Introduction
Pancreatic adenocarcinoma (PDAC) is a leading cause
of cancer-related mortality worldwide, with a 5-year survival rate
of ~5% (1). Because of delayed
diagnosis and early metastasis, only 15-20% of patients have a
chance to undergo surgery (2).
Therefore, systemic chemotherapy is applied in the majority of
patients with locally advanced and metastatic PDAC. The first-line
systemic chemotherapies for locally advanced or metastatic PDAC
include fluorouracil, leucovorin, irinotecan and oxaliplatin
(FOLFIRINOX) and gemcitabine plus nab-paclitaxel (3), both of which have been proven to
improve the clinical outcome. FOLFIRINOX is associated with a
better clinical outcome but has greater toxicity than gemcitabine,
which limits its application in patients with poor performance
status (4). However, oxaliplatin
can be offered as a replacement therapy for these patients as well
(5). Unfortunately, after several
weeks of chemotherapy, an initially sensitive tumor may become
resistant to platinum-based chemotherapy (6). Therefore, there remains an urgent
need for novel therapies to overcome resistance and increase the
efficacy of oxaliplatin in PDAC treatment.
DNA double-strand breaks (DSBs) are modified through
either homologous recombination (HR) or nonhomologous end joining
(NHEJ) pathways. The choice of these two DSB repair pathways is
determined by a number of factors, such as DSB end structure and
cell cycle. The DNA damage response protein p53-binding protein 1
(53BP1) regulates these DSB repair pathways; it promotes NHEJ and
suppresses HR (7).
Hepatocyte nuclear factor 1 homeobox A (HNF1A) is
enriched in the liver, and is expressed in the kidney, intestine
and pancreas (8). Previous studies
have shown that HNF1A regulates lipid and carbohydrate metabolism
in hepatocytes and pancreatic islet cells (9,10).
HNF1A gene variants have been reported to cause maturity-onset
diabetes of the young, type 3 (MODY3) and are associated with the
risk of PDAC (11,12). In addition, previous studies
(13-15) have shown that HNF1A regulates
gemcitabine resistance by targeting ABCB1, and that it serves a
tumor suppressor role in PDAC (14). HNF1A has been proven to regulate a
series of genes associated with glucose metabolism and multidrug
resistance proteins to mediate oxaliplatin resistance (16). However, to the best of our
knowledge, the role of HNF1A in oxaliplatin chemotherapy resistance
in PDAC has not been determined. The present study explored the
effect of HNF1A on oxaliplatin resistance in PDAC.
Materials and methods
Patients and clinical samples
A total of 76 locally advanced PDAC specimens were
collected from patients (median age, 61.2 years; age range, 32-79
years; 43 male patients, 33 female patients) who received a
platinum-based chemotherapy program at Guangdong Provincial
People's Hospital and Sun Yat-sen Memorial Hospital (both
Guangzhou, China) between July 2015 and June 2021. Patients with
locally advanced PDAC underwent laparoscopic biopsy or endoscopic
ultrasound-guided sample acquisition from a solid pancreatic mass.
The samples were confirmed as PDAC by two certified pathologists.
Patients were included if they i) were 18-80 years of age; ii) had
advanced PDAC stage III or IV, according to the pathology results;
iii) had received oxaliplatin chemotherapy; and iv) had provided
written informed consent. Patients were excluded if they i) had
received other specific pancreatic cancer treatments (surgery,
preoperative chemotherapy or chemoradiation); ii) had a pathologic
diagnosis of a benign tumor; iii) had a history of other
malignancies; or iv) had a mental illness. In accordance with the
Guangdong Provincial People's Hospital's Protection of Human
Subjects Committee (approval no. KY-H-2022-011-01), the protocol
was approved, and all of the patients provided written informed
consent before the biopsy sample collection. Progression-free
survival (PFS) was measured from the date of chemotherapy to the
occurrence of an event (progressive disease, death or diagnosis of
a second malignant neoplasm). The response of oxaliplatin
chemotherapy was assessed 4 months after the start of chemotherapy,
complete response (CR) and partial response (PR) were classified as
oxaliplatin-sensitive (35 cases), while stable disease (SD) and
progressive disease (PD) were classified as oxaliplatin-resistant
(41 cases).
Cell culture
MiaPaCa-2 [cat. no. CRM-CRL-1420; Research Resource
Identifier (RRID): CVCL_0428] and Panc-1 (cat. no. CRL-1469MET;
RRID: CVCL_A4BT) cells were obtained from the American Type Culture
Collection. Cells were maintained in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin, and were
cultured at 37°C in a humidified atmosphere with 5%
CO2.
Construction of oxaliplatin-resistant
cell lines
Panc-1 and MiaPaCa-2 cells with resistance to
oxaliplatin were named Panc-1-R and MiaPaCa-2-R, respectively.
First, 2 mg/ml oxaliplatin (cat. no. S1224; Selleck Chemicals,
dissolved to storage concentration with DMSO and diluted to working
concentrations with DMEM) solution was added to Panc-1 and
MiaPaCa-2 cells and incubated at 37°C for 24 h; subsequently, the
medium was replaced with oxaliplatin-free medium and cell
proliferation was observed using a CCK-8 assay (cat. no. K1018;
APExBIO Technology LLC). Panc-1 and MiaPaCa-2 cells were repeatedly
stimulated in this manner until proliferation was no longer
inhibited. The concentration of oxaliplatin was then increased to
3, 4, 6, 8 and 10 mg/ml to develop Panc-1-R and MiaPaCa-2-R
cells.
Panc-1 and MiaPaCa-2 cells (5×103/well)
were seeded in 96-well plates to detect the drug response. After 24
h, fresh medium containing oxaliplatin at a gradient concentration
of 0, 0.001, 0.01, 0.1, 1, 10, 100 and 1,000 µM was added to
the cells and incubated for 72 h at 37°C. The cells were then
incubated with 10 µl CCK-8 solution at 37°C for 2 h and the
absorbance was measured using a microplate reader (Thermo Fisher
Scientific, Inc.) at 450 nm. The degree of drug response for tumor
cells was determined by the half-maximal inhibitory concentration
(IC50), which was calculated with GraphPad Prism 8.0
(Dotmatics).
Immunohistochemistry (IHC)
Paraffin-embedded human tissue samples (5 µm)
were deparaffinized using the following steps: 3×10 min in xylol,
2×5 min in 100% ethanol, 1×5 min in 95% ethanol, 1×5 min in 85%
ethanol and 1×5 min in 75% ethanol. Antigen retrieval was performed
in a microwave oven with sodium citrate buffer (0.01 M, pH 6.0;
Beyotime Institute of Biotechnology) for 20 min. Subsequently, 3%
H2O2 was used to remove endogenous peroxidase
for 15 min at room temperature. Slides were incubated with normal
goat serum (Beyotime Institute of Biotechnology) for 30 min at room
temperature to block unspecific background staining. The tissues
were then incubated at 4°C overnight with primary rabbit anti-human
antibodies against HNF1A (1:200; cat. no. ab96777), Ki-67 (1:200;
cat. no. ab15580), 53BP1 (1:200; cat. no. ab87097) and γ-H2AX
(1:200; cat. no. ab81299) (all from Abcam), followed by incubation
withbiotin-conjugated secondary antibodies at room temperature for
20 min. Subsequently, each slide was incubated at room temperature
with streptavidin-HRP conjugate (Beyotime Institute of
Biotechnology) for 20 min. The staining procedure was performed
using a DAB Kit (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. Mayer's hematoxylin (Beyotime
Institute of Biotechnology) was used for counterstaining all slides
at room temperature for 20-30 sec. Staining was observed and images
were captured with an inverted microscope (Olympus IX-71; Olympus
Corporation).
A semi-quantitative assessment of HNF1A expression
levels was performed using the immunoreactive score and staining
intensity. The scoring of the staining intensity was as follows: 0,
no staining; 1, light; 2, intermediate; and 3, strong. The scoring
of the proportion of HNF1A- or 53BP1 positive-cells was as follows:
1, <25%; 2, ≥25-<50%; 3, ≥50-<75%; and 4, ≥75-100%. The
final score was obtained by multiplying the staining intensity and
the proportion of positively stained cells. Cases with a score of
>4 were considered to have high expression, whereas those with a
score of ≤4 were considered to have low expression. Discrepancies
in staining were re-evaluated by two pathologists until a consensus
was reached after the staining was professionally assessed using
the scoring criteria.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA from tissues and cells was isolated by
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions and was reverse
transcribed using HiScript Reverse Transcriptase (cat. no. R101-01;
Vazyme Biotech Co., Ltd.). qPCR was performed using the SYBR Premix
Ex Taq™ kit (cat. no. DRR420A; Takara Bio, Inc.), GAPDH was used as
the universal control. RT-qPCR was conducted using the CFX96 Touch
Real-Time PCR detection system (Bio-Rad Laboratories, Inc.). The
thermocycling conditions include initial denaturation at 95°C for
30 sec, followed by 35 cycles at 95°C for 5 sec, annealing at 60°C
for 30 sec. All reactions were examined in technical triplicate.
Gene expression analysis was performed using the 2−ΔΔCq
method (17). The primer sequences
are listed in Table SI.
Establishment of knockdown and
overexpression cell lines
Human HNF1A and 53BP1 coding sequences were
subcloned into the pMKO.1-puro vector (Guangzhou IGE Biotechnology
Ltd.) and the empty vector was used as a control. siRNA against
HNF1A and a scrambled negative control (si-NC) were obtained from
Guangzhou IGE Biotechnology Ltd. According to the manufacturer's
protocol, 2 µg plasmid/50 nM siRNA, 10 µl
Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) and
500 µl Opti-MEM Medium (Gibco; Thermo Fisher Scientific,
Inc.) were mixed gently and then incubated at room temperature for
15 min. The mixture was then slowly added to Panc-1 and MiaPaCa-2
cells. The cells transfected with plasmids/siRNA were incubated at
37°C for 24 h, and then the culture medium was subsequently
replaced with fresh DMEM supplemented with 10% FBS. Panc-1 and
MiaPaCa-2 cells were harvested at 48 h after transfection. The
siRNA sequences are listed in Table
SII.
Western blotting
Panc-1 and MiaPaCa-2 cells were collected and lysed
for 15 min on ice in RIPA buffer (Beyotime Institute of
Biotechnology). Quantification of protein concentration was
measured by bicinchoninic acid kit (Beyotime Institute of
Biotechnology). A total of 30-50 µg protein was loaded per
lane, separated by SDS-PAGE on 8-12% gels and transferred to
0.45-µm polyvinylidene difluoride (PVDF) membranes
(Immobilon-P; MilliporeSigma). Subsequently, 5% skim milk (cat. no.
70166; Sigma-Aldrich; Merck KGaA) was used to block PVDF membranes
for 1 h at room temperature. The membranes were incubated with
primary antibodies overnight at 4°C. Horseradish
peroxidase-conjugated secondary antibodies were then used to
incubate membranes at room temperature for 1 h. Protein expression
was detected using an enhanced chemiluminescence detection system
(Thermo Fisher Scientific, Inc.). The following primary antibodies
were used: Rabbit anti-human HNF1A (1:1,000; cat. no. ab96777;
Abcam), 53BP1 (1:1,000; cat. no. ab87097; Abcam), mouse anti-human
GAPDH (1:2,000; cat. no. abs830030; Absin Bioscience, Inc.) The
secondary antibodies were goat anti-rabbit IgG-HRP (1:10,000;
abs20002) and goat anti-mouse IgG-HRP (1:10,000; cat. no. abs20001)
(both from Absin Bioscience, Inc.).
Immunofluorescence staining
Panc-1 and MiaPaCa-2 cells were cultured in 6-well
plates overnight. Cells were fixed in 4% paraformaldehyde for 10
min at room temperature and permeabilized with 0.1% Triton X-100
(Thermo Fisher Scientific, Inc.) for 15 min. After blocking with 5%
bovine serum albumin (Beyotime Institute of Biotechnology) at room
temperature for 60 min, cells were incubated with the following
antibodies overnight at 4°C: HNF1A (1:1,000; cat. no. ab96777;
Abcam), γ-H2AX (1:1,000; cat. no. ab81299; Abcam). The cells were
then incubated for 1 h in the dark at room temperature with the
following secondary antibodies: Goat Anti-Rabbit IgG H&L (Alexa
Fluor® 488) (1:2,000; cat. no. ab150077; Abcam)
Subsequently, the cells were stained with DAPI (5 µg/ml;
Beyotime Institute of Biotechnology) for 5 min at room temperature.
The results were examined by laser scanning confocal microscopy
(Leica Microsystems GmbH).
Cell proliferation assay
Panc-1 and MiaPaCa-2 were seeded in 96-well plates
in duplicate at 3,000 cells/well. Oxaliplatin (5
µM)/cisplatin (5 µM; cat. no. S1166; Selleck
Chemicals)/carboplatin (4 µM; cat. no. S1215; Selleck
Chemicals) was added to the cells after 24 h and incubated for 4
days. According to the manufacturer's protocol, cell proliferation
was determined daily using the Cell Counting Kit-8 (CCK-8) assay
(cat. no. K1018; APExBIO Technology LLC) over 4 days. The
absorbance was measured at 450 nm using a multiwell plate reader
(Spark; Tecan Group, Ltd.).
Colony formation assay
Panc-1 and MiaPaCa-2 (2,000/well) were seeded in
6-well plates and treated with oxaliplatin (5 µM)/cisplatin
(5 µM)/carboplatin (4 µM) at 37°C for 48 h. The
colonies were fixed for 15 min with 4% paraformaldehyde at room
temperature and stained for 10 min with 0.25% crystal violet at
room temperature after 2 weeks of incubation. The visible colonies
(containing >50 cells) were counted and images were captured
under an optical microscope.
Sphere formation assay
MiaPaCa-2 and Panc-1 cells (1,000 cells/well) were
plated in 96-well extremely low attachment plates (Corning, Inc.)
and treated with oxaliplatin (5 µM)/cisplatin (5
µM)/carboplatin (4 µM) at 37°C for 48 h. Fibroblast
growth factor (20 ng/ml), human recombinant epidermal growth factor
(20 ng/ml) and serum substitute B27 (1X) were mixed into serum-free
DMEM/F-12 and were added to each well (all from Invitrogen; Thermo
Fisher Scientific, Inc.). The number of spheres >50 mm in
diameter was counted under a light microscope after 2 weeks of
incubation at 37°C and 5% CO2.
Annexin V/PI apoptosis assay
Panc-1 or MiaPaCa-2 cells were treated with 5
µM oxaliplatin for 48 h. The cells were stained with an
Annexin V-PI staining kit (cat. no. BMS500FI; eBioscience; Thermo
Fisher Scientific, Inc.) to detect apoptosis. After being
harvested, the cells were resuspended in FITC-conjugated Annexin V
binding solution for 15 min at 37°C and then stained with PI at
room temperature for 5 min. Within 1 h, flow cytometry was
performed using a FACSCanto II instrument (BD Biosciences) and
FlowJo V10.0.7 software (FlowJo LLC).
Neutral comet assay
After treatment of MiaPaCa-2 and Panc-1 cells
(2×105) with 5 µM oxaliplatin for 1 h at room
temperature, time-lapse imaging was performed. A comet assay kit
(cat. no. 4250-050-K; Trevigen, Inc.) was used in accordance with
the manufacturer's instructions. SYBR gold stain kit (cat. no.
S11494; Invitrogen; Thermo Fisher Scientific, Inc.) was used to
stain the samples for 45 min at room temperature, which were
observed under an Olympus FluoView 500 fluorescence microscope. The
images were analyzed using CASP software (version no.
2013062811125300; Beijing Bio-launching Technologies Co.,
Ltd.).
pimEJ5-GFP reporter and pDR-GFP reporter
assays
Transfection of MiaPaCa-2 and Panc-1 cells
(4×104) with pimEJ5-GFP (ID 44026; Addgene, Inc.) or
pDR-GFP (ID 26475; Addgene, Inc.) plasmids was performed using
Lipofectamine 3000. According to the manufacturer's protocol, 1
µg plasmid, 10 µl Lipofectamine 3000 and 500
µl Opti-MEM were mixed gently and then incubated at room
temperature for 15 min. The mixture was then slowly added to Panc-1
and MiaPaCa-2 cells. After incubating at 37°C for 24 h, the culture
medium was subsequently replaced with fresh DMEM supplemented with
10% FBS. Puromycin (2 µg/ml; MedChemExpress) was used to
select stable clones of MiaPaCa-2 and Panc-1 cells that expressed
pimEJ5-GFP or pDR-GFP. MiaPaCa-2 and Panc-1 cells that stably
expressed pimEJ5-GFP or pDR-GFP were transfected with pCBASceI
(I-SceI) plasmids (Addgene plasmid no. 26477; Addgene, Inc.) using
Lipofectamine 3000 according to the aforementioned protocol. The
GFP-positive cells were analyzed by FACSCanto II instrumentand
FlowJo V10.0.7 software 48-72 h after transfection. A pair of
plasmids carrying pimEJ5-GFPand pDR-GFPwere provided by Dr Jeremy
Stark.
Chromatin immunoprecipitation (ChIP)
assay
For 10 min at 37°C, 1% formaldehyde was used to
cross-link the proteins of Panc-1 cells. ChIP experiments were
carried out using an anti-HNF1A antibody (5 µl/1 mg total
protein; cat. no. ab96777; Abcam), with ChIP-grade rabbit IgG (1
µg/ml; cat. no. ab171870; Abcam) used as a negative control.
ChIP assays were performed using an EZ-Magna ChIP Chromatin
Immunoprecipitation kit (cat. no. 17-408. MilliporeSigma) according
to the manufacturer's instructions. Immunoprecipitated DNA was
analyzed by qPCR as aforementioned.
53BP1 promoter deletion constructs and
luciferase reporter assay
The 53BP1 promotor deletion constructs were
generated by Guangzhou IGE Biotechnology Ltd. Predictive binding
site mutagenesis of the 53BP1 promotor was performed using a Quick
Change Site-Directed Mutagenesis kit (Agilent Technologies, Inc.).
Subsequently, the luciferase reporter plasmids [pGL3-53BP1,
pGL3-53BP1-P (-5000-+200), pGL3-53BP1-P (-4000-+200), pGL3-53BP1-P
(-3000-+200), pGL3-53BP1-P (-2000-+200), pGL3-53BP1-P (-1000-+200),
pGL3-53BP1-P (+1-+200), pGL3-basic; Guangzhou IGE Biotechnology
Ltd.] containing the 53BP1 promoter deletions were cotransfected
with the HNF1A overexpression vector or empty vector into Panc-1
cells (3×105 cells) using 1 µl Lipofectamine 3000
at 37°C for 24 h. A total of 24 h after transfection, the cells
were collected, washed, and harvested for firefly and Renilla
luciferase assays (cat. no. 16816; Thermo Fisher Scientific,
Inc.).
Animal experiments
For orthotopic xenograft models, the BALB/c nude
mice (female; age, 4-6 weeks; body weight, 18-20 g; Shulaibao
Biotech) were housed at 5 per cage under specific pathogen-free
conditions (temperature, 21±2°C; humidity, 40-60%; 12-h light/dark
cycle; free access to standard sterile food and water), and were
randomly divided into the following three groups (n=5/group): i)
luc-Panc-1 cells/empty vector, ii) luc-Panc-1 cells/HNF1A and iii)
luc-Panc-1 cells/53BP1. Full-length HNF1A and 53BP1 were subcloned
into pCDH-PGK-luciferase-EF1a-mcherry-T2a-puro vector (Guangzhou
IGE Biotechnology Ltd.) and an empty vector was used as a control.
The plasmids (2 µg) were transfected into Panc-1 cells using
Lipofectamine 3000 according to the manufacturer's protocol. After
24 hours, the culture medium was replaced with fresh DMEM
supplemented with 10% FBS. Puromycin (2 µg/ml;
MedChemExpress) was used to select stable clones of Panc-1 cells
that expressed HNF1A or 53BP1. A total of 48-72 h after
transfection, the cells were harvested for mouse experiments.
BALB/c nude mice were anesthetized with isoflurane inhalation (4-5%
induction, 1-2% maintenance). A 1-cm left subcostal incision was
made to expose the pancreas, which was injected with a 50-µl
PBS suspension of luc-Panc-1 cells using a 30-G needle.
Monofilament sutures were used to close the wound following
orthotopic implantation. In a 15-day treatment period, oxaliplatin
(5 mg/kg) was administered intraperitoneally once every 3 days,
followed by IVIS imaging and tumor removal. D-Luciferin (150 mg/kg;
cat. no. 161055-47-6; MilliporeSigma) and potassium salt
(40902ES01; Shanghai Yeasen Biotechnology Co. Ltd.) were injected
intravenously during the IVIS study, and orthotopic fluorescence
images were recorded in vivo using an FXPRO system (Bruker
Corporation). At each time point in the in vivo study, the
mice were assessed by investigators who were blinded to the group
allocation. All of the mice were sacrificed 30 days later.
A patient-derived xenograft (PDX) model was
developed by propagating primary tumor specimens obtained from the
aforementioned patients with oxaliplatin-resistant PDAC as
subcutaneous tumors in 4-week-old NOD-SCID IL-2 receptor γ-null
mice (F1, n=5/group; female; age, 4-6 weeks; body weight, 18-20 g;
Shulaibao Biotech) were housed at 5 per cage under specific
pathogen-free conditions (temperature, 21±2°C; humidity, 40-60%;
12-h light/dark cycle; free access to standard sterile food and
water). The NOD-SCID IL-2 receptor γ-null mice were anesthetized
with 1% sodium pentobarbital (intraperitoneal injection, 30 mg/kg).
The xenografts from F1 were implanted into other mice (F2) after
separating them into small pieces. When the tumor volumes reached
1,500 mm3, they were excised, cut into small pieces
again and transplanted into other mice (F3). A combination of AVV
virus (Guangzhou IGE Biotechnology Ltd.) and oxaliplatin
chemotherapy (intraperitoneal injection, 10 mg/kg) was administered
when the xenograft volume reached ~200 mm3. Throughout
the treatment period, tumor volume was monitored every week. All of
the mice were sacrificed 30 days later and IHC was performed on
each tumor.
All mice were maintained in a specific pathogen-free
environment. When reaching the following humane endpoints, the mice
were euthanized: i) Tumor burden was >10% of body weight, and
the diameter of tumor was >20 mm; ii) tumor ulceration,
infection or necrosis was observed; iii) tumor interferes with
eating or walking; iv) rapid weight loss (loss of 15-20% of
original weight). Notably, none of these endpoints were reached
during the experiments. Animal health and behavior were monitored
twice a day (at the beginning and end of the day) by an experienced
individual. The mice were euthanized by cervical dislocation under
anesthesia (1% sodium pentobarbital intraperitoneal injection, 30
mg/kg). A comprehensive judgment on death was made by observing
signs of respiration, heartbeat, and pupil and nerve reflexes.
Animal experiments were conducted according to guidelines approved
by the Animal Experimental Research Ethics Committee of South China
University of Technology (Guangzhou, China; approval no.
2021042).
Database analysis
To predict the binding site of HNF1A at the 53BP1
promoter region, the JASPAR database (https://jaspar.genereg.net/) was used.
Statistical analysis
SPSS version 20.0 software (IBM Corp.) was used to
conduct all statistical analyses. All experiments were repeated at
least three times. For parametric data, one way ANOVA with a
Tukey's post-hoc test or unpaired Student's t-test was used. Unless
otherwise stated, all values are expressed as the mean ± standard
deviation. Kaplan-Meier method was used to assess differences in
patient survival and the log-rank test was applied to analyze the
data. Spearman's correlation analysis was used for the correlation
analysis. P<0.05 was used to indicate a statistically
significant difference.
Results
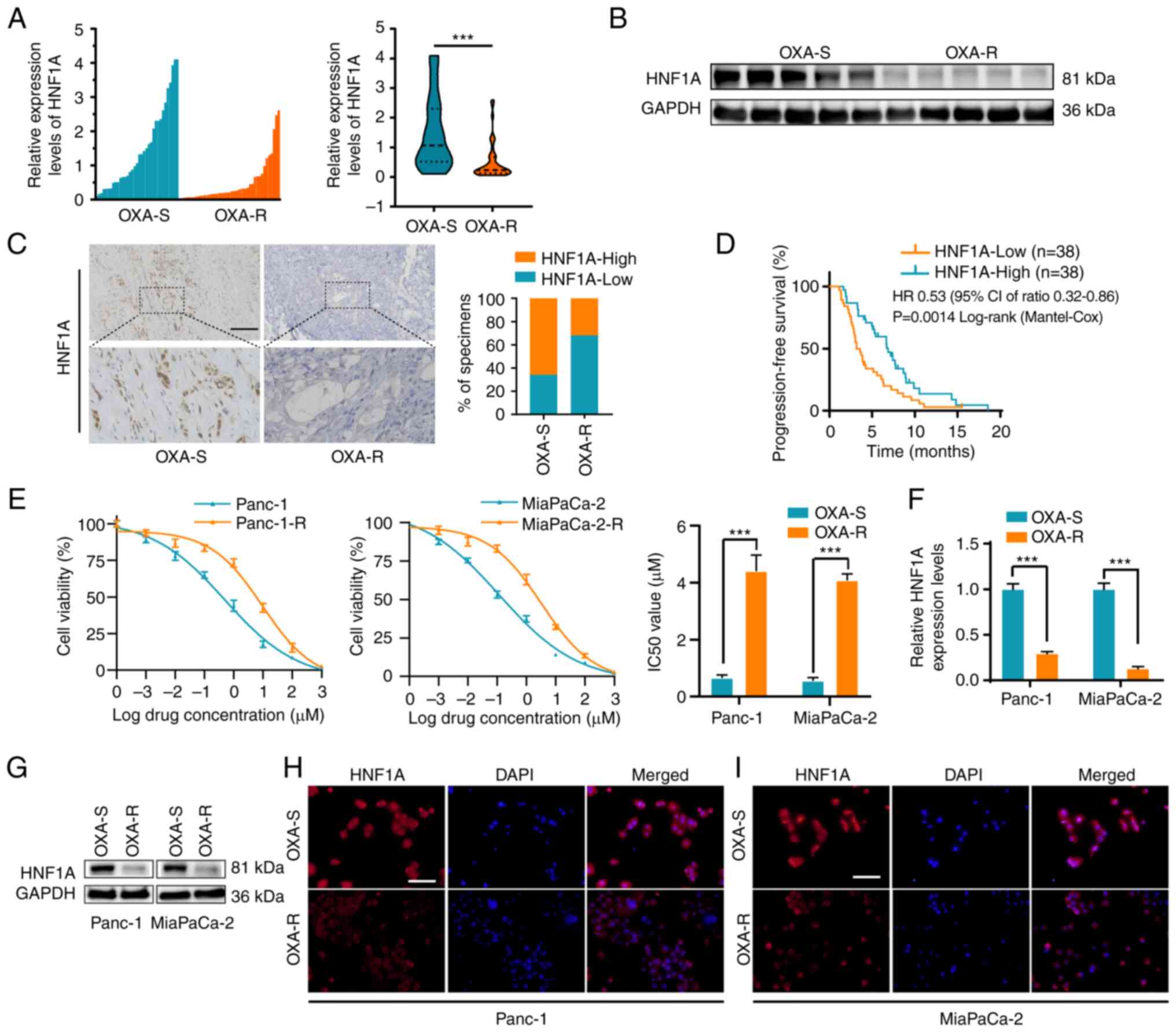
HNF1A expression is associated with
oxaliplatin resistance in PDAC tissues and cell lines
Given that HNF1A is a critical suppressor of
chemotherapy resistance in pancreatic cancer (14), the mRNA expression levels of HNF1A
were detected in 76 patients with PDAC who accepted platinum-based
chemotherapy. The results demonstrated that the expression levels
of HNF1A were reduced in oxaliplatin-resistant patients compared
with those in oxaliplatin-sensitive patients which were described
in the methods (Fig. 1A). In
addition, significantly lower expression levels of HNF1A were
detected in oxaliplatin-resistant patients compared with those in
oxaliplatin-sensitive patients, as determined by western blotting
and IHC (Fig. 1B and C). Those
with low HNF1A expression levels, according to RT-qPCR (cut-off
according to the median expression level, there were 38 patients in
the low HNF1A expression group and 38 in the high HNF1A expression
group), had poorer PFS than those with high HNF1A expression levels
(Fig. 1D).
Panc-1-R and MiaPaCa-2-R cells were treated with
escalating concentrations of oxaliplatin in vitro. Panc-1
and MiaPaCa-2 cells were repeatedly stimulated in this manner until
proliferation was no longer inhibited. As shown by the elevated
IC50 values, Panc-1-R and MiaPaCa-2-R cells responded
poorly to oxaliplatin compared with the parental cells (Fig. 1E). To clarify the relationship
between HNF1A expression and oxaliplatin chemotherapy resistance,
the mRNA and protein expression levels of HNF1A were detected in
oxaliplatin-resistant and oxaliplatin-sensitive cells. RT-qPCR,
western blotting and immunofluorescence assays indicated that the
expression levels of HNF1A were markedly lower in the Panc-1-R and
MiaPaCa-2-R cell lines compared with those in the
oxaliplatin-sensitive cell lines (Fig.
1F-I).
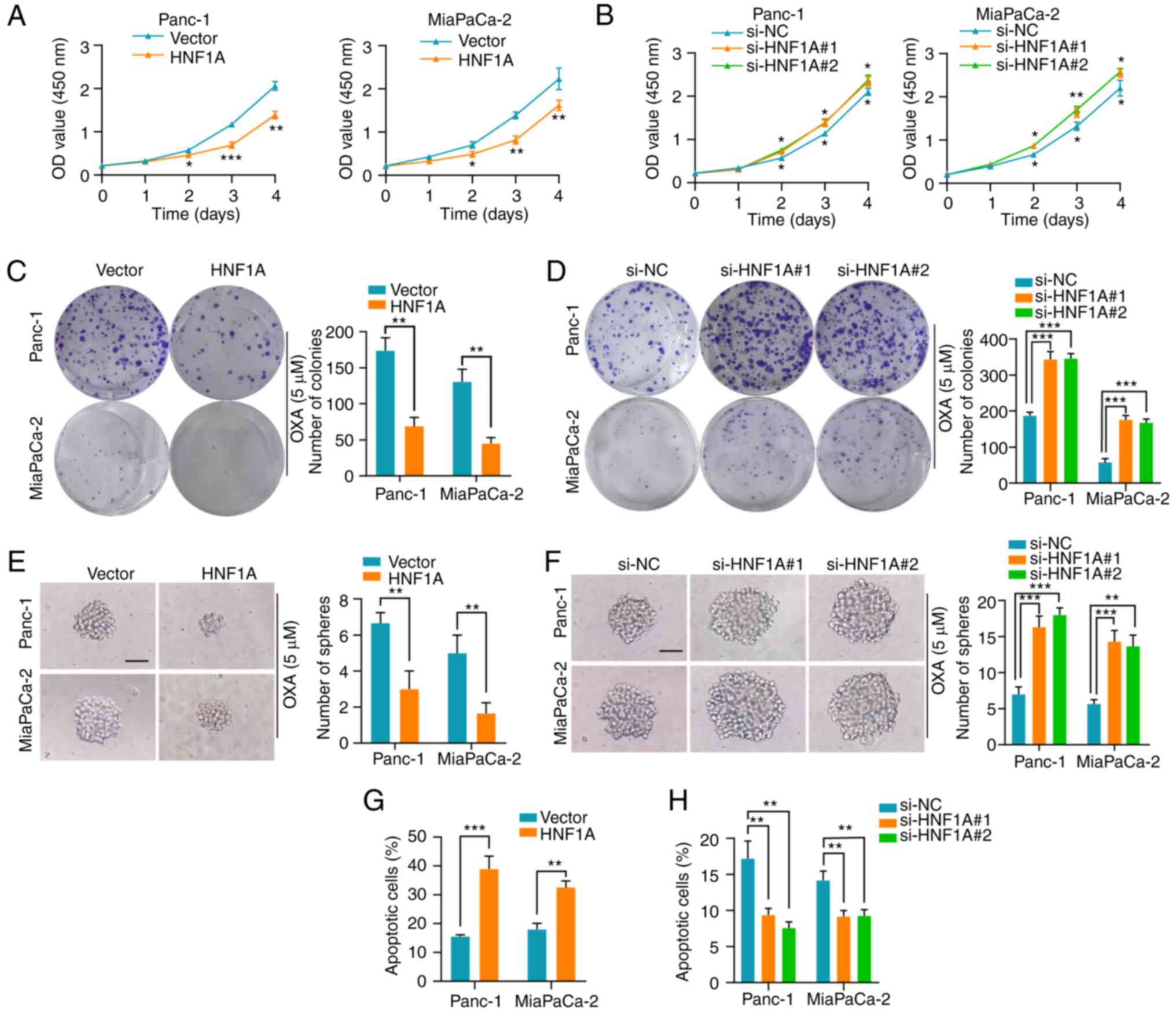
HNF1A mediates platinum-based drug
resistance in PDAC cell lines in vitro
To explore the role of HNF1A in oxaliplatin,
cisplatin and carboplatin resistance, HNF1A was overexpressed or
depleted in Panc-1 and MiaPaCa-2 cells. (Fig. S1A-D). HNF1A overexpression
decreased the proliferation rate of Panc-1 and MiaPaCa-2 PDAC
cells, as confirmed by the CCK-8 cytotoxicity assay. Conversely,
silencing HNF1A enhanced platinum-based drug resistance in PDAC
cells (Figs. 2A and B, S2A and B, and S3A and B). After exposure
to platinum-based drugs, PDAC cells overexpressing HNF1A had a
lower survival rate than control cells, as shown by colony
formation assays, whereas HNF1A knockdown enhanced their survival
(Figs. 2C and D, S2C and D, and S3C and D). Cancer stem
cells have a pivotal role in chemoresistance (18). Notably, HNF1A overexpression in
PDAC cells significantly reduced the self-renewal ability of the
cells, as demonstrated by a sphere formation assay. By contrast,
HNF1A knockdown significantly attenuated this effect (Figs. 2E and F, S2E and F, and S3E and F). Additionally,
overexpression of HNF1A in tumor cells promoted cell early and late
apoptosis under oxaliplatin treatment conditions, as determined by
flow cytometry (Figs. 2G and
S4A). Conversely, knockdown of
HNF1A in PDAC cells markedly reversed this effect (Figs. 2H and S4B).
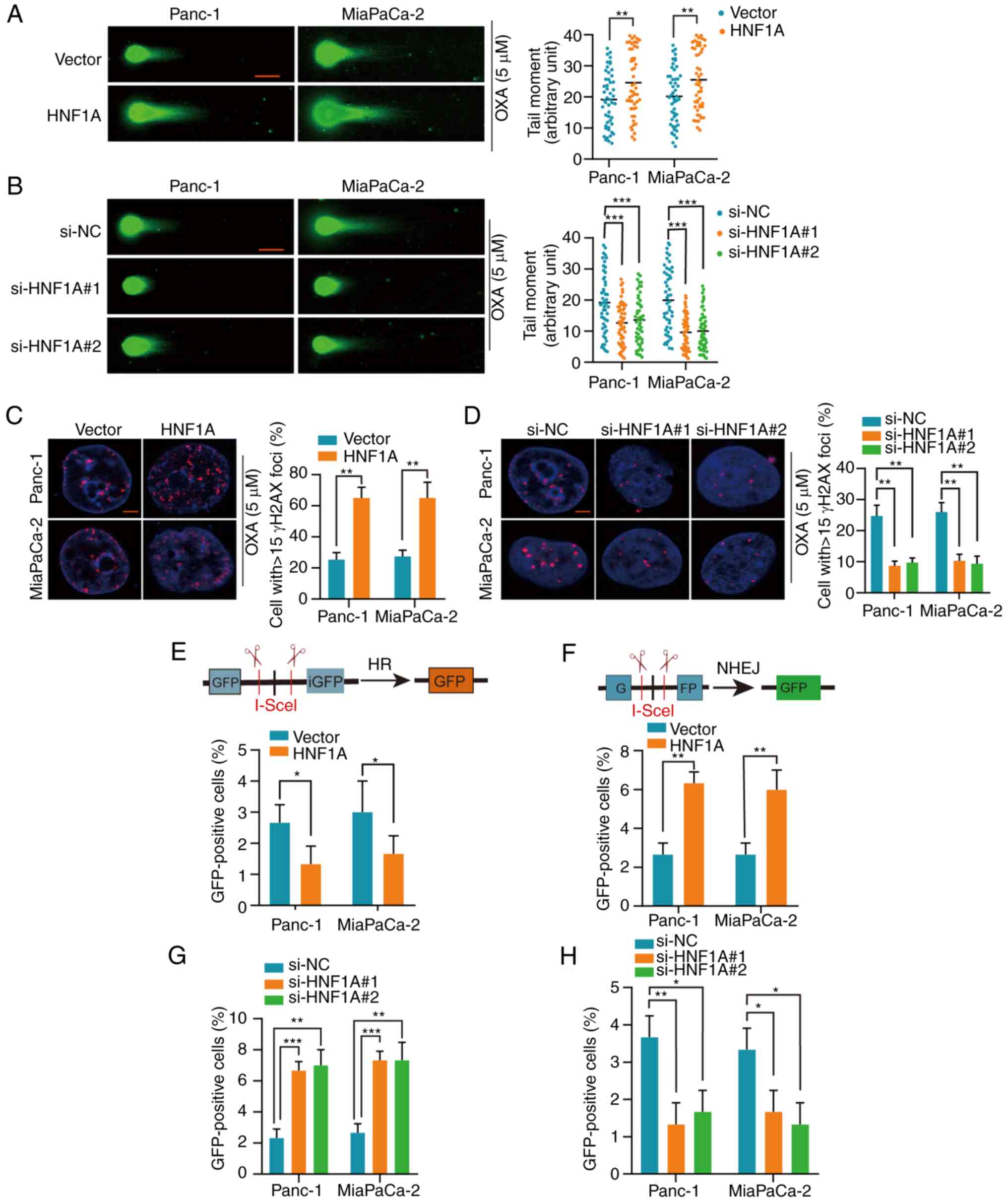
HNF1A switches HR to NHEJ
To determine the mechanism by which HNF1A
downregulation mediates oxaliplatin resistance in PDAC cells, the
present study detected whether DSB repair was altered by HNF1A. A
neutral comet assay was performed and the expression levels of
γH2AX were detected to assess damage to the genetic material
(DSBs). Longer comet tails were present in the HNF1A-overexpressing
groups, whereas HNF1A knockdown shortened the lengths of the comet
tails following treatment with oxaliplatin, as determined by the
neutral comet assay (Fig. 3A and
B). In addition, following oxaliplatin treatment, the
HNF1A-overexpressing groups had higher levels of γH2AX than the
control group, indicating a higher number of DSBs. However, the
expression levels of γH2AX were lower in the HNF1A knockdown group
compared with in the si-NC group (Fig.
3C and D).
NHEJ and HR are the main pathways for DSB repair
(19). To explore the effect of
HNF1A on NHEJ and HR, the pimEJ5-GFP and pDR-GFP reporters were
stably transfected into PDAC cells. The percentage of GFP-positive
PDAC cells post-transfection with the pDR-GFP reporter or the
pimEJ5-GFP reporter represented HR repair efficiency and NHEJ
repair efficiency, respectively. HNF1A overexpression significantly
suppressed HR-dependent DSB repair and promoted NHEJ-dependent DSB
repair, whereas HNF1A knockdown had the opposite effect (Figs. 3E-H and S5A-D).
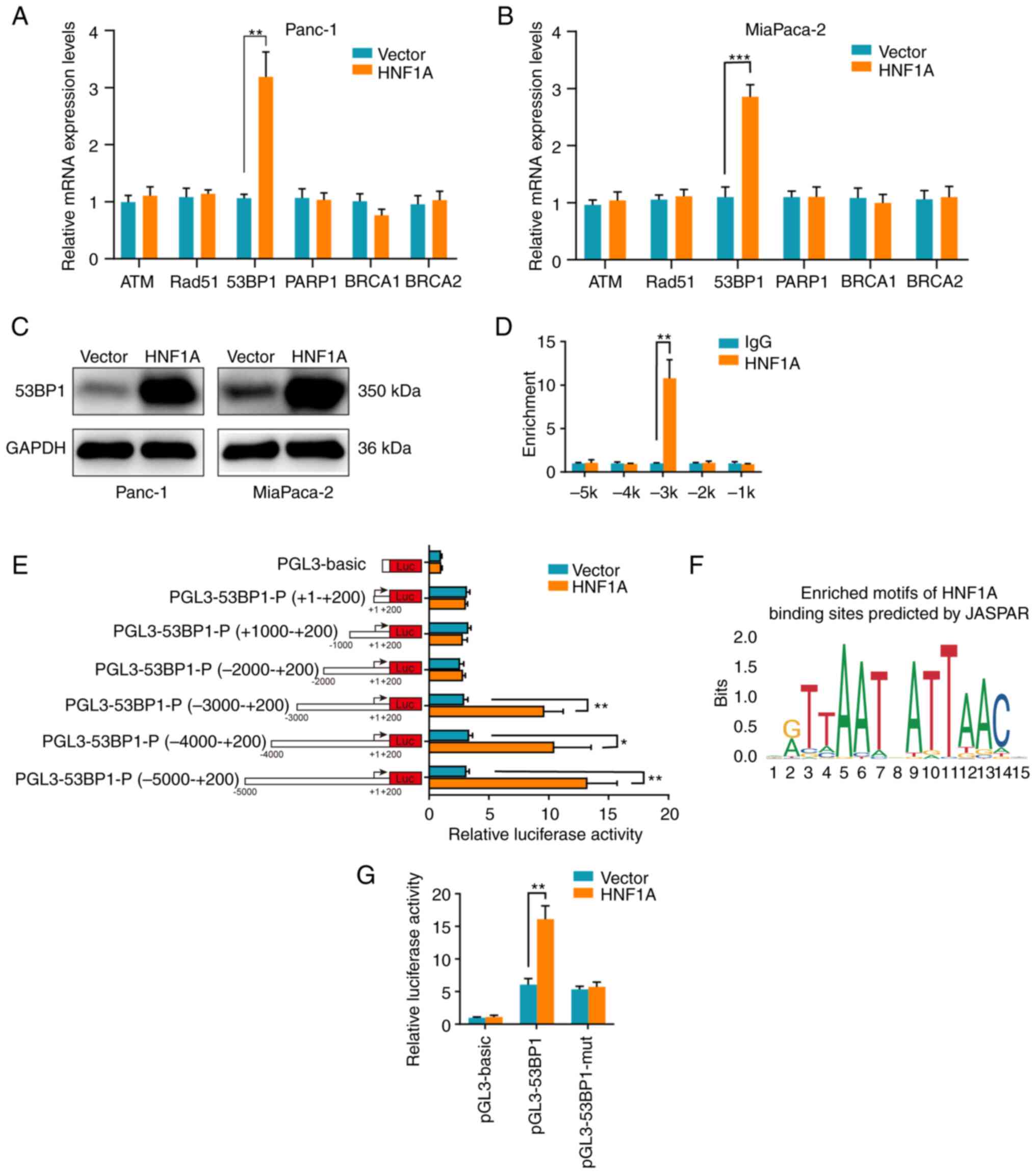
HNF1A regulates 53BP1 expression by
binding to the 53BP1 promoter
To further reveal the downstream target of HNF1A in
mediating HR/NHEJ, the mRNA expression levels of the key factors in
HR and NHEJ were detected. Notably, only the mRNA expression levels
of 53BP1 were significantly upregulated following overexpression of
HNF1A in PDAC Panc-1 and MiaPaCa-2 cells (Fig. 4A and B). In addition, western
blotting confirmed that HNF1A overexpression markedly increased the
protein expression levels of 53BP1 (Fig. 4C). Given that HNF1A is known as a
transcriptional activator, a ChIP assay was performed to determine
whether HNF1A transcriptionally upregulated 53BP1. The results
demonstrated that HNF1A interacted with the 5′ regulatory region of
53BP1 at the -3 kb location (from the transport start site) instead
of the 5' regulatory regions at the −2, −1, −4 and -5 kb locations
(Fig. 4D). Furthermore, a series
of 53BP1 promoter deletion reporter constructs were generated, and
luciferase assays showed that the −3 to −5 kb region of the 53BP1
promoter led to a significant increase in transcriptional activity
in HNF1A-overexpressing tumor cells (Fig. 4E). Using JASPAR, it was predicted
that HNF1A may bind to the promoter region of 53BP1 from −2,817 to
−2,803 bp (5′-AATTAATCATTAAAT-3′) (Fig. 4F). Luciferase assays showed that
mutation of the predicted binding site in the 53BP1 promoter
abrogated the increase in luciferase intensity induced by HNF1A
overexpression (Fig. 4G).
Collectively, these results indicated that HNF1A was bound to the
specific promoter region (−2,817 to −2,803 bp) of 53BP1 and
promoted 53BP1 expression to sensitize PDAC cells to
chemotherapy.
53BP1 reverses the effects of HNF1A
inhibition
The present study explored whether 53BP1 rescued the
effect of HNF1A on oxaliplatin resistance. 53BP1was successfully
overexpressed in Panc-1 and MiaPaCa-2 cells (Fig. S1E and F). The CCK-8 cytotoxicity
and colony formation assays indicated that 53BP1 overexpression
abrogated the promoting effect of HNF1A knockdown on tumor cells
(Fig. 5A and B). In addition, the
increase in tumor stemness induced by HNF1A knockdown was blocked
by 53BP1 overexpression (Figs.
5C), and apoptosis resistance induced by HNF1A knockdown was
also blocked by 53BP1 overexpression (Figs. 5D and S6). Consistently, neutral comet assays
and γH2AX analysis revealed that 53BP1 overexpression weakened the
enhancing effect of HNF1A knockdown on DNA repair efficiency
(Fig. 5E and F). Furthermore, the
pimEJ5-GFP reporter assay showed that the increase in the ratio of
HR-dependent DSB repair caused by HNF1A knockdown was attenuated by
53BP1 overexpression (Figs. 5G and
S7A). Similarly, the pDR-GFP
reporter assay indicated that 53BP1 overexpression enhanced
NHEJ-dependent DSB repair in HNF1A-depleted tumor cells (Figs. 5H and S7B).
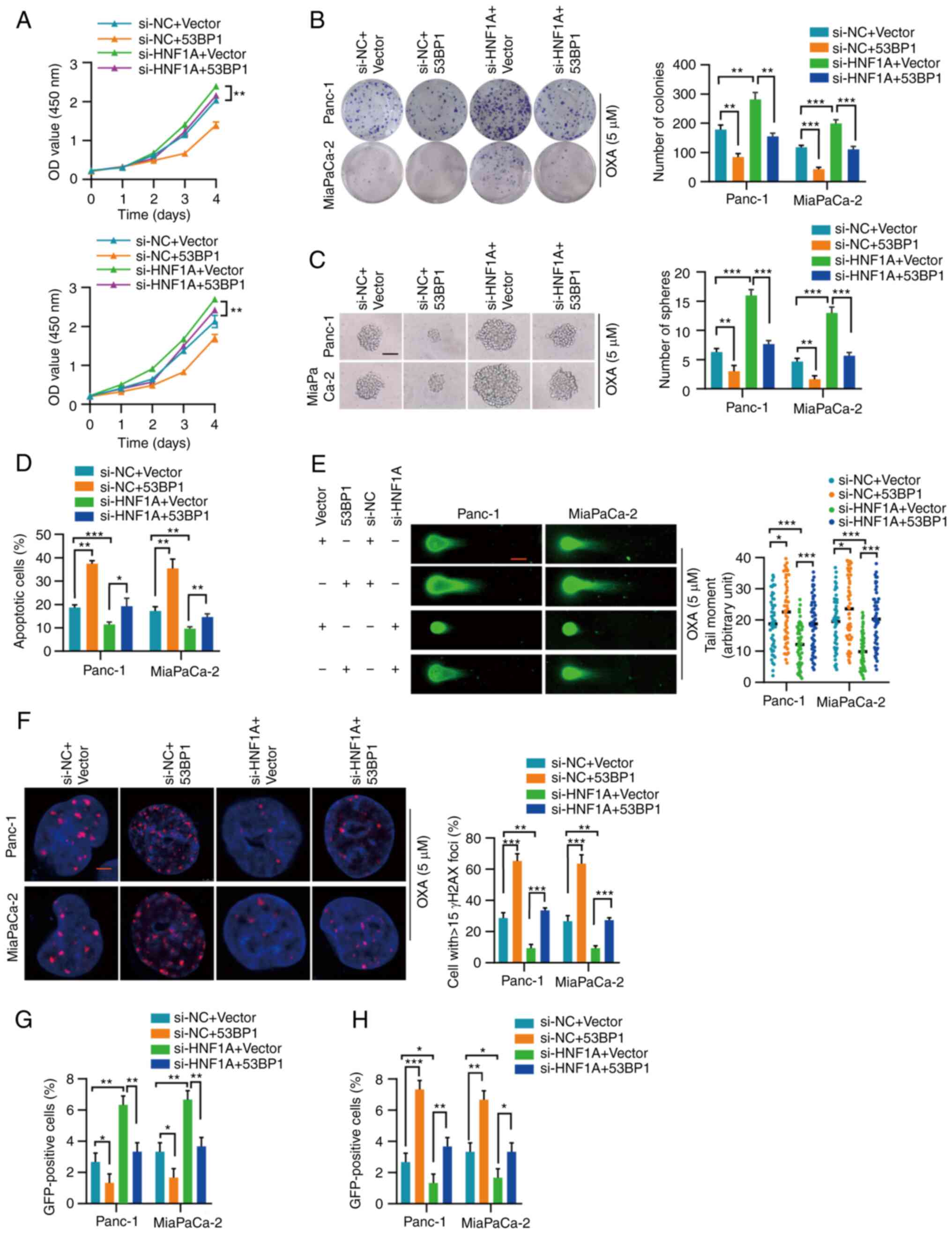 | Figure 553BP1 reverses the effect of HNF1A
knockdown. (A) Proliferation of Panc-1 and MiaPaCa-2 cells with the
indicated treatment was measured by Cell Counting Kit-8 assay. (B)
Representative images and semi-quantification of colony formation
in Panc-1 and MiaPaCa-2 cells treated with OXA (5 µM). (C)
Representative images and semi-quantification of sphere formation
in Panc-1 and MiaPaCa-2 cells with the indicated treatment under
OXA incubation (5 µM). Scale bars, 50 µm. (D) Flow
cytometry was performed to assess the apoptosis of Panc-1 and
MiaPaCa-2 cells treated with OXA (5 µM). (E) Representative
images and semi-quantification of the neutral comet assay in Panc-1
and MiaPaCa-2 cells. In total, 50 cells per group were counted.
Scale bar, 10 µm. (F) Semi-quantification and representative
images of γH2AX-positive foci in each group. More than 40 cells
from each group were counted. (G and H) HR-mediated DNA repair
efficiency was detected by the pimEJ5-GFP reporter assay, whereas
NHEJ-mediated DNA repair efficiency was detected by the pDR-GFP
reporter assay. Three independent replicates were performed. Data
are expressed as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. 53BP1,
p53-binding protein 1; HNF1A, hepatocyte nuclear factor 1 homeobox
A; HR, homologous recombination; NC, negative control; NHEJ,
nonhomologous end joining; OXA, oxaliplatin; si, small
interfering. |
HNF1A loss promotes oxaliplatin
resistance in vivo
For the in vivo experiments, the pancreases
of nude mice were orthotopically implanted with the indicated
luciferase-labeled Panc-1 cells, and oxaliplatin chemotherapy was
administered 12 days after pancreatic implantation. The mice in the
HNF1A-overexpression and 53BP1-overexpression groups exhibited
lower fluorescence intensity in the pancreas and had smaller tumors
than the control mice (Fig. 6A-C).
IHC further confirmed that HNF1A/53BP1 markedly reduced γH2AX and
Ki-67 expression (Fig. 6D).
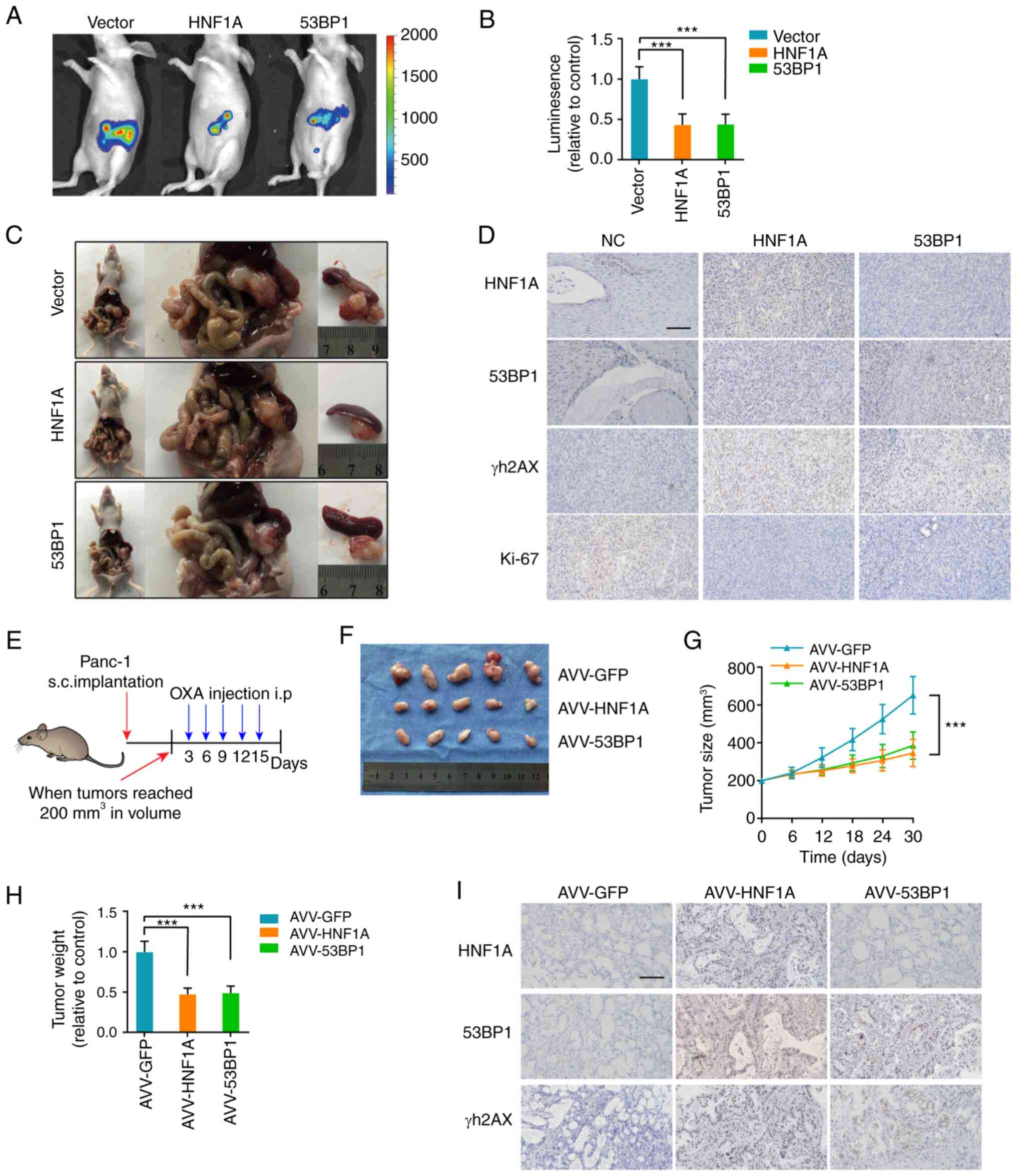 | Figure 6HNF1A knockdown promotes OXA
resistance in vivo. (A) Representative IVIS images of an
orthotopic xenograft model, n=5/group. (B) Calculation of the
luminescence intensity of the orthotopic xenograft model. (C)
Representative pancreatic tumor images of the orthotopic xenograft
model. (D) Representative IHC images for HNF1A, 53BP1, γH2AX and
Ki-67 in the orthotopic xenograft model. Scale bars, 50 µm.
(E) Once the tumor volume reached 200 mm3, the xenograft
subcutaneous model received oxaliplatin (5 mg/kg) once every 3
days, n=5/group. Changes in (F and G) tumor volume and (H) weight
were monitored. (I) Representative IHC images for HNF1A, 53BP1 and
γH2AX in the patient-derived xenograft model. The results are
presented as the mean ± SD. ***P<0.001, 53BP1,
p53-binding protein 1; HNF1A, hepatocyte nuclear factor 1 homeobox
A; IHC, immunohistochemistry; NC, negative control; OXA,
oxaliplatin. |
To further simulate the situation in the human body,
a PDX model was established. Animals were treated with oxaliplatin
and AVVs (AVV-GFP, AVV-HNF1A, AVV-53BP1) when the tumor reached 200
mm3 (Fig. 6E). The
tumors of the AVV-HNF1A and AVV-53BP1 groups were substantially
smaller and lighter than those in the AVV-GFP group (Fig. 6F-H). In addition, the data
demonstrated that tumors from the AVV-HNF1A and AVV-53BP1 groups
exhibited a marked decrease in the positive rate of γH2AX staining
(Fig. 6I). Collectively, these
results support the idea that HNF1A transcriptionally activates
53BP1 expression and sensitizes tumors to oxaliplatin.
Clinical significance of the HNF1A/53BP1
axis in PDAC
To better understand the clinical significance of
the HNF1A/53BP1 axis in oxaliplatin resistance, the expression
levels of 53BP1 and HNF1A were examined in 76 patients with PDAC
who received platinum-based chemotherapy. The findings revealed
that the expression levels of 53BP1 were significantly lower in
oxaliplatin-resistant patients compared with those in
oxaliplatin-sensitive patients (Fig.
7A and B). Moreover, 53BP1 expression was positively correlated
with HNF1A expression (Fig. 7C).
Furthermore, patients with PDAC and low expression levels of 53BP1
had a significantly shorter PFS than those with high expression
levels of 53BP1, as shown by Kaplan-Meier analysis (Fig. 7D). Overall, the present study
indicated that HNF1A may have an important role in oxaliplatin
resistance by regulating 53BP1 expression, and mediating HR and
NHEJ imbalances in patients with PDAC (Fig. 7E).
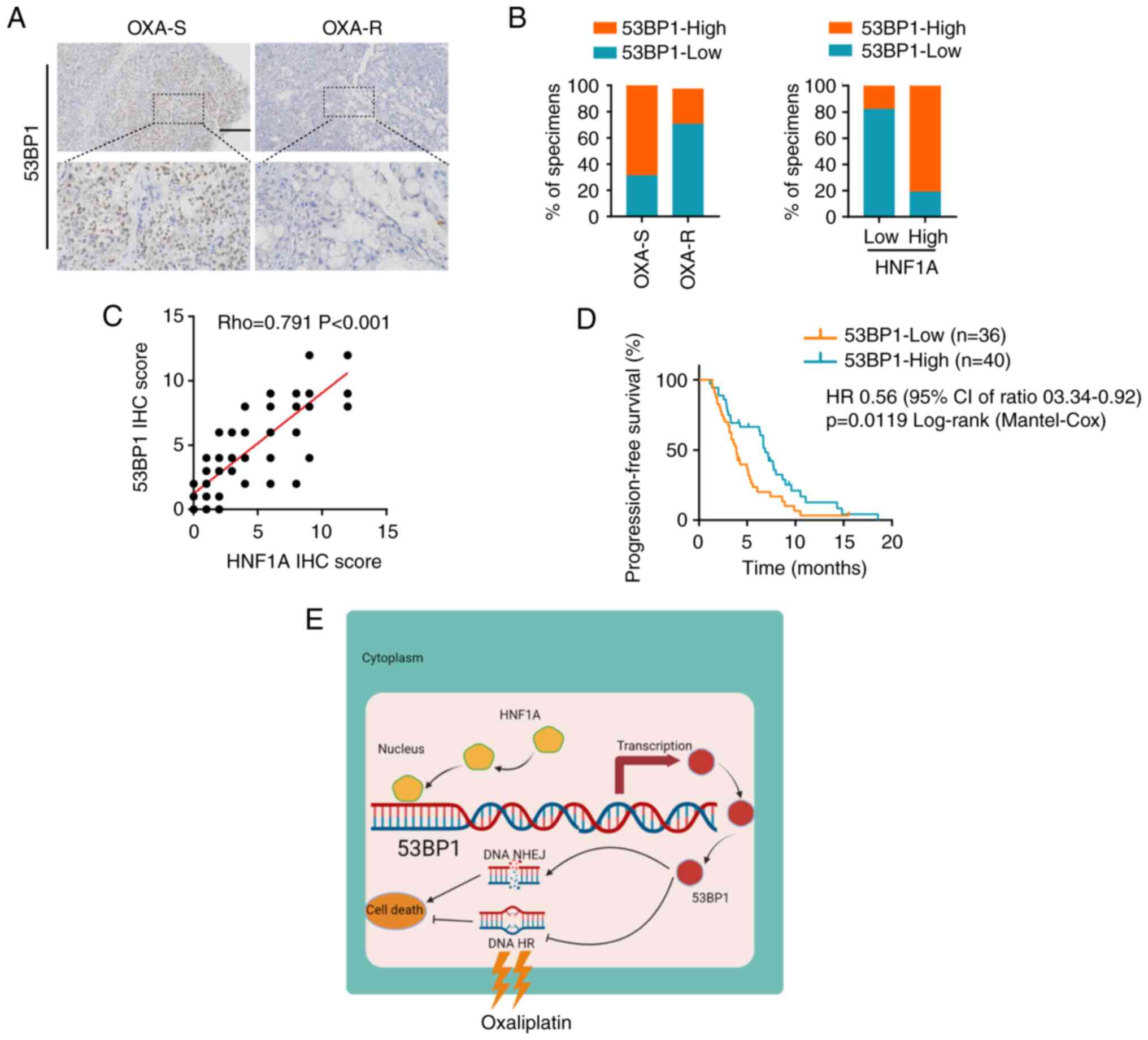 | Figure 7Clinical significance of the
HNF1A/53BP1 axis in PDAC. (A) Immunohistochemistry analysis of
53BP1 in OXA-S and OXA-R PDAC tissues. Scale bars, 50 µm.
(B) Semi-quantification of the percentage of OXA-S and OXA-R PDAC
specimens with low or high 53BP1 expression. (C) Correlation
between HNF1A expression and 53BP1 protein levels in PDAC tissues.
(D) Kaplan-Meier curves of progression-free survival associated
with 53BP1 expression evaluated by IHC in patients with PDAC who
received platinum-based chemotherapy. (E) Graphical illustration of
the mechanism by which HNF1A mediates 53BP1 activation in PDAC
cells to reduce oxaliplatin resistance. 53BP1, p53-binding protein
1; HNF1A, hepatocyte nuclear factor 1 homeobox A; HR, homologous
recombination; IHC, immunohistochemistry; NHEJ, nonhomologous end
joining; OXA, oxaliplatin; OXA-R, OXA-resistant; OXA-S,
OXA-sensitive; PDAC, pancreatic ductal adenocarcinoma. |
Discussion
Pancreatic cancer is a lethal disease with a poor
survival rate, due to a poor response to chemotherapy (20). Platinum-based chemotherapy is a
first-line chemotherapy treatment for patients with advanced
pancreatic cancer; however, the overall response rate is only 30%
worldwide (21). Our previous
study reported that cancer-associated fibroblasts promote the
acquired oxaliplatin resistance of pancreatic cancer cells via
paracrine signaling (22).
However, there are few studies on the inherent oxaliplatin
resistance of pancreatic cancer tumor cells (23). The present study revealed that low
HNF1A expression was associated with platinum-based chemotherapy
resistance and a poor prognosis. Gain- and loss-of-function assays
indicated that HNF1A serves an important role in oxaliplatin
resistance by affecting DNA DSB repair. In addition, HNF1A was
shown to mediate cisplatin and carboplatin resistance in PDAC
cells, which expands the therapeutic value of HNF1A in
platinum-based chemotherapy. Mechanistically, HNF1A directly binds
to the 53BP1 promoter region and transcriptionally activates 53BP1
expression, which switches HR-dependent DNA repair to
NHEJ-dependent DNA repair. Pancreatic cancer orthotopic and PDX
models revealed the potential value of HNF1A as a therapeutic
target for over-coming oxaliplatin resistance in pancreatic
cancer.
Conflicting roles of HNF1A in anticancer drug
resistance of different types of cancer have been reported. Fujino
et al (16) and Wang et
al (24) reported that HNF1A
inhibition significantly reduced the proliferation and anticancer
drug resistance of non-small cell lung cancer and colorectal cancer
via glucose metabolism. Conversely, in another study, inhibition of
HNF1A enhanced gemcitabine resistance by regulating multidrug
resistance genes in pancreatic cancer (14). The present study revealed that
HNF1A influenced the balance of HR and NHEJ to enhance the
oxaliplatin sensitivity of PDAC in vivo and in vitro.
It was hypothesized that the function of HNF1A may differ in
different molecular subtypes of cancer (25). Germline HNF1A mutations have been
linked to MODY3 through impaired insulin secretion and decreased
β-cell mass (12,26). According to a genome-wide
association study, genetic variants in the HNF1A locus predispose
patients to PDAC (27). Kalisz
et al (28) reported that
KDM6A mutations led to HNF1A deficiency in non-classical PDAC (also
defined as quasimesenchymal, basal and squamous-like PDAC). The
present study revealed low expression of HNF1A in patients with
oxaliplatin-resistant PDAC, which may be caused by HNF1A mutations.
These observations and results indicated the value of HNF1A as a
biomarker for guiding the use of platinum-based chemotherapy and
even some targeted therapies.
Platinum-based chemotherapy resistance is an
important obstacle in improving the prognosis of pancreatic cancer
and is exemplified by a low objective response rate (29). Tumor cells repair double-strand
breaks caused by platinum-based drugs via HR and NHEJ (30). Compared with NHEJ, HR-dependent DNA
repair is more precise, and NHEJ-dependent DNA repair often causes
genome instability and cell death (19,31).
A recent clinical trial suggested that the combination of PARP
inhibitors in patients with BRCA1/2 mutations greatly improved
patient outcomes (32). A
promising research direction involves the use of PARP inhibitors in
tumor cells with HR deficiency. The combination of platinum-based
therapy and PARP inhibitors may benefit patients with pancreatic
cancer and high HNF1A expression, and requires further
exploration.
The regulators 53BP1, ATM, Rad51, PARP1, BRCA1 and
BRCA2 are important in DSB repair (29,33-35).
The present study identified 53BP1 as the downstream target of
HNF1A via RT-qPCR and western blotting. 53BP1 is a
chromatin-binding protein, which regulates DSB repair by
suppressing the nucleolytic resection of DNA termini (36). 53BP1 serves a crucial role in
maintaining the balance of HR and NHEJ, which is important for
genomic stability. The upstream regulatory mechanism of 53BP1 has
attracted increasing attention. Parnandi et al (37) reported that Tudor-interacting
repair regulator interacted with the 53BP1 Tudor domain and
prevented its recruitment to DSBs (37). AMPK is directly associated with
53BP1 and phosphorylates it at Ser1317, promoting 53BP1 recruitment
to DSBs (38). There is no
evidence of 53BP1 transcriptional activation. Given that HNF1A is a
known transcription factor, the present study demonstrated that
HNF1A could directly enhance the transcription of 53BP1. ChIP and
luciferase assays confirmed that HNF1A interacted with the 53BP1
promoter region. The present study provided a novel mechanism for
regulating 53BP1, which may improve the understanding of DNA repair
and oxaliplatin resistance. There are some limitations to the
present study that could be addressed in future research. First,
the clinical sample size was small, which limits the application of
53BP1 and HNF1A in clinical practice. The clinical value of HNF1A
and 53BP1 should be further confirmed in a larger cohort in future
studies. Second, further research is required before this may be of
use clinically. Finally, the mechanism of HNF1A in mediating
oxaliplatin was not fully clarified. Other regulatory mechanisms
may exist, which require further study.
In conclusion, the present study suggested that the
HNF1A/53BP1 axis is critical for oxaliplatin resistance in PDAC.
Therefore, HNF1A may be a potential therapeutic target in patients
with PDAC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RC, XW and SZ conceived the idea for the project,
coordinated the research and modified the manuscript. RX, CH and YY
conducted most of the experiments. RC and XW confirmed the
authenticity of all the raw data. XZ, TLi and RH collected the
clinical samples and information, and also performed some
experiments. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
This research was approved by the Ethical Committee
of Guangdong Provincial People's Hospital (approval no.
KY-H-2022-011-01). All of the patients provided written informed
consent before the biopsy sample collection. Animal experiments
were carried out according to guidelines approved by the Animal
Experimental Research Ethics Committee of South China University of
Technology (approval no. 2021042).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was supported by grants from the National Natural
Science Foundation of China (grant nos. 82203691 and 82072639), the
Guangdong Science and Technology Department (grant no.
2021A1515011089), the Special Fund of 'Dengfeng Plan' of Guangdong
Provincial People's Hospital, China (grant nos. KJ012019509 and
DFJH2020027) and the National Key Clinical Specialty Construction
Project (2021-2024, grant no. 2022YW030009).
Abbreviations:
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
HR
|
homologous recombination
|
|
NHEJ
|
nonhomologous end-joining
|
|
53BP1
|
p53-binding protein 1
|
|
DSB
|
DNA double strand break
|
|
CCK-8
|
Cell Counting Kit-8
|
|
IF
|
immunofluorescence
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neoptolemos JP, Kleeff J, Michl P,
Costello E, Greenhalf W and Palmer DH: Therapeutic developments in
pancreatic cancer: Current and future perspectives. Nat Rev
Gastroenterol Hepatol. 15:333–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sohal DPS, Kennedy EB, Khorana A, Copur
MS, Crane CH, Garrido-Laguna I, Krishnamurthi S, Moravek C,
O'Reilly EM, Philip PA, et al: Metastatic pancreatic cancer: ASCO
clinical practice guideline update. J Clin Oncol. 36:2545–2556.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wattenberg MM, Asch D, Yu S, O'Dwyer PJ,
Domchek SM, Nathanson KL, Rosen MA, Beatty GL, Siegelman ES and
Reiss KA: Platinum response characteristics of patients with
pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or
PALB2 mutation. Br J Cancer. 122:333–339. 2020. View Article : Google Scholar :
|
|
7
|
Iwabuchi K, Bartel PL, Li B, Marraccino R
and Fields S: Two cellular proteins that bind to wild-type but not
mutant p53. Proc Natl Acad Sci USA. 91:6098–6102. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pontoglio M: Hepatocyte nuclear factor 1,
a transcription factor at the crossroads of glucose homeostasis. J
Am Soc Nephrol. 11(Suppl 16): S140–S143. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Odom DT, Zizlsperger N, Gordon DB, Bell
GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA,
Gifford DK, et al: Control of pancreas and liver gene expression by
HNF transcription factors. Science. 303:1378–1381. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Servitja JM, Pignatelli M, Maestro MA,
Cardalda C, Boj SF, Lozano J, Blanco E, Lafuente A, McCarthy MI,
Sumoy L, et al: Hnf1alpha (MODY3) controls tissue-specific
transcriptional programs and exerts opposed effects on cell growth
in pancreatic islets and liver. Mol Cell Biol. 29:2945–2959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Duell EJ, Yu K, Risch HA, Olson SH,
Kooperberg C, Wolpin BM, Jiao L, Dong X, Wheeler B, et al: Pathway
analysis of genome-wide association study data highlights
pancreatic development genes as susceptibility factors for
pancreatic cancer. Carcinogenesis. 33:1384–1390. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Firdous P, Nissar K, Ali S, Ganai BA,
Shabir U, Hassan T and Masoodi SR: Genetic testing of
maturity-onset diabetes of the young current status and future
perspectives. Front Endocrinol (Lausanne). 9:2532018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo Z, Li Y, Wang H, Fleming J, Li M, Kang
Y, Zhang R and Li D: Hepatocyte nuclear factor 1A (HNF1A) as a
possible tumor suppressor in pancreatic cancer. PloS One.
10:e01210822015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu Y, Xu D, Peng J, Luo Z, Chen C, Chen Y,
Chen H, Zheng M, Yin P and Wang Z: HNF1A inhibition induces the
resistance of pancreatic cancer cells to gemcitabine by targeting
ABCB1. EBioMedicine. 44:403–418. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hoskins JW, Jia J, Flandez M, Parikh H,
Xiao W, Collins I, Emmanuel MA, Ibrahim A, Powell J, Zhang L, et
al: Transcriptome analysis of pancreatic cancer reveals a tumor
suppressor function for HNF1A. Carcinogenesis. 35:2670–2678. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fujino S, Miyoshi N, Ito A, Yasui M,
Matsuda C, Ohue M, Uemura M, Mizushima T, Doki Y and Eguchi H:
HNF1A regulates colorectal cancer progression and drug resistance
as a downstream of POU5F1. Sci Rep. 11:103632021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells-perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ceccaldi R, Rondinelli B and D'Andrea AD:
Repair pathway choices and consequences at the double-strand break.
Trends Cell Biol. 26:52–64. 2016. View Article : Google Scholar :
|
|
20
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meneses-Medina MI, Gervaso L, Cella CA,
Pellicori S, Gandini S, Sousa MJ and Fazio N: Chemotherapy in
pancreatic ductal adenocarcinoma: When cytoreduction is the aim. A
systematic review and meta-analysis. Cancer Treat Rev.
104:1023382022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Zheng S, Hu C, Li G, Lin H, Xia
R, Ye Y, He R, Li Z, Lin Q, et al: Cancer-associated
fibroblast-induced lncRNA UPK1A-AS1 confers platinum resistance in
pancreatic cancer via efficient double-strand break repair.
Oncogene. 41:2372–2389. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiu CF, Park JM, Chen HH, Mau CZ, Chen
PS, Su YH, Chen HA, Liu YR, Hsieh TH, Chiu CC, et al: Organic
cation transporter 2 activation enhances sensitivity to oxaliplatin
in human pancreatic ductal adenocarcinoma. Biomed Pharmacother.
153:1135202022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Huang H and Liu F: DNAJC12
activated by HNF1A enhances aerobic glycolysis and drug resistance
in non-small cell lung cancer. Ann Transl Med. 10:4922022.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Collisson EA, Bailey P, Chang DK and
Biankin AV: Molecular subtypes of pancreatic cancer. Nat Rev
Gastroenterol Hepatol. 16:207–220. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farrelly AM, Wobser H, Bonner C,
Anguissola S, Rehm M, Concannon CG, Prehn JH and Byrne MM: Early
loss of mammalian target of rapamycin complex 1 (mTORC1) signalling
and reduction in cell size during dominant-negative suppression of
hepatic nuclear factor 1-alpha (HNF1A) function in INS-1 insulinoma
cells. Diabetologia. 52:136–144. 2009. View Article : Google Scholar
|
|
27
|
Klein AP, Wolpin BM, Risch HA,
Stolzenberg-Solomon RZ, Mocci E, Zhang M, Canzian F, Childs EJ,
Hoskins JW, Jermusyk A, et al: Genome-wide meta-analysis identifies
five new susceptibility loci for pancreatic cancer. Nat Commun.
9:5562018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kalisz M, Bernardo E, Beucher A, Maestro
MA, Del Pozo N, Millá I, Haeberle L, Schlensog M, Safi SA, Knoefel
WT, et al: HNF1A recruits KDM6A to activate differentiated acinar
cell programs that suppress pancreatic cancer. EMBO J.
39:e1028082020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laurini E, Marson D, Fermeglia A, Aulic S,
Fermeglia M and Pricl S: Role of Rad51 and DNA repair in cancer: A
molecular perspective. Pharmacol Ther. 208:1074922020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou J, Kang Y, Chen L, Wang H, Liu J,
Zeng S and Yu L: The drug-resistance mechanisms of five
platinum-based antitumor agents. Front Pharmacol. 11:3432020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brandsma I and Gent DC: Pathway choice in
DNA double strand break repair: Observations of a balancing act.
Genome Integr. 3:92012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reiss KA, Mick R, O'Hara MH, Teitelbaum U,
Karasic TB, Schneider C, Cowden S, Southwell T, Romeo J, Izgur N,
et al: Phase II study of maintenance rucaparib in patients with
platinum-sensitive advanced pancreatic cancer and a pathogenic
germline or somatic variant in BRCA1, BRCA2, or PALB2. J Clin
Oncol. 39:2497–2505. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao Y, Simon M, Seluanov A and Gorbunova
V: DNA damage and repair in age-related inflammation. Nat Rev
Immunol. 23:75–89. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ray Chaudhuri A and Nussenzweig A: The
multifaceted roles of PARP1 in DNA repair and chromatin
remodelling. Nat Rev Mol Cell Biol. 18:610–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cleary JM, Aguirre AJ, Shapiro GI and
D'Andrea AD: Biomarker-guided development of DNA repair inhibitors.
Mol Cell. 78:1070–1085. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Noordermeer SM, Adam S, Setiaputra D,
Barazas M, Pettitt SJ, Ling AK, Olivieri M, Aacute;lvarez-Quilón A,
Moatti N, Zimmermann M, et al: The shieldin complex mediates
53BP1-dependent DNA repair. Nature. 560:117–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Parnandi N, Rendo V, Cui G, Botuyan MV,
Remisova M, Nguyen H, Drané P, Beroukhim R, Altmeyer M, Mer G and
Chowdhury D: TIRR inhibits the 53BP1-p53 complex to alter cell-fate
programs. Mol Cell. 81:2583–2595.e2586. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang Y, Dong Y, Luo Y, Jiang S, Meng FL,
Tan M, Li J and Zang Y: AMPK-mediated phosphorylation on 53BP1
promotes c-NHEJ. Cell Rep. 34:1087132021. View Article : Google Scholar : PubMed/NCBI
|