Introduction
Burkitt lymphoma, a B-cell neoplasm, is typically
characterized by a t(8;14)(q24;q32) translocation. There can be
other rare variant cases commonly identified as ‘double hit’
lymphoma or ‘triple hit’ lymphoma. Currently this terminology is
most often referred to a combination of translocations involving
the c-myc gene located on chromosome 8 combined with t (8;14) (q24;
q32) translocation involving antiapoptotic protein BCL2 or other
antiapoptotic proteins (such as BCL6). The updated WHO
classification refers to such lymphomas as ‘high-grade B cell
lymphoma with MYC and BCL2 and/or BCL6 translocations’ (1). Due to highly aggressive nature of
disease classical Burkitt lymphoma is relatively rarely accompanied
by other cytogenetic abnormalities. However, when such
abnormalities are present it usually entails an even more
aggressive behavior/resistance to existing therapy (2). We will present and discuss a case of a
patient with additional cytogenetic abnormality
(47,XY,+1,i(1)(q10),t(8;14)(q24;q32)[2]/46,XY[18]) which results in
an unusual ‘blastoid’ morphology of the tumor cells and an
extremely aggressive clinical course.
Case report
We report a case of a 59 year old male who presented
in July 2016 to the emergency department because of a one week
history of nausea, vomiting with episodic abdominal pain and a
decrease in urination with hematuria and dysuria. He has associated
weight loss but no recent fevers, chills or a decrease in appetite.
He was then admitted for leukocytosis and thrombocytopenia with a
possible diagnosis of leukemia, tumor lysis syndrome (elevated
potassium and hyperuricemia), pre-renal and post-renal acute kidney
injury from dehydration, BPH and renal stone obstruction. A CT of
the abdomen and pelvis revealed calculi in the distal right ureter
and right ureteral vesicle junction as well as the posterior left
aspect of the urinary bladder versus a left ureteral vesicle
junction causing bilateral hydroureter and hydronephrosis.
Splenomegaly was noted, 22.7 cm in its greatest diameter. No
significant lymphadenopathy was seen. Peripheral blood smear showed
numerous large blastoid like cells. He then underwent bilateral
ureteral stent placement and a bone marrow biopsy and aspirate
procedure.
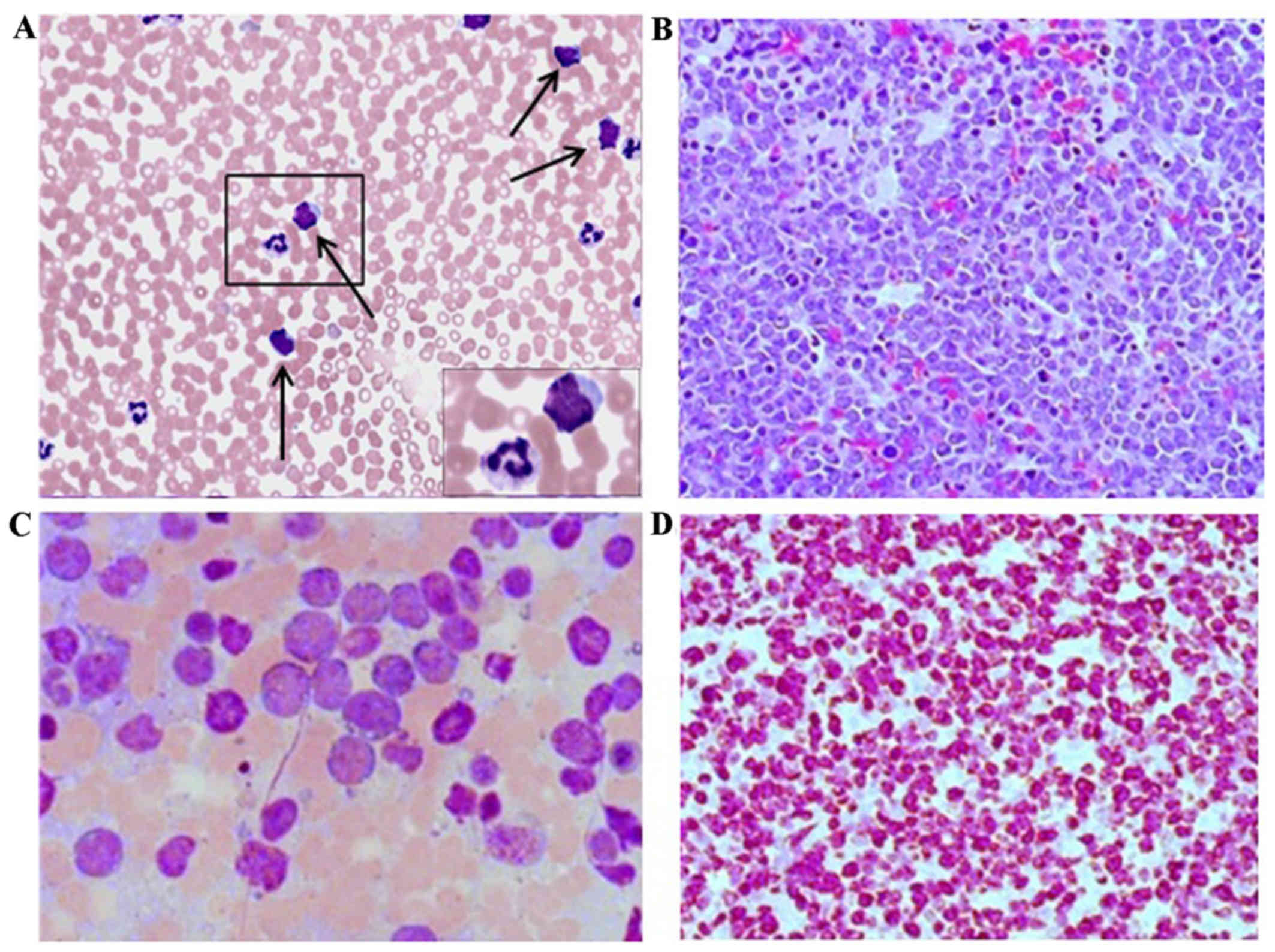
The peripheral blood smear was performed and it
demonstrated a normocytic and normochromic anemia, thrombocytopenia
and leukocytosis with numerous large atypical lymphocytes
exhibiting a ‘blastoid’ morphology (prominent nucleoli, basophilic
cytoplasm and rare occasional cytoplasmic vacuoles) (Fig. 1A). Flow cytometry demonstrated tumor
cells positive for B cells marker CD79A, CD22, CD10 (dim), CD45.
Cytoplasmic light chain staining demonstrated monoclonal expression
of cKappa light chain. Cells were negative for surface light chain
expression, TdT, cCd3 and MPO, CD20, BCL2, BCL6, CD34, CD43. FISH
for IgH/BCL2 t(14;18) and BCL6 (3q27) were negative. This profile
is consistent with a CD10-positive kappa light chain restricted
B-cell lymphoproliferative disorder with a differential that
includes follicular lymphoma, Burkitt lymphoma and large B-cell
lymphoma including double hit lymphoma. Concurrent FISH analysis
demonstrated t(8;14)(q24;q32) translocation characteristic for
Burkitt lymphoma. The sample was also sent for cytogenetic analysis
and it revealed a 47,XY,+1,i(1)(q10),t(8;14)(q24;q32)[2]/46,XY[18]
with formation of an isochromosome from long arm of chromosome 1,
with two normal chromosome 1 homologues, yielding partial tetrasomy
1q. Thereafter a bone marrow biopsy was performed showing 95%
hypercellularity with normal hematopoiesis largely replaced by
proliferation of atypical medium to large size lymphocytes
(Fig. 1B). Atypical lymphocytes
demonstrated irregular, often banded, nuclei with notable nucleoli
(Fig. 1C). The cytoplasm is deeply
basophilic with occasional lipid vacuoles. Multiple mitotic figures
were noted. Ki67 index approached 100% (Fig. 1D). Diagnosis of Burkitt lymphoma was
established.
After initial treatment with hydrea, rasburicase and
allopurinol, his labs showed improvement. Imaging was negative for
marked lymphadenopathy however there was evidence of splenomegaly.
Physical exam findings demonstrated multiple petechiae on the lower
extremities.
MRI showed possible meningeal enhancement. An LP was
obtained and cytology and flow cytometry showed findings consistent
with CNS involvement with lymphoma. He received multiple
intrathecal chemotherapies. The patient received chemotherapy as
well as intrathecal treatment with methotrexate alternating with
cytarabin.
Patient finished cycle 1 of Hyper-CVAD and the
course was complicated by sepsis from which he eventually died.
Discussion
Burkitt lymphoma is an aggressive B cell neoplasm
characterized by the translocation and deregulation of the c-MYC
gene on chromosome 8 (8q24) along with one of three locations on
immunoglobulin (Ig) genes; Ig heavy chain gene on chromosome 14
(~80%) - t(8;14), kappa light chain gene on chromosome 2 (~15%) -
t(2;8), and the lambda light chain gene on chromosome 22 (~5%) -
t(8;22) (1). Classification of cases
with additional copies of cMYC as opposed to translocation remain
controversial (3). Present case
demonstrated classic t(8;14)(q24;q32) Burkitt lymphoma
translocation with additional cytogenetic abnormality
(supernumerary isochromosome 1q, i(1)(q10) aberration) resulted in
unusual immature ‘blastoid’ morphology and immature immunophenotype
(lack of CD20 and surface immunoglobulin expression) of the tumor
cells. Those factors made initial diagnosis in the present case
challenging. Peripheral blood so closely mimicked acute leukemia
that during initial assessment the peripheral blood smear was
demonstrated to medical students, residents and oncology attendings
as typical example of acute leukemia, likely ALL, much to the
embarrassment of pathologist when Flow and cytogenetic data become
available and the very next day the ‘text book blasts’ turn out to
be lymphocytes.
Gain of chromosome 1q has been frequently noted in
pediatric malignancies including Wilms tumor, neuroblastoma, Ewing
sarcoma and brain tumors such as ependymoma and high grade gliomas
and the presence of this anomaly is usually associated with disease
recurrence and poor prognosis. However the candidate genes on
chromosome 1q that could be involved in tumorigenesis remain
unidentified (4). An extra copy 1q
was demonstrated before in 10 patients with hematologic
malignancies out of 3786 studied: 5 with myelodysplastic syndrome,
3 with acute myeloid leukemia, and a single patient with acute
lymphoblastic leukemia and myeloproliferative syndrome (5).
To our knowledge 12 other cases of Burkitt lymphoma
with supernumery idic (1)(p12) or
i(1)(q10) were described in literature before with >80% cases
presented with immature B-cell immunophenotype and >60% of
patients relapsed or died (6). Our
case demonstrated a very similar pattern with ‘blastoid’ immature
phenotype and immunophenotype and highly aggressive course
resulting in rapid death. This case combined with literature data
allows us to view those cases of Burkitt lymphoma with
t(8;14)(q24;q32) combined with polysomy of 1q as possible separate
category of Burkitt lymphoma similar to ‘double hit’ and ‘triple
hit’ lymphomas. Such cases are characterized by immature ‘blastoid’
tumor cells morphology and immunophenotype, leukemic presentation
without lymph nodes involvement as well as highly aggressive
clinical behavior.
Acknowledgements
The authors would like to thank the hematology
department of Truman Medical Centers-Hospital Hill (KansasCity, MO,
USA) for their cooperation.
Funding
Not applicable.
Availability of data and materials
All data analyzed during this study are included in
the article.
Authors' contributions
All authors contributed equally to this manuscript.
This is further indicated by the asterisk marks with author names
in the title of the paper.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
In accordance with regulations in effect with the
University of Missouri-Kansas City all the identifying information
was removed, publication of data does not compromise anybody
anonymity or confidentiality or breach local data protection laws.
Any minimal risk of breaking of confidentiality if at all present
is outweighed by public interest consideration. The patient is long
time deceased and therefore at this time it is impossible to obtain
permission from him or trace his significant others. All the
identifying material has been thoroughly removed from the
manuscript therefore no reasonable individual would object to
publication.
Competing Interests
The authors declare that there are no competing
interests regarding the publication of this paper.
References
|
1
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD, et al: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Onciu M, Schlette E, Zhou Y, Raimondi SC,
Giles FJ, Kantarjian HM, Medeiros LJ, Ribeiro RC, Pui CH and
Sandlund JT: Secondary chromosomal abnormalities predict outcome in
pediatric and adult high-stage Burkitt lymphoma. Cancer.
107:1084–1092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang QA, Qasem A, Madhusudhana S and
Glazyrin A: The t(14;18)(q32;q21) with extra MYC signal - is it a
gray zone lymphoma? Int J Clin Exp Pathol. 8:9602–9608.
2015.PubMed/NCBI
|
|
4
|
Puri L and Saba J: Getting a clue from 1q:
Gain of chromosome 1q in cancer. J Cancer Biol Res. 2:10532014.
|
|
5
|
Djordjević V, Dencić-Fekete M, Jovanović
J, Drakulić D, Stevanović M, Janković G and Gotić M: Pattern of
trisomy 1q in hematological malignancies: a single institution
experience. Cancer Genet Cytogenet, Oct. 186:12–18. 2008.
View Article : Google Scholar
|
|
6
|
Roug AS, Wendtland P, Bendix K and
Kjeldsen E: Supernumerary isochromosome 1, idic(1)(p12), leading to
tetrasomy 1q in Burkitt lymphoma. Cytogenet Genome Res. 142:7–13.
2014. View Article : Google Scholar : PubMed/NCBI
|