Introduction
Granular cell tumor (GCT) is a clinically rare
neoplasm composed of characteristic cells that contain numerous
eosinophilic cytoplasmic granules, as shown on hematoxylin and
eosin (H&E) staining (1). GCT
may be divided into the neural type, which exhibits S-100
reactivity (conventional GCT), and non-neural GCT, which does not
exhibit S-100 reactivity (2).
Although non-neural GCT was first described in 1991 (3), it has not been fully characterized, as
it has a rather unique immunophenotype, unlike conventional GCT,
and reports of this entity in the literature are scarce (2–4). GCT may
only be definitively diagnosed postoperatively based on detailed
pathological and immunohistochemical examinations. Complete
resection is recommended for malignant or metastatic GCT (1,5,6).
GCTs most commonly arises in dermal or subcutaneous
regions, or the tongue (1); they may
also develop in other parts of the body, but uterine GCT is
extremely rare. To date, only three cases of uterine cervical GCT
have been documented, all involving conventional GCTs (6–8);
however, to the best of our knowledge, there have been no reports
of GCT of the uterine corpus in the literature to date.
We herein describe the first reported case of
non-neural GCT arising from the uterine corpus, which mimicked
uterine leiomyoma and was treated with complete surgical resection,
and discuss other related cases that have been reported in the
literature.
Case report
A 55-year-old perimenopausal woman, gravida 1, para
1, was referred to the Department of Obstetrics and Gynecology of
Hashimoto Municipal Hospital (Wakayama, Japan) with a suspected
uterine tumor in October 2015. The patient had no history of lower
abdominal or pelvic discomfort, pelvic surgery, or other relevant
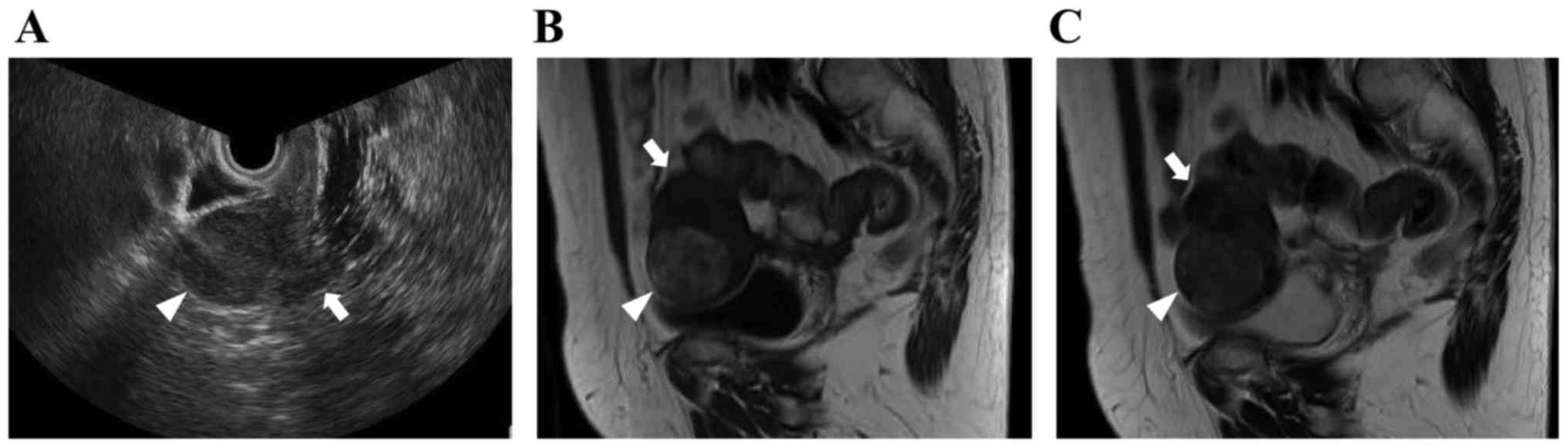
medical conditions. Transvaginal ultrasound examinations revealed a
4-cm well-defined uterine tumor, which exhibited iso-echogenicity
(Fig. 1A). On magnetic resonance
imaging (MRI), the tumor had a round shape, with heterogeneous
signal intensity, including areas of isointensity or hyperintensity
on T1-weighted imaging (WI) (Fig.
1B) and isointensity relative to muscle on T2WI (Fig. 1C). Based on these radiological
findings, the mass was suspected to be a leiomyoma of the uterine
corpus. A laboratory analysis of the patient's peripheral blood
revealed normal tumor marker levels [cancer antigen (CA)125, CA19-9
and carcinoembryonic antigen] and an elevated lactate dehydrogenase
level (215 IU/l). Total abdominal hysterectomy and bilateral
salpingo-oophorectomy were planned. On intraoperative examination,
a tumor was identified arising from the anterior part of the
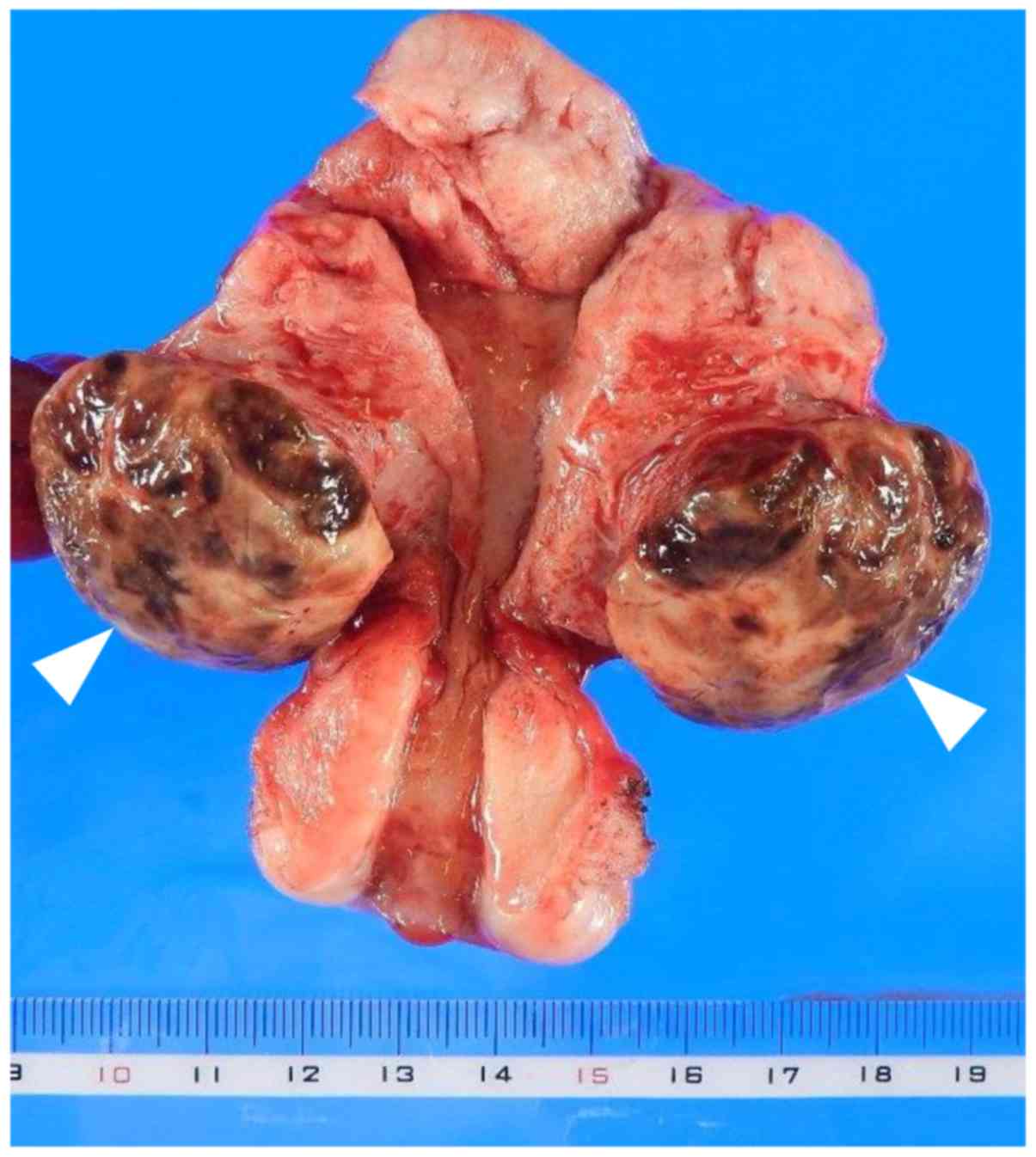
uterine corpus, and it was completely resected. The mass was 3.7 cm
in greatest diameter and elastic-hard in consistency; the cut
surface of the surgical specimen was yellowish-brown (Fig. 2). Microscopically, histological
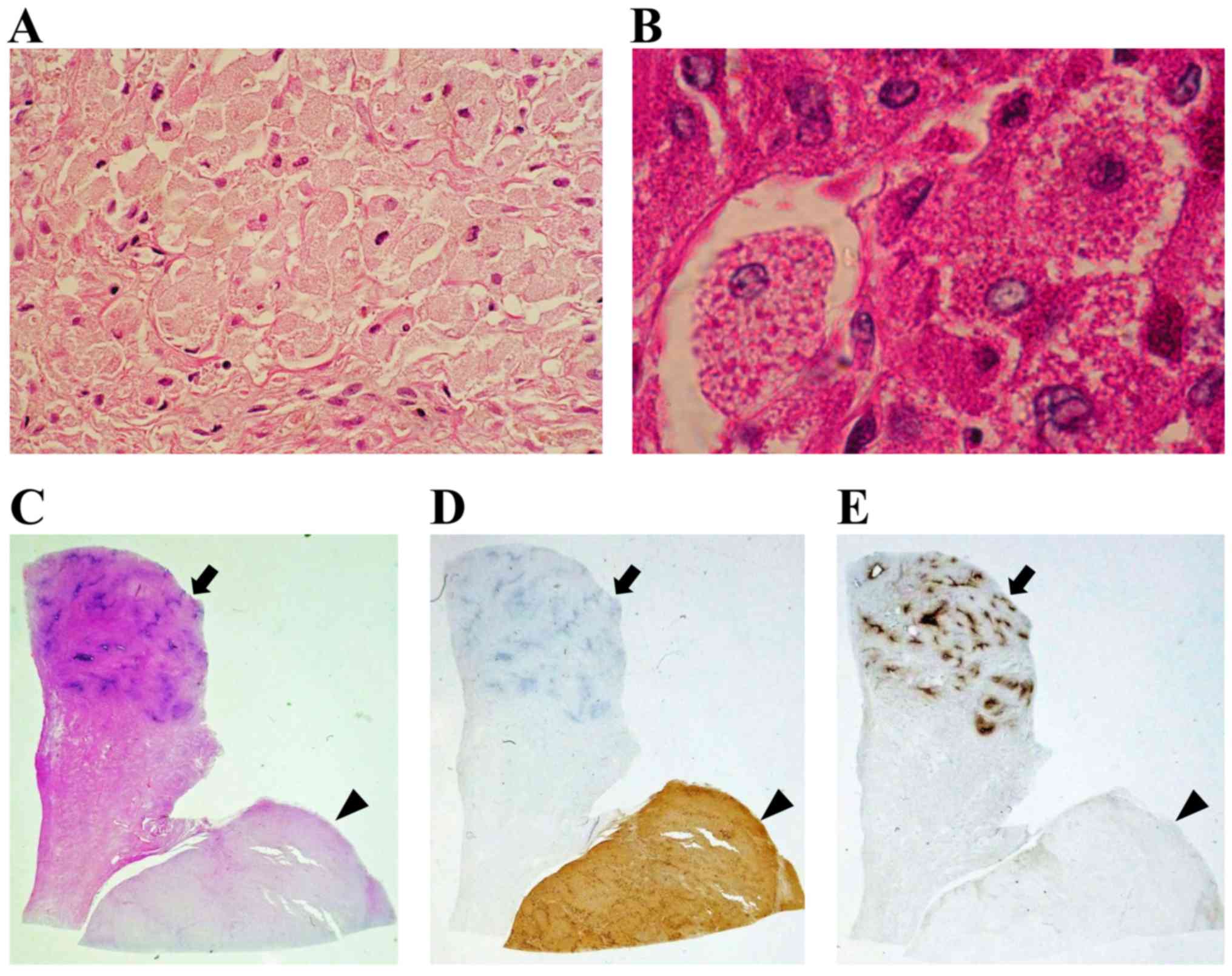
examination of the surgical specimen revealed large polygonal cells
with abundant eosinophilic granular cytoplasm and round to oval
nuclei (Fig. 3A). The tumor was
located mainly in the muscle layer of the uterine body (Fig. 3C). The mitotic activity of the tumor
cells were <5/10 high-power fields (HPFs) and the tumor had not
invaded the mucosal layer (Fig. 3C).
Immunohistochemistry revealed positive periodic acid-Schiff (PAS)
staining of the cytoplasmic granules, which was resistant to
diastase (Fig. 3B). In addition, the
tumor cells stained positive for CD68 (Fig. 3D), but negative for S-100,
neuron-specific enolase (NSE), cytokeratin, CD34, α-smooth muscle
actin (SMA), desmin, estrogen receptor (ER) and progesterone
receptor (PR) (Fig. 3E). The Ki-67
labeling index was ~6% in 20 random HPFs (data not shown). The
immunohistochemistry findings excluded leiomyoma, leiomyosarcoma,
malignant schwannoma, gastrointestinal stromal tumor and solitary
fibrous tumor. The diagnosis of non-neural GCT of the uterine
corpus was confirmed based on the pathological and
immunohistochemical findings. The postoperative course was
uneventful, and the patient was discharged from the hospital on
postoperative day 7. At 12 months after the diagnosis, there was no
evidence of local recurrence or systemic disease. Written informed
consent was obtained from the patient regarding the publication of
this case report and associated images.
Discussion
GCT is a rare slow-growing neoplasm that accounts
for ~0.5% of all soft tissue tumors. Women are twice as likely to
develop GCT as men. Furthermore, GCT mainly occurs between 40 and
60 years of age, and generally arises in the subcutaneous tissues
of the head and neck region, although it may occur at several other
sites (1). The majority of GCTs are
considered to have undergone neural crest differentiation and
exhibit positivity for S-100. These tumors are referred to as
conventional GCTs (1). However, GCTs
that do not display S-100 protein expression have been described in
certain case series since 1991, and these tumors are referred to as
non-neural GCTs (2–4). Therefore, GCT is classified into
conventional GCT (S-100-positive) and non-neural GCT
(S-100-negative) types. GCT rarely occurs as a gynecological tumor,
although when it does it mainly arises in the vulva (6,7). As
regards the uterus, only three cases of uterine cervical GCT have
been reported to date, all involving conventional GCTs. However, to
the best of our knowledge, no cases of non-neural GCT of the
uterine corpus have been reported in the English literature (PubMed
and MEDLINE databases) to date (6–8).
The diagnosis of non-neural GCT of the uterine
corpus is difficult, as surgery is required for a definitive
diagnosis. In a previous case of GCT, it was demonstrated that the
results of imaging analysis are non-specific (5). On MRI, GCTs are usually located
superficially and display a round or oval shape and heterogeneous
signal intensity, including areas of isointensity or hyperintensity
relative to muscle or suppressed fat signals on T1WI, whereas they
frequently exhibited isointensity or hyperintensity relative to
muscle on T2WI. In a case of GCT involving a premenopausal female
patient, leiomyoma was initially suspected due to the round shape
and smooth surface and the fact that it appeared isointense on both
T1WI and T2WI, which made the diagnosis of GCT difficult.
Macroscopically, GCT surgical specimens are usually
composed of yellowish-brown tissue with a nodular yellow-grey cut
surface (1), as in the present case.
Microscopically, GCT is composed of ovoid or polygonal cells with
abundant eosinophilic granular cytoplasm, as shown on H&E
staining. Immunohistochemical examination may be used to confirm
the pathological diagnosis. In non-neural GCT, some of the
cytoplasmic granules stain positively for PAS and are resistant to
diastase, and the tumor cells stain positive for CD68, but are
negative for S-100, NSE, cytokeratin, CD34 and other mesenchymal
markers (α-SMA and desmin) or sex hormone receptors, namely ER and
PR (2–4). In case of gynecological tumors,
clinicians must be able to differentiate GCT from granular cell
variants of leiomyoma. Such granular cell changes usually only
affect part of the lesion, which allows GCT to be diagnosed using
conventional morphological and immunohistochemical criteria based
on mesenchymal markers and sex hormone receptors. In the present
case, immunohistochemistry revealed that the tumor stained positive
for PAS and CD68, whereas staining for S-100, NSE, cytokeratin,
CD34, α-SMA, desmin, ER and PR was negative. Thus, a final
diagnosis of non-neural GCT of the uterine corpus was made.
The most appropriate clinical management strategy
for non-neural GCT of the uterine corpus is not always clear, as
our experience with such cases is limited. GCT is usually a
clinically and histologically benign tumor, but some malignant
forms of GCT have been reported (1,5,6). Malignant GCTs are relatively uncommon,
constituting only 1–2% of all GCTs. The characteristics of
malignant GCT include nuclear pleomorphism, necrosis, and the
presence of any mitotic activity combined with an aggressive
clinical course and the destruction of neighboring structures.
Thus, the differentiation between benign and malignant GCTs is only
possible postoperatively based on detailed pathological and
immunohistochemical examinations of the tumor. However, even in
benign GCTs, local recurrence and secondary lymph node invasion
have been reported (5). A wide,
complete excisional margin is always preferred due to the lesion's
infiltrative pattern and potential for recurrence, and to ensure
that a correct pathological diagnosis is obtained. Metastasis and
recurrence usually occurs within 2 years; thus, GCT should be
followed up for at least 2 years postoperatively. According to
three previous reports on GCT of the uterine cervix (6–8), uterine
GCT appears to have a better prognosis following complete
resection, as in the present case, although one of the cases
involved a malignant GCT (6).
Further data on uterine GCT are needed, particularly regarding its
natural history, diagnosis and treatment.
To the best of our knowledge, this is the first
reported case of non-neural GCT arising from the uterine corpus.
The tumor was successfully treated with complete surgical
resection. It is important for gynecologists to be aware of the
existence of non-neural GCT of the uterine corpus, which requires
accurate diagnosis, complete resection and long-term follow-up,
combined with the findings on clinical presentation and pathology,
as it may be misdiagnosed as uterine leiomyoma.
References
|
1
|
Elkousy H, Harrelson J, Dodd L, Martinez S
and Scully S: Granular cell tumors of the extremities. Clin Orthop
Relat Res. 1–198. 2000.
|
|
2
|
Lazar AJ and Fletcher CD: Primitive
nonneural granular cell tumors of skin: Clinicopathologic analysis
of 13 cases. Am J Surg Pathol. 29:927–934. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
LeBoit PE, Barr RJ, Burall S, Metcalf JS,
Yen TS and Wick MR: Primitive polypoid granular-cell tumor and
other cutaneous granular-cell neoplasms of apparent nonneural
origin. Am J Surg Pathol. 15:48–58. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chaudhry IH and Calonje E: Dermal
non-neural granular cell tumour (so-called primitive polypoid
granular cell tumour): A distinctive entity further delineated in a
clinicopathological study of 11 cases. Histopathology. 47:179–185.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Behzatoğlu K and Bahadir B: Malignant
granular cell tumor with unusual histological features. Pathol Int.
57:115–119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo N, Peng Z, Yang K and Lou J: Uterine
cervical malignant granular cell tumor. J Obstet Gynaecol Res.
38:944–947. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang F, He YM, Yao XY and Yang KX:
Granular cell tumor of the uterine cervix. A case report. J Reprod
Med. 58:177–180. 2013.PubMed/NCBI
|
|
8
|
Haberal A, Turgut F, Ozbey B, Küçükalï T
and Sapmaz M: Granular cell myoblastoma of the cervix in a 14 year
old girl. Cent Afr J Med. 41:298–300. 1995.PubMed/NCBI
|