Introduction
Wilms' tumor or nephroblastoma, named by the German
surgeon Carl Max Wilhelm Wilms' in the 19th century, is the second
most common intra-abdominal tumor in young children, with a peak
incidence between 2 and 5 years. The incidence of Wilms' tumor in
adults is extremely low, and the total cases presented to date
account for <1% (1). However, the
precise number of cases of Wilms' tumor in adults remains unknown,
as a significant number of cases are either insufficiently
documented or misdiagnosed. The stage-for-stage prognosis in adults
is poorer compared with that in children. Due to the improved
practicable treatments, the survival of adult patients has improved
significantly from <30 to >90% (2). To the best of our knowledge, 9 cases of
adult Wilms' tumor were previously published (Table I) and our patient, aged 51 years, is
the second oldest patient reported to date. A radical nephrectomy
was performed with subsequent chemotherapy. The present study was
approved by the Ethics Committee of Peking University Shenzhen
Hospital (Shenzhen, China) and written informed consent was
obtained from the patient regarding the publication of his
clinicopathological data.
 | Table I.Reported cases of adult Wilms
tumor. |
Table I.
Reported cases of adult Wilms
tumor.
| Case | First
author/Refs. | Year | Age (years),
gender | Main complaint | Size (cm) | Localization | Microscopic
characteristics | Immunohistochemical
staining |
|---|
| 1 | Huang/(3) | 2015 | 20, female | Left flank pain and
back pain | 6.4×6.2 | Left kidney, lower
pole | Triphasic pattern of
blastemal, epithelial and stromal components | Vimentin+,
CD99+, CD117+, WT1+ |
| 2 | Varma/(4) | 2006 | 48, male | Flank pain and
hematuria | 11×10 | Right kidney, upper
pole | Highly cellular,
comprising epithelial, blastemal and stromal elements | No description |
| 3 | Thevendran/(5) | 2010 | 37, female | Left flank mass | 9.5×14.2 | Right kidney, upper
pole | Triphasic tumor
composed of epithelial, blastematous and stromal elements | WT1+ |
| 4 | Morabito/(6) | 2014 | 38, male | Abominal pain and
macroscopic hematuria | 10 | Right kidney | Triphasic cellular
pattern with undifferentiated blastemal cells and cells
differentiating toward epithelial and stromal lineages | Vimentin+,
desmin+, WT1+ |
| 5 | Patnayak/(7) | 2012 | 19, male | Low backache and
colicky left loin pain | 15×10 | Right kidney | Monomorphous tumor
cells presenting as nests, islands and sheets, with intervening
necrosis and lymphoid collections | S-100+,
CD117+, NSE+ |
| 6 | Guo/(8) | 2011 | 54, male | Low backache and
colicky left loin pain | 2.5×2.3 | Right kidney | Triphasic pattern of
blastemal, epithelial, and stromal components | WT1+ |
| 7 | Masuda/(9) | 2004 | 22, male | Right flank pain | 4.2×1.8 | Right kidney, upper
pole | Predominantly
epithelial histology | No description |
| 8 | Seifert/(10) | 2012 | 26, female | Fever | 12 | Left kidney, | Primarily
undifferentiated blastemal cells | PAN-CK+,
vimentin+, |
| 9 | Present case | 2015 | 51, male | Identified on checkup
examination | 4.0×4.5 | Left kidney, upper
pole | Undifferentiated
blastemal cells differentiating to various degrees and epithelial
and stromal lineages in different proportions | WT1+ |
Case report
A 51 year-old man was diagnosed with a left kidney
tumor on routine examination and was admitted to the Department of
Urology of Peking University Shenzhen Hospital (Shenzhen, China)
for further evaluation. The patient had no urinary or respiratory
symptoms and had not undergone previous surgery. The general
examination revealed no significant findings. The patient had a
heart rate of 88 beats/min, a blood pressure of 118/67 mmHg, a
temperature of 36.8°C and a respiratory rate of 22 breaths/min.
Laboratory examination revealed a haemoglobin level
of 11.3 g/dl, and a white blood cell count of
6.86×109/l, with 59.8% granulocytes. The glucose level
was 4.98 mmol/l, the blood urea nitrogen was 7.95 mmol/l and the
serum creatinine was 79.2 µmol/l. The liver function tests and
serum electrolyte levels were normal. Urine examination revealed
several erythrocytes per high-power field. The chest X-ray, renal
function tests, cardiovascular and neurological investigations were
largely normal. The non-contrast computed tomography (CT) scan of
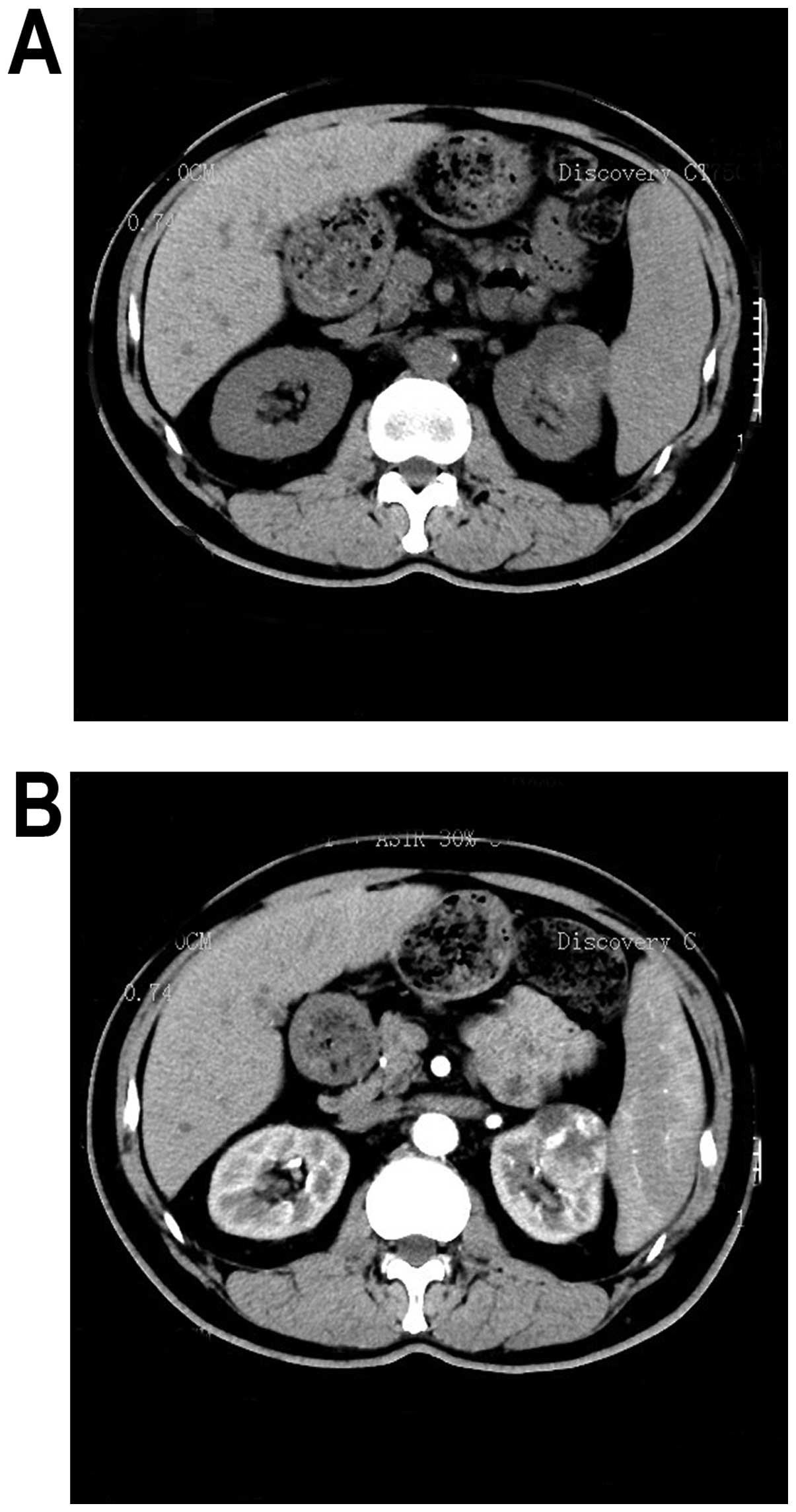
the kidneys revealed a 4.0×4.0×4.5-cm round hypodense mass [39
Hounsfield units (HU)] arising from the upper pole of the left
kidney (Fig. 1A). The
contrast-enhanced CT revealed a heterogeneously enhanced lesion (51
HU) (Fig. 1B).
The patient underwent left radical nephrectomy and
macroscopically the tumor was a well-encapsulated mass that did not
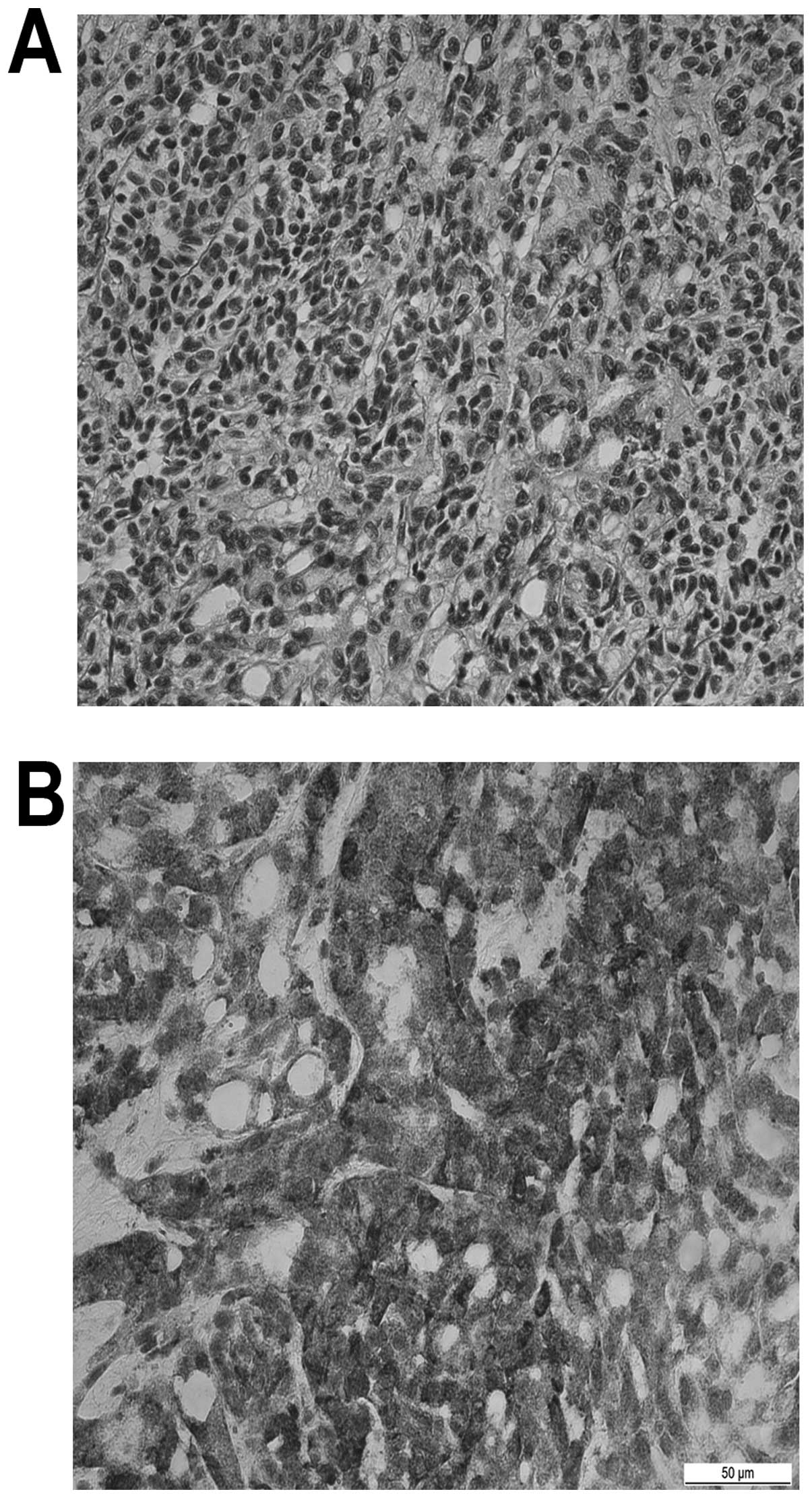
appear to invade neighboring tissue. The microscopic examination
identified blastemal cells, undifferentiated or differentiated to
various degrees, and epithelial and stromal lineages in different
proportions (Fig. 2A). However, the
most typical characteristic was the presence of undifferentiated
blastemal cells. On immunohistochemical staining, Wilms' tumor 1
antibody (WT1) was found to be positive (Fig. 2B) and the renal neoplasm was confirmed
as Wilms' tumor. Positron emission tomography-CT revealed no
metastases. Chemotherapy was performed regularly and there was no
evidence of cancer on medical examination at the 2-year follow-up
(11).
Discussion
Nephroblastoma, also referred to as Wilms' tumor, is
a embryonal neoplasm originating from nephrogenic blastemal cells,
which replicates the histology of the kidneys and usually exhibits
various patterns of differentiation (12). Approximately one in every 7,000
children suffer from this disease (12). There are no gender differences and
Wilms' tumor occurs in equal frequency in both kidneys. The mean
age is 35 and 45 months for males and females, respectively, and
98% of the cases occur at ages <10 years (12). Wilms' tumor has also been reported in
adults, but it is extremely rare (13). The most common main complaints are
local pain and painless hematuria, but in children a palpable mass
is more common (14). The tumor is
often detected by abdominal palpation, CT and ultrasound scan
(15). Compared with pediatric
counterparts, adult patients with advanced clinical stage exhibit
metastatic manifestations more frequently (10 vs. 29%,
respectively) (12). The prognosis of
adult Wilms' tumor is worse compared with that in the pediatric
population, and there is no detailed explanation for that
phenomenon to date. Approximately 5% of Wilms' tumors are
associated with an unfavorable outcome and are recognized
pathologically as having unfavourable histology, mainly due to the
presence of nuclear anaplasia (12).
In pediatric patients as well as in adults, Wilms'
tumors present as triphasic embryonic kidney tumors, and their
histological appearance has the characteristics of marked
structural diversity (5). Although,
classic Wilms' tumor consists of three types of cells (blastemal,
stromal and epithelial), the presence of all three types is
uncommon in the same case (16). The
presence of blastemal cells is the predominant histological
component of Wilms' tumors, and it appears in distinctive patterns.
Blastemal cells exhibit scant cytoplasm and are very small,
mitotically active, with rotund and overlapping nuclei containing
coarsely distributed chromatin and evenly small nucleoli (17). An epithelial component is present in
several Wilms' tumors, and this feature may be represented by
primitive structures merely recognizable as tubular formations.
Other Wilms' tumors are composed of papillary and tubular elements
that are easily recognized, recapitulating normal stages of
nephrogenesis (18). Various stromal
patterns may occur and cause diagnostic difficulties when there is
lack of blastemal and epithelial differentitation. The common
undifferentiated RCC has been divided into clear-cell and
non-clear-cell categories, with papillary RCC forming the most
common subtype of non-clear-cell RCC (19,20). To
the best of our knowledge, various undifferentiated tumors in
adults should be considered in the differential diagnosis,
particularly when the carcinoma is predominantly monophasic.
From a genetics aspect, ~10% of Wilms' tumors
develop in association with one of several well-characterized
dysmorphic syndromes. The molecular events of adult Wilms'
tumorigenesis have not been fully elucidated (21). However, similar to other tumor genes,
the location of Wilms' tumor may be detected by cytogenetic
analysis of DNA from the patients whose abnormalities were
genetically determined, enabling the prediction of a number of
chromosomal disruptions significantly associated with phenotypic
abnormalities. The constitutional loss of band 13 of the short arm
of chromosome 11 (11p13 or WT1) was significantly associated with
adult Wilms' tumors (22). The
deletion of genetic material from chromosome 11p13 was clearly
associated with tumorigenesis, indicating that some critical
deletions may involve the tumor suppressor genes. The candidate
genes were entirely selected from the deleted regions of chromosome
11p13 and the target gene WT1 or 11p13 was isolated and cloned
(23). The sequence analysis
demonstrated that 11p13 acts as a transcriptional regulator whose
protein product significantly affects specific DNA motifs (23). However, the accurate function of the
WT1 protein remains unknown. Recent research suggests that patterns
of WT1 expression may play an extremely significant role in cell
differentiation of the metanephric stem, which may explain the
finding of associated genitourinary abnormalities. The gene for
Beckwith-Wiedemann syndrome has been localized to the chromosome
11p15 and named WT2, but the precise genetic mechanism has not yet
been fully elucidated (24). Efforts
to identify the precise genetic event at this locus have identified
the presence of clusters of imprinted genes. At the locus, a
preferential deletion of its maternal allele in several cases of
Wilms' tumor reveals that genomic imprinting is associated with the
pathogenesis of certain neoplasms (24). Furthermore, additional genetic loci
are involved in familial Wilms' tumor in patients carrying WT1 and
WT2. A proportion of patients with nephroblastoma usually have a
positive family history of identical tumors. It remains unclear
whether the aberration of genes results in Wilms' tumor or other
tumors. Therefore, this issue requires further investigation.
There are currently no adequate treatment guidelines
for adult Wilms' tumor (25). The
pediatric regimen, which includes radical nephrectomy and adjuvant
chemotherapy, with or without radiotherapy, is recommended for the
treatment of the adult counterpart. However, there is limited
information available for adult Wilms' tumor treatment if the
initial chemotherapy fails or if the tumor recurs (25).
In the present case, our patient is the second
oldest reported to date and he had received no prior chemotherapy.
Furthermore, all three types of cells were histopathologically
identified in the tumor. Therefore, we consider this to be an
extremely rare case of adult Wilms' tumor.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81101922), the
Guangdong Province Natural Science Foundation of China (no.
S2012010008365), the Science and Technology Development Fund
Project of Shenzhen (nos. JCYJ20130402114702124 and
JCYJ20150403091443329) and the fund of Guangdong Key Medical
Subject.
References
|
1
|
Özyörük D, Demir HA, Emir S, Karakuş E and
Tunç B: Occurrence of Wilms' tumor in a child with hereditary
spherocytosis. Turk J Pediatr. 57:206–209. 2015.PubMed/NCBI
|
|
2
|
Sakai K, Shimodaira S, Maejima S, et al:
Dendritic cell-based immunotherapy targeting Wilms' tumor 1 in
patients with recurrent malignant glioma. J Neurosurg. 123:989–97.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang JL, Liao Y, An Y and Qiu MX:
Spontaneous rupture of adult Wilms' tumor: A case report and review
of the literature. Can Urol Assoc J. 9:E531–E534. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Varma AV, Malukani K, Rihal P and
Nandedkar SS: Adult Wilms' tumor: A case report with review of
literature. J Cancer Res Ther. 11:934–936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thevendran G, Farne HA and Kaisary AV:
Wilms' tumor in a 37-yeal-old. J Clin Med Res. 2:194–197.
2010.PubMed/NCBI
|
|
6
|
Morabito V, Guglielmo N, Melandro F,
Mazzesi G, Alesini F, Bosco S and Berloco PB: Adult Wilms tumor:
Case report. Int J Surg Case Rep. 6:273–276. 2015. View Article : Google Scholar
|
|
7
|
Patnayak R, Rambabu DV, Jena A, Vijaylaxmi
B, et al: Rare case of blastemal predominant adult Wilms' tumor
with skeletal metastasis case report and brief review of
literature. Indian J Urol. 28:447–449. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
The reference of case 6: Guo A, Wei L,
Song X and Liu A: Adult wilms tumor with intracaval and
intracardiac extension: report of a case and review of literature.
J Cancer. 2:132–135. 2011.PubMed/NCBI
|
|
9
|
Masuda H, Azuma H, Nakajima F, Watsuji T
and Katsuoka Y: Adult Wilms' tumor with calcification untreated for
5 years-a case report. BMC Urol. 4:52004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seifert RP, McNab P, Sexton WJ, Sawczyn
KK, Smith P, Coppola D and Bui MM: Rhabdomyomatous differentiation
in Wilms tumor pulmonary metastases: a case report and literature
review. Ann Clin Lab Sci. 42:409–416. 2012.PubMed/NCBI
|
|
11
|
Huszno J, Starzyczny-Słota D, Jaworska M
and Nowara E: Adult Wilms tumor-diagnosis and current therapy. Cent
European J Urol. 66:39–44. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
PDQ Pediatric Treatment Editorial Board:
Wilms' tumor and other childhood kidney tumors treatment (PDQ®).
Bethesda MD: 2002.
|
|
13
|
Ali EM and Elnashar AT: Adult Wilms'
tumor: Review of literature. J Oncol Pharm Pract. 18:148–151. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yadav SC, Sathe PA, Ghodke RK and
Fernandes GC: Giant untreated Wilms' tumor with intracardiac
extension: A rare case. Indian J Pathol Microbiol. 56:68–69. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu J, Zhu Q, Zhu W and Chen W: CT and MRI
imaging features and long-term follow-up of adult Wilms tumor. Acta
Radiol. Oct 8–2015.(Epub ahead of print). View Article : Google Scholar
|
|
16
|
Shukrun R, Pode-Shakked N, Pleniceanu O,
Omer D, Vax E, Peer E, Pri-Chen S, Jacob J, Hu Q, Harari-Steinberg
O, et al: Wilms tumor blastemal stem cells dedifferentiate to
propagate the tumor bulk. Stem Cell Reports. 3:24–33. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Patnaik N, Mishra K, Saini P and Agarwal
N: Primitive neuroectodermal tumor of the kidney in a young male:
Case report and review of literature. Urol Ann. 7:236–239. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hussong JW, Perkins SL, Huff V, McDonald
JM, Pysher TJ, Beckwith JB and Coffin CM: Familial Wilms tumor with
neural elements: Characterization by histology,
immunohistochemistry and genetic analysis. Pediatr Dev Pathol.
3:561–567. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sircar K, Rao P, Jonasch E, Monzon FA and
Tamboli P: Contemporary approach to diagnosis and classification of
renal cell carcinoma with mixed histologic features. Chin J Cancer.
32:303–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ehrlich PF, Anderson JR, Ritchey ML, et
al: Clinicopathologic findings predictive of relapse in children
with stage III favorable-histology Wilms tumor. J Clin Oncol.
31:1196–1201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kohler B, Biebermann H, Friedsam V,
Gellermann J, Maier RF, Pohl M, Wieacker P, Hiort O, Grüters A and
Krude H: Analysis of the Wilms tumor suppressor gene (WT1) in
patients 46,XY disorders of sex development. J Clin Endocrinol
Metab. 96:E1131–E1136. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akpa MM, Iglesias DM, Chu LL, Cybulsky M,
Bravi C and Goodyer PR: Wilms' tumor suppressor, WT1, suppresses
epigenetic silencing of the β-catenin gene. J Biol Chem.
290:2279–2288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wagner KD, Cherfils-Vicini J, Hosen N,
Hohenstein P, Gilson E, Hastie ND, Michiels JF and Wagner N: The
Wilms tumour suppressor Wt1 is a major regulator of tumour
angiogenesis and progression. Nat Commun. 5:58522014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park M, Choi Y, Choi H and Roh J: Wilms'
tumor suppressor gene (WT1) suppresses apoptosis by
transcriptionally downregulating BAX expression in immature rat
granulosa cells. J Ovarian Res. 7:1182014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Provenzi M, Saettini F, Conter V,
Chinaglia D, Vai P, Bruno A, Cavalleri L, Foglia C, Giraldi E,
Collini P and Spreafico F: Is there a role for FDG-PET for the
assessment of treatment efficacy in Wilms tumor? A case report and
literature review. Pediatr Hematol Oncol. 30:633–639. 2013.
View Article : Google Scholar : PubMed/NCBI
|