that TRIB2 confers
resistance to anticancer therapy by activating the serine/threonine
protein kinase AKT. Therefore, investigating the role played by
TRIB2 in tumor drug resistance is urgent and meaningful. The
present study investigated the relationship between TRIB2 and IM
resistance in CML and identified that reversal of CML resistance
was through regulation of miR-33a-5p.
MicroRNAs (miRNAs) are a family of small non-coding
RNAs consisting of 18-25 nucleotides (16). miRNA plays important roles in
numerous cellular and biological processes, such as proliferation,
apoptosis, differentiation and metabolism (17). Concurrently, a largely
relationship exists between miRNA and drug resistance of tumor
cells (18). Diaz-Martinez et
al (19) found that
miR-204-5p and miR-211-5p contribute to melanoma resistance against
BRAF inhibitors. In addition, exosomal transfer of miR-1238
contributes to temozolomide resistance in glioblastoma (20). Thus, miRNA plays an important role
in reversing tumor drug resistance. Thus, in the present
experiment, the function of miR-33a-5p in CML-resistant cells was
investigated. Although miRNA expression plays an important role in
tumorigenesis and drug resistance, little is known about the
mechanisms leading to aberrant miRNA regulation in cancer. Numerous
upstream factors affect miRNAs, including proteins that exercise
various functions and non-coding RNAs (21,22). Meanwhile, transcription factors
(TFs) are the most intensely studied factors. The role played by
TFs in regulating miRNAs needs to be further explored.
TFs are key regulators controlling gene expression
in the process of carcinogenesis and development (23). C-Fos is a well-known oncogene and
a member of AP-1 TF family (24).
It can be used as a TF to regulate the development of tumor cells.
C-myc is a major transcriptional regulator of RhoA, and alkaline
phosphatase downregulation promotes lung adenocarcinoma metastasis
via the c-myc/RhoA axis (25).
C-Fos was predicted as a possible TF of miR-33a-5p using in
silico bioinformatics analysis (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3).
Similar to c-myc/RhoA, the mechanism by which c-Fos
acts as a TF to affect miR-33a-5p remains to be investigated.
Increasing clinical and biological evidence indicated that the ERK
pathway plays a key role in the progression of tumors (26). The mechanism of ERK pathway
involvement in the present study was investigated using ERK
signaling pathway blockers and activators. The specific mechanism
through which TRIB2 may reverse the IM resistance process of CML
cells by regulating miR-33a-5p was investigated. The present study
provided a new therapeutic target for CML treatment and acquired
resistance to IM and a new strategy for clinical treatment.
Materials and methods
Cell culture and reagents
The cell line K562 was kindly provided by the Cell
Bank of The Chinese Academy of Sciences (Shanghai, China). IM
(Aladdin Industrial Corporation) was added to the logarithmic
growth phase K562 cell suspension at a final concentration of 100
ng/ml. The cells were observed so that the drug dose could be
gradually escalated over the time of incubation and the cells were
finally allowed to survive on a concentration of 10 μM IM in
the culture system. Monoclonal screening by limiting dilution
yielded an IM resistant KG cell line. The cells were incubated in
basic RPMI-1640 medium with 10% fetal bovine serum (both from
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Beyotime Institute of Biotechnology) at
37°C in a 5% CO2 humidified atmosphere. KG cells were
maintained in the presence of 10 μM IM. Prior to the
experiment, the cells were cultured in drug-free medium for 2
weeks. All cell experiments were repeated three times. Honokiol
(HNK) was purchased from Aladdin Industrial Corporation, and U0126
was obtained from Promega Corporation. At 50-60% of KG cells, 20
μM U0126 or 20 μM HNK at 37°C for 24 h for subsequent
experiments.
Cell viability assay
Cell viability was determined by Cell Counting Kit-8
(CCK-8) assays. Briefly, the cells were grouped and treated
differently. Then, KG cells were seeded in 96-well plates
(4×103 cells/well) and cultured in an incubator at 37°C
with 5% CO2. A volume of 10 μl CCK-8 (Beyotime
Institute of Biotechnology) was added into the wells for 2 h at 0,
24, 48 and 72 h. The absorbance of each well at 450 nm (A450)
wavelength was measured by a microplate reader (Multiskan FC;
Thermo Fisher Scientific, Inc.). The IM resistance index was
calculated as IC50-KG/IC50-K562; i.e., drug
resistance index=IC50 of drug resistant
cells/IC50 of parental cells. In the present study that
was IC50-KG/IC50-K562=24.12/0.98=24.61.
Cell infection
The KG cells were routinely cultured in six-well
plates. A total of 12 μg lentiviral expression vectors
(pCDH-NC or pCDH-TRIB2; KeyyBio Scientific, Inc.) and
second-generation lentiviral packaging vectors pSPAX2 and pMD2.G
(from Addgene) were transfected into 293T (Shanghai Cell Bank of
the Chinese Academy of Sciences, Shanghai, China) cells at a ratio
of 4:3:2 and cultured at 37°C. The virus was three times collected
at an interval of 24 h between collections. KG cells were infected
using polybrene (Sigma) and lentiviral particles at an MOI of 20.
KG cells were seeded within six well plates and cultured in a 37°C
5% CO2 incubator until transfection was performed at a
cell density of 50-60%. GV141-Fos (2 μg) and 6 μl
Lipofectamine 2000® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) were added at the time of transfection and empty
plasmid served as a negative control. Fluid changes were performed
after 6-8 and 48 h for subsequent experiments.
Negative control siRNA was used as a negative
control (NC). The sequences were as follows: si-NC sense, 5'-UUC
UCC GAA CGU GUC ACG UTT-3' and si-NC antisense, 5'-ACG UGA CAC GUU
CGG AGA ATT-3'; si-TRIB2 sense, 5'-UAG CGA GAU AUG GGA GAU CTT-3'
and si-TRIB2 antisense, 5'-GAU CUC CCA UAU CUC GCU ATT-3'. The
sequences of the miR-33a-5p-mimics were as follows: sense, 5'-GUG
CAU UGU AGU UGC AUU GCA-3' and antisense, 5'-UGC AAU GCA ACU ACA
AUG CAC UU-3'. The sequences of the miR-33a-5p-NC were as follows:
sense, 5'-CAG UAC UUU UGU GUA GUA CAA-3' and antisense, 5'-GUA CUA
CAC AAA AGU ACU GUU-3'. The sequences of the miR-33a-5p-inhibitor
were as follows: 5'-UGC AAU GCA ACU ACA AUG CAC UU-3'. After 24 h,
the changes in GFP expression were detected by fluorescence
microscopy and flow cytometry and images were captured. After 48 h,
G418 (300 μg/ml) was used to select and maintain stably
transfected cells. After 4 weeks, the cells expressing GFP were
selected using a BD FACSAria II flow cytometer (BD FACSAria II; BD
Biosciences) to obtain a stably transfected cell line.
Vector construction
The possible binding sites of c-Fos in the promoter
of miR-33a-5p were analyzed using JASPAR (http://jaspardev.genereg.net/). All miR-33a-5p
promoter sequences were segmented according to c-Fos binding sites
(-534 ~+69 nt, -1124 ~+69 nt, -1575 ~+69 nt, and -2,000 ~+69 nt),
and they were connected to pGL3 basic luciferase reporter gene
vector (Promega Corporation) to construct promoter luciferase
segmented vector. The promoter binding site was point-mutated,
amplified by PCR and cloned into pGL3 basic luciferase reporter
gene vector to construct luciferase mutation vector. PCR
amplification was performed using a Takara LA Taq DNA polymerase
(Takara Biotechnology Co., Ltd.). The PCR conditions were as
follows: initial denaturation at 94°C for 1 min; 30 cycles of 94°C
for 30 sec, 60°C annealing for 30 sec and extension at 72°C for 2
min. The amplification was completed with a final step of 72°C for
5 min. PCR products were separated by electrophoresis on a 1.25%
agarose gel followed by visualization under the Tanon 2500 gel
imaging system (Tanon Science and Technology Co., Ltd.). A pair of
primers was designed on both ends of the miR-33a-5p promoter
truncation fragments based on their size and location (i.e., -534,
-1,124, -1,575 and -2,000). A protective base and a specific
enzymatic cleavage site before the primer sequence were added. The
primer sequences are listed in Table
I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Vector name | Primer sequences
(5'→3') |
|---|
| -2000 ~ +69 nt | F:
CTAGCTAGCCAAGTTGGGCCAGCAG |
| R:
CCCAAGCTTCTGTGATGCACTGTGGAA |
| -1575 ~ +69 nt | F:
CTAGCTAGCTGATGGGCAGCCCTG |
| R:
CCCAAGCTTCTGTGATGCACTGTGGAA |
| -1124 ~ +69 nt | F:
CTAGCTAGCCCTGCAGGCACTGCT |
| R:
CCCAAGCTTCTGTGATGCACTGTGGAA |
| -534 ~ +69 nt | F:
CTAGCTAGCCCCTGGGAAAGCAGATG |
| R:
CCCAAGCTTCTGTGATGCACTGTGGAA |
| Mutation (A) | F:
GGAATGATGTCGACACGTTGGAATGA |
| R:
TTACCTATAAAGTGGCCACAATAAGGT |
| Mutation (B) | F:
GCACCACTGGATTAGTGACCTGCCTCT |
| R:
GTGACAGGCAGAGGGGCCCAAGCCA |
Luciferase activity assay
Cancer cells (6x103/well) were seeded in
six-well plates and luciferase vectors containing the miR-33a-5p
promoter (miR-33a-5p-luc) were co-transfected with TRIB2 and c-Fos
expression vectors using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h of
transfection, the cells were collected and lysed on ice. The
luciferase activity was detected according to the manufacturer's
instructions using a double luciferase detection kit (Promega
Corporation). Firefly luciferase activity was normalized to
Renilla luciferase activity. Targetscan7.1 (http://www.targetscan.org/vert_71/) was used to
predict the downstream target genes of miR-33a-5p.
Western blot analysis and
immunoprecipitation
Differently treated KG cells were collected, washed
twice with cold PBS and lysed on ice in RIPA lysis buffer (Beyotime
Institute of Biotechnology) for 30 min. The BCA method was used to
determine protein concentration, and 20 μg protein were
loaded per lane. Proteins were separated using 10% SDS-PAGE and
transferred to a PVDF membrane. The membrane was blocked in 5% milk
powder for 2 h at 37°C. The membrane was incubated with primary
antibodies at 4°C overnight. The next day, the membrane was washed
with TBST (0.1% Tween-20) three times and incubated with goat
anti-rabbit IgG (H+L) HRP conjugate (1:5,000; cat. no. BS13278;
Bioworld Technology, Inc.) for 2 h at 4°C and detected by
chemiluminescence (ECL reagent; Biosharp Life Sciences) method. The
densities of the bands were analyzed using iBright 1500 v1.4.3
software (Thermo Fisher Scientific, Inc.) The information for
antibodies are as follows: TRIB2 (1:1,000; cat. no. ab272544;
Abcam), c-Fos (1:500; cat. no. 48283; SAB Technology, Inc.),
p-c-Fos (1:750; cat. no. 12599; SAB Technology, Inc.), ERK (1:750;
cat. no. BS1112; Bioworld Technology, Inc.), p-ERK (1:500; cat. no.
BS65784; Bioworld Technology, Inc.) and GAPDH (1:10,000; cat. no.
BS65483M; Bioworld Technology, Inc.). For immunoprecipitation, the
cells were lysed on ice with NP-40 lysis buffer (Beyotime Institute
of Biotechnology) for 30 min and total cell lysates were incubated
with appropriate antibodies overnight at 4°C and subsequently
rotated with protein A/G beads (50 μl; cat. no. 20421;
Thermo Fisher Scientific, Inc.) for 4 h at 4°C. The information for
antibodies are as follows: IgG (1:5,000; cat. no. ab181236; Abcam),
p-c-Fos (1:100; cat. no. 5348; Cell Signaling Technology), TBP
(1:100; cat. no. 44059; Cell Signaling Technology). Beads were
washed three times using NP-40 lysis buffer, mixed with 2X SDS
sample buffer and boiled for 10 min. The co-precipitates were
analyzed by western blot analysis.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed using a ChIP assay kit
(Beyotime Institute of Biotechnology). Briefly, differently treated
KG cells were collected and treated with 1% formaldehyde for 10 min
at 37°C for cross-linking. Chromatin was sheared by sonication on
ice to produce DNA fragments with a size of 200-1,000 bp. Chromatin
was immunoprecipitated with 5 μg of anti-p-c-Fos (1:100;
cat. no. 5348; Cell Signaling Technology) or rabbit IgG (1:5,000;
cat. no. ab181236; Abcam). The immunoprecipitated DNA was amplified
with a primer pair specific for the miR-33a-5p promoter (forward,
5'-CCT GCA GGC ACT GCT-3' and reverse, 5'-CTG TGA TGC ACT GTG
GAA-3'). PCR amplification was performed using a TaKaRa LA Taq DNA
polymerase (Takara Biotechnology Co., Ltd.). The PCR conditions
were as follows: initial denaturation at 94°C for 1 min; 35 cycles
of 95°C for 20 sec, 60°C annealing for 30 sec and extension at 72°C
for 2 min. The amplification was completed with a final step of
72°C for 3 min.
Reverse transcription-quantitative (RT-q)
PCR
KG cells transfected with TRIB2 and miR-33a-5p
expression vectors were collected and RNA was extracted using
TRIzol® reagent (Thermo Fisher Scientific, Inc.).
Reverse transcription of RNA was performed to generate cDNA using
PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Inc.)
according to the manufacturer's protocol. Reaction system was
determined through RT-qPCR by employing the 7500 Real-Time PCR
System using TB Green (Takara Biotechnology Co., Ltd.). The qPCR
conditions were as follows: initial denaturation at 95°C for 30
sec; 40 cycles of 95°C for 10 sec, 60°C annealing for 20 sec and
extension at 72°C for 20 sec. The 2-ΔΔCq method was used
to calculate relative expression and fold change. Primers were
purchased from Guangzhou RiboBio Co., Ltd. The sequences of the
primers were as follows: TRIB2 forward, 5'-CTC CGA ACT TGT CGC ATT
G-3' and reverse, 5'-CAC ATA GGC TTT GGT CTC AC-3'; GAPDH forward,
5'-GAC AGT CAG CCG CAT CTT CTT-3' and reverse, 5'-AAT CCG TTG ACT
CCG ACC TTC-3'; miR-33a-5p forward, 5'-GTG CAT TGT AGT TGC ATT-3'
and reverse, 5'-AAC ATG TAC AGT CCA TGG ATG-3'; and 5s rRNA
forward, 5'-GCC ATA CCA CCC TGA ACG-3' and reverse, 5'-AAC ATG TAC
AGT CCA TGG ATG-3'. GAPDH and 5S rRNA were used as the reference
genes.
Animal model
The animal experiments were reviewed and approved
(approval no. 2019-11-06) by the ethics committee of Binzhou
Medical University (Yantai, China). Cell lines were injected
subcutaneously into the armpits of approximately 20 g female nude
mice at 4-5 weeks of age. A total of five mice were injected for
each group of cells. They were kept in a laminar airflow cabinet
under specific pathogen-free conditions with a controlled
temperature (23±2°C), humidity (40-70%) with free access to food
and water. The number of injected cells was 5x106. Cells
were resuspended with PBS. Mice were anesthetized with 50 mg/kg
sodium pentobarbital. A total of 70 μl cells and 30
μl Matrigel (BD Biosciences) mixed together were injected
subcutaneously into nude mice. When the tumors became visible to
naked eyes after ~a week, tumor volume was calculated every 4 days
and a tumor growth curve was plotted. Finally, the mice were
anesthetized by inhalation of 3% isoflurane and sacrificed by
cervical dislocation. The mice were euthanized on day 28 after
cancer cell injection. The weight of tumors was then measured and
the results were analyzed.
KG cells stably transfected with TRIB2, NC and
si-TRIB2 were directly inoculated into the NOD scid gamma (NSG)
mice caudal vein. A total of four mice were injected for each group
of cells. The number of injected cells was 2x106/100
μl. Peripheral blood was collected from the tail vein
regularly for blood cell count and blood smear observation in order
to monitor the successful establishment of the model. After
successful modeling, one group was treated with 50 mg/kg IM and one
with 0.9% normal saline.
Determination of blood parameters
One week after constructing leukemia model mice,
mice were subjected to tail cutting for blood collection. The blood
was collected in EP tubes containing EDTA-K2 anticoagulant for
upper machine analysis within 4 h. Blood cell counts and
classification were determined with a fully automated blood cell
analyzer (Hemavet®950, Drew Scientific, Inc.).
Statistical analysis
Normality tests were conducted on all data with
Shapiro-Wilk test, and all data were assessed for normality of
distribution. All experimental data are expressed as the mean ±
standard deviation (mean ± SD). Graphs and statistical analyses
were performed using GraphPad Prism 6 (GraphPad Software, Inc.).
The mRNA expression datasets of leukemias were obtained from
Oncomine. The Kaplan-Meier method was used to evaluate the
association between OS and TRIB2 and draw the survival curve.
Patients were separated into two groups using median gene
expression, and the log-rank test was used to compare the
differences between groups. Statistical analysis was performed
using an appropriate analysis of variance (ANOVA) when more than
two groups were compared. Tukey's post hoc test was used following
ANOVA. Comparisons between the control and experimental groups were
performed using the Student's t-test. For unpaired samples,
unpaired t-tests were used. For paired datasets, paired t-test were
used. P<0.05 was considered to indicate a statistically
significant difference.
Results
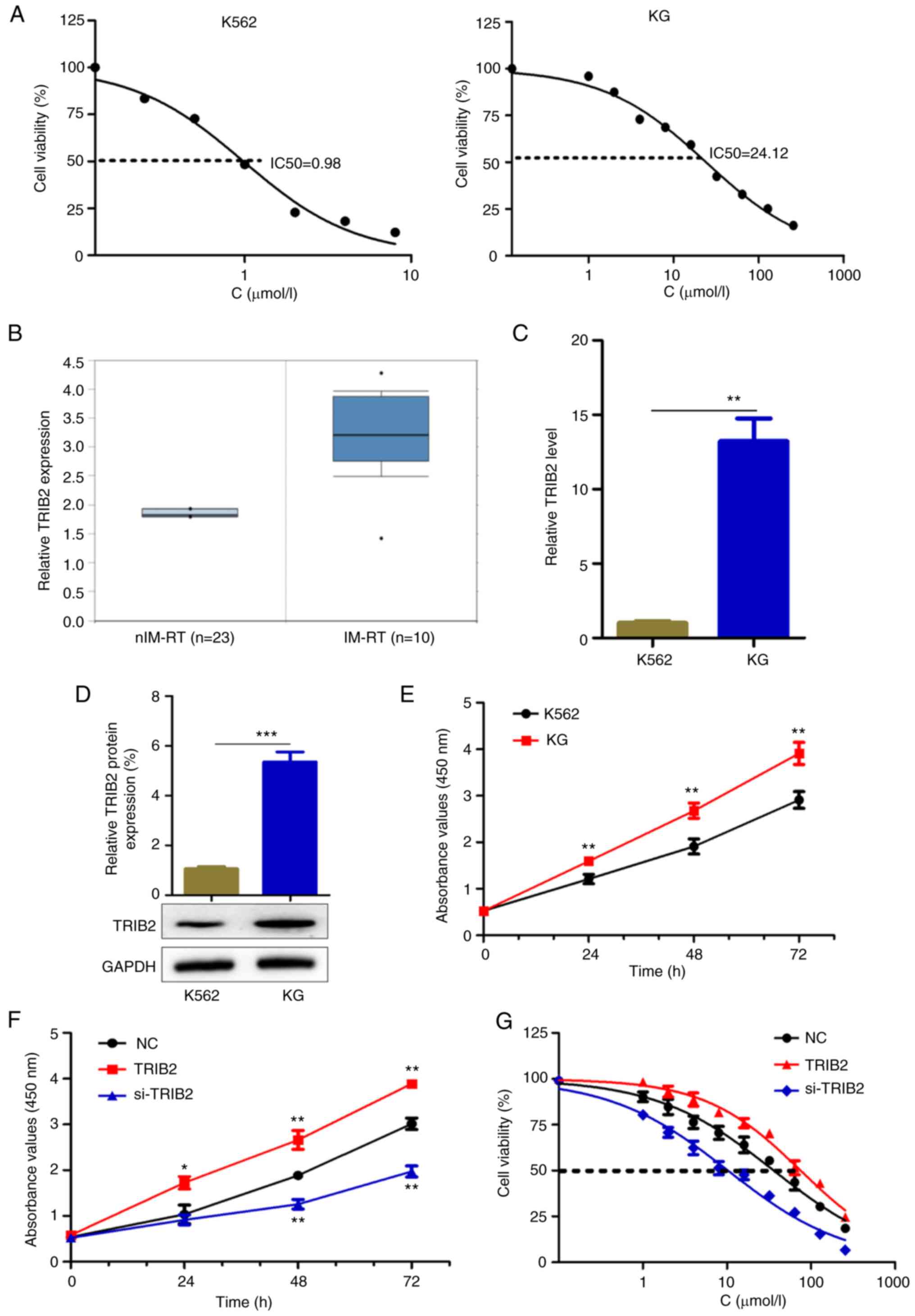
TRIB2 promotes the proliferation of KG
cells and increases the IC50 of IM
The IM resistance index of KG cells was 24.61
(Fig. 1A). The protein expression
levels of TRIB2 in IM-resistant CML and CML specimens were compared
using the Oncomine database. The results revealed that the
expression level of TRIB2 was higher in IM-resistant specimens than
in normal CML specimens (Fig.
1B). Subsequently, the expression of TRIB2 in cells was
examined at the mRNA and protein levels and was found to be higher
in KG cells than in K562 cells (Fig.
1C and D). KG cells proliferated faster than K562 cells, as
evidenced CCK-8 assay (Fig. 1E),
which suggested that TRIB2 might promote the growth of KG cells. To
further investigate the role played by TRIB2 in KG cells, TRIB2 was
successfully overexpressed or knocked down in KG cells, which was
verified experimentally using RT-qPCR (Fig. S1A), Western blot (Fig. S1B) and fluorescence microscopy
(Fig. S1C). TRIB2 was found to
promote cell proliferation (Fig.
1F), and increase the IC50 of IM (Fig. 1G). Therefore, TRIB2 promotes
proliferation of IM-resistant cells and increases the
IC50 of IM in these cells.
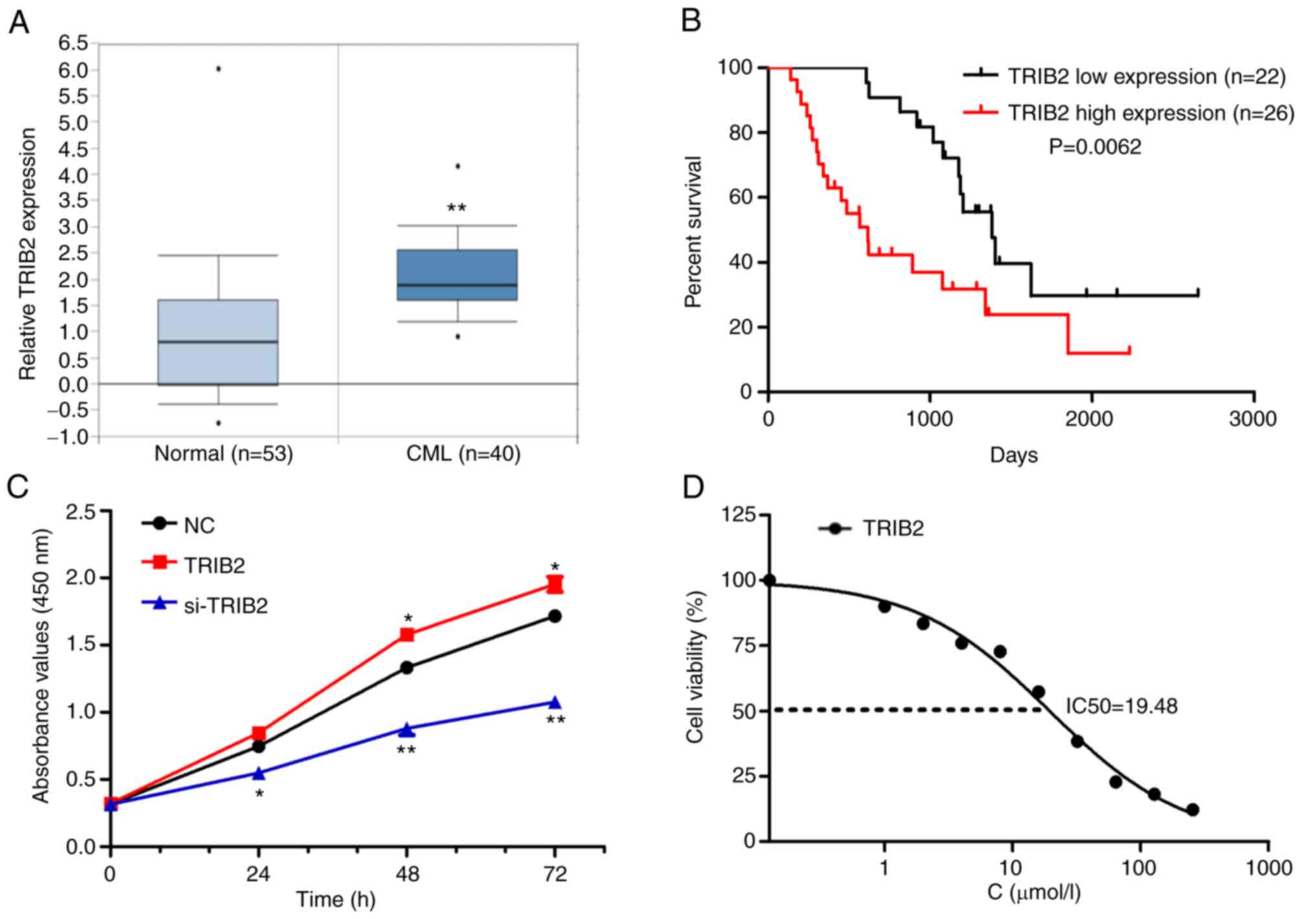
TRIB2 promotes K562 cell proliferation
and increases IM resistance
The Oncomine database showed that TRIB2 was highly
expressed in patients with CML (Fig.
2A). Further analysis of CML datasets from the Oncomine
database revealed that patients with high expression of TRIB2
experienced significantly worse overall survival (Fig. 2B). To further investigate the role
played by TRIB2 in K562 cells, TRIB2 was successfully overexpressed
or knocked down in K562 cells, which was verified using RT-qPCR
(Fig. S1D), Western blot
(Fig. S1E), and photographs by
green fluorescence microscope (Fig.
S1F). TRIB2 was found to promote cell proliferation (Fig. 2F), but also increased the
IC50 of the cells (Fig.
2G). Therefore, TRIB2 promoted K562 cell proliferation and
increased IM resistance.
TRIB2 promotes proliferation and IM
resistance of KG cells in vivo
To investigate the role played by TRIB2 in
leukemia-bearing mice after intravenous injection, a KG leukemia
mice model was established (Fig.
3A). The TRIB2-related stable transgenic cell line was injected
into the mice through the tail vein, and blood smear and count were
used to confirm the successful modeling of leukemia model mice
(Fig. 3B and C). It was found
that TRIB2 could promote the proliferation of KG cells by a blood
cell sorter (Fig. 3D). In
addition, the TRIB2 overexpression group had worse survival in the
model mice (Fig. 3E). The
aforementioned experiments revealed that TRIB2 promoted the
proliferation of KG cells within the model mice. The role of TRIB2
on KG tumor generation was further confirmed in vivo by
transplanting tumors in mice. Plotting the growth curve indicated
that the volume of tumor overexpressing TRIB2 showed a faster
growth than the control group, whereas knockdown of TRIB2 revealed
a slower growth (Fig. 3F and G).
Overexpression of TRIB2 promoted tumors with high weight, whereas
low-expression TRIB2 promoted tumors with low weight (Fig. 3H). Thus, these xenograft
experiments demonstrated that TRIB2 can promote the proliferation
of IM-resistant cells.
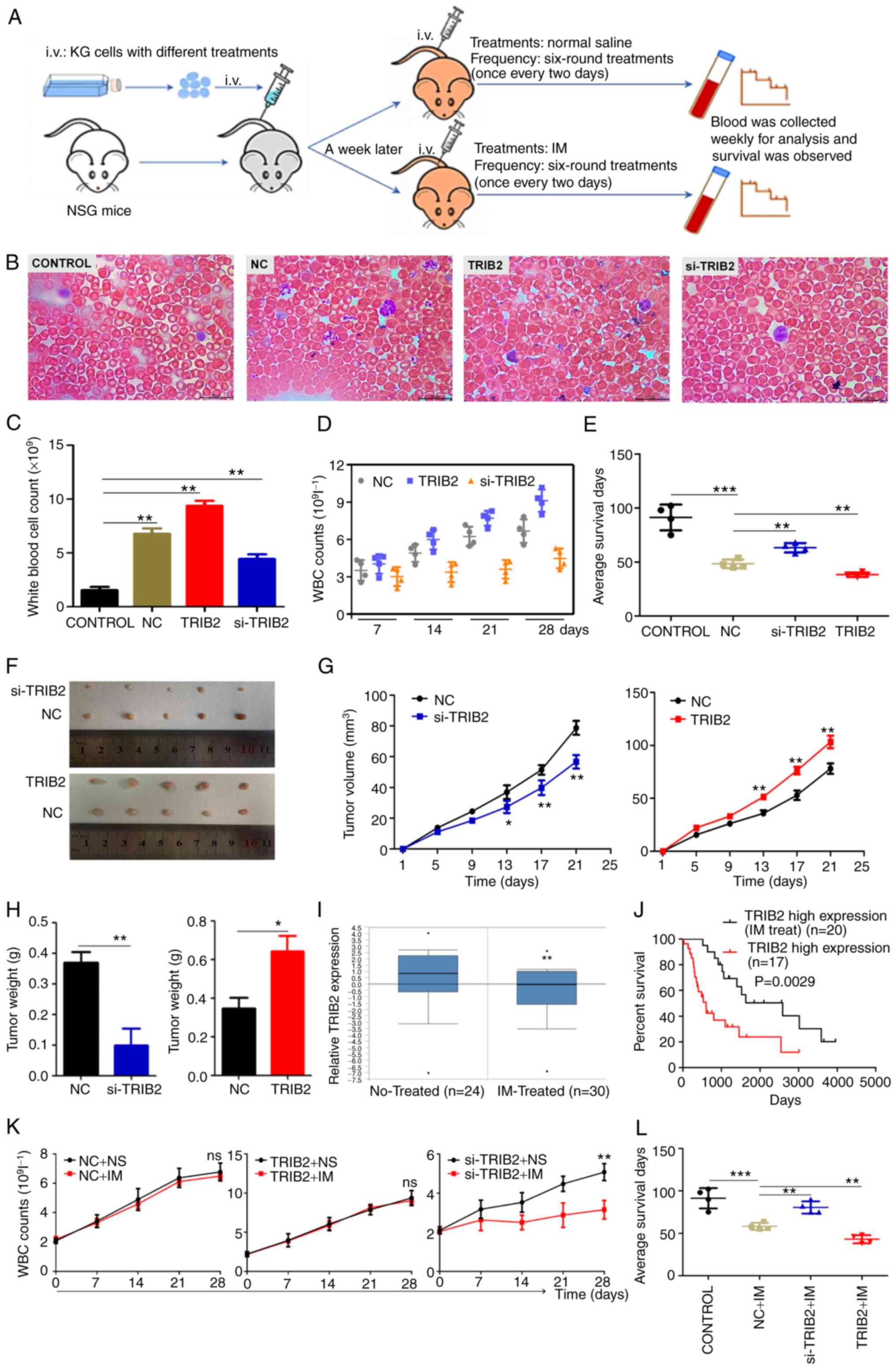 | Figure 3TRIB2 promotes the proliferation and
IM resistance of KG cells in vivo. (A) Illustration of
experimental design for assessing the effect of TRIB2 in
vivo. (B and C) Successful modeling of chronic myeloid leukemia
model mice was confirmed by blood counts and microscopic
observation of blood smears (magnification, x1,000). (D) WBC counts
of leukemia-bearing mice at different time points after various
treatments. The number of WBC from mice overexpressing TRIB2
leukemia model grew at the fastest rate. (E) Compared with NC
group, TRIB2 overexpression led to poor survival in the model mice,
whereas TRIB2 knockdown resulted in improved survival. (F and G) In
tumor-bearing nude mice, the volume of xenograft tumors
overexpressing TRIB2 showed faster growth, while knockdown TRIB2
exhibited slower growth. (H) In tumor-bearing nude mice,
overexpression TRIB2 promoted xenograft tumors with high weight,
and TRIB2 knockdown promoted those with low weight. (I and J)
Analysis of the Oncomine database showed decreased TRIB2 expression
and improved survival after IM treatment. (K and L) si-TRIB2
increased the sensitivity of KG cells to IM in leukemia model mice,
prolonging their survival. *P<0.05,
**P<0.01 and ***P<0.001. i.v.,
intravenous; TRIB2, tribbles pseudokinase 2; IM, imatinib; WBC,
white blood cell; si-, small interfering; NC, negative control. |
The relationship between TRIB2 and IM resistance was
investigated using the Oncomine database to explore the drug
sensitivity of TRIB2 on KG cells in vivo. It was revealed
that TRIB2 expression decreased after IM treatment and survival was
improved (Fig. 3I and J). Cell
proliferation was examined following IM treatment, and the results
identified that there was no change in cell proliferation after
overexpression of TRIB2. Both the NC group and si-TRIB2 group
showed slower cell proliferation under IM treatment, and the
si-TRIB2 group revealed the slowest cell proliferation. This
indicated that knockdown of TRIB2 rendered KG cells more sensitive
to IM (Fig. 3K). In agreement
with database studies, IM was used to treat leukemia mice. It was
found that drug treatment could prolong the survival time of mice
in each group (Fig. 3L), but the
life prolonging rate of the low-expression TRIB2 group was
significantly higher than that of the high-expression TRIB2 group
(Table II). This finding
indicated that TRIB2 promoted proliferation and resistance to IM of
KG cells in vivo.
| Table IISurvival time and life extension rate
of model mice. |
Table II
Survival time and life extension rate
of model mice.
| Group | Average survival,
days | Extension rate,
% |
|---|
| Control | 91.25±2.533 | |
| TRIB2-NC | 48.25±3.750 | 19.71 |
| TRIB2-NC + IM | 58.25±2.345 | |
| si-TRIB2 | 63.25±3.473 | 27.17 |
| si-TRIB2 + IM | 80.50±2.598 | |
| TRIB2 | 38.25±2.016 | 12.40 |
| TRIB2 + IM | 43.00±2.380 | |
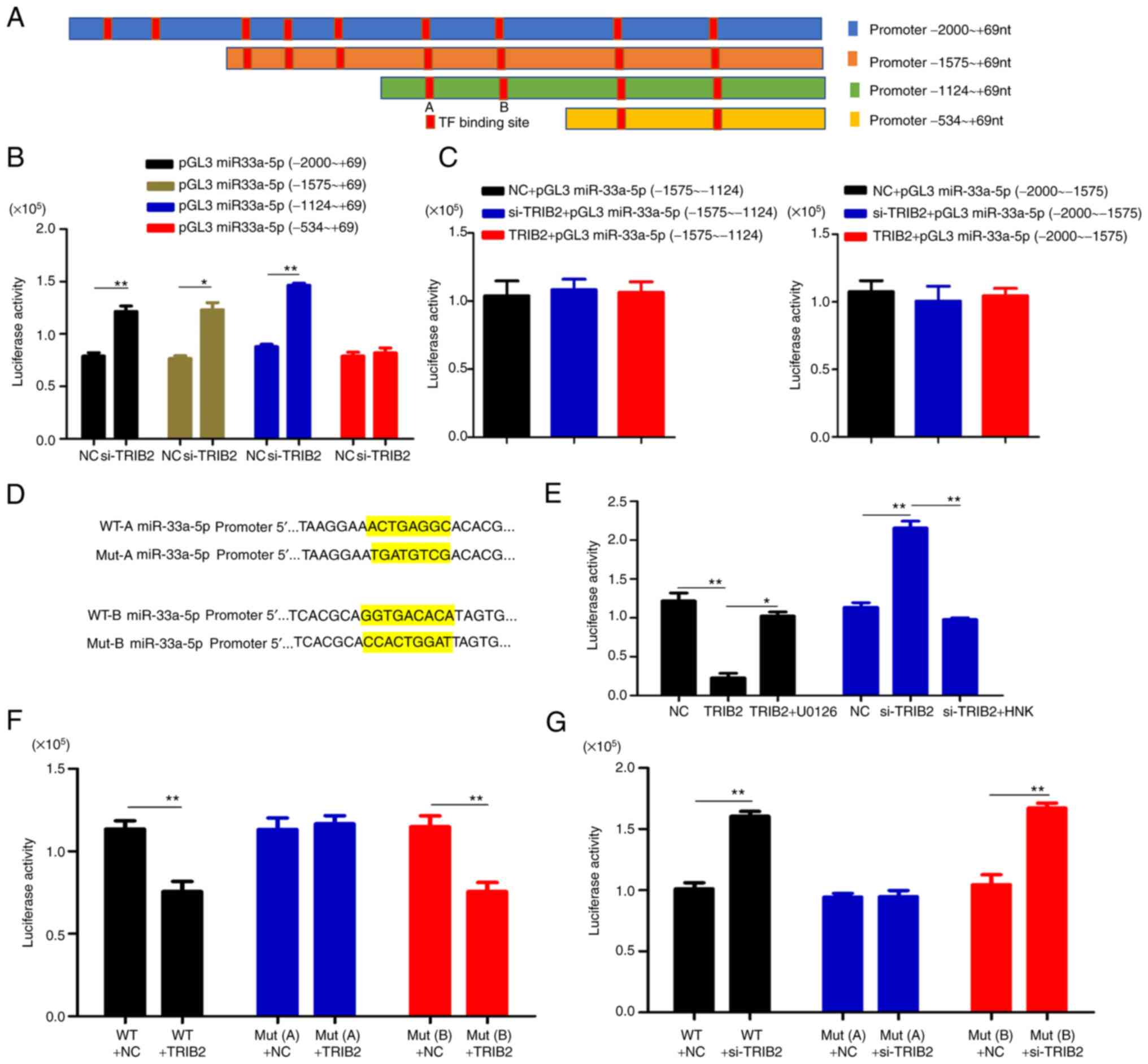
TRIB2 regulates the expression of
miR-33a-5p through the ERK/c-Fos signaling pathway
The aforementioned results indicated that TRIB2 can
promote KG cell proliferation and enhance cell drug resistance. The
mechanism by which TRIB2 exerts its effects was the focus of the
next experiment. It was found that TRIB2 could downregulate the
promoter activity of miR-33a-5p by dual-luciferase reporter assay,
which was again further confirmed by RT-qPCR (Fig. 4A). Potential TFs that bind to the
miR-33a-5p promoter were predicted via JASPAR and c-Fos was
screened by prophase experiments to investigate the effect of TRIB2
on miR-33a-5p expression (Fig.
4B). The website prediction results were validated by ChIP
assay, which demonstrated c-Fos as a TF for miR-33a-5p promoter
(Fig. 4C). c-Fos was successfully
transfected and overexpressed in KG and K562 cells (Fig. S1G-H). Subsequently, it was found
that c-Fos inhibited miR-33a-5p expression and therefore played an
inhibitory role in transcriptional effects (Fig. 4D). In addition, c-Fos was found to
bind to TATA box binding protein (TBP) and suppress transcription
(Fig. 4E). Notably,
co-precipitation and immunoblotting experiments were performed to
verify the inability of binding interaction between TRIB2 and c-Fos
(Fig. 4F). Through further
experiments, it was identified that the ERK signaling pathway could
decrease miR-33a-5p promoter activity and then affect the
expression of miR-33a-5p (Fig.
4G). The activation of the ERK signaling pathway can be
regulated by TRIB2, which was confirmed experimentally by western
blotting (Fig. 4H). Finally, the
ERK pathway blocker U0126 and activator HNK were applied to the
cells and detected the expression of related proteins. The results
showed that UO126 inhibited and HNK increased ERK and c-Fos
activity. It was further revealed that TRIB2 could activate the ERK
pathway and then promote c-Fos expression (Fig. 4I-L). It was similarly concluded
that TRIB2 could promote c-Fos expression by western blot analysis
of transplanted tumors in nude mice from in vivo experiments
(Fig. 4M). The aforementioned
results suggested that TRIB2 regulated c-Fos expression through the
ERK signaling pathway (Fig.
4N).
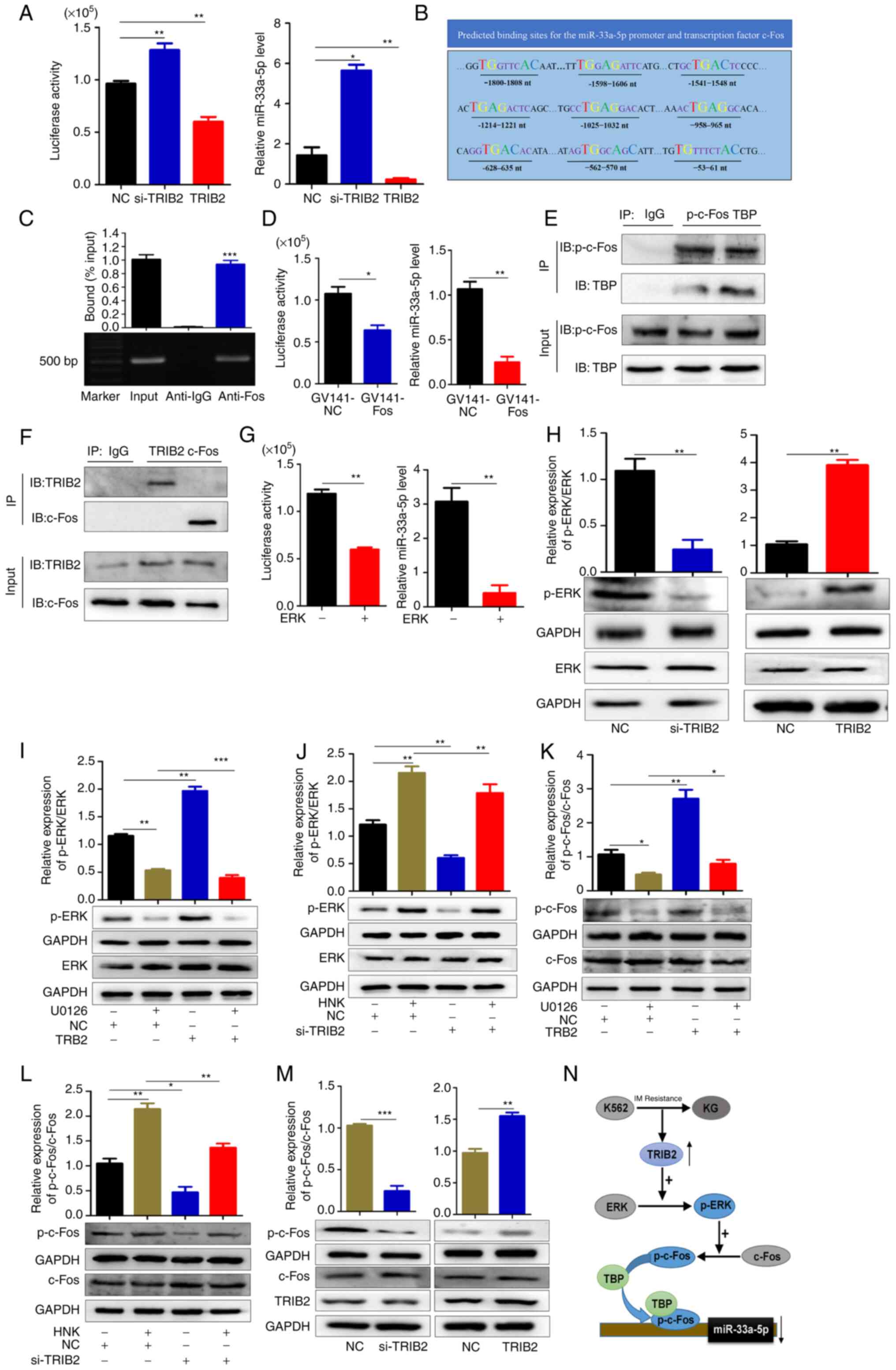 | Figure 4TRIB2 regulates the expression of
miR-33a-5p through the ERK/c-Fos pathway in KG cells. (A) TRIB2
downregulated miR-33a-5p expression as revealed by luciferase
activity assay and RT-qPCR. (B) Prediction of TF binding sites of
miR-33a-5p promoter by JASPAR. (C) The ability of c-Fos as a TF and
miR-33a-5p promoter region to bind was verified using chromatin
immunoprecipitation experiments. (D) GV141-NC and GV141-Fos were
transfected within KG cells, and the expression of miR-33a-5p after
overexpression of c-Fos was detected by luciferase activity assay
and RT-qPCR. (E) Co-IP verified that p-c-Fos and TBP were able to
physically bind. (F) Co-IP verified that c-Fos and TRIB2 could not
physically associate. (G) Overexpression of ERK decreased
miR-33a-5p promoter activity, then downregulated miR-33a-5p
expression. (H-J) TRIB2 promoted the phosphorylation of ERK, and
this effect could be blocked by U0126. Knockdown of TRIB2 inhibited
ERK phosphorylation, and this inhibitory effect could be relieved
by HNK. TRIB2 can function through the ERK signaling pathway as
revealed by western blotting. TRIB2 promoted the phosphorylation of
ERK and this effect could be blocked by U0126. Knockdown of TRIB2
inhibited ERK phosphorylation and this inhibitory effect could be
relieved by HNK. It was therefore hypothesized that TRIB2 might
function through ERK signaling pathway. (K and L) TRIB2 could
activate the ERK pathway and then promote c-Fos expression as
revealed by western blotting. (M) The expression and
phosphorylation levels of c-Fos and the expression of TRIB2 in mice
xenografts were detected by western blotting. (N) Proposed model:
Elevated TRIB2 expression in KG cells promoted phosphorylation of
ERK, which in turn activated c-Fos expression, and c-Fos binding to
TBP inhibited miR-33a-5p and transcriptionally repressed miR-33a-5p
expression. *P<0.05, **P<0.01 and
***P<0.001. TRIB2, tribbles pseudokinase 2; miR,
microRNA; RT-qPCR, reverse transcription-quantitative PCR; si-,
small interfering; NC, negative control; TF, transcription factor;
TBP, TATA box binding protein. |
Specific site of action of c-Fos on the
miR-33a-5p promoter
The presence of nine c-Fos possible binding sites on
the miR-33a-5p promoter was predicted by JASPAR. To investigate
which binding site specifically plays a role, by location of the
nine binding sites, truncations of the miR-33a-5p promoter were
made and constructed into a luciferase vector. A total of four
segments of vectors were constructed -2000 ~+69 nt, -1575 ~+69 nt,
-1124 ~+69 nt, and -534 ~+69 nt (Fig.
5A). The site of action was determined by the luciferase assay
to be in the −534 to 1124 region (Fig. 5B and C). Two binding sites A
(-958-965 nt) and B (-628-635 nt) were available for this fragment,
and both sites were mutated (Fig.
5D). Further studies revealed that the luciferase activity was
not altered when point mutation A was used, but it was altered when
point mutation B was used (Fig.
5E-G). Description -958-965 nt is the specific site where c-Fos
binds the miR-33a-5p promoter.
miR-33a-5p inhibits proliferation of KG
cells and decreases IC50 by suppressing HMGA2
expression
The expression of miR-33a-5p in cells was detected
by RT-qPCR to investigate the role played by miR-33a-5p in KG
cells. The results showed that miR-33a-5p expression was decreased
in KG cells compared with that in K562 cells (Fig. 6A). To verify the role of
miR-33a-5p in drug-resistant cells, miR-33a-5p was successfully
overexpressed and knocked down in cells, which was verified
experimentally by RT-qPCR (Fig.
S1I-J) and fluorescence microscopy (Fig. S1K). It was found that miR-33a-5p
could inhibit KG cell proliferation and reduce IC50
(Fig. 6B and C). A KG cell
xenograft mouse model was established to evaluate the role of
miR-33a-5p in KG cell proliferation in vivo. Plotting the
growth curve indicated that the volume of tumor knockdown
miR-33a-5p showed a faster growth than the control group, whereas
overexpression of miR-33a-5p showed a slower growth (Fig. 6D and E). The tumor weight was then
measured and similar conclusions were reached (Fig. 6F and G). These results supported
the conclusion that miR-33a-5p inhibits KG cell proliferation in
vivo.
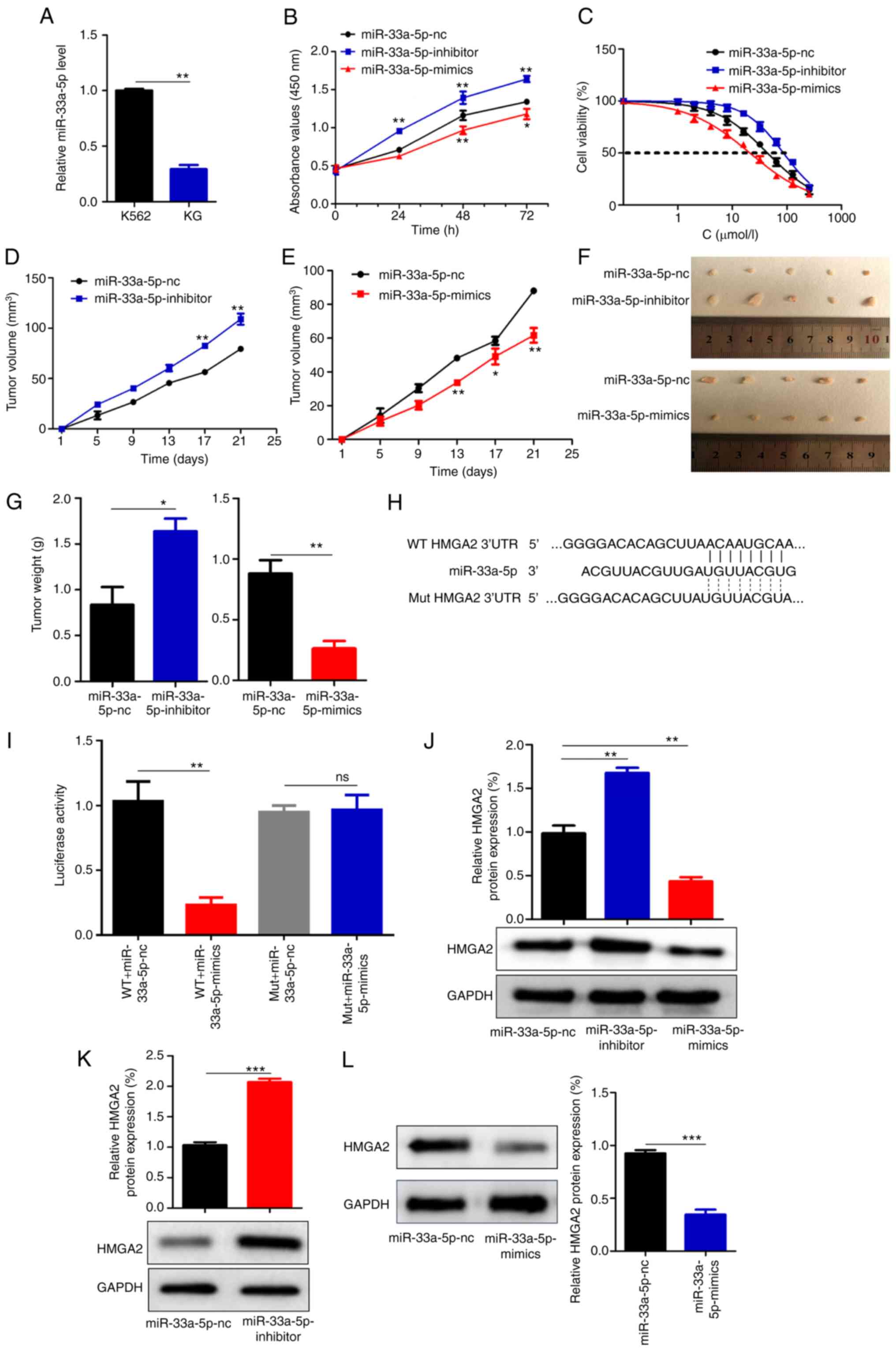 | Figure 6miR-33a-5p inhibits the proliferation
of KG cells and decreases the IC50 of IM. (A) miR-33a-5p
expression was decreased in KG cells compared with that in K562
cells. (B and C) miR-33a-5p inhibits KG cell proliferation and
reduces the IC50 of IM. (D and E) In tumor bearing nude
mice, the volume of xenograft tumor knockdown miR-33a-5p showed
faster growth, while overexpression miR-33a-5p showed slower
growth. (F and G) In tumor-bearing nude mice, transfection of the
miR-33a-5p inhibitors increased tumor weight, whereas transfection
of the miR-33a-5p reduced tumor weight. (H) The target binding
sites between miR-33a-5p and HMGA2 were indicated by TargetScan
prediction, a binding site was found at positions 262-269 of HMGA2.
(I) The dual-luciferase assay showed that miR-33a-5p could
specifically bind to HMGA2, and HMGA2 was the target gene of
miR-33a-5p. (J) Western blot analysis showed that miR-33a-5p was
able to downregulate HMGA2 expression levels in KG cells. (K and L)
Western blotting showed that miR-33a-5p was able to downregulate
HMGA2 expression levels in xenografts of nude mice.
*P<0.05, **P<0.01 and
***P<0.001. miR, microRNA; NC, negative control; ns,
no significance; WT, wild-type; Mut, mutant. |
To investigate the mechanism of miR-33a-5p, the
target gene of miR-33a-5p was predicted on the prediction website
TargetScan. The results showed that there was a target binding site
between miR-33a-5p and HMGA2 (Fig.
6H). Moreover, the target gene of miR-33a was detected by
dual-luciferase reporter gene assay (Fig. 6I): miR-33a-5p could specifically
bind to HMGA2, and HMGA2 was the target gene of miR-33a-5p. Further
investigation by western blotting revealed that miR-33a-5p
inhibited HMGA2 expression in KG cells (Fig. 6J). The same results were also
found in nude mice xenografts (Fig.
6K and L). Through the aforementioned studies, it was
identified that miR-33a-5p can slow the proliferation of KG cells
and increase the drug sensitivity of KG cells to IM by decreasing
the expression of HMGA2.
Discussion
CML is a clinical hematological disease, and
aberrant BCR-ABL fusion transcripts are a hallmark of the disease.
Thus, IM is currently the first-line therapy to target the BCR-ABL
fusion gene in CML (27).
Although the application of IM is expected to overcome CML, a large
body of clinical data indicated that patients develop acquired IM
resistance as treatment duration increases (28,29). In response to the problem of drug
resistance in leukemia, Zheng et al (30) found that chloroquine combined with
IM overcame IM resistance through the MAPK/ERK pathway. S100A8
promotes drug resistance by promoting autophagy in leukemia cells,
and S100A8 may be a novel target to improve leukemia drug
resistance (31). At the same
time ST6GAL1 had a reversal effect on multidrug resistance in human
leukemia by regulating the PI3K/Akt pathway and the expression of
P-gp and MRP1 (32). Research
data treatment against leukemia drug resistance has focused on
reversing drug resistance by targeting certain drug resistance
proteins. However, whether these target proteins and their
signaling pathways can be smoothly applied in the clinic and then
exert clinical efficacy requires a long time of clinical
experiments and exploration.
Tribbles are members of the pseudokinase family and
were shown to regulate numerous cellular functions and were
involved in various cellular processes including proliferation,
drug resistance, metabolism and oncogenic transformation (33). In the present study, it was
confirmed that TRIB2 promoted the proliferation of IM-resistant CML
cells and increased their resistance to drugs. Furthermore, the
role of TRIB2 in vivo was investigated to determine whether
TRIB2 can serve as a potential target for the clinical treatment of
IM resistance in CML. An IM-resistant CML model mice was
established to mimic clinical resistant patients and it was
experimentally confirmed that TRIB2 promoted the proliferation of
IM-resistant cells and reduced drug sensitivity in model mice,
while knockdown of TRIB2 reversed its drug resistance. Illustrating
the important clinical role of TRIB2 in IM-resistant CML patients
suggested that TRIB2 may be a novel target for the treatment of
IM-resistant CML patients. TKIs are well known as kinase inhibitors
that inhibit the activity of ABL tyrosine kinases (34,35). Similar to TKIs, whether a
'pseudokinase inhibitor' interferes with TRIB2 expression and then
IM resistance in CML is unknown. Thus, addressing the question of
clinical resistance is important. This aspect may require extensive
basic research and clinical experiments for validation. Therefore,
TRIB2 has potential as a resistance gene to treat IM resistance in
CML.
A series of experiments was performed in the cell
lines to further explore the oncogenic and drug sensitivity
reducing roles played by TRIB2 in IM-resistant CML cells. It was
found that TRIB2 acted by affecting miR-33a-5p. Accumulating
evidence has indicated that miR-33a-5p and chemoresistance of
tumors are associated and are downregulated in numerous different
types of cancers to participate in the progression of tumors. For
example, miR-33a-5p-based therapy may be a promising strategy for
overgrowing the chemoresistance of TNBC (36). A possible mechanism of
chemoresistance in liver cancer is downregulation of miR-33a-5p
(37). In the present study, it
was similarly found that miR-33a-5p could inhibit KG cells
proliferation and enhance the sensitivity of KG cells to IM. The
possible mechanism by which miR-33a-5p functions is through
inhibiting HMGA2 expression. It has been reported that reducing
HMGA2 expression levels can render tumor resistant cells more
sensitive to the drug (38,39). Thus, miR-33a-5p has emerged as a
key factor in the study of drug resistance in IM-resistant CML
cells. ~20 clinical trials using miRNA and siRNA-based therapies
have been initiated to date. Therapeutic miRNAs and siRNAs have
moved from the laboratory to the clinic as the next generation of
medicine. MiR-33a-5p-based therapies hold broad clinical promise
for overcoming IM resistance in CML.
C-Fos, as an upstream factor of miR-33a-5p,
regulates its expression and may provide a novel strategy to
overcome IM resistance in CML clinically. It was experimentally
confirmed that TRIB2 could affect the expression of miR-33a-5p by
regulating c-Fos, which was a TF for miR-33a-5p. Given that the TF
c-Fos can inhibit the activity of the miR-33a-5p promoter, it was
hypothesized that c-Fos can physically bind to the TBP, which
prevents the binding of TF IID to the promoter to inhibit
transcription. The aforementioned hypothesis was verified by co-IP
experiments and a literature review by Metz et al (40) that c-Fos can physically associate
with TBP. PARP inhibitors (PARPi) were the first approved
anticancer drugs, and they can improve the sensitivity of AML to
chemotherapeutic agents as protein inhibitors (41). Similar to PARPi, inhibiting the
expression of c-Fos may become a new direction and strategy for the
clinical treatment of IM resistance in CML. In addition, Li et
al (42) reported that the
ERK/c-Fos signaling pathway plays a role in the proliferation and
migration of colon cancer. This is consistent with the present
study, in which it was also demonstrated that ERK/c-Fos signaling
promoted IM resistance in KG cells.
In conclusion, TRIB2 can regulate c-Fos expression
through the ERK signaling pathway and c-Fos, as a transcriptional
suppressor of miR-33a-5p, could inhibit the transcription of
miR-33a-5p. Thus, TRIB2 regulates the expression of miR-33a-5p
through the ERK/c-Fos pathway to affect the IM-resistance of CML
cells. Ultimately miR-33a-5p exerts its effect by suppressing HMGA2
expression. The present study is promising for the clinical
treatment of IM resistance in CML and can broaden the ideas of
clinical treatment of IM resistance, find improved clinical
treatment strategies for IM resistance, and address the problem of
clinical patient resistance. Due to the limitations of experiments
at the cellular or animal level, there is still a certain distance
from cellular and animal studies to clinical applications.
Therefore, this needs to be addressed by future collaborative
research efforts between clinicians and scientists.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS, YL and SX performed study concept and design.
HS, YL and XW performed development of methodology and writing,
review and revision of the manuscript. HS, RW, YY, XZ, SR, DL, GS
and HC provided acquisition, analysis and interpretation of data
and conducted statistical analysis. HS, SX and YS confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The animal experiments were reviewed and approved
(approval no. 2019-11-06) by the ethics committee of Binzhou
Medical University (Yantai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by The National Natural Science
Foundation of China (grant nos. 81800169, 31371321 and 82002604),
The Natural Science Foundation of Shandong (grant no. ZR2019MH022),
The Support Plan For Youth Entrepreneurship and Technology of
Colleges and Universities in Shandong (grant no. 2019KJK014), The
Shandong Province Taishan Scholar Project (grant no. ts201712067)
and The Yantai Science and Technology Committee (grant no.
2018XSCC051).
References
|
1
|
Luo Z, Gao M, Huang N, Wang X, Yang Z,
Yang H, Huang Z and Feng W: Efficient disruption of bcr-abl gene by
CRISPR RNA-guided FokI nucleases depresses the oncogenesis of
chronic myeloid leukemia cells. J Exp Clin Cancer Res. 38:2242019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heaney NB, Pellicano F, Zhang B, Crawford
L, Chu S, Kazmi SM, Allan EK, Jorgensen HG, Irvine AE, Bhatia R and
Holyoake TL: Bortezomib induces apoptosis in primitive chronic
myeloid leukemia cells including LTC-IC and NOD/SCID repopulating
cells. Blood. 115:2241–2250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen SH, Hsieh YY, Tzeng HE, Lin CY, Hsu
KW, Chiang YS, Lin SM, Su MJ, Hsieh WS and Lee CH: ABL genomic
editing sufficiently abolishes oncogenesis of human chronic myeloid
leukemia cells in vitro and in vivo. Cancers (Basel). 12:13992020.
View Article : Google Scholar :
|
|
4
|
Tauer JT, Hofbauer LC, Jung R, Gerdes S,
Glauche I, Erben RG and Suttorp M: Impact of long-term exposure to
the tyrosine kinase inhibitor imatinib on the skeleton of growing
rats. PLoS One. 10:e01311922015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cortes J, Hochhaus A, Hughes T and
Kantarjian H: Front-line and salvage therapies with tyrosine kinase
inhibitors and other treatments in chronic myeloid leukemia. J Clin
Oncol. 29:524–531. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang XY, Zhang XH, Peng L, Liu Z, Yang YX,
He ZX, Dang HW and Zhou SF: Bardoxolone methyl (CDDO-Me or RTA402)
induces cell cycle arrest, apoptosis and autophagy via
PI3K/Akt/mTOR and p38 MAPK/Erk1/2 signaling pathways in K562 cells.
Am J Transl Res. 9:4652–4672. 2017.PubMed/NCBI
|
|
7
|
Fan Z, Luo H, Zhou J, Wang F, Zhang W,
Wang J, Li S, Lai Q, Xu Y, Wang G, et al: Checkpoint kinase1
inhibition and etoposide exhibit a strong synergistic anticancer
effect on chronic myeloid leukemia cell line K562 by impairing
homologous recombination DNA damage repair. Oncol Rep.
44:2152–2164. 2020.PubMed/NCBI
|
|
8
|
Mayoral-Varo V, Jimenez L and Link W: The
critical role of TRIB2 in cancer and therapy resistance. Cancers
(Basel). 13:27012021. View Article : Google Scholar
|
|
9
|
Hou Z, Guo K, Sun X, Hu F, Chen Q, Luo X,
Wang G, Hu J and Sun L: TRIB2 functions as novel oncogene in
colorectal cancer by blocking cellular senescence through AP4/p21
signaling. Mol Cancer. 17:1722018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim HS, Oh SH, Kim JH, Sohn WJ, Kim JY,
Kim DH, Choi SU, Park KM, Ryoo ZY, Park TS and Lee S: TRIB2
regulates the differentiation of MLL-TET1 transduced myeloid
progenitor cells. J Mol Med (Berl). 96:1267–1277. 2018. View Article : Google Scholar
|
|
11
|
Liang Y, Yu D, Perez-Soler R, Klostergaard
J and Zou Y: TRIB2 contributes to cisplatin resistance in small
cell lung cancer. Oncotarget. 8:109596–109608. 2017. View Article : Google Scholar
|
|
12
|
Liu Q, Zhang W, Luo L, Han K, Liu R, Wei S
and Guo X: Long noncoding RNA TUG1 regulates the progression of
colorectal cancer through miR-542-3p/TRIB2 axis and Wnt/β-catenin
pathway. Diagn Pathol. 16:472021. View Article : Google Scholar
|
|
13
|
Link W: Tribbles breaking bad: TRIB2
suppresses FOXO and acts as an oncogenic protein in melanoma.
Biochem Soc Trans. 43:1085–1088. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Zuo J, Wahafu A, Wang MD, Li RC
and Xie WF: Combined elevation of TRIB2 and MAP3K1 indicates poor
prognosis and chemoresistance to temozolomide in glioblastoma. CNS
Neurosci Ther. 26:297–308. 2020. View Article : Google Scholar :
|
|
15
|
Hill R, Madureira PA, Ferreira B, Baptista
I, Machado S, Colaco L, Dos Santos M, Liu N, Dopazo A, Ugurel S, et
al: TRIB2 confers resistance to anti-cancer therapy by activating
the serine/threonine protein kinase AKT. Nat Commun. 8:146872017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang HY, Lin YC, Li J, Huang KY, Shrestha
S, Hong HC, Tang Y, Chen YG, Jin CN, Yu Y, et al: MiRTarBase 2020:
Updates to the experimentally validated microRNA-target interaction
database. Nucleic Acids Res. 48(D1): D148–D154. 2020.
|
|
17
|
Ren D, Yang Q, Dai Y, Guo W, Du H, Song L
and Peng X: Oncogenic miR-210-3p promotes prostate cancer cell EMT
and bone metastasis via NF-ĸB signaling pathway. Mol Cancer.
16:1172017. View Article : Google Scholar
|
|
18
|
Li X, Strietz J, Bleilevens A, Stickeler E
and Maurer J: Chemotherapeutic stress influences
epithelial-mesenchymal transition and stemness in cancer stem cells
of triple-negative breast cancer. Int J Mol Sci. 21:4042020.
View Article : Google Scholar
|
|
19
|
Diaz-Martinez M, Benito-Jardon L, Alonso
L, Koetz-Ploch L, Hernando E and Teixido J: MiR-204-5p and
miR-211-5p contribute to BRAF inhibitor resistance in melanoma.
Cancer Res. 78:1017–1030. 2018. View Article : Google Scholar :
|
|
20
|
Yin J, Zeng A, Zhang Z, Shi Z, Yan W and
You Y: Exosomal transfer of miR-1238 contributes to
temozolomide-resistance in glioblastoma. EBioMedicine. 42:238–251.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Z, Zhou G, Tao F, Cao Y, Han W and Li
Q: circ-ZUFSP regulates trophoblasts migration and invasion through
sponging miR-203 to regulate STOX1 expression. Biochem Biophys Res
Commun. 531:472–479. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tu J, Zhao Z, Xu M, Chen M, Weng Q and Ji
J: LINC00460 promotes hepatocellular carcinoma development through
sponging miR-485-5p to up-regulate PAK1. Biomed Pharmacother.
118:1092132019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen J, Chen J, Sun B, Wu J and Du C:
ONECUT2 accelerates tumor proliferation through activating ROCK1
expression in gastric cancer. Cancer Manag Res. 12:6113–6121. 2020.
View Article : Google Scholar :
|
|
24
|
Racca AC, Prucca CG and Caputto BL: Fra-1
and c-Fos N-Terminal deletion mutants impair breast tumor cell
proliferation by blocking lipid synthesis activation. Front Oncol.
9:5442019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lou Z, Lin W, Zhao H, Jiao X, Wang C, Zhao
H, Liu L, Liu Y, Xie Q, Huang X, et al: Alkaline phosphatase
downregulation promotes lung adenocarcinoma metastasis via the
c-Myc/RhoA axis. Cancer Cell Int. 21:2172021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ban MJ, Byeon HK, Yang YJ, An S, Kim JW,
Kim JH, Kim DH, Yang J, Kee H and Koh YW: Fibroblast growth factor
receptor 3-mediated reactivation of ERK signaling promotes head and
neck squamous cancer cell insensitivity to MEK inhibition. Cancer
Sci. 109:3816–3825. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guilhot F, Druker B, Larson RA, Gathmann
I, So C, Waltzman R and O'Brien SG: High rates of durable response
are achieved with imatinib after treatment with interferon alpha
plus cytarabine: Results from the International randomized study of
interferon and STI571 (IRIS) trial. Haematologica. 94:1669–1675.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ciftci HI, Radwan MO, Ozturk SE, Ulusoy
NG, Sozer E, Ellakwa ED, Ocak Z, Can M, Ali TFS, Abd-Alla IH, et
al: Design, synthesis and biological evaluation of pentacyclic
triterpene derivatives: Optimization of Anti-ABL kinase activity.
Molecules. 24:35352019. View Article : Google Scholar
|
|
29
|
Nardi V, Naveiras O, Azam M and Daley GQ:
ICSBP-mediated immune protection against BCR-ABL-induced leukemia
requires the CCL6 and CCL9 chemokines. Blood. 113:3813–3820. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zheng S, Shu Y, Lu Y and Sun Y:
Chloroquine Combined with imatinib overcomes imatinib resistance in
gastrointestinal stromal tumors by inhibiting autophagy via the
MAPK/ERK pathway. Onco Targets Ther. 13:6433–6441. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang M, Zeng P, Kang R, Yu Y, Yang L, Tang
D and Cao L: S100A8 contributes to drug resistance by promoting
autophagy in leukemia cells. PLoS One. 9:e972422014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma H, Cheng L, Hao K, Li Y, Song X, Zhou H
and Jia L: Reversal effect of ST6GAL 1 on multidrug resistance in
human leukemia by regulating the PI3K/Akt pathway and the
expression of P-gp and MRP1. PLoS One. 9:e851132014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rome KS, Stein SJ, Kurachi M, Petrovic J,
Schwartz GW, Mack EA, Uljon S, Wu WW, DeHart AG, McClory SE, et al:
Trib1 regulates T cell differentiation during chronic infection by
restraining the effector program. J Exp Med. 217:e201908882020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pulte D, Jansen L and Brenner H: Changes
in long term survival after diagnosis with common hematologic
malignancies in the early 21st century. Blood Cancer J. 10:562020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bernardi S, Malagola M, Zanaglio C,
Polverelli N, Dereli Eke E, D'Adda M, Farina M, Bucelli C, Scaffidi
L, Toffoletti E, et al: Digital PCR improves the quantitation of
DMR and the selection of CML candidates to TKIs discontinuation.
Cancer Med. 8:2041–2055. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guan X, Gu S, Yuan M, Zheng X and Wu J:
MicroRNA-33a-5p overexpression sensitizes triple-negative breast
cancer to doxorubicin by inhibiting eIF5A2 and
epithelial-mesenchymal transition. Oncol Lett. 18:5986–5994.
2019.PubMed/NCBI
|
|
37
|
Meng W, Tai Y, Zhao H, Fu B, Zhang T, Liu
W, Li H, Yang Y, Zhang Q, Feng Y and Chen G: Downregulation of
miR-33a-5p in hepatocellular carcinoma: A possible mechanism for
chemotherapy resistance. Med Sci Monit. 23:1295–1304. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deng X, Kong F, Li S, Jiang H, Dong L, Xu
X, Zhang X, Yuan H, Xu Y, Chu Y, et al: A KLF4/PiHL/EZH2/HMGA2
regulatory axis and its function in promoting
oxaliplatin-resistance of colorectal cancer. Cell Death Dis.
12:4852021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han S, Han B, Li Z and Sun D:
Downregulation of long noncoding RNA CRNDE suppresses drug
resistance of liver cancer cells by increasing microRNA-33a
expression and decreasing HMGA2 expression. Cell Cycle.
18:2524–2537. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Metz R, Bannister AJ, Sutherland JA,
Hagemeier C, O'Rourke EC, Cook A, Bravo R and Kouzarides T:
c-Fos-induced activation of a TATA-box-containing promoter involves
direct contact with TATA-box-binding protein. Mol Cell Biol.
14:6021–6029. 1994.PubMed/NCBI
|
|
41
|
Rose M, Burgess JT, O'Byrne K, Richard DJ
and Bolderson E: PARP Inhibitors: Clinical relevance, mechanisms of
action and tumor resistance. Front Cell Dev Biol. 8:5646012020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li T, Li Z, Wan H, Tang X, Wang H, Chai F,
Zhang M and Wang B: Recurrence-associated long non-coding RNA
LNAPPCC facilitates colon cancer progression via forming a positive
feedback loop with PCDH7. Mol Ther Nucleic Acids. 20:545–557. 2020.
View Article : Google Scholar : PubMed/NCBI
|