Case report
A 66 year-old woman presented to the Emergency Room
of Tor Vergata University Hospital with lower back pain. Her past
medical history was negative. Blood counts (CBC) revealed
leukocytosis (55 109/l; differential: 87% neutrophils,
5.4% lymphocytes, 6.8% monocytes) and anemia (10 gr/dl).
Splenomegaly was present at physical examination (18 cm
longitudinal diameter at the abdomen ultrasound). The peripheral
blood smear showed presence of immature granulocytes (>10%
promyelocytes, myelocytes and metamyelocytes) and 6% myeloblasts,
in the absence of monocytosis.
Bone marrow (BM) smears and BM biopsy were
hypercellular with left shift of the granulocyte maturation curve,
less than 5% CD34+ blasts, dyserythropoiesis and
presence of small hypolobated megakaryocytes. Molecular analysis
for the BCR-ABL fusion gene (p210, p190, p230), as well as
locus-specific FISH for t(9;22) were negative. The standard
karyotype analysis gave the initial result of a Xq23 deletion. To
better characterize the molecular lesion, array CGH, and a
locus-specific FISH were performed, as well as mutation studies on
the diagnostic BM sample (Fig. 1 and
Table I). The only detectable
clonality marker was a X isochromosome i(X)(p10) in 7 of the 15
metaphases analyzed. The diagnosis of aCML was made.
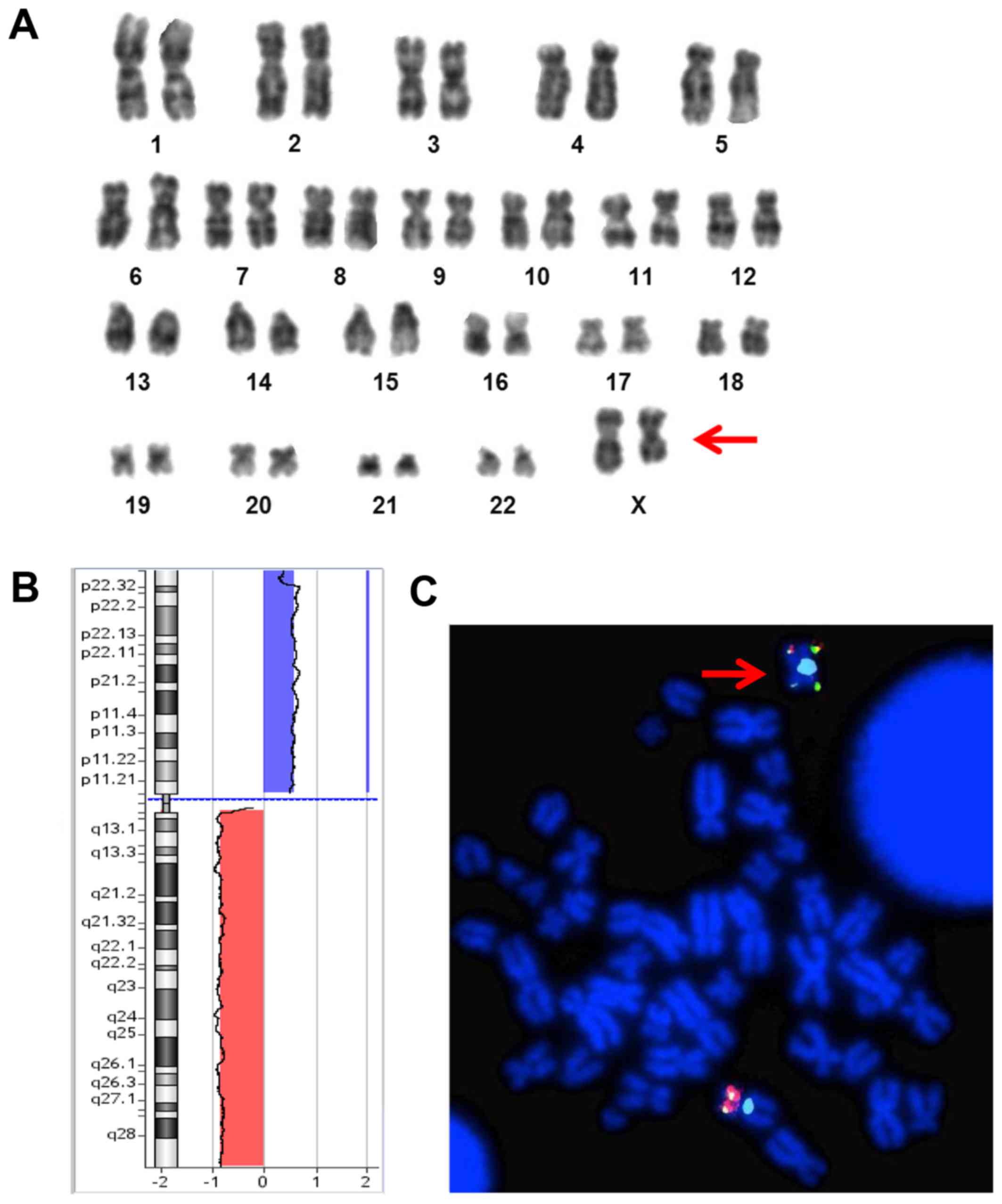 | Figure 1.(A) GTG-banded karyotype from cultured
bone marrow of the patient showing a del(X)(q23) in ~47% (7/15) of
the metaphases analyzed. (B) X chromosome Array-CGH (oligo 180K;
Agilent Technologies, Santa Clara, CA, USA) profile showing one
duplication of the short arm at Xp22.33q11.1 (blue bar), 62,2 Mb in
size and one deletion of the long arm at Xq11.1q28 (red bar), 92,7
Mb in size. (C) FISH analysis, using subtelomeric probes (Abbott
Molecular, Des Plaines, IL, USA) specific for chromosome Xpter and
Xqter, showing and confirming an isochromosome Xp (red arrow). |
 | Table I.Somatic mutations studied on the
diagnostic BM sample by Sanger sequencing. |
Table I.
Somatic mutations studied on the
diagnostic BM sample by Sanger sequencing.
| Gene locus | Mutation status |
|---|
| IDH1 R132 | WT |
| IDH2 R140 | WT |
| IDH2 R172 | WT |
| DNMT3A R882 | WT |
| SRSF2 P95-96 | WT |
| SF3B1 exons
13–16 | WT |
| U2AF1 | WT |
| SETBP1 | WT |
| ASXL1 | Silent mutation
S1253S; 3′UTR A/G, position Chr20:32437360 |
| ETNK1 exon 3 | WT |
The patient started 5-hydroxyurea (1 g per day),
with modest control on leucocytosis. Five months after the primary
diagnosis, the patient presented with left hypochondrium pain. A
CT-scan was diagnostic for a spleen infarction, with intracapsular
hematoma. Thus, she underwent urgent splenectomy. The
hystopatological examination of the spleen showed 5%
MPO+ and CD34+ cells with hematopoietic
invasion.
Almost one year after the primary diagnosis,
exacerbation of leukocytosis (≥100×109/l), anemia and
thrombocytopenia were observed, and the patient became
transfusion-dependent. The BM examination showed leukemic evolution
of aCML, with the presence of giant myeloid granular blasts (30% M2
FAB subtype), erythroid dysplasia and absence of megakariocytes.
Immunophenotyping confirmed the progression to sAML.
Cytogenetic and molecular studies for recurrent
mutations confirmed i(X)(p10) as the sole abnormality.
Discussion
We report on a patient with aCML and a i(X)(p10) as
the sole molecular abnormality. Little is known on the mechanism of
isochromosome formation and its pathogenetic impact (1). Frequency varies in different cancers
types, with the highest incidence in germ cell neoplasms (60%), and
the lowest in chronic myeloproliferative disorders (2.3%) (1).
Isochromosomes could result from transverse, instead
of longitudinal meiotic mis-division of the centromere; another
mechanism could be chromatid exchange involving two homologous
chromosomes. In both cases, it leads to loss and gain of genetic
material, likely resulting in deletion of tumor suppressor genes
and amplification of oncogenes (2).
Of note, in hematologic neoplasms isochromosome
formation is more frequent in lymphoid disorders (three times as
common), while in myeloid neoplasms the highest incidence is
observed in CML (18%) (2). In
particular, i(X)(p10) isochromosome has been described as the sole
abnormality in 11 patients, mostly in myeloid neoplasms (4 MDS, 3
AML, 2 chronic myelomonocytic leukemia) while in lymphoid
malignancies is usually part of a complex karyotype and seems to be
a secondary event (3,4).
The pathogenetic importance of i(X)(p10) is
underscored by its presence as the sole acquired abnormality in
these disorders. Its formation leads to the loss of the long-arm
and gain of short-arm of chromosome X, resulting in a state of
genetic imbalance. Costitutional i(X)(p10) is incompatible with
life (2). Since i(X)(p10) is found
also in males with hematologic disorders, it may reasonably arise
from the active X chromosome. Thus, its formation in females may
randomly derive from both the active or inactive X chromosome
(4).
To the best of our knowledge, this is the first case
of i(X)(p10) in aCML. Werner-Favre et al described in 1985 a
case of aCML carrying a structural rearrangement of X chromosome,
del (X)(q23) (5). The cytogenetic
distinction between del(Xq) and i(Xp) is known to be difficult due
to the similarity of X p- and q-arm band pattern, extending from
the centromere to band Xq24 (6).
Thus, it is not clear whether the previosuly reported case could be
a i(Xp), due to the lack of data from advanced cytogenetic
techniques as locus-specific FISH and CGH-array, not available at
that time (Fig. 1).
Deletion of the long arm of X chromosome has been
reported as recurrent karyotypic abnormality in patients with AML
or MDS as well as with solid tumors. Previously, two patients with
AML and three with MDS, and del (X)(q23) had been described
(7,8). In these cases, only conventional
G-banding and dual-color FISH had been performed to rule out the
presence of a iso-dicentric (X)(q13). Of note all these patients
were females between 46 and 65 years old, with breakpoints regions
apparently restricted to q13~q24. The median time to AML
progression reported by Olshanskaya for MDS patients was 19 months
with a prevalence of FAB M2 AML subtype.
In conclusion, our case highlights the importance of
using X p-arm FISH probes to characterize abnormal karyotypes with
structural abnormalities of the X chromosome. Furthermore, these
results, together with the previously reported i(Xp) and Xq-,
indicate that genetic imbalance resulting haploinsufficiency of
genes located on the long arm of the X chromosome could drive
neoplastic transformation. Of note, the prevalence in females, aged
between 46 and 65 years, the myeloid phenotype, and the poor
prognosis of all these cases indicate the need for specific
molecular biology studies to better characterize the genetic
lesions on the X chromosome, and the putative genes involved in the
leukemic evolution.
Acknowledgements
The authors would like to acknowledge research
funding from Associazione Italiana Ricerca sul Cancro (A.I.R.C, IG
16952 to MTV and 5916 to FLC).
References
|
1
|
Mertens F, Johansson B and Mitelman F:
Isochromosomes in neoplasia. Genes Chromosom. Cancer. 10:221–230.
1994.
|
|
2
|
Adeyinka A, Smoley S, Fink S, Sanchez J,
Van Dyke DL and Dewald G: Isochromosome (X)(p10) in hematologic
disorders: FISH study of 14 new cases show three types of
centromere signal patterns. Cancer Genet Cytogenet. 179:25–30.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitelman F, Johansson B and Mertens F:
Mitelman database of chromosome aberration and gene fusions in
cancer. https://cgap.nci.nih.gov/Chromosomes/MitelmanApril
15–2017
|
|
4
|
Gindina T: i(X)(p10) in female patients.
Atlas genes Cytogenet Oncol Haematol. http://AtlasGeneticsOncology.org/Anomlies/iXp10FemaleID1500.htmlApril
15–2017
|
|
5
|
Werner-Favre C, Beris P and Engel E: X
chromosome rearrangements and leukemia. Cytogenet Cell Genet.
39:801985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wolff DJ, Miller AP, Van Dyke DL, Schwartz
S and Willard HF: Molecular definition of breakpoints associated
with human Xq isochromosomes: Implications for mechanisms of
formation. Am J Hum Genet. 58:154–160. 1996.PubMed/NCBI
|
|
7
|
Wong KF, Siu LL and So CC: Deletion of
Xq23 is a recurrent karyotypic abnormality in acute myeloid
leukemia. Cancer Genet Cytogenet. 122:33–36. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Olshanskaya YV, Udovichenko AI, Vodinskaya
LA, Glasko EN, Parovitchnikova EN, Lorie YY, Dvirnik VN, Savchenko
VG and Domracheva EV: Myelodysplastic syndromes with isolated
deletion of the long arm of the chromosome X as a sole cytogenetic
change. Cancer Genet Cytogenet. 167:47–50. 2006. View Article : Google Scholar : PubMed/NCBI
|














